

Abstract
Antiviral drugs are a class of medicines particularly used for the treatment of viral infections. Drugs that combat viral infections are called antiviral drugs. Viruses are among the major pathogenic agents that cause number of serious diseases in humans, animals and plants. Viruses cause many diseases in humans, from self resolving diseases to acute fatal diseases. Developing strategies for the antiviral drugs are focused on two different approaches: Targeting the viruses themselves or the host cell factors. Antiviral drugs that directly target the viruses include the inhibitors of virus attachment, inhibitors of virus entry, uncoating inhibitors, polymerase inhibitors, protease inhibitors, inhibitors of nucleoside and nucleotide reverse transcriptase and the inhibitors of integrase. The inhibitors of protease (ritonavir, atazanavir and darunavir), viral DNA polymerase (acyclovir, tenofovir, valganciclovir and valacyclovir) and of integrase (raltegravir) are listed among the Top 200 Drugs by sales during 2010s. Still no effective antiviral drugs are available for many viral infections. Though, there are a couple of drugs for herpesviruses, many for influenza and some new antiviral drugs for treating hepatitis C infection and HIV. Action mechanism of antiviral drugs consists of its transformation to triphosphate following the viral DNA synthesis inhibition. An analysis of the action mechanism of known antiviral drugs concluded that they can increase the cell’s resistance to a virus (interferons), suppress the virus adsorption in the cell or its diffusion into the cell and its deproteinisation process in the cell (amantadine) along with antimetabolites that causes the inhibition of nucleic acids synthesis. This review will address currently used antiviral drugs, mechanism of action and antiviral agents reported against COVID-19.
Keywords: antiviral drugs, integrase inhibitors, mechanism of action, nucleoside and nucleotide reverse transcriptase inhibitors, protease inhibitors, viral infections
Introduction
Infectious diseases are well known since ancient time to human civilisation. Infectious disease are caused due to different microorganisms (bacteria, viruses and fungi).1 Viral structure is simple and consists of a protein coat, nucleic acid, viral enzymes and, sometimes, a lipid envelope, unlike the complex structure of fungi, helminths and protozoa. Additionally, viruses use the host’s cellular machinery for replication, hence are obligate intracellular pathogens. Such characteristics create the difficulties in developing drugs with selective toxicity against viruses.2 Viruses are ultra microscopic agents having either DNA or RNA as the genetic material and are known to cause variety of diseases in humans, animals and plants. The fight between humans and viruses is continuous process, as both will adopt different strategies to fight against each other. Antiviral drugs development is a tedious process involving many stages such as target identification and screening, lead generation and optimisation, clinical studies and the drug registration, etc.3 Dynamic antiviral drug development is a pressing need, as viral infections have caused millions of human fatalities worldwide over the course of human civilisation. The approval of first antiviral drug ‘idoxuridine’ in June 1963 has opened a new era in antiviral drug development. Since then, number of drugs with antiviral potential have been developed for clinical use for the treatment of millions of human beings worldwide.4 Antiviral drugs are a class of medicines particularly used for the treatment of viral infections. Specific antiviral drugs are used for treating specific viruses just like the antibiotics for bacteria. Antiviral drugs, unlike the most antibiotics, do not destroy their target pathogens; rather inhibit their development. As the viruses use the host’s cells to replicate, hence makes it difficult to design a safe and effective antiviral drug. Therefore, it is difficult to find the drug targets that would interfere with the virus without damaging the host’s cells. Furthermore, the major complications in developing anti-viral drugs and vaccines are because of viral variation.5 One of the important ways of finding antiviral drugs is the computer based drug discovery and for this approach nelfinavir is an example discovered in the 1990s for the treatment of human immunodeficiency virus (HIV) infection.6
In spite of modern tools and stringent measures for the quality control only a few antiviral drugs are getting approved for the use of human either due to the side effects or resistance to antiviral drugs. With increase in the awareness about the viruses, their mechanism of infection and the rapid evolvement of novel strategies and techniques for antiviral will speed up the novel antiviral drugs development.7 The current scenario all over the world indicates that continuous emergence of microbial threats at an accelerating pace, mainly due to unprecedented climate change and globalisation.8
DNA virus
Viruses such as poxviruses, herpes, adenoviruses and papilloma viruses usually contain double-stranded DNA, leaving single-digit DNA. DNA virus enters the cell centre and leads to new viruses.
RNA virus
RNA viruses include influenza, measles, mumps, colds, meningitis, polio, retroviruses (AIDS, T-cell leukaemia), arena viruses, all considered, single descriptor RNA (ssRNA). RNA virus does not enter the cell centre (in addition to the cold virus contamination this season). Viral RNA is then used to make a DNA copy of the viral RNA, which is organised by the host genome followed by a retroviruses.
Steps of viral infections
Viral infection involves the entry of viral DNA into a host cell, replication of that DNA and releasing the new viruses. The six steps of viral replication include viral attachment, invasion, uncoating, replication, assembly and release. The steps of virus life cycle highlighting the entry and exit of the virus are described below9 (Figure 1 and Table 1).
Figure 1.

Common inhibitory actions of antiviral drugs.
Table 1.
Mechanism of action of antiviral drugs used for the treatment of COVID-19.
| Group | Drugs | Mechanism of action |
|---|---|---|
| Viral RNA polymerase inhibitors | Remdesivir (GS-5734) | RdRp inhibitor, prodrug, analogue of adenosine nucleotide |
| Favipiravir | RdRp inhibitor, prodrug, analogue of guanosine nucleotide | |
| Viral protein synthesis inhibitors | Ritonavir/Lopinavir | Inhibitor of protease |
| Inhibitors of viral entry | Hydroxychloroquine | Increase in endosomal pH needed for the virus/cell fusion. Interfere with cellular receptor glycosilation of SARS CoV (ACE-2) |
| Chloroquine | ||
| Immunomodulators | Nitazoxanide | Interfere with host regulated pathways of virus replication, amplification of type 1 IFN pathways and cytoplasmic RNA sensing |
| Ivermectin | Inhibition of importin 1 heterodimer to inhibit the nuclear import of host and viral proteins |
The virus attaches to a host cell injecting its genetic material into the host cell during attachment and penetration stage.
