

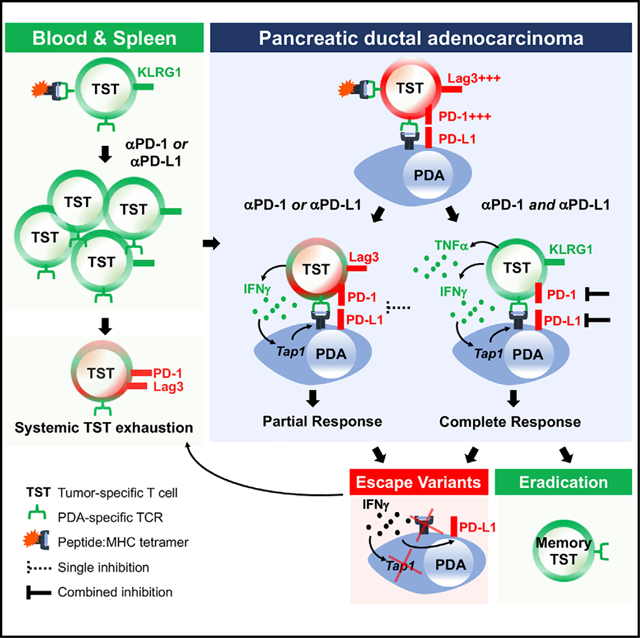
SUMMARY
Pancreatic ductal adenocarcinoma (PDA) is a lethal cancer resistant to immunotherapy. We create a PDA mouse model and show that neoantigen expression is required for intratumoral T cell accumulation and response to immune checkpoint blockade. By generating a peptide:MHC tetramer, we identify that PDA induces rapid intratumoral, and progressive systemic, tumor-specific T cell exhaustion. Monotherapy PD-1 or PD-L1 blockade enhances systemic T cell expansion and induces objective responses that require systemic T cells. However, tumor escape variants defective in IFNγ-inducible Tap1 and MHC class I cell surface expression ultimately emerge. Combination PD-1 + PD-L1 blockade synergizes therapeutically by increasing intratumoral KLRG1+Lag3−TNFα+ tumor-specific T cells and generating memory T cells capable of expanding to spontaneous tumor recurrence, thereby prolonging animal survival. Our studies support that PD-1 and PD-L1 are relevant immune checkpoints in PDA and identify a combination for clinical testing in those patients with neoantigen-specific T cells.
Graphical Abstract
In Brief
Burrack et al. investigate tumor-specific T cells during immunotherapy of pancreas cancer. T cells accumulate intratumorally yet rapidly exhaust. Combined PD-1 + PD-L1 blockade promotes peripheral T cell expansion, TNFα production, and eradication of spontaneous tumor recurrence in 50% of animals. Tumor variants defective in IFNγ-inducible Tap1 and MHC class I ultimately emerge.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA) is a lethal malignancy with a dismal 5-year survival rate of 9% (Chiaravalli et al., 2017). Lethality is due to late diagnosis, early metastasis, and therapeutic resistance. Some immune-based treatment options are showing clinical promise (Johnson et al., 2017; Vonderheide, 2018). However, a better understanding of the requirements necessary to achieve antitumor T cell-mediated immunity will inform T cell engineering approaches and new immune-based strategies for durable patient benefit.
Immune checkpoint blockade (ICB) is demonstrating remarkable clinical success in several solid tumors (Hu-Lieskovan and Ribas, 2017). Some patients respond robustly, whereas others either fail to respond or respond transiently before acquiring resistance. The extent of cancer nonsynonymous mutations often correlates with clinical responses following an immune checkpoint blockade (Le et al., 2015; Rizvi et al., 2015), presumably by creating new epitopes (neoepitopes) that have a high binding affinity to MHC and are likely recognized as foreign by the immune system. Common epithelial cancers, such as breast, ovarian, and pancreatic, often have low to moderate mutational burdens and are resistant to an immune checkpoint blockade (Brahmer et al., 2012; Foley et al., 2016; Hamanishi et al., 2015; Royal et al., 2010; Solinas et al., 2017). However, adoptive transfer of mutation-specific T cells can result in clinical antitumor activity in some epithelial cancers (Tran et al., 2014, 2016), with as few as 62 coding mutations (Zacharakis et al., 2018), suggesting the potential to harness rare tumor-specific T cells.
The genetically engineered KrasG12D/+;Trp53R172H/+;p48-Cre (KPC) mouse model of PDA is commonly used to study the human disease. There have been no spontaneous neoepitopes identified in KPC mice, because of the few coding mutations reported (Evans et al., 2016; Kinkead et al., 2018). Combination therapies appear necessary for transient benefit in this model (Jiang et al., 2016; Steele et al., 2016; Winograd et al., 2015), presumably by enhancing priming of T cells specific to self and tumor antigens. In contrast to KPC mice, human PDAs harbor ~30–60 somatic coding mutations (Balachandran et al., 2017; Biankin et al., 2012). A minor fraction (<1%) of PDA patients have abnormally high tumor mutational load because of defects in mismatch-repair genes (Hu et al., 2018b). Although few patients have been treated, a subset of these patients (five of eight) exhibited a clinical response following PD-1 blockade, yet most responses were transient, consistent with acquired resistance (Hu et al., 2018a, 2018b).
Despite a moderate mutational burden, we found that ~40% of resected human PDAs are enriched for CD8+ T cells, and many of these T cells exhibited features of recent or prolonged T cell receptor (TCR) signaling (Stromnes et al., 2017). These data are consistent with both T cell-mediated recognition of tumor antigens and the genomic identification of a putative immunogenic subset of PDAs (Bailey et al., 2016). Analyses of tumor neoepitopes in a long-term survivor (LTS) cohort of PDA patients found that neoepitope quality, including similarity to identified microbial epitopes, was a stronger predictor of survival than neoepitope quantity (Balachandran et al., 2017). We thus set out to investigate the role for specific antigen in pancreatic cancer immunotherapy outcomes.
Aside from our previous work on an engineered T cell therapy targeting the self and tumor antigen mesothelin in KPC mice (Stromnes et al., 2015), there are few models to investigate endogenous tumor antigen-specific T cell longitudinally in PDA. Therefore, the goal of this study is to assess the fate of neoantigen-specific T cells in PDA to uncover why immune therapies fail. Here, we develop a mouse model and a peptide:MHC tetramer that binds endogenous PDA-specific CD8+ T cells and track the fate of these cells during immune checkpoint blockade. The study is a substantive advance in our ability to investigate and model tumor-specific T cell dysfunction in PDA and supports the idea that combination PD-1 + PD-L1 blockade is advantageous therapeutically compared with manipulating either pathway alone.
RESULTS
Generation of an Immunotherapy-Responsive Pancreas Cancer Animal Model
The continual expression of mutant Kras and Trp53 causes multifocal disease in KPC mice (Hingorani et al., 2005). To develop a model that harbors a single tumor, we isolated primary tumor epithelial cells (TECs) from KPC mice, previously backcrossed to C57BL/6 (Stromnes et al., 2015). We identified TECs that induced the accumulation of tumor-associated macrophages (TAM), Ly6G+ granulocytes, and αSMA+ cancer-associated fibroblasts (CAFs) with few vessels following orthotopic implantation (Figure 1A; Figure S1A). Both autochthonous and orthotopic KPC tumors contained few CD8+ T cells and negligible PD-L1 (Figure 1A).
Figure 1. Generation of an Immunotherapy-Responsive Pancreas Cancer Animal Model.
