

Abstract
Summary: We report a case of GM2 gangliosidosis revealed by MR imaging of an isolated brain stem abnormality in a 3-year-old girl referred for gait difficulties related to ataxia and pyramidal signs. Brain MR imaging displayed a brain stem lesion with high signal intensity on fluid-attenuated inversion recovery and T2-weighted images, suggesting either a tumor or an inflammatory process. Stereotactic biopsy findings showed the presence of swollen neurons with storage material in lysosomes. Enzyme study revealed deficiency of hexosaminidase A, variant B1. Gangliosidoses should be considered in the differential diagnosis of isolated infiltrating brain stem lesions in childhood.
The major differential diagnoses of brain stem mass lesions in children are brain stem glioma, resolving hematoma, vascular malformations, encephalitis, and tuberculoma (1). MR imaging features can readily exclude hemorrhage or vascular malformations, although in some patients it is difficult to differentiate encephalitis or tuberculoma from brain stem glioma. Clinical symptoms and laboratory data can contribute to the diagnosis when there is evidence of inflammation in blood or in CSF. Nevertheless, it is sometimes necessary to perform a stereotactic biopsy to establish the final diagnosis and propose the adequate treatment. The clinical course of this 3-year-old girl was strongly suggestive of an involvement of the posterior fossa, particularly a brain stem infiltrative neoplasm. MR imaging findings confirmed an abnormal brain stem, suggesting a neoplastic process or an inflammation. This brain stem image unexpectedly proved to be the hallmark of a metabolic disease, namely, late-onset GM2 gangliosidosis.
Case Report
A 3-year-old girl referred for gait difficulties related to ataxia and pyramidal signs was the first child of nonconsangiuneous parents. Pregnancy was uneventful, and delivery occurred at 40 weeks’ gestation. Birth weight was 3650 g (25th centile); length, 50 cm (50th centile); and head circumference, 33 cm (25th centile). Psychomotor development was characterized by the ability to walk at 13 months and pronunciation of first words at 20 months.
At 2.5 years of age, she was referred for gait difficulties with frequent falls, which began 3 months before. Clinical examination showed a waddling gait with proximal weakness of the lower limbs and difficulty running and jumping. A muscular disease was suspected, but nerve conduction velocities, electromyography, and creatine kinase were normal.
At the age of 3 years, clinical status deteriorated. General examination revealed normal growth and absence of liver or spleen enlargement. Head circumference was at the 25th centile. Neurologic examination showed an ataxic gait with falls after a few steps and the inability to toe walk. Deep tendons reflexes were increased with a bilateral Babinski sign. There was a right sixth nerve palsy.
Brain MR imaging showed an ill-defined and asymmetric abnormal signal intensity in the brain stem and cerebellar peduncles with slight hypointensity on a T1-weighted image and hyperintensity on T2-weighted fluid-attenuated inversion recovery (FLAIR) images (Fig 1A and B) with slight mass effect, suggesting an inflammatory or a tumoral process. There was no contrast enhancement of the lesions (data not shown). White matter and basal ganglia appeared normal.
Fig 1.

Brain MR imaging.
A, Axial T2-weighted MR image, showing asymmetric involvement of the brain stem and in cerebellar peduncles with high signal intensity.
B, Axial FLAIR-imaging, which confirms a severe diffuse involvement of the medulla.
Routine blood analyses—including blood cell count and inflammatory markers, glucose, calcium, and phosphate, and assessment of liver and kidney function—were normal. CSF analysis showed normal cell count, protein, and glucose levels, without evidence of local immunoglobulin synthesis. Findings at abdominal sonography were normal, as was optic fundus.
Stereotactic biopsy of the cerebellar peduncle was performed. Microscopic examination revealed the presence of swollen neurons filled with granular storage material (Fig 2). The accumulation of abnormal material pushed nuclei and Nissl bodies to the cell periphery. Glial cells also showed evidence of storage. The granular material in neurons and glial cells stained strongly with Luxol Fast Blue and with Sudan Black, suggesting a lipid storage disorder. Reactive gliosis was also found.
Fig 2.