In the next step, the viral DNA or RNA is itself incorporated into the genetic material of the host cell inducing it to replicate the viral genome. This step involves the uncoating, replication and assembly during the virus life cycle.
During release, the host cell releases the newly created viruses, either through the breakage of the cell, waiting cell death or by budding off through the cell membrane.9,10
Antiviral medication and its mechanism of action
Acyclovir
Acyclovir is the basis of 2′-deoxiguanosin which applies antiviral effects after manipulation on acyclovir triphosphate. The hidden development of this methodology, an increase in acyclovir monophosphate, is catalysed by thymidine kinase caused by cells contaminated by herpes simplex infection11,12 or varicella zoster infection or phosphotransferase made by cytomegalovirus. Cellular protein then adds phosphate to produce acyclovir diphosphate and acyclovir triphosphate. Acyclovir triphosphate slows the mixing of viral DNA by countering 2′-deoxy guanosin triphosphate as a substrate for viral DNA polymerase.11,12 After acyclovir (not 2′-deoxiguanosin) was implanted in a duplicate of viral DNA, fusion stopped. The acyclovir monophosphate circuit into viral DNA is irreversible, given the way exonuclease bound to polymerases 3′, 5′ cannot separate them.13 In this technique, viral DNA polymerase is inactivated in the same way. Acyclovir triphosphate is 30 times greater than herpes simplex type 1 DNA polymerase inhibitors than human alpha-DNA polymerase cells.14 The small formation of acyclovir triphosphate in uninfected cells and its expression for DNA viral load results in harmless cellular toxic effects. In addition, more than 80% of acyclovir that appears during diffusion is unaffected in the urine.15 The 50% central acyclovir inhibitory group in contradiction of herpes simplex disease type 1 is 0.1 μM, and 0.4 μM against herpes simplex disease type 216 and 47.1 µM against cytomegalovirus.17 Even with reduced oral bioavailability, obsession with plasma acyclovir exceeds 50% inhibitory concentration for type 1 and 2 herpes simplex contamination that grows in adults after a combination of 200 mg d ‘Acyclovir, on the other hand, 800 mg is very important to provide plasma obsession over the centre 50% inhibitory concentration for varicella zoster virus. Acyclovir with a fairly short half-life of plasma, 7.7 mg should be given every 4–6 h for patients damaged by varicella-zoster infection. Acyclovir has been shown to be suitable for the treatment of pollution resulting from contamination with herpes simplex types 1 and 218 and varicella-zoster virus and to disguise specific types of cytomegalovirus.16
Valacyclovir
Valacyclovir, L-valyl ester from acyclovir, is also available in oral form. After swallowing, drug is immediately changed to acyclovir by the substance valacyclovir hydrolase in the digestive tract and liver. The original bioavailability is three to several times that of acyclovir.19 Valacyclovir has proven exceptional in treatment of pollution obtained by the herpes simplex virus and varicella-zoster virus and in prophylaxis against cytomegalovirus. Ganciclovir, which starts overseeing the Journal late, contrasts with acyclovir by extending a hydroxymethyl group in position 3′ from a non-cyclic side chain. The assimilation and arrangement of its action are similar to acyclovir, on the other hand, it actually has carbon 3′ with a hydroxyl package that can allow the widening of the foundation design similar to levelled DNA chain terminators. Ganciclovir is replaced by ganciclovir monophosphate by viral encoded phosphotransferase sent to cells contaminated with cytomegalovirus. This is a substrate that is superior to acyclovir for this phosphotransferase, and half the presence of intracellular ganciclovir triphosphate in any case is 12 h, compared to 1–2 h for acyclovir. This difference is the reason why ganciclovir is better than acyclovir for the treatment of cytomegalovirus. Peak plasma fixation after intravenous administration in common portions is much higher than 3 μM, which should inhibit most cytomegalovirus strains.20 Intravenous ganciclovir is very powerful for hiding and treating cytomegalovirus. Oral ganciclovir has also been found to be beneficial in hiding cytomegalovirus 28, but its value is limited by its low bioavailability (8%–9%).21
Penciclovir
Penciclovir is basically like ganciclovir, in contrast only by replacing the methylene connection for oxygen either in the non-cyclic ribose portion of the particle. Its digestive component and activity are similar to acyclovir, so again, it is only a DNA chain terminator that is bound. The inhibitory effect of in vitro penciclovir on herpes simplex 1 and 2 types and varicella-zoster infection is alike to acyclovir.22 Now, it has claimed only as topical plan for the treatment of cold sores. Intravenous preparations are considered as treatment for mucocutaneous herpes in immunocompromised patients.