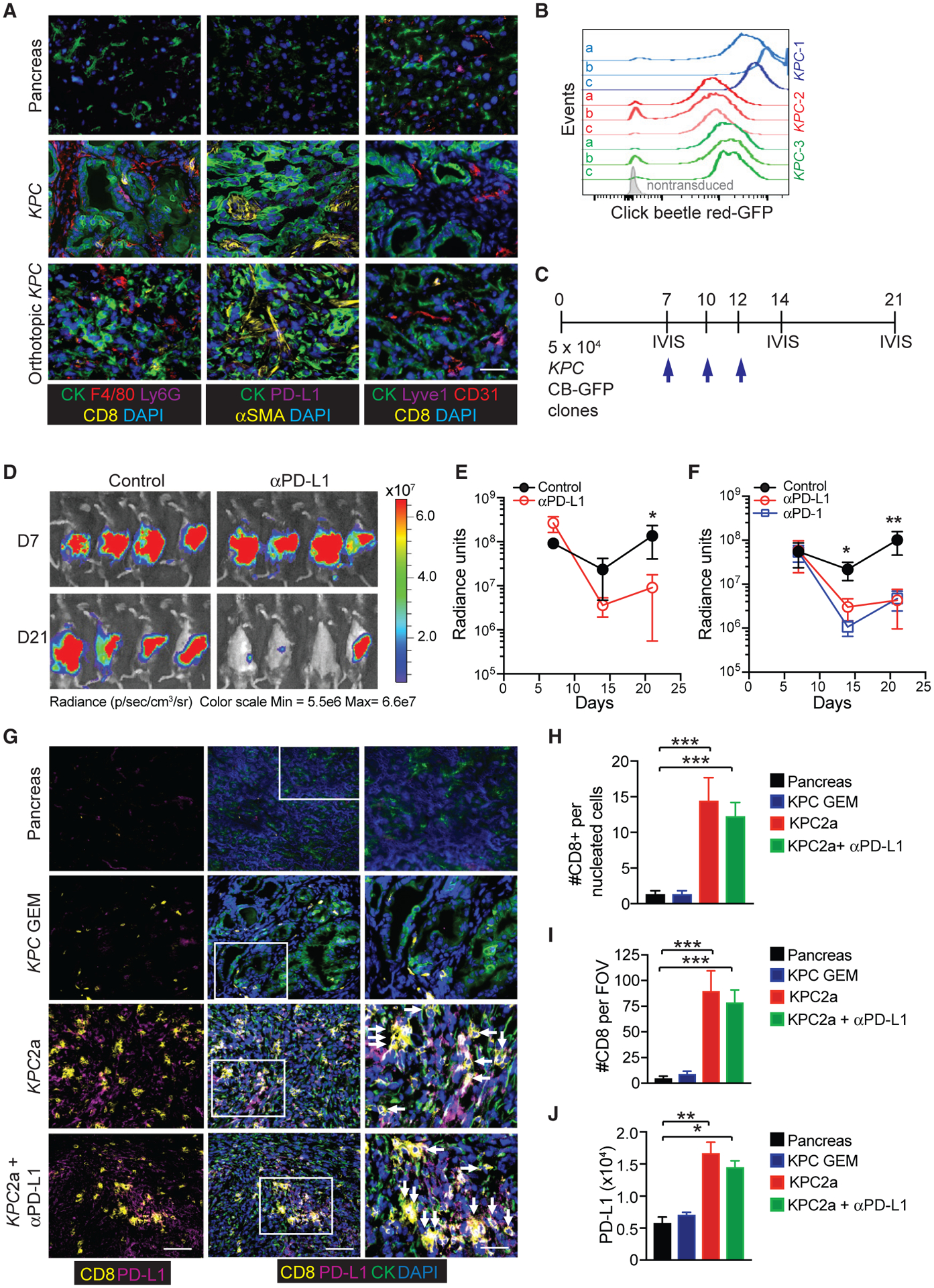
(A) Representative immunofluorescent (IF) staining of wild-type mouse pancreas (top row), primary tumors isolated from a genetically engineered KPC mouse (middle row), and a polyclonal KPC TECs following orthotopic implantation into B6 pancreas at 3 weeks post-implantation (bottom row). Images depict cytokeratin+ (CK) tumor cells, CD8+ T cells, F4/80+ macrophages, Ly6G+ neutrophils, PD-L1, αSMA cancer-associated fibroblast (CAFs), CD31+ endothelial cells, and Lyve-1+ lymphatic venules. Scale bar, 25 μM.
(B) Histogram overlay of CB-eGFP+ KPC clones generated from three independent KPC mice. Shown are representative clones (n = 3) from the three independent polyclonal KPC tumor epithelial cell lines (KPC1–3).
(C) Timeline for orthotopic tumor cell injection, αPD-L1 treatments (blue arrows), and IVIS imaging.
(D) Representative IVIS imaging of tumor bioluminescence at 7 days (prior to treatment) and 14 days following the initial αPD-L1 injection (day 21 post-tumor).
(E) Radiance of tumor bioluminescence following orthotopic injection of a CB+KPC clone (KPC3b) and following αPD-L1 treatment. Data are mean ± SEM. *p < 0.05 (unpaired two-tailed Student’s t test). n = 5 mice per group.
(F) Radiance of tumor bioluminescence following orthotopic injection of a CB+KPC clone (KPC2a) and following treatment with either αPD-1 or αPD-L1. Data are mean ± SEM. *p < 0.05 and **p < 0.005 (unpaired two-tailed Student’s t test). n = 5 mice per group.
(G) Representative IF of CD8+ T cells, cytokeratin (CK+) tumor cells, and PD-L1 in wild-type mouse pancreas, primary tumor isolated from a genetically engineered KPC mouse, and KPC2a (CB+) orthotopic tumors isolated from B6 mice ± αPD-L1. Scale bars, 50 μM (left and middle columns) and 10 μM (right column, inset).
(H) CD8+ T cell number per all nucleated cells in IF sections. Data are mean ± SEM. n = 3–5 mice per group.
(I) CD8+ T cell number per field of view in IF sections. Data are mean ± SEM. n = 3–5 mice per group.
(J) PD-L1 intensity was quantified through pixel intensity measurement by Fiji 2.0.
Data in (H)–(J) are mean ± SEM. *p < 0.05, **p < 0.005, and ***p < 0.005 (one-way ANOVA, with a post hoc test to correct for multiple comparisons). Data are representative of n = 3–6 mice per group. See also Figure S1.
We previously validated a click beetle red (CB) luciferase linked to eGFP for imaging engineered T cells in PDA (Stromnes et al., 2015). CB is widely used for deep in vivo imaging (Branchini et al., 2017; Hall et al., 2018; Serganova et al., 2016; Smith et al., 2017; Ur Rahman et al., 2017). To quantify orthotopic tumor growth in real time, we transduced three independently derived polyclonal KPC TECs with a retrovirus that expressed CB-eGFP. Next, we single cell-sorted CB-eGFP+ tumor cell clones (Figure 1B). Because KPC mice are refractory to PD-1 or PD-L1 blockade (Steele et al., 2016; Winograd et al., 2015), we tested if CB+ tumor clones respond to αPD-L1 (Figure 1C). Unexpectedly, αPD-L1 significantly decreased tumor size 7 days after the initial treatment (clone KPC3b; Figures 1D and 1E). We observed similar results with another CB+ clone (KPC2a; Figure 1F). Because αPD-L1 can signal directly to both tumor cells (Clark et al., 2016) and macrophages (Hartley et al., 2018), the requirement for T cells was tested. T cell depletion abrogated αPD-L1 antitumor activity (Figures S1B–S1E), and αPD-1 worked similarly (Figure 1F), indicating that T cells are required.
Intratumoral CD8+ T cell abundance is predictive of LTS in PDA patients (Balachandran et al., 2017) and clinical responses following immune checkpoint inhibitors (Cristescu et al., 2018; Tumeh et al., 2014). Tumor cell expression of CB-eGFP significantly increased intratumoral CD8+ T cell frequency >20% (Figures S1F and S1G), resulting in a >1 log increase in intratumoral CD8+ T cell number (Figure S1H). Most PD-1+ T cells infiltrating CB+ tumors also co-expressed the integrin LFA-1 (Figure S1I). αPD-L1 did not affect the proportion or number of intratumoral CD8+ T cells (Figures S1F–S1H). However, in mice harboring CB-eGFP+ tumors and treated with αPD-L1, splenic CD8+ T cell frequency significantly increased (Figure S1J). Although splenic CD8+ T number remained unchanged (Figure S1K), αPD-L1 significantly increased the proportion of splenic CD8+ T cells that co-expressed PD-1 and LFA-1 (Figure S1L), likely reflecting enhanced T cell priming or expansion.
Flow cytometric analysis of nonlymphoid tissues can underestimate lymphocyte number (Steinert et al., 2015). We therefore performed immunofluorescence (IF) staining for CD8+ T cells and PD-L1. Normal pancreas and autochthonous KPC PDA contained few CD8+ T cells (Figure 1G). In contrast, CD8+ T cells were abundant in CB-eGFP+ KPC2a tumors, resulting a ~1 log increase in CD8+ T cell number (Figures 1G–1I). PD-L1 was undetectable in normal ductal cells and autochthonous tumors and was significantly increased in KPC2a tumors (Figures 1G and 1J). Thus, this tumor model may reflect a subset of human PDAs with CD8+ T cells and PD-L1 (Stromnes et al., 2017).