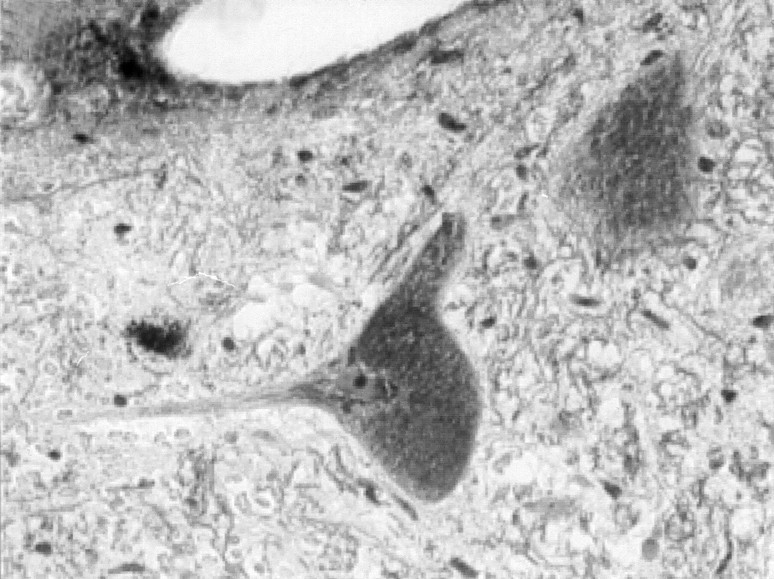
Stereotactic brain biopsy.
Swollen neurons filled with granular material stain strongly with Luxol Fast Blue (×400).
Enzyme studies on white blood cells showed a profound defect of hexosaminidase A activity (10 nmol/h/mg protein [normal values between 250–750 nmol/h/mg protein). Electrophoretic pattern of hexosaminidases was suggestive of a B1 variant hexosaminidase A deficiency. Enzyme levels of both parents were intermediate between those of affected subjects and normal controls. Molecular study is pending.
Discussion
The GM2 gangliosidoses are inherited disorders associated with an accumulation of GM2 gangliosides in neurons and, to a much lesser extent, in other cell types. Deficiency of hexosaminidase A causes Tay-Sachs disease and late-onset GM2-gangliosidosis (2, 3). Tay-Sachs disease is the classic infantile form, seen mainly in Ashkenazi Jews. The clinical hallmarks include an abnormal startle response and psychomotor deterioration within the first months of life, together with blindness with macular cherry red spots. The disease is fatal before 5 years of age. Neuroimaging reports disclose thalamic and white matter abnormalities. CT shows increased attenuation of the thalami (4). At MR imaging, thalami are hypointense on T2-weighted images and hyperintense on T1-weighted-images, probably secondary to calcium deposition. In addition, MR imaging may show high signal intensity on T2-weighted images in other basal ganglia or patchy high signal intensity in the white matter. In later stages, cerebral and cerebellar atrophy ensues (5, 6).
Late-onset GM2 gangliosidoses include subacute and chronic forms (2, 3). In childhood subacute GM2 gangliosidosis, onset is between the ages of 3 and 6 years, with signs of diffuse encephalopathy: gait difficulties associated with pyramidal signs and cerebellar ataxia as well as progressive loss of speech. An abnormal startle response is seen occasionally, and retinal changes are inconstant. After a period of approximately 3–10 years, the patient becomes bedridden and demented (2, 3, 7, 8). A number of these patients belong to the so-called B1 variant, which results in an altered substrate specificity of hexosaminidase A (the mutated enzyme retains the ability to degrade most conventional artificial substrates, but not sulfated substrate or the natural substrate [8]). Onset of chronic GM2 gangliosidosis occurs between childhood and adulthood. Signs of motor neuron and spinocerebellar dysfunction are prominent. Psychiatric disturbances are present in half the patients (2, 9, 10).
There are few reports of neuroradiologic findings in late-onset GM2 gangliosidosis (7–10). A preferential involvement of infratentorial structures has been described with marked cerebellar atrophy with typical enlargement of the fourth ventricle and outer CSF space. There are no signs of pontine or supratentorial involvement (7–9). This cerebellar atrophy, particularly of the vermis, was also the prominent feature in a CT and MR imaging study of 10 patients with chronic GM2 gangliosidosis reported by Streifler (9). The cerebellar involvement did not correlate with the age at onset and the severity of the clinical signs. In these previously described patients, the clinical course suggested a brain stem dysfunction, as evidenced by marked disturbance of ocular movements, but brain MR imaging did not reveal any brain stem involvement.