Famciclovir
Famciclovir is a simple diacetyl-6-deoxy from penciclovir. All this is assimilated after oral organisation and is quickly used for penciclovir by deacetylation in digestive tract, blood and liver, next it is oxidised by liver in position 6 of purine cycle. Half of the presence of a dynamic intracellular drug, penciclovir triphosphate, is very long, offering the possible for a dose once a day. Famciclovir works against genital herpes and the shingles virus.23
Foscarnet
Foscarnet (trisodium phosphonoformate) is a simple and natural inorganic pyrophosphate. This building structure with DNA, DNA polymerase at the site which limits the pyrophosphate, maintains the division of the pyrophosphate from the nucleoside triphosphate and along this line blocks a further increase in base format. Foscarnet should be administered intravenously, as fair oral details have not yet been made. It is not treated at a clear level and is destroyed by glomerular filtration and removal of the cylinder. Clinical examination shows that foscarnet is identical to ganciclovir for the treatment of cytomegalovirus and better than vidarabine for the treatment of contamination caused by an acyclovir-resistant herpes simplex infection.24
Ribavirin
Ribavirin is a simple guanosine that has an inadequate purine cycle as opposed to a serving of non-cyclic ribose. After intracellular phosphorylation, ribavirin triphosphate interferes with the initial timeliness of virus translation, for example, by supplementing and expanding the birther’s RNA and suppressing ribonucleoprotein synthesis. It has a wide range of in vitro movements against RNA infections. The significant convergence of the metabolites – 1,2,4-triazole-3-carboxamide – is higher when urinating after oral administration than after intravenous administration, which indicates that drug is lowered in digestive tract and the liver. Ribavirin aerosol is assimilated on an elementary basis, as indicated by proximity of fixation which can be measured in plasma. Clinical suitability has been demonstrated for treatment of contamination caused by dengue (with details oral and intravenous ribavirin) and hepatitis C (by mouth) ribavirin mixed with interferon.25
Lamivudine
Lamivudine is a pyrimidine nucleoside that was initially manufactured as an antiretroviral drug. It is simple cytidine that is converted intracellularly to lamivudine triphosphate which contains hepatitis B DNA polymerase as well as HIV reverse transcriptase. Lamivudine is a prescription nucleoside reverse transcriptase inhibitor (NRTI) that is used in combination with other drugs as antiviral treatment for human immunodeficiency virus type-1 (HIV-1) and as a monotherapy for hepatitis B virus (HBV).26 The high oral bioavailability and generally long half-life (5–7 h) of lamivudine allow once every day dose up in patients with hepatitis B.
Amantadine and rimantadine
Amantadine hydrochloride is an amine having a special ring of 10 carbon atoms; Rimantadine hydrochloride is a pair prepared by combining an ethyl carbon linkage with ammunition and a C10 cycle. Both drugs appear to suppress influenza infection replication by blocking the particle channel of the M2 protein virus, which reduces the effect of this viral protein on virus release and pH guidelines in contaminated cells. Amantadine has a high oral bioavailability and a number of symptoms, especially in patients with 60 years of age or older, who have approximately several times higher plasma concentrations than young adults receiving one and a half doses – plasma life is approximately 12 h longer. Amantadine is eliminated by glomerular filtration and non-drug cylindrical release, so the altered pharmacokinetics in the elderly is likely to be due to decreased renal capacity. Rimantadine is also well consumed; 75% of the dose is processed in the liver, mainly by hydroxylation. Elderly people need a dose reduction, probably due to age-related decreases in liver capacity. These two drugs are active in the inhibition and treatment of influenza infection.27
Interferon alpha
Normal interferon is a glycoprotein that has the proposed antiviral effect due to the registration of cellular chemicals that inhibit the incorporation of viral proteins. The commercial arrangement of interferon alpha is slightly smaller than that of ordinary proteins (subatomic mass, approximately 19,000) and is produced in microbes by recombinant DNA strategy.11 Interferon is not available orally and should be administered by intramuscular or subcutaneous infusion. Insufficient information is available on the inhibition of viral replication in vitro, presumably because interferons inhibit their antiviral activity by suppressing and interpreting viral RNA and retaining cells. Interferon alpha has been shown to be effective in the treatment of diseases caused by human herpesvirus 8, papillomavirus (Kaposi’s sarcoma) virus, hepatitis B and C virus.
Antiviral drugs and COVID-19
The worldwide outbreak of COVID-19 virus infection is associated with the unavailability of specific drug(s) to combat with this viral infection. To date, nearly 10 million people are infected and about 500,000 people die worldwide due to COVID-19 viral infection. To find the solutions for this viral infection, great efforts have been made and are continued to develop vaccines, small molecule drugs or monoclonal antibodies that can prevent the infection spread to avoid the expected human, social and economic devastation related to this infection. Several FDA approved drugs have been reported in the literature and in hospitals during clinical trials to treat or reduce the COVID-19 severity.
Remdesivir (GS-5734)
Remdesivir is a novel antiviral drug originally used for treating Marburg virus and Ebola virus infections and this drug was developed by Gilead Sciences. The chemical formula of remdesivir is C27H35N6O8P with a molecular mass of 602.6 g/mol. This is a prodrug of a nucleotide analogue metabolised intracellularly to adenosine triphosphate analogue inhibiting the viral RNA polymerases (Figure 2). It acts as an inhibitor of RNA dependant RNA polymerase and its characteristics and pharmacokinetics have been studied in MERS-CoV and SARS-CoV infections. This drug causes decline in the replication of viral genome and its production due to the alterations in the viral exonuclease function and disturbed proof reading. It can be recommended to prevent the disease progression severity in COVID-19 patients since it prevents the replication of the virus. To confirm its therapeutic potential against COVID-19, double blind randomised clinical trials with such patients are underway in phase 3.28 In vitro studies have shown that in addition to its efficacy against COVID-19 in epithelial cells of the human airways, remdesivir has virologic as well as clinical efficacy in a non human primate model.29
Figure 2.