Neoantigen Is Required for Response to Immune Checkpoint Blockade
To test the hypothesis that the antitumor response following αPD-L1 required CB, we administered αPD-L1 to mice with parental (KPC2), CB-eGFP+ (KPC2a), or KPC2 TECs transduced to express eGFP only. αPD-L1 induced objective responses to KPC2a tumors but not parental (KPC2) or eGFP+ clones (KPC2-eGFP) (Figure 2A). αPD-L1 also significantly decreased KPC2a tumor growth (Figure 2B), prolonged animal survival (Figure 2C), and decreased tumor weights (Figure 2D). In contrast, αPD-L1 had no effect on parental or eGFP+ tumor growth and animal survival (Figures 2E–2G). CB+ KPC clones were similar to parental cells with respect to MHC class I, PD-L1 and mesothelin expression ± IFNγ (Figures 2H and 2I).
Figure 2. CB Expression Is Required for Immune Checkpoint Blockade Response.
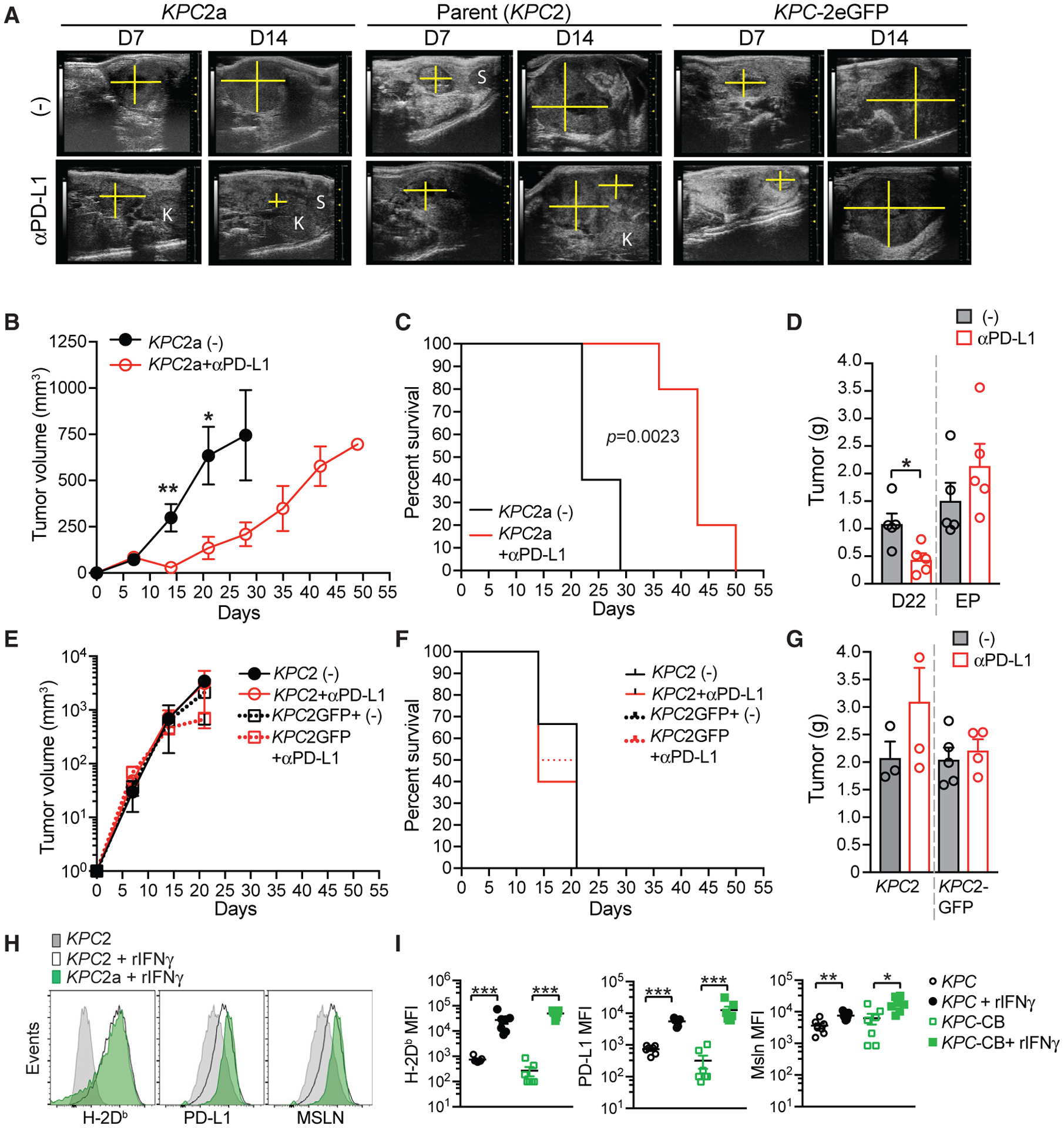
(A) Representative ultrasound images of pancreatic tumors in B6 mice orthotopically implanted with KPC2a (CB-eGFP+), KPC2 parental, or KPC2-eGFP tumor cells. Pancreas mass, yellow cross section; K, kidney; S, spleen.
(B) Mean tumor volume ± SEM. n = 5 mice per cohort. *p < 0.05 and **p < 0.005 (unpaired two-tailed Student’s t test).
(C) Kaplan-Meier survival curve of mice bearing orthotopic tumors. Significance was determined using a log rank (Mantel-Cox) test. n = 5 per group.
(D) Mean tumor weight in grams ± SEM at day 22 or at endpoint (EP; tumors > 500 mm3). Each dot is an independent mouse. n = 5 mice per group. *p < 0.05 (unpaired two-tailed Student’s t test).
(E) Mean tumor volume ± SEM of KPC2 parental or KPC-eGFP+ cells. n = 3–6 mice per group.
(F) Kaplan-Meier survival curve of mice bearing KPC2 parental or KPC-eGFP+ orthotopic tumors ± αPD-L1. n = 3–6 mice per group.
(G) Mean tumor weight in grams ± SEM at day 22 or at endpoint (EP; tumors > 500 mm3). Each dot is an independent mouse. n = 3–6 mice per group.
(H) Representative histograms of the indicated cell surface proteins in KPC2 parental and KPC2a clones ± 48 h pre-treatment with recombinant mouse IFNγ.
(I) Mean fluorescence intensity (MFI) of H-2Db, PD-L1, and mesothelin (Msln) expression by independent KPC parental tumor cells (n = 3) and their respective CB+ clones (KPC-CB) ± IFNγ. Each dot is an independent clone. Statistical significance was determined using Student’s t test to compare the induction of protein expression following IFNγ. *p < 0.05, **p < 0.005, and ***p < 0.005. Representative of two independent experiments.
Identification of an Immunodominant CB H-2Db Restricted Epitope in B6 Mice
We questioned the rationale for further study of the CB-specific T cell response because other model antigens such as ovalbumin (Ova) are widely used. However, Pdx1-Cre-driven KPC tumors that express Ova fail to establish tumors in B6 mice, because of T cell-dependent rejection independent of therapy (Evans et al., 2016). To determine if this was also the case in our B6 KPC cells, which originate from a p48-Cre-driven KPC model, we transduced similar parental KPC2 cells with Ova-eGFP, single cell-sorted to >99% purity, and measured tumor growth following orthotopic injection. Ova+ tumors grew but at a significantly reduced rate compared with the parental tumors (Figures S2A and S2B), resulting in prolongation of mouse survival (Figure S2C). KPC-Ova+ clones that eventually grew no longer expressed Ova-eGFP (Figure S2D). In contrast, in untreated mice, KPC-CB+ cells grew unabated until reaching euthanasia criteria (tumor volume > 500 mm3) and did not confer a significant difference in mouse survival compared with mice with parental or eGFP+ tumors (Figures S2E and S2F). Therefore, in contrast to CB, Ova expression drove robust immune editing and antigen-loss variants independent of immunotherapy.
We therefore pursued identification of a CB-derived immunotherapy responsive epitope (IRE). The H-2Db peptide binding motif contains an anchoring Asn residue is at position 5, and Met, Ile, or Leu is at the C terminus (Hudrisier et al., 1996). We identified CB peptides predicted to strongly bind H-2Db and synthesized the top five 9-mer peptides (Table S1). We then vaccinated B6 mice with peptides+αCD40+PolyI:C, which expands antigen-specific T cells (Ahonen et al., 2004) via IL-15 and IL-27 (Klarquist et al., 2018). CB101–109+αCD40+PolyI:C elicited significantly more IFNγ-producing CD8+ T cells compared with naive mice (Figures 3A and 3B), whereas T cell responses to other peptides were not detected. Notably, CB101–109 was the top-ranked peptide by all algorithms (Table S1), with an estimated binding affinity of 3.6 nM to H-2Db. To test if expanding CB101–109:H-2Db-specific T cells could control KPC2a growth, we vaccinated mice with CB101–109+αCD40+PolyI:C to elicit T cells that use mitochondrial metabolism (Klarquist et al., 2018), which may boost T cell antitumor activity (Ho and Kaech, 2017). Tumor size decreased in four of six vaccinated mice (Figure 3C). Similar to αPD-L1, vaccine failed to control tumor growth (not shown). Thus, CB101–109 is processed and presented in vivo.