The clinical presentation of our case was suggestive of a brain stem lesion (tumor or inflammatory process) because of the presence of progressive pyramidal signs, ataxia, and right sixth nerve palsy. It was compatible with subacute childhood gangliosidosis but was not specific in the absence of typical retinal changes. An exaggerated startle response was recognized in retrospect. Enzyme analysis, prompted by the results of stereotactic biopsy, confirmed a B1-variant phenotype of hexosaminidase A deficiency. The peculiar feature in this case was the MR imaging pattern, with localized high signal intensity in the brain stem on FLAIR and T2-weighted-images, without any involvement of basal ganglia, white matter, or cerebellum. In particular, there was no cerebellar atrophy, in contrast to previously reported cases of late-onset gangliosidosis (7–10). Nevertheless, this brain stem localization agrees with previous neuropathologic reports of late-onset forms of GM2 gangliosidoses, in which pathologic changes predominantly affect the anterior horn cells of the spinal cord, the cerebellar cortical neurons, the brain stem nuclei, and basal ganglia with prominent neuronal storage and degeneration (11). The cerebral cortex is less severely or minimally involved.
In retrospect, the presence of an abnormal startle response to sounds in our patient could have suggested the diagnosis. This acoustico-motor response is virtually pathognomonic of GM2 gangliosidosis. It can be occasionally seen in Krabbe leukodystrophy or pyridoxine dependency, but not in brain stem tumoral or inflammatory processes (3). Similarly, some MR imaging features of this case may be considered atypical for a neoplastic process, among them the lack of enlargement of the pons and the patchy involvement of the brain stem. Absence of gadolinium enhancement provides no information as to the presence of a tumor process but is not in favor of an inflammation.
Conclusion
Gangliosidoses should be considered in the differential diagnosis of isolated infiltrating brain stem lesions in childhood. Biochemical confirmation using blood tests could help to avoid invasive stereotactic biopsy.
References
- 1.Barkovich AJ. Pediatric Neuroimaging. 2d ed. Philadelphia: Lippincott-Raven;1996;321–437
- 2.Gravel RA, Clarke JTR, Kaback MM, et al. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly VS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 7th ed. New York: McGraw-Hill;1995. :2839–2879
- 3.Lyon G, Adams RD, Kolodny EH. Neurology of Hereditary Metabolic Diseases of Children. 2d ed. New York: McGraw-Hill;1996;141:235–237 [Google Scholar]
- 4.Brismar J, Brismar G, Coates R, et al. Increased density of the thalamus on CT scans in patients with GM2 gangliosidosis. AJNR Am J Neuroradiol 1990;11:125–130 [PMC free article] [PubMed] [Google Scholar]
- 5.Mugikura S, Takahashi S, Higano S, et al. MR findings in Tay-Sachs disease. J Comput Assist Tomogr 1996;20:551–555 [DOI] [PubMed] [Google Scholar]
- 6.Fukumizu M, Yoshikawa H, Takashima S, et al. Tay-Sachs disease: progression of changes on neuroimaging in four cases. Neuroradiology 1992;34:483–486 [DOI] [PubMed] [Google Scholar]
- 7.De Gasperi R, Gama Sosa MA, Battistini S, et al. Late-onset GM2 gangliosidosis: Ashkenazi Jewish family with an exon 5 mutation (Tyr180-His) in the Hex A α-chain gene. Neurology 1996;47:547–552 [DOI] [PubMed] [Google Scholar]
- 8.Goebel HH, Stolte G, Kustermann-Kuhn B, Harzer K. B1 variant of GM2 gangliosidosis in a 12-year-old patient. Pediatric Res 1989;25:89–93 [DOI] [PubMed] [Google Scholar]
- 9.Streifler JY, Gornish M, Hadar H, Gadoth N. Brain imaging in late-onset GM2 gangliosidosis. Neurology 1993;43:2055–2058 [DOI] [PubMed] [Google Scholar]
- 10.Hund E, Grau A, Fogel W, et al. Progressive cerebellar ataxia, proximal neurogenic weakness and ocular motor disturbances: hexosaminidase A deficiency with late clinical onset in four siblings. J Neurol Sci 1997;145:25–31 [DOI] [PubMed] [Google Scholar]
- 11.Sandhoff K, Conzelmann E, Neufeld EF, et al. The GM2 gangliosidoses. In Scriver CR, Beaudet AL, Sly VS, Valle D, eds. The metabolic basis of inherited disease. 6th ed. New York: McGraw-Hill;1989. :1807–1842