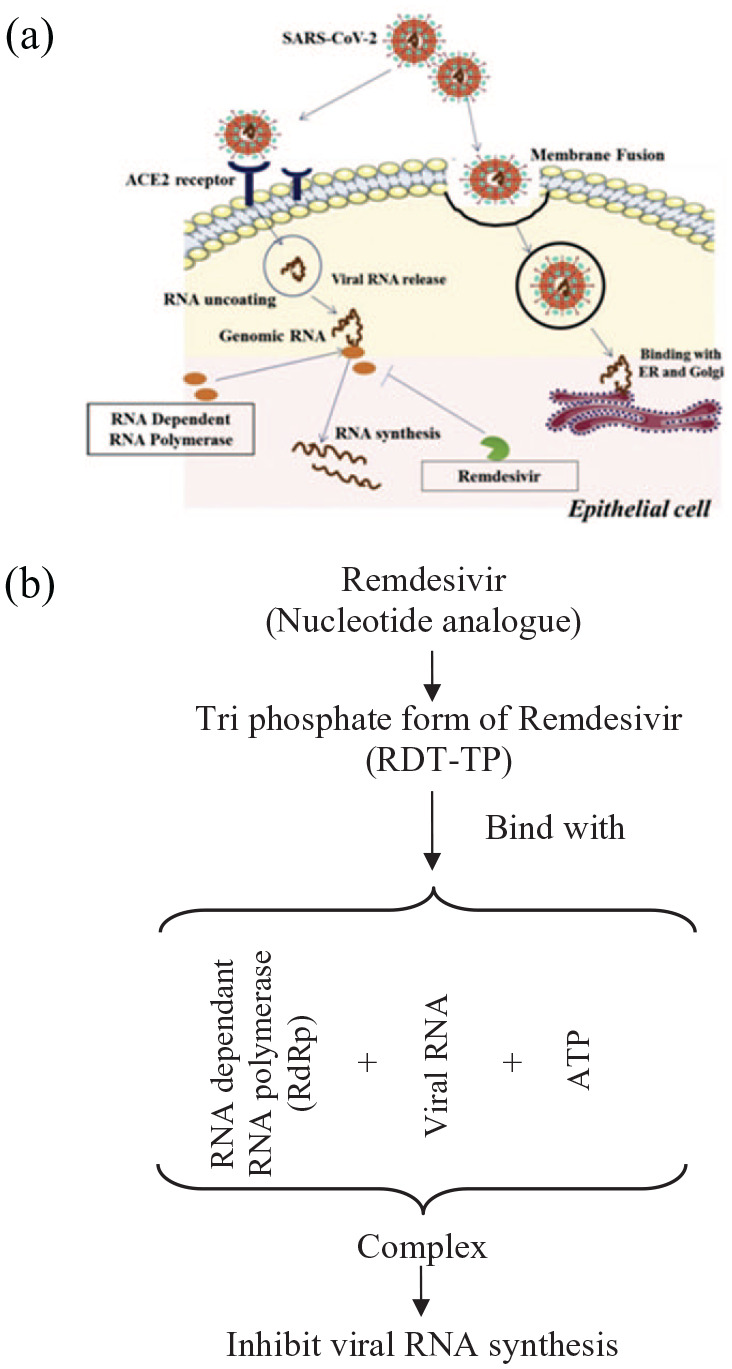
Possible mechanism of Remdisivir against SARS-CoV-2 at molecular level. (a) Diagram shows the entry of SARS-CoV-2 virus and the synthesis of its RNA that can be blocked by Remdisivir. (b) Molecular mechanism of viral RNA synthesis inhibition by Remdisivir.30
Remdesivir has broadspectrum antiviral activity against several virus family members including the coronaviruses for example, Middle East respiratory syndrome coronavirus (MERSCoC) and SARSCoV, and filoviruses for example, Ebola and has shown therapeutic and prophylactic efficacy in these coronaviruses when used as non clinical models. Remdesivir when tested through in vitro studies using the Vero E6 cells showed an EC50 value of 1.76 µM that revealed its activity against SARS-CoV-2 suggesting its working concentration probably be achieved in nonhuman primate models.31 Intravenous remdesivir treatment showed significant improvement for the first COVID-19 patient in US32 and then a trial has been started to rapidly evaluate the safety and efficacy of remdesivir in nCoV-19 infected hospitalised patients. In a cohort of hospitalised patients with severe COVID-19 treated with remdesivir, improvements in the clinical finding were observed in 68% patients.33 Without any placebo or active comparator in the study, it is difficult to draw any solid conclusion about the efficacy of remdesivir therapy. Currently in the United States, four clinical trials are enrolling the patients and two additional trials in China only have been registered on ClinicalTrials.gov, NCT04252664 (mild-moderate disease) and NCT04257656 (severe disease).34
Nitazoxanide
Nitazoxanide and its active constituent, tizoxanide showed the potential against MERS CoV and SARS CoV-2 in an in vitro study using Vero E6 cells with EC50 of 0.92 and 2.12 µM, respectively.31 It also showed broad spectrum activity against certain viruses including norovirus, rotavirus, parainfluenza, respiratory syncytial virus and influenza virus in addition to coronaviruses. This antiviral activity is due to the fact that action mechanism is based on interfering with the host regulated pathways of virus replication rather than the specific pathways of the virus.35 The innate antiviral mechanisms are upregulated by nitazoxanide through the amplification of cytoplasmic RNA sensing and type 1 IFN pathways. Nitazoxanide upregulate the precise host mechanisms interfering with the viral infection and the viruses target to bypass the host cellular defences.36 Studies have shown that nitazoxanide when used against influenza viruses block the maturation of viral hemagglutinin at post translational stage.35 This drug is being evaluated in randomised controlled clinical trials for the management of some acute respiratory infections such as influenza, even though the results are yet unavailable or not encouraging. Although encouraging results are found through the in vitro activity of nitazoxanide against SARS-CoV-2 and more studies are required to clearly determine its role in managing the COVID-19.37
Antagonistic impacts of antiviral drugs
Because infections involve intracellular pathogens that have cellular capacity, cynics once accepted that no specific inhibitor of viral reproduction could be found. This confidence was strengthened by the disappointments of the first antivirals like idoxuridine and cytarabine essential, and moderately late with fialuridine. Fortunately, drugs have been developed that affect viral replication to a greater extent than cells. All antiviral drugs, whether alone or not, can have effects and some are unexplained, such as thrombotic microangiopathy linked to valaciclovir in patients with immunodeficiency syndrome.38
Virus inactivating agents
Some compound operators have been performed which use a fairly attractive antiviral movement by straight disabling infection. Calcium elenolate, a monoterpene gained from corrosive liquid concentrates hydrolysed from various pieces of the olive tree, uses a virucidal effect in vitro against a variety of RNA and DNA infections, clearly by communicating with the protein layer of the infecting molecule.39 In a creature study, intranasal administration reduced yields of parainfluenza infection without significant adverse effects. Human preparations with this compound have only demonstrated viability if treatment is started immediately after infection. Certain dihydroisoquinolines have shown an inactivating effect on influenza A and B infections and parainfluenza infections; these infections had a strong antiviral effect in cell culture and were later found to have a moderate effect in animal tests. The mixtures have in any case been neglected in order to obtain the antiviral effect required in humans.40
Restraint of viral attachment, entrance and uncoating
Because the infection first contaminated a eukaryotic cell, certain general stages of the disease process occur that can be spots of outbreak by potential antiviral drugs. At these stages, the contaminating virion binds to receptors on the cell film, enters the cell layer and once in the cell’s cytoplasm, the virion’s protein layer is emptied and the viral nucleus corrodes the substance.