Figure 3. Identification of an Immunodominant CB H-2Db Restricted Epitope.
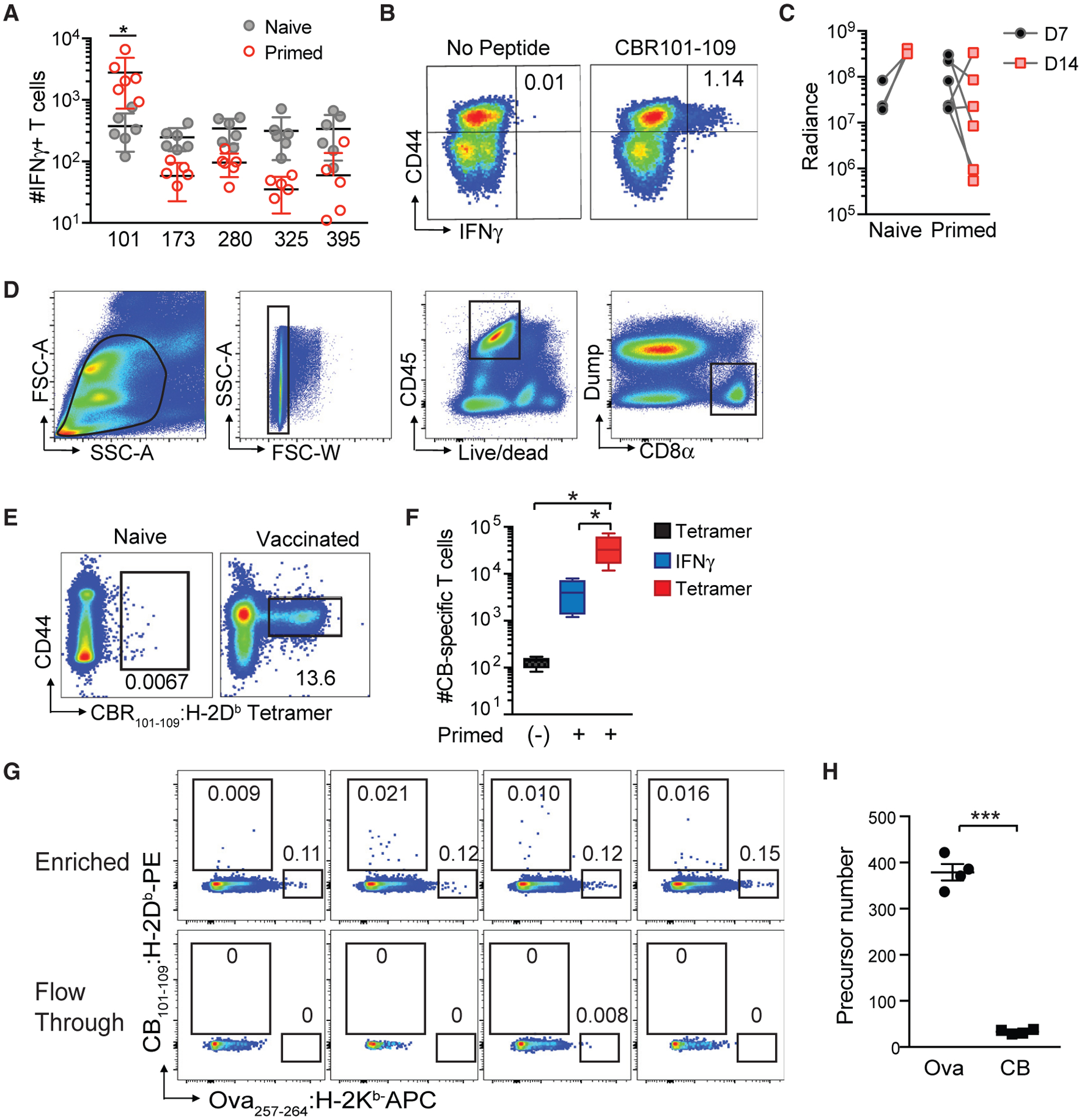
(A) Number of IFNγ-producing T cells per spleen from naive or peptide/αCD40/PolyI:C vaccinated C57BL/6 mice following ex vivo re-stimulation with the indicated peptide. Each dot is an independent mouse. n = 6 per group. *p < 0.05 (unpaired two-tailed Student’s t test).
(B) Representative CD44 and IFNγ staining of splenic CD8+ T cells following ex vivo stimulation with CB101–109 peptide. Plots are gated on live, CD45+CD8+ T cells.
(C) Tumor radiance from control or vaccinated B6 mice on days 7 (D7) and 14 (D14).
(D) Gating strategy for validating fluorescently labeled CB101–109:H-2Db -tetramer-binding CD8+ T cells.
(E) Representative CD44 and CB101–109:H-2Db -tetramer staining of splenocytes isolated from naive and CB101–109-immunized mice at day 7 post-vaccination. Plots are gated on live, CD45+ CD8+ Dump− T cells as shown in Figure 3D. n = 6 mice per group.
(F) Mean number of CB101–109-specific T cells per spleen in naive (−) or vaccinated B6 mice was determined by tetramer staining or measuring IFNγ-producing T cells in response to CB101–109 peptide ex vivo ± SEM. n = 4 mice per group. *p < 0.05 (one-way ANOVA, with a post hoc test to correct for multiple comparisons).
(G) Representative plots of CB101–109:H-2Db and Ova257–264 :H-2Kb tetramer staining of naive B6 splenocytes following tetramer staining and magnetic bead-based tetramer enrichment. Gates are on live CD45+ CD8+ Dump− T cells.
(H) Number of CB101–109:H-2Db- and Ova257–264:H-2Kb-specific T cells in naive B6 mice. Each dot is an independent mouse, and data are mean ± SEM. ***p < 0.0005 (unpaired two-tailed Student’s t test). See also Table S1 and Figure S2.
We next generated a CB101–109:H-2Db fluorescently labeled tetramer to detect T cells that express CB101–109:H-2Db-specifc TCRs. To validate the tetramer, we vaccinated mice with CB101–109+αCD40+PolyI:C and 7 days later performed flow cytometry (Figure 3D). We detected significantly more CD44high tetramer-binding CD8 T cells in vaccinated mice (Figure 3E). Vaccination increased CB-specific T cell number more than 2 logs, and tetramer detected more CB-specific T cells compared with IFNγ (Figure 3F).
We next investigated if T cell precursor frequency influenced disparate immune editing outcomes in KPC clones expressing CB or Ova using a tetramer magnetic bead-based enrichment protocol (Moon et al., 2007). The mean precursor number of Ova257–264:H-2Kb-specific T cells in B6 mice was 379 cells (Figures 3G and 3H), consistent with prior studies (Jenkins and Moon, 2012; Obar et al., 2008). In contrast, the naive precursor number of CB101–109:H-2Db-specific T cells averaged 33. To support the rigor of this approach, the two tetramer-binding T cell populations were non-overlapping and not detected in the flow-through. Thus, the naive precursor frequency of neoantigen-specific T cells may impact resistance mechanisms in cancer, and neoepitopes that remain targets for immunotherapy may be those that elicit antigen-specific T cells that have a low naive precursor frequency.
Immune Checkpoint Blockade Efficacy Requires Peripheral CB101–109:H-2Db-Specific T Cells
We next investigated intratumoral CB101–109:H-2Db-specific T cell kinetics using the gating strategy shown in Figure S3A. On day 7, tetramer+ T cells were detected at highest frequency in PDA compa (Figures 4A and 4B). By day 14, 30%–40% of the tumor-infiltrating CD8+ T cells bound tetramer. αPD-L1 significantly increased the frequency of both circulating and splenic CB-specific T cells on day 12 (Figures 4C and 4D). In contrast, αPD-L1 did not alter intratumoral CB-specific T cell frequency (Figure 4E). αPD-L1 significantly increased circulating CB-specific T cell number on day 15 (Figure 4F), a trend also observed in spleen (Figure 4G). However, total circulating CD8+ T cell quantity was unchanged (Figures S3B and S3C), indicating a selective expansion of tumor-specific T cells. αPD-L1 did not significantly affect the number of intratumoral CB-specific T cells, which decreased 5-fold between 21 and 43 days (Figure 4H). To investigate if intratumoral antigen-specific T cell maintenance was due to enhanced T cell proliferation, we analyzed Ki67 expression in tetramer-binding T cells. A significantly higher frequency of intratumoral CB-specific T cells expressed Ki67 compared with splenic tetramer+ T cells (Figure S3D). Unexpectedly, αPD-L1 significantly decreased the proportion of Ki67+ CB-specific T cells (Figures 4I–4K; Figure S3D). Because absolute numbers of tetramer+ T cells did not increase, PDA may drive both T cell proliferation and death.