Contact or viral adsorption was the least viable site to attack antiviral agents, without discovering substances that were still dynamic enough to warrant a clinical trial. The sulfated polysaccharide is thought to communicate with infectious particles, thereby reducing the rate of cell binding in vitro.41 Affected infections include encephalomyocarditis, reverberation, flu, dengue fever and rabies. A moderate effect in vivo has also been observed against dengue infection in mice. Heparin, an unfavourably charged mucopolysaccharide, clearly forms a non-infectious complex with a herpes infection that prevents it from being secreted into the host cell. An action against herpes infection was observed both in vitro and in the analysis of creatures, in the latter case a heparin infusion was injected into the skin of the rabbit before or as a whole. Because of the ionic concept of communication, in all respects, heparin would have an impressive degree of non-specificity.
Inhibitors of enzymes associated with virions
DNA polymerases
Countless substances accept antiviral movement due to the inhibition of DNA polymerases associated with virions. Antivirals of this type can be widely collected in pyrophosphate analogues and analogues of conventional nucleoside polyphosphates. This latter collection is regularly distinguished in the sweet portion of the particle or in the particles of purine or pyrimidine, although hardly in both. There are two interesting mixtures in main classification: trisodium phosphonooformate (PF An) and trisodium phosphonoacetate (PA). PFA removes half of DNA polymerase type I from herpes simplex infection at 3.5 p.M. The effect of eukaryotic DNA polymerase on α can reduce protein expansion. For cell expansion (HeLa cells), a more notable requirement of 100 µM PFA in the medium was to achieve half inhibition. PFA is generally dynamic in vitro against DNA-containing herpes simplex 1 and 2 infections and infection in simulated animals. Like PF A, P may give the impression that a potent inhibitor of herpes simplex infection depends on DNA polymerase, but has no effect on the polymerase of the host cell (WI-38). Exceptionally, point-to-point reactions to the polymerisation and trade of nucleoside triphosphate pyrophosphates using DNA polymerase activated by infection with turkey herpes.42 PA appeared to communicate with DNA polymerase at the level of the site limiting polyphosphates.43 Overby et al.43 have shown that resistance to PA infection is rightly linked to a similar relative obstruction of the comparison without cellular DNA polymerase.
RNA polymerases
Various substances are recognised to prevent DNA and RNA-mediated RNA polymerase in vitro, and this activity is repeatedly believed to be responsible for antiviral activity. For example, in a careful report, Ericsson et al. reported that a very important class of malaria, ribavirin triphosphate (RTP), is a potent antioxidant that promotes RNA polymerase. The polarisation of viral polymers is strong for ATP and GTP, but not for UTP or CTP. RNA interference polymers have been identified as more complex than guanine-containing dinucleotides, and Plotch and Krug have shown that ApG or GpC is inserted at the 5′ end of the AcG gene. Ericsson et al. discovered that RTP abolished ApG and GpC-mediated enhancement of the virtual polymerase. It is not well understood that this approach may reflect the unique effects of influenza ribavirin infection. Ericsson et al. stated that a more important goal is that RTP blockade of viral RNA polymerase inhibitors extends from the formation of cellular polymers to non-functional eukaryotic RNAs. Jamieson et al. showed that RTP does not inhibit eukaryotic RNA polymerases I and II and does not affect eukaryotic polymerases (A) Deoxypyrimidine nucleoside kinase and thymidine kinase. Deoxypyrimidine kinase initiates the virus. There are two ways to do this, of course: the first is immediate competition with conventional substrates, and the second is catalytic restriction by allosteric modulators.44 Kit et al., pointed out that pseudorabies and viral growth are phosphorylated, while stimulating a kinase ready to phosphorylate another thymidine, deoxycytidine, which phosphorylates thymidine. It has been described in detail and compared with human and mouse mitochondrial chemistry in some embodiments, especially phosphorylated extractions, although dCTP does not control thymidine virus infection.45 All thymidine kinases are critically involved in dTTP. Cheng et al.46 showed that thymidine analogues have antiviral activity, whereas herpes simplex virus can activate thymidine kinase and Declercq and Torrence47 (10S) showed some of the thyroid analogues, which is especially true for herpesviruses. Cheng et al found in a cautious report that many 5-subdeoxyuridine-rich companies are herpes simplex 1 and 2-thymidine kinase have been shown to be a strong driving force. 5-IdC and 5-BrdC are more and more active, attractive inhibitors of thymidine kinase. Herpes simplex class 1 fights only thymidine kinase. The above combinations are herpes simplex type 1 or herpes simplex type 2. It is an active ingredient in the regeneration of but not a specific type of herpes simplex virus that has rapidly acquired the ability to stimulate thymidine kinase.48
Viral neuraminidase
There are different views on the work of virion-associated neuraminidases, but whether they are infiltrated or agglomerated, the severity of influenza side effects increases among volunteers and increases the immune response to neuraminidase against plasma. Concentration is declining. 2-Deoxy-2,3-dehydro-N-trifluorocetylneuramine caustic is an inhibitor of influenza infection. This involves the enzymatic removal of neuramine caustic from the infected envelope, as well as the widespread collection of infectious particles and, ultimately, the inhibition of viral replication. mRNA guanylyl transferase and mRNA methyl transferase mRNAs consist of 7 methylguanose structures associated with 2′× triphosphate hybrids from 5′ locations of various viruses and eukaryotes. The structure contains ‘O’ methylribonucleoside and a suitable chemical containing the ‘upper’ structure, which was found in the Vaccine and Reovirus Centres. Subsequent tests in this area showed that infections containing various RNAs and DNAs had a ‘superior’ structure, while poliomyelitis was not an infection and ribavirin was not dynamic against polio infection. Therefore, the effect on the polishing procedure was studied.49 Show that RTP is a potent and severe inhibitor of vaccine-infected mRNA guanyltransferase (Kj = 32 p.M and GTP Km = 22 p.M). Furthermore, in the absence of GTP, 1 mm RTP inhibits vaccine mRNA methylation, but synepungin increases success even if it is an antifungal operator. The peptides in influenza viruses do not bind rapidly to the ribavirin field, but that peptide synthesis in host kidney cells is not regulated. This recognition may be due to the formation of a viral RNA mixture or a ‘top’. The replication of influenza infection in reticulocytes is approximately 15 terminal nucleotides generated from globin mRNA, as well as 5′ ‘top‘ effects requires additional synchronisation of host cell mRNA.
Inhibitors of the translational processes of viral mRNA
mRNA translation
This suggests that the interpretation of various mRNAs in the wheat germ range is restricted to 7-methylguanosine-5′-monophosphate (m7-GMP). However, guanosine nucleotides are released rapidly upon entering the 7-methyl collection or other methyl collections. Not enough, surprisingly, m7-GMP suppresses RNA interpretation of satellite tobacco spoilage infections in the wheat germ range. This could be part of another recognition section of the ‘upper’ restriction site. Additional studies using reovirus mRNA in wheat germ have shown by Adams et al.50 Regarding the mRNA interpretation of vesicular stomatitis infection in reticulocytes.51 In a subsequent report, Bergman and Rodish determined the amount of mRNA infection in vesicular stomatitis infection by binding K+ low wheat embryo ribosomes.52 They added that the interpretation of mRNA in the reticulocyte range was less important 5′ ‘up’ under any response condition.
Early viral polypeptide chains
Parafluorophenylalanine (pFPhe) was first used in 1951 in a simple, non-corrosive manner and along these lines has been shown to have broad spectrum antiviral activity against RNA and DNA infections. The way it works is to replace the protein phenylalanine, which does not stimulate antiviral peptides well. Continuously, an entirely new method destroys cells that are contaminated with Calascovirus, so methylene GTP inhibits encephalomyocarditis protein synthesis and enters these cells (not yet normal). Contreras et al. further showed that other explanatory inhibitors were some toxic cells rather than simple cells.53
Inhibitors of the synthesis of viral DNA
Many exacerbations that inhibit the binding of viral DNA occur either by direct blocking of the polymerase (and were hidden by previous regions), while, on the other hand, due to the impedance in the previous binding or binding. Square DNA replication or in collaboration with the layout, which ultimately makes defective material work.
Fused to DNA
The fusion of 5-ldU with viral DNA instead of thymine and its subsequent delicacy and distortion of this DNA were investigated, and an extensive variety of halogenated deoxypyrimidine nucleosides was rather widely illuminated. The fusion of these substances can lead to non-functional DNA along these lines that destroy the nose of genetic data. In addition, there are other DNA-related deoxythymine analogues that have been specifically tested by De Clerk and Torrens. It is interesting to note that 5-AlddU is associated with herpes simplex DNA, and the authors draw attention to the pronounced corrosion instability of P-N bonds along these lines. However, the organic effects of this binding can be quite intimidating, since DNA usually does not cause corrosion. Ara-AMP binds to herpes simplex infected DNA, as well as the DNA of L5178 Y cells and mouse fibroblasts.
Inhibitors of non-viral enzymatic processes involved in DNA synthesis
Among the procedures that can change the proportion or volume of DNA mixtures, antiviral specialists mainly influence the estimates of thymidylate synthetase and deoxynucleoside triphosphate pools either directly or bypassing. Countless deoxyuridine subsidiaries show incredible barriers to synthetic TMP. In the model, 5-iodoacetamido-methyldeoxyuridine and 5-ethyldeoxyuridine, as well as 5-fluorodeoxyuridine and 5-trifluoromethyldeoxyuridine 5′-monophosphate. Linking to the Intercalation pattern ensures successful DNA replication due to the presence of various substances that interact with DNA. Although a significant number of these substances have been demonstrated to be dynamic antiviral experts, these effects also affect cell DNA replication. Muller recently investigated these substances for their antiviral effects. In addition, Kersten and Kersten recently completed an amazing study of these experts. In addition, daunomycin interacts only with adriamycin, which is essentially the same as in real life with DNA infections, especially with the herpes simplex virus and vaccinia, as well as with carcinogenic RNA infections that mimic the middle of the DNA pathway. Both of these agents are considered implant specialists and are generally toxic, since both of them inhibit nuclear-corrosion combinations, including DNA mockups.
Inhibitors of the biosynthesis and assembly of viral glycoprotein
Both DNA and RNA infections include membranes with glycopeptides integrated into the infection, recommended by another possible direction of antiviral drugs. Influenza infections include hemagglutinin spikes. This is an important part of the envelope glycoprotein of the infection and is suitable for connecting infectious molecules with their cellular receptors. Another important part of the influenza infection film is chemical neuraminidase (N-acetylneuraminic acid glycohydrolase), which is outside the infection and appears to be associated with the lipid membrane of the infection like hemagglutinin. The effects of the mixture on neuraminidase have been investigated in previous areas.
Conclusion
The fight between human and viruses in on and both are rapidly improving the strategies of attacking and defence. In recent years, there has been tremendous progress in understanding the genetic basis and molecular mechanism of diseases. Various new drugs have been formulated and the development of a lot more is in underway. Though, the new infectious diseases caused by viruses such as COVID-19 remain a challenge. Furthermore, the drugs failure in human trials is a general process that requires to be worked out and addressed. The promising results are expected through the emergence of many new technologies. A greater help in the development of new drugs with antiviral activities is provided by the growing knowledge about viruses and the rapidly developing techniques and tools. The better understanding about viruses will make it possible to establish useful measures for fighting against the viral diseases and the researchers around the globe are putting their possible efforts to control the spread of viral diseases and we hope that we live in the world free from viral diseases.