Figure 4. Immune Checkpoint Blockade Efficacy Requires the Recruitment of Systemic CB101–109:H-2Db-Specific T Cells.
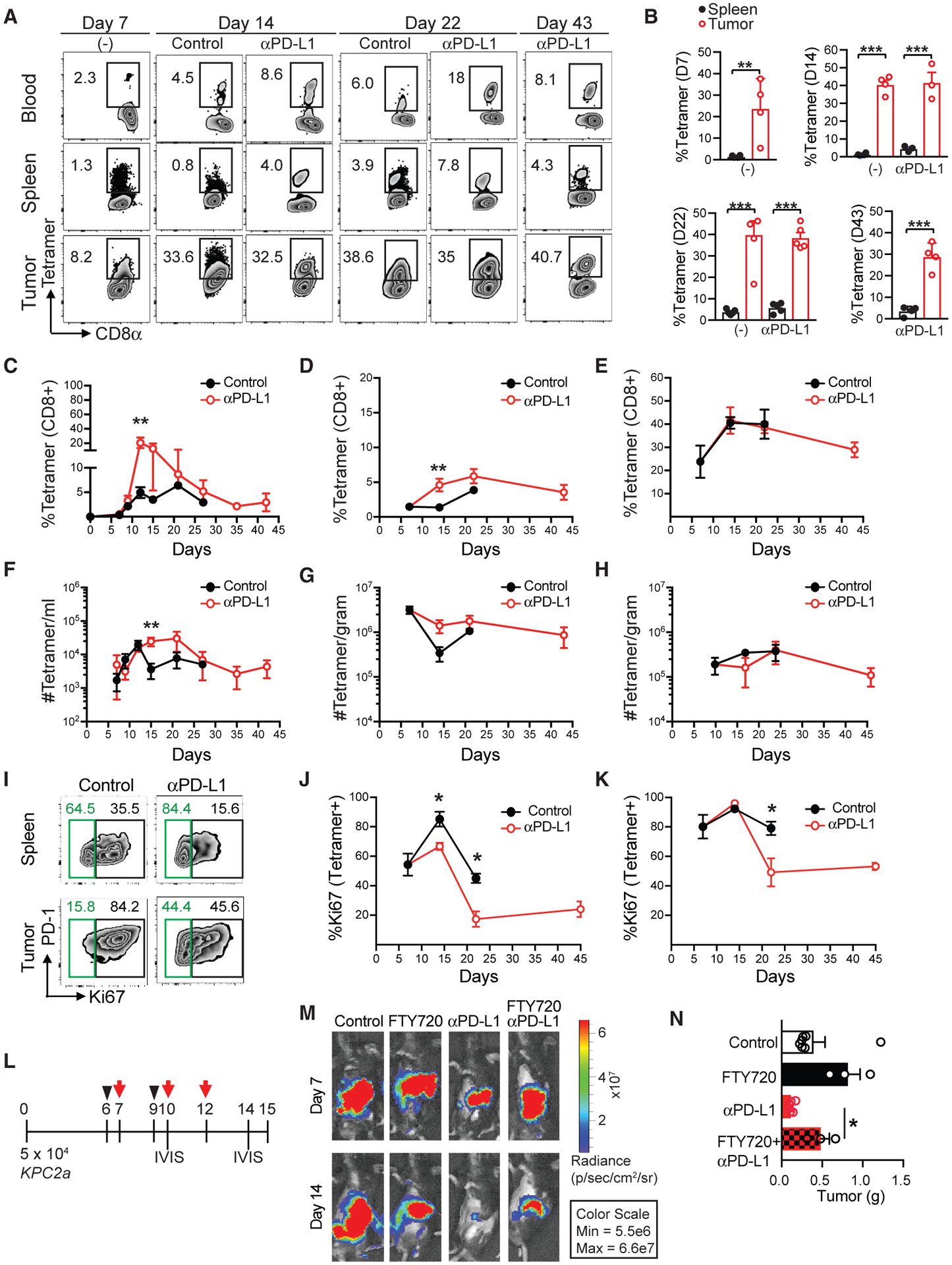
(A) Representative CB101–109:H-2Db-tetramer staining of blood, splenic, or intratumoral CD8+ T cells ± αPD-L1 at the indicated time points post-tumor implantation.
(B) Mean frequency of tetramer+ T cells of total CD8+ T cells in spleen (spl) or tumor (Tu) ± SEM on days 7, 14, 22, and 43 post-tumor implantations. n = 4 or 5 mice per group. **p < 0.005 and ***p < 0.0005 (unpaired two-tailed Student’s t test).
(C–E) Mean frequency of tetramer+ T cells of total CD8+ T cells ± αPD-L1 in blood (C), spleen (D), and tumor (E). Error bars are SEM. n = 3–6 mice per group. *p < 0.05 and **p < 0.005 (unpaired two-tailed Student’s t test).
(F–H) Mean CB101–109:H-2Db-specific T cell number in blood (F), spleen (G), and tumor (H) following tumor implantation. Error bars are SEM. n = 3–6 mice per group. **p < 0.005 (unpaired two-tailed Student’s t test).
(I) Representative plots of PD-1 and Ki67 staining gated on tetramer+CD8+ T cells at day 14.
(J and K) Proportion of CD8+tetramer+ T cells that express Ki67 in spleen (J) and tumor (K). Error bars are SEM. n = 4 or 5 mice per group. *p < 0.05 (unpaired two-tailed Student’s t test).
(L) Timeline for FTY720 treatment (black arrowhead) and αPD-L1 (red arrow).
(M) Representative images of tumor radiance at 7 and 14 days post-KPC2a injection. n = 3 mice per group.
(N) Tumor weight in grams at 14 days post-orthotopic KPC2a injection. n = 3 mice per group. *p < 0.05 (one-way ANOVA, with a post hoc test to correct for multiple comparisons). Data are mean ± SEM.
See also Figure S3.
To test if αPD-L1 required recruitment of systemic T cells, we treated mice with FTY720, which causes S1P1R internalization, promoting lymphocyte sequestration in lymph nodes and preventing T cell trafficking into tissues (Chun and Hartung, 2010; Penaranda et al., 2010) prior to PD-L1 blockade (Figure 4L). FTY720 alone had no effect on tumor growth (Figure 4M). However, tumor weights were significantly greater in FTY720+αPD-L1-treated cohorts compared with αPD-L1 (Figure 4N). Therefore, “resident” intratumoral lymphocytes appear insufficient for therapeutic effects of αPD-L1, and instead recruitment of peripheral T cells is required for full therapeutic benefit.
Rapid Intratumoral and Progressive Systemic Dysfunction of Neoantigen-Specific T Cells
A significantly higher frequency of intratumoral tetramer+ T cells expressed activating and inhibitory receptors PD-1, Lag3, Tim-3, and TIGIT compared with splenic tetramer+ T cells (Figures 5A and 5B), consistent with a subset of human PDAs (Stromnes et al., 2017) and chronic TCR signaling (McLane et al., 2019). αPD-L1 significantly decreased the proportion of tetramer+ T cells that expressed Lag-3 (Figures 5A and 5B) and that co-expressed multiple inhibitory receptors (Figure 5C; Figure S4A). We observed a similar trend in splenic tetramer+ T cells (Figures 5C). Whereas ~75% of tetramer+ T cells expressed Lag-3, only ~25% expressed Tim-3 (Figures 5A and 5B). In contrast, most PD-1+tetramer+ T cells from orthotopic tumors derived from a different KPC CB+ tumor cell clone preferentially expressed Tim-3 rather than Lag-3 (Figure S4B), suggesting tumor cell intrinsic factors may affect the hierarchy of co-inhibitory receptor expression.
Figure 5. Rapid Intratumoral and Progressive Systemic Dysfunction of Neoantigen-Specific T Cells.