Acknowledgments
We acknowledge Dr Abid Rashid for guiding us in writing this article.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs: Fahad Said khan  https://orcid.org/0000-0002-4497-9770
https://orcid.org/0000-0002-4497-9770
Muhammad Riaz
https://orcid.org/0000-0002-5524-7735
References
- 1. Balloux F, van Dorp L. (2017) Q&A: What are pathogens, and what have they done to and for us? BMC Biology 15: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Champe HRAPC, Fisher BD. (2007) Lippincott’s Illustrated Reviews: Microbiology. Philadelphia:Lippincott Williams & Wilkins. [Google Scholar]
- 3. Saxena SK, Saxena S, Saxena R, et al. (2010) Emerging trends, challenges and prospects in antiviral therapeutics and drug development for infectious diseases. Electronic Journal of Biology 6: 26–31. [Google Scholar]
- 4. De Clercq E, Li G. (2016) Approved antiviral drugs over the past 50 years. Clinical Microbiology Reviews 29: 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He H. (2013) Vaccines and antiviral agents. Current Issues in Molecular Virology: Viral Genetics and Biotechnological Applications 2013: 239–250. [Google Scholar]
- 6. Parks JM, Smith JC. (2020) How to discover antiviral drugs quickly. The New England Journal of Medicine 382(23): 2261–2264. [DOI] [PubMed] [Google Scholar]
- 7. Shin W-J, Seong BL. (2019) Novel antiviral drug discovery strategies to tackle drug-resistant mutants of influenza virus strains. Expert Opinion on Drug Discovery 14: 153–168. [DOI] [PubMed] [Google Scholar]
- 8. Asiri YI, Alsayari A, Muhsinah AB, et al. (2020) Benzothiazoles as potential antiviral agents. Journal of Pharmacy and Pharmacology 72: 1459–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryu W-S. (2017) Virus life cycle. Molecular Virology of Human Pathogenic Viruses 2017: 31–45. [Google Scholar]
- 10. Connolly SA, Jackson JO, Jardetzky TS, et al. (2011) Fusing structure and function: a structural view of the herpesvirus entry machinery. Nature Reviews Microbiology 9: 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balfour HH., JR (1983) Resistance of herpes simplex to acyclovir. Annals of Internal Medicine 98: 404–406. [DOI] [PubMed] [Google Scholar]
- 12. Fyfe J, Keller P, Furman P, et al. (1978) Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl) guanine. The Journal of Biological Chemistry 253: 8721–8727. [PubMed] [Google Scholar]
- 13. Derse D, Cheng Y, Furman P, et al. (1981) Inhibition of purified human and herpes simplex virus-induced DNA polymerases by 9-(2-hydroxyethoxymethyl) guanine triphosphate. Effects on primer-template function. The Journal of Biological Chemistry 256: 11447–11451. [PubMed] [Google Scholar]
- 14. Furman PA, St, Clair M, Spector T. (1984) Acyclovir triphosphate is a suicide inactivator of the herpes simplex virus DNA polymerase. The Journal of Biological Chemistry 259: 9575–9579. [PubMed] [Google Scholar]
- 15. de Miranda P, Blum MR. (1983) Pharmacokinetics of acyclovir after intravenous and oral administration. The Journal of Antimicrobial Chemotherapy 12: 29–37. [DOI] [PubMed] [Google Scholar]
- 16. Balfour HH, Jr, Chace BA, Stapleton JT, et al. (1989) A randomized, placebo-controlled trial of oral acyclovir for the prevention of cytomegalovirus disease in recipients of renal allografts. The New England Journal of Medicine 320: 1381–1387. [DOI] [PubMed] [Google Scholar]
- 17. Fletcher C, Englund J, Edelman C, et al. (1991) Pharmacologic basis for high-dose oral acyclovir prophylaxis of cytomegalovirus disease in renal allograft recipients. Antimicrobial Agents and Chemotherapy 35: 938–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meyers JD, Wade JC, Mitchell CD, et al. (1982) Multicenter collaborative trial of intravenous acyclovir for treatment of mucocutaneous herpes simplex virus infection in the immunocompromised host. The American Journal of Medicine 73: 229–235. [DOI] [PubMed] [Google Scholar]
- 19. Soul-Lawton J, Seaber E, On N, et al. (1995) Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrobial Agents and Chemotherapy 39: 2759–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Erice A, Jordan MC, Chace BA, et al. (1987) Ganciclovir treatment of cytomegalovirus disease in transplant recipients and other immunocompromised hosts. JAMA 257: 3082–3087. [PubMed] [Google Scholar]
- 21. Anderson RD, Griffy KG, Jung D, et al. (1995) Ganciclovir absolute bioavailability and steady-state pharmacokinetics after oral administration of two 3000-mg/d dosing regimens in human immunodeficiency virus—and cytomegalovirus-seropositive patients. Clinical Therapeutics 17: 425–432. [DOI] [PubMed] [Google Scholar]
- 22. Boyd MR, Bacon TH, Sutton D, et al. (1987) Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl) guanine (BRL 39123) in cell culture. Antimicrobial Agents Chemotherapeutics 31: 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tyring S, Barbarash RA, Nahlik JE, et al. (1995) Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia: A randomized, double-blind, placebo-controlled trial. Annals of Internal Medicine 123: 89–96. [DOI] [PubMed] [Google Scholar]
- 24. Safrin S, Crumpacker C, Chatis P, et al. (1991) A controlled trial comparing foscarnet with vidarabine for acyclovir-resistant mucocutaneous herpes simplex in the acquired immunodeficiency syndrome. The New England Journal of Medicine 325: 551–555. [DOI] [PubMed] [Google Scholar]
- 25. Huggins JW, Hsiang CM, Cosgriff TM, et al. (1991) Prospective, double-blind, concurrent, placebo-controlled clinical trial of intravenous ribavirin therapy of hemorrhagic fever with renal syndrome. The Journal of Infectious Diseases 164: 1119–1127. [DOI] [PubMed] [Google Scholar]
- 26. Taylor K, Fritz K, Parmar M. (2020) Lamivudine. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. [Google Scholar]