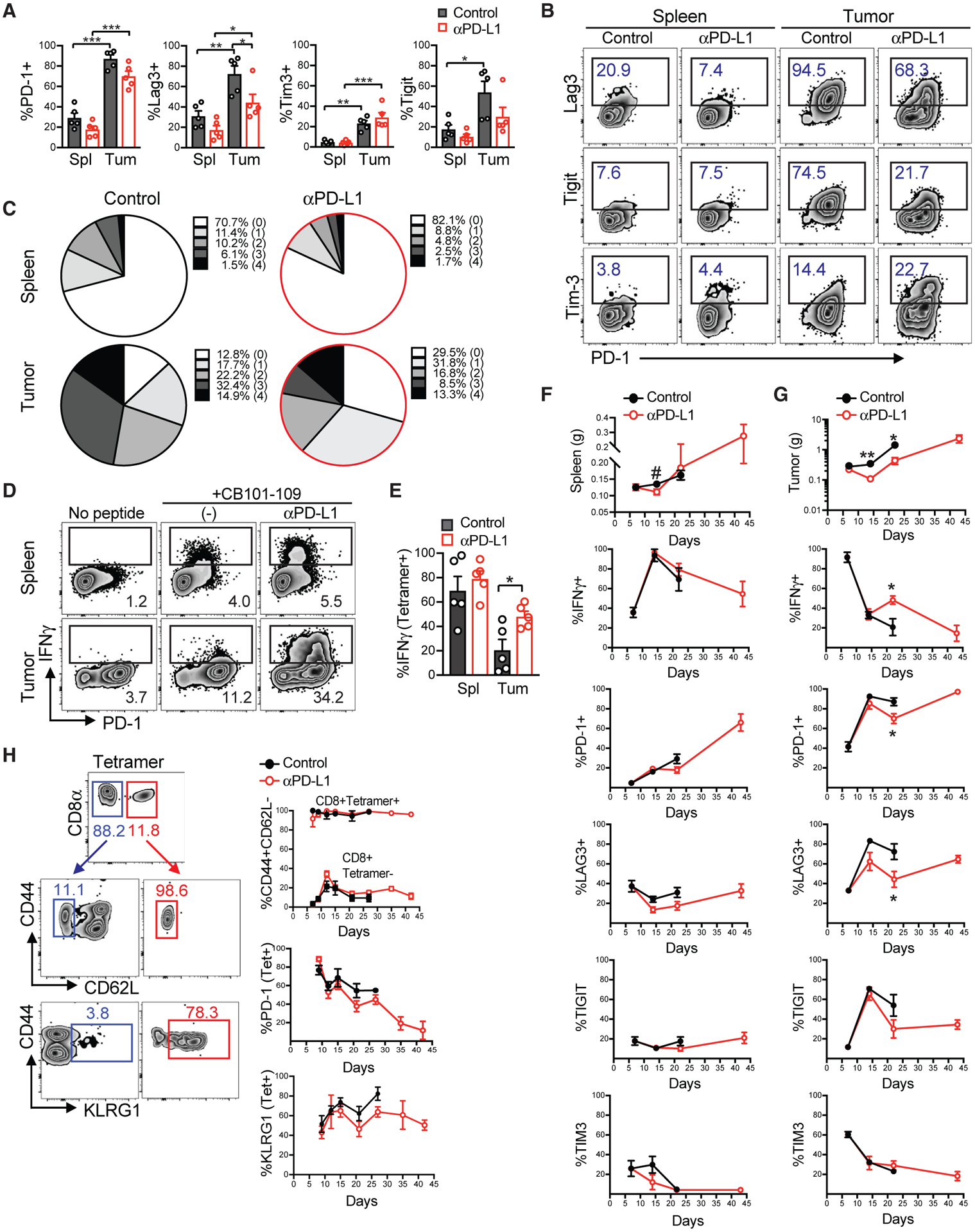
(A) Frequency of PD-1, Lag3, Tim3, and TIGIT expression by tetramer+ T cells in the spleen (Spl) and tumor (Tum) at day 22 post-tumor implantation. Each dot is an independent mouse, and data are mean ± SEM. *p < 0.05, **p < 0.05, and ***p < 0.005 (one-way ANOVA, with a post hoc test to correct for multiple comparisons).
(B) Representative plots of markers quantified in (A).
(C) Mean frequency of CB101–109:H-2Db-specific CD8+ T cells that express none (0), one (PD-1), or co-express 2, 3, or 4 inhibitory receptors at day 22.
(D) Proportion of total CD8+ T cells producing IFNγ following ex vivo re-stimulation with CB101–109 peptide at day 22.
(E) Number of tetramer+ T cells producing IFNγ at day 22. Data are mean ± SEM, and each dot is an independent mouse. *p < 0.05 (one-way ANOVA, with a post hoc test to correct for multiple comparisons).
(F) Progressive phenotypic and functional changes in splenic tetramer+ CD8+ T cells in mice bearing KPC2a tumors ± αPD-L1. Data are mean ± SEM. n = 4 or 5 mice per group.
(G) Progressive phenotypic and functional changes in intratumoral tetramer+ CD8+ T cells. Data are mean ± SEM. *p < 0.05 (unpaired two-tailed Student’s t test). n = 4 or 5 mice per group.
(H) Analysis of the indicated antigens on circulating tetramer+ T cells following αPD-L1 treatment. Data are mean ± SEM. n = 4 or 5 mice per group. See also Figure S4.
αPD-L1 increased the proportion (Figures 5D and 5E) and number (Figures S4C and S4D) of tetramer+ T cells that produced IFNγ following ex vivo re-stimulation with CB101–109 peptide. Intratumoral IFNγ-producing CB-specific T cells included both PD-1high and PD-1-intermediate (PD-1int) cell subpopulations, whereas splenic IFNγ-producing T cells were PD-1int (Figure 5D). αPD-L1 did not affect the amount of IFNγ produced per T cell (Figure S4E).
We next asked how the phenotype and function of tumor-specific T cells evolved over time. The spleen steadily increased in weight, likely reflecting increased tumor growth and myeloid-promoting cytokines (Stromnes et al., 2014). Splenic CB-specific T cells became progressively dysfunctional over time, which correlated with an increase in the proportion of tetramer+ T cells that expressed PD-1 and to a lesser extent, Lag-3 (Figure 5F). Notably, although the absolute numbers of both splenic PD-1+ and PD-1− tetramer+ T cells slightly increased over time (Figure S4C), the numbers of both intratumoral PD-1+ and PD-1− tetramer+ T cells tended to decrease (Figure S4D). Thus, systemic antigen-specific T cell dysfunction may present a barrier to immunotherapy. Intratumoral CB-specific T cells rapidly lost the ability to produce IFNγ between days 7 and 14 (Figure 5G). αPD-L1 caused a significant decrease in tumor weight at days 14 and 22 (Figure 5G) and increased IFNγ production by tumor-specific T cells at day 22. At day 22, increased intratumoral CB-specific T cell function correlated with a reduction in the proportion of tetramer-binding T cells that expressed Lag-3 or PD-1 (Figure 5G). Although the proportion of PD-1+, Lag3+, Tim-3+, or Tigit+ tetramer+ T cells increased progressively, absolute number of tetramer + T cells expressing these markers instead decreased over time (Figures S4C and S4D).
Circulating neoantigen-specific T cells may identify patients who benefit from immunotherapy (Gros et al., 2016). We therefore further investigated tetramer+ T cell phenotype in circulation. Circulating tetramer+ T cells were CD44hiCD62L−, an effector phenotype (Figure 5H). PD-1 decreased progressively over time on circulating tetramer+ T cells, and more than 60% of circulating tetramer+ T cells continually expressed KLRG1 (Figure 5H). In contrast, most intratumoral CB-specific T cells were KLRG1− (Figure S4F). Circulating tetramer-binding T cells from vaccinated mice transiently expressed KLRG1 and instead progressively differentiated toward a central memory phenotype (Figure S4G).
Dual PD-L1 + PD1 Blockade Alters Intratumoral T Cell Function, Phenotype, Antitumor Activity, and Longevity
To test whether PDA evaded therapy or if dosing was insufficient, we treated tumor-bearing mice with prolonged PD-1 or PD-L1 antibody alone or in combination. Tumor size immediately decreased in all mice treated with αPD-L1 or αPD-1 (Figures 6A and 6B; Figure S5A). Following an initial objective response, most αPD-L1-treated mice (four of five) showed tumor stasis while on therapy. In contrast, two of five αPD-1-treated mice clearly rebounded while on therapy. Tumors gradually returned in four of five αPD-L1-treated and four of five αPD-1-treated mice (median survival 43 versus 48 days, respectively; Figure 6C). Tumors remained bioluminescent, indicating that prolonged immunotherapy did not select for antigen-loss variants (Figures 6A and 6B; Figure S5A). Prolonged αPD-L1 treatment did not increase median survival compared with short-term treatment (Figure S5B). However, one of five of the mice treated with αPD-1 and one of five of the mice treated with αPD-L1 survived indefinitely (>80 days) with no detectable tumor on the basis of bioluminescent imaging (Figure 6C; Figure S2A), necropsy, and flow cytometry of eGFP+CD45− cells in pancreas (Figure S5C). Three of five αPD-1-treated mice exhibited mild signs of vitiligo during therapy (Figure S5D), suggesting that PD-1 inhibition may promote autoimmunity to skin self-antigens and consistent with dermatologic complications in patients (Sibaud et al., 2016).
Figure 6. Combined PD-L1 + PD1 Blockade Promotes Tumor Eradication and T Cell Persistence in the Absence of Antigen.
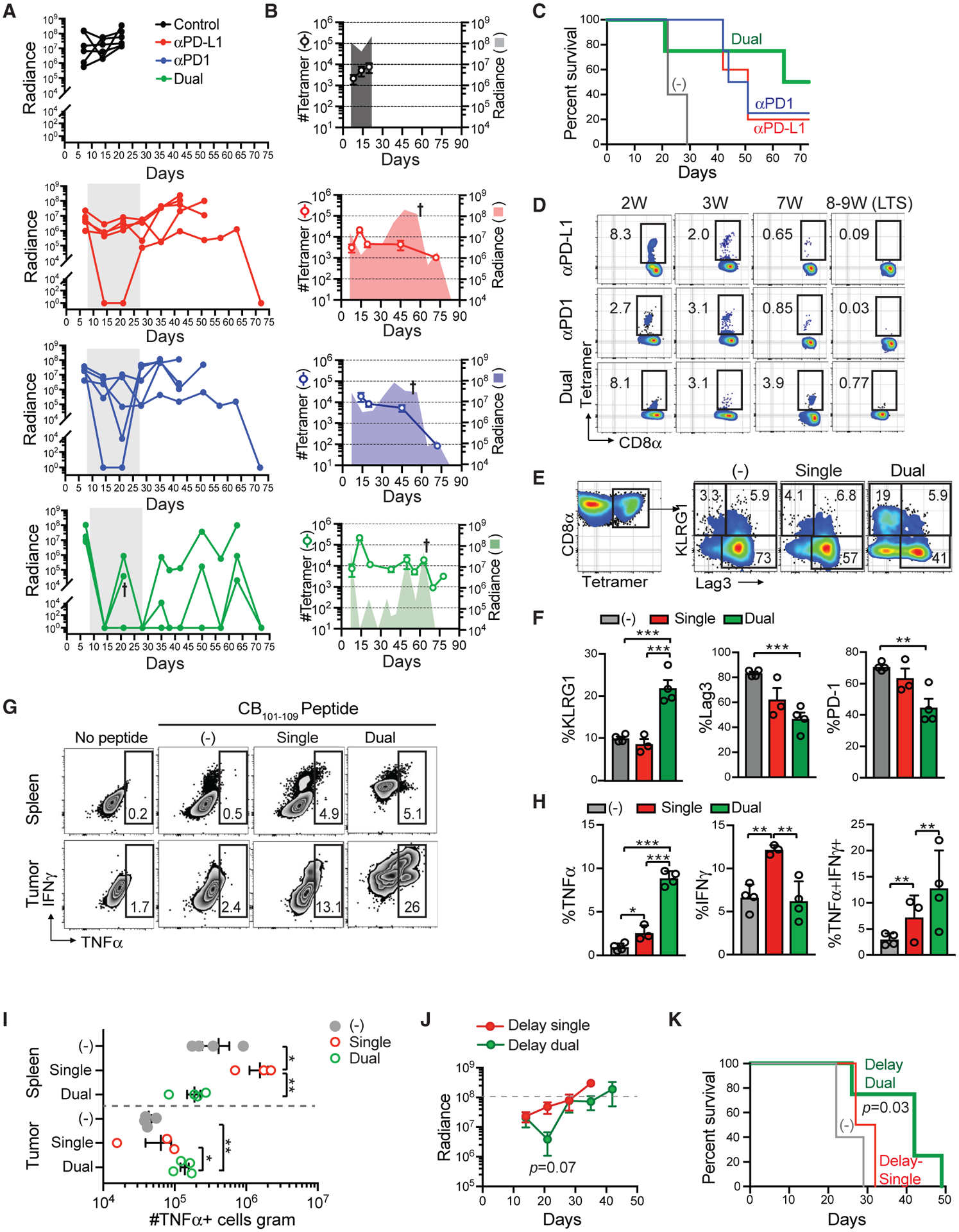
(A) Radiance of tumor growth in mice treated with either αPD-L1 or αPD1, combination αPD-L1 + αPD1 (dual). Gray box indicates treatment period. Each line is an independent mouse. Arrow indicates unexpected death.
(B) Number of tetramer+ T cells per milliliter in blood and mean tumor radiance. Symbol indicates that some mice were euthanized because of tumor growth. Data are mean ± SEM for tetramer number. Data are mean for tumor radiance.
(C) Kaplan-Meier survival curve of mice in (A) and (B). n = 4 or 5 mice per group.
(D) Proportion of tetramer+ T cells (gated on live CD45+CD8+ T cells) in blood at the weeks (W) post-tumor. LTS, long-term survivors.
(E) Proportion of intratumoral tetramer+ T cells that express KLRG1 and/or Lag3 at day 14. Single, αPD-L1 only.
(F) Quantification of groups in (E). Data are mean ± SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005 (one-way ANOVA with a Tukey post hoc test).
(G) Proportion of CD8+ T cells that produce TNFα following CB101–109 peptide re-stimulation ex vivo.
(H) Proportion of CD8+ T that produce the indicated cytokines from tumors in mice treated with single (αPD-L1) or dual (αPD-L1 + αPD-1) blockade at day 14. Data are mean ± SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005 (one-way ANOVA with a Tukey post hoc test).
(I) Number of CD8+ T cells per gram that produce TNFα from mice treated with single or dual therapy at day 14. Each dot is an independent mouse. Data are mean ± SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005 (one-way ANOVA with a Tukey post hoc test). See also Figure S5.
(J) Tumor radiance following delayed single (αPD-L1) or delayed dual (αPD-L1 + αPD-1) blockade beginning day 14 post-tumor implantation. Significant difference in tumor size at day 21 was assessed using an unpaired two-tailed Student’s t test. Data are mean ± SEM.
(K) Kaplan-Meier survival curve of mice in treated as in (J). Control group is the same animals shown in (C). Survival prolongation for delay dual (MST 42 days) versus untreated (MST 22 days) was determined by a log rank test. See also Figure S5.
In contrast to monotherapy, all mice treated with dual PD-1 + PD-L1 blockade had undetectable tumor on day 14 (Figures 6A and 6B; Figure S5A). One of the dual-treated mice died on day 22, which was not due to tumor growth. Following cessation of therapy, tumor radiance fluctuated, reflecting a cycle of tumor recurrence and immunological control (Figures 6A and 6B; Figure S5A). Overall, 50% of the dual-treated mice survived indefinitely (Figure 6C), suggesting synergy with dual blockade and consistent with results in a melanoma animal model (Hartley et al., 2018).
We next analyzed if tumor control correlated with changes in CB-specific T cells in circulation. In contrast to control mice in which circulating tetramer+ T cell number remained <1 × 104 T cells/ml, all treated mice increased circulating tetramer+ T cell number >1 × 104 T cells/ml at 2 weeks (Figure 6B). Notably, in the two LTSs that received monotherapy, tetramer+ T cells became undetectable in circulation following tumor clearance (Figure 6D), suggesting that these T cells required antigen for persistence. In dual-treated mice, however, circulating tetramer+ T cells fluctuated with tumor burden (Figure 6B) and persisted in the LTS in the absence of tumor antigen (Figures 6D and S5C). Around day 50, 10%–15% of the circulating tetramer+ T cells began to re-express the central memory marker CD62L in dual-treated mice (Figure S5E). Thus, circulating neoantigen-specific T cell fluctuations may identify cyclical patterns of tumor control and recurrence.
We did not observe a significant increase in frequency or number of tetramer+ T cells following dual blockade at day 14 (not shown). However, dual blockade significantly increased the proportion of intratumoral KLRG1+Lag3− tetramer+ T cells while decreasing the proportion of PD-1+tetramer+ T cells (Figures 6E and 6F), resulting in a 2.5-fold increase in intratumoral KLRG1+tetramer+ T cell number compared with monotherapy (not shown). Significant alterations in Lag3 and PD-1 expression on splenic tetramer+ T cells following dual blockade were also noted (Figure S5F). Dual blockade significantly increased the proportion (Figures 6G and 6H) and number (Figure 6I) of intratumoral TNFα-producing CB-specific T cells. In contrast, the number of IFNγ-producing CB-specific T cells following single or dual blockade was similar in PDA (Figure S5F).