- 27. Dolin R, Reichman RC, Madore HP, et al. (1982) A controlled trial of amantadine and rimantadine in the prophylaxis of influenza A infection. The New England Journal of Medicine 307: 580–584. [DOI] [PubMed] [Google Scholar]
- 28. Frediansyah A, Tiwari R, Sharun K, et al. (2020) Antivirals for COVID-19: A critical review. Clinical Epidemiology and Global Health 9: 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jomah S, Asdaq SMB, Al-Yamani MJ. (2020) Clinical efficacy of antivirals against novel coronavirus (COVID-19): A review. Journal of Infection and Public Health 13(9): 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saha A, Sharma AR, Bhattacharya M, et al. (2020) Probable molecular mechanism of Remdesivir for the treatment of COVID-19: Need to know more. Archives of Medical Research 51(6): 585–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yavuz S, Ünal S. (2020) Antiviral treatment of COVID-19. Turkish Journal of Medical Sciences 50: 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guan W-j, Ni Z-y, Hu Y, et al. (2020) Clinical characteristics of coronavirus disease 2019 in China. The New England Journal of Medicine 382: 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grein J, Ohmagari N, Shin D. (2020) Original: Compassionate use of remdesivir for patients with severe covid-19. The New England Journal of Medicine 382: 2327–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCreary EK, Pogue JM. (2020) Coronavirus disease 2019 treatment: A review of early and emerging options. Open Forum Infectious Diseases 7(4): ofaa105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rossignol J-F (2016) Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. Journal of Infection and Public Health 9: 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jasenosky LD, Cadena C, Mire CE, et al. (2019) The FDA-approved oral drug nitazoxanide amplifies host antiviral responses and inhibits Ebola virus. iScience 19: 1279–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choy K-T, Wong AY-L, Kaewpreedee P, et al. (2020) Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Research 178: 104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bell WR, Chulay JD, Feinberg JE. (1997) Manifestations resembling thrombotic microangiopathy in patients with advanced human immunodeficiency virus (HIV) disease in a cytomegalovirus prophylaxis trial (ACTG 204). Medicine 76: 369–380. [DOI] [PubMed] [Google Scholar]
- 39. Renis HE. (1969) In vitro antiviral activity of calcium elenolate. Antimicrobial Agents and Chemotherapy 9: 167. [PubMed] [Google Scholar]
- 40. Stark J, Heath R, Oswald N, et al. (1970) A trial of chemoprophylaxis of natural influenza infection with UK 2371. Thorax 25: 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takemoto K, Liebhaber H. (1961) Virus-polysaccharide interactions: I. An agar polysaccharide determining plaque morphology of EMC virus. Virology 14: 456–462. [DOI] [PubMed] [Google Scholar]
- 42. Leinbach SS, Reno JM, Lee LF, et al. (1976) Mechanism of phosphonoacetate inhibition of herpesvirus-induced DNA polymerase. Biochemistry 15: 426–430. [DOI] [PubMed] [Google Scholar]
- 43. Overby L, Duff R, Mao J-H. (1977) Antiviral potential of phosphonoacetic acid. Annals of New York Academy of Sciences 284: 310. [DOI] [PubMed] [Google Scholar]
- 44. Jamieson A, Gentry G, Subak-Sharpe J. (1974) Induction of both thymidine and deoxycytidine kinase activity by herpes viruses. The Journal of General Virology 24: 465–480. [DOI] [PubMed] [Google Scholar]
- 45. Kit S, Leung WC, Trkula D, et al. (1974) Gel electrophoresis and isoelectric focusing of mitochondrial and viral-induced thymidine kinases. International Journal of Cancer 13: 203–218. [DOI] [PubMed] [Google Scholar]
- 46. Cheng Y-C, Domin BA, Sharma RA, et al. (1976) Antiviral action and cellular toxicity of four thymidine analogues: 5-Ethyl-, 5-vinyl-, 5-propyl-, and 5-allyl-2′-deoxyuridine. Antimicrobial Agents and Chemotherapy 10: 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Declercq Et, Torrence P. (1978) Nucleoside analogs with selective anti-viral activity. Journal of Carbohydrates-Nucleosides-Nucleotides 5: 187–224. [Google Scholar]
- 48. Cheng YC. (1977) A rational approach to the development of antiviral chemotherapy: Alternative substrates of herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) thymidine kinase (TK). Annals of New York Academy of Sciences 284: 594–598. [DOI] [PubMed] [Google Scholar]
- 49. Goswami BB, Borek E, Sharma OK, et al. (1979) The broad spectrum antiviral agent ribavirin inhibits capping of mRNA. Biochemical Biophysical Research Communications 89: 830–836. [DOI] [PubMed] [Google Scholar]
- 50. Adams B, Morgan M, Muthukrishnan S, et al. (1978) The effect of “cap” analogs on reovirus mRNA binding to wheat germ ribosomes. Evidence for enhancement of ribosomal binding via a preferred cap conformation. The Journal of Biological Chemistry 253: 2589–2595. [PubMed] [Google Scholar]
- 51. Lodish HF, Rose J. (1977) Relative importance of 7-methylguanosine in ribosome binding and translation of vesicular stomatitis virus mRNA in wheat germ and reticulocyte cell-free systems. The Journal of Biological Chemistry 252: 1181–1188. [PubMed] [Google Scholar]
- 52. Bergmann J, Lodish H. (1979) Translation of capped and uncapped vesicular stomatitis virus and reovirus mRNA’S. Sensitivity to m7GpppAm and ionic conditions. The Journal of Biological Chemistry 254: 459–468. [PubMed] [Google Scholar]
- 53. Contreras A, Carrasco L. (1979) Selective inhibition of protein synthesis in virus-infected mammalian cells. Journal of Virology 29: 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]