CB-specific T cell accumulation plateaued at day 14 post-tumor implantation (Figure 4H), and continual encounter with persistent antigen can render T cells refractory to PD-1 blockade because of epigenetic mechanisms (Pauken et al., 2016; Philip et al., 2017; Schietinger et al., 2016; Sen et al., 2016). Therefore, we next compared the impact of dual blockade that may be more clinically relevant by enrolling mice at a later time point (day 14). Although delayed single blockade (αPD-L1) had a minimal impact on tumor growth, delayed dual blockade (αPD-1 + αPD-L1) caused objective responses in 100% of the treated mice (Figure 6J; Figure S5H). Delayed single blockade had no benefit on median survival time (MST) compared with untreated mice (Figure 6K). In contrast, delayed dual blockade significantly prolonged animal survival compared with untreated controls (p = 0.0392, MST 42 versus 22 days).
Tumor Escape Variants Fail to Express MHC I because of a Defect in Tap1 following IFNγ Signaling
A previous study showed that at the time of tumor relapse, CD8 T cells were restricted to the tumor margin (Zaretsky et al., 2016). However, CD8+ T cells remained prominent in PDA, whereas PD-L1 expression became undetectable at later time points (Figure 7A). To identify mechanisms of immune evasion, we re-derived tumor cells that evaded immunotherapy. KPC2a re-derived tumor cells were low for MHC class I and PD-L1 without exogenous cytokine, similar to the parental line (KPC2) and to the KPC2a cells prior to orthotopic injection (Figure 7B). In response to IFNγ, PD-L1 was similarly upregulated in all cell lines tested (Figure 7B). However, whereas IFNγ induced expression of both MHC class I alleles (H-2Db and H-2Kb) on both the parental and KPC2a cells pre-transfer, neither MHC class I allele was expressed on the cell surface of all escape variants following IFNγ incubation (Figure 7B).
Figure 7. Tumor Escape Variants Fail to Express MHC I because of a Defect in Tap1 following IFNγ.
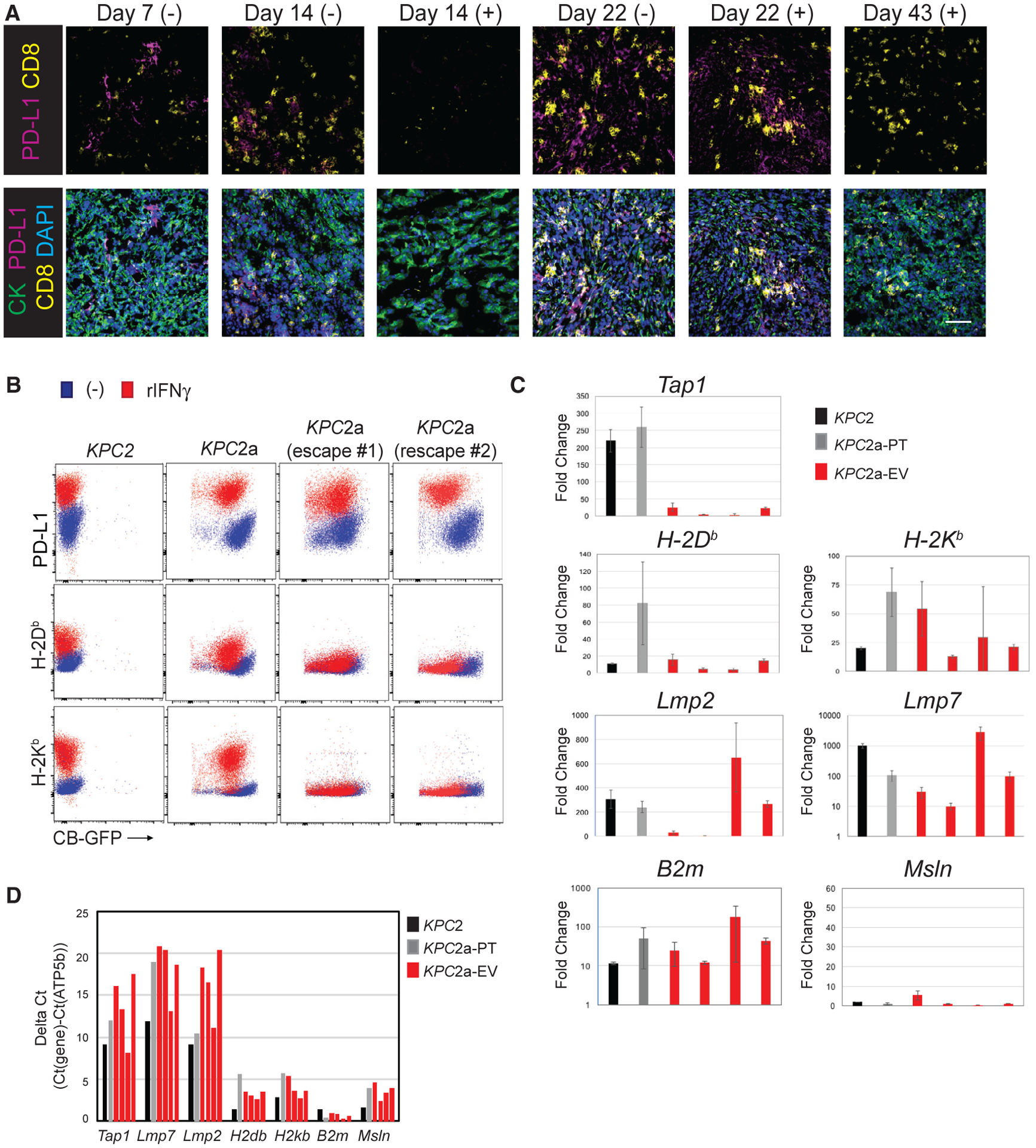
(A) IF staining for tumor cells (CK+), PD-L1, and CD8 in KPC2a tumors in control (−) or αPD-L1 (+) at the indicated time points. Tumors were not easily identifiable in the mice that received αPD-L1 at day 14. Scale bar, 50 μM.
(B) Cell surface expression of indicated proteins ± IFNγ treatment in parental KPC2 cells, KPC2a clone prior to implantation (pre-transfer), and two independent KPC2a escape variants. Representative of n = 2 independent experiments.
(C) Fold induction of antigen processing and/or presentation genes following IFNγ treatment in parental KPC2 cells, KPC2a pre-transfer (KPC2a-PT), and four independently re-derived KPC2a escape variants (KPC2a-EV). Data are mean ± SEM.
(D) Gene expression in cell lines from Figure 7C without IFNγ treatment normalized to housekeeping gene ATP5b. Note that a higher the delta Ct indicates lower target gene expression relative to ATP5b. All qPCR data were performed in triplicate.
To investigate the mechanism of defective cell surface MHC class I molecules, we performed qPCR for genes involved in antigen processing and presentation in cell lines cultured in the presence or absence IFNγ. Although we observed minor variability among the independent escape variants with respect to MHC alleles, all (four of four) had a defect in Tap1 expression following IFNγ (Figure 7C). Two of four tumor escape variants also had a defect in immunoproteosome subunits Lmp2 and, to a lesser extent, Lmp7. In contrast, B2m and a control gene, Msln (not induced by IFNγ), were unchanged. Gene expression was not different in tumor escape variants in the absence of cytokine (Figure 7D). These data suggest that defects in Tap1 following IFNγ signaling may promote immunotherapy resistance in PDA.
DISCUSSION
Pancreas cancer is considered a poorly immunogenic cancer. However, the genetic and immunologic heterogeneity of human PDA is emerging (Bailey et al., 2016; Balachandran et al., 2017; Stromnes et al., 2017; Tiriac et al., 2018). Indeed, a subset of human PDAs contain a distinct T cell infiltrate (Bailey et al., 2016; Stromnes et al., 2017). Tumor cell intrinsic differences underlie some variation in intratumoral T cell infiltration in KPC mice (Li et al., 2018). However, no neoantigens have yet been identified in KPC mice to investigate endogenous tumor-specific T cells, presumably because of the paucity of coding mutations in this model (Evans et al., 2016; Kinkead et al., 2018). Here, we develop a system to track the longevity and functionality of endogenous PDA-specific T cells. Consistent with our prior study of engineered T cells in KPC PDA (Stromnes et al., 2015), we show that tumor antigen-specific T cells preferentially accumulate in PDA.
We show that CB is immunogenic in B6 mice and identify the immunodominant H-2Db-restricted CB101–109 epitope. CB luciferases are commonly used for robust in vivo imaging in mice (Branchini et al., 2017; Hall et al., 2018; Serganova et al., 2016; Smith et al., 2017; Stromnes et al., 2015; Ur Rahman et al., 2017). CB induces priming, intratumoral accumulation, and rapid intratumoral dysfunction of CB-specific T cells in PDA. Results that contrast to the Panc02 model, which harbors >800 mutations and detection of neoepitope-specific T cells requires therapy (Kinkead et al., 2018). Despite producing an epitope that has a 3.6 nM estimated binding affinity to H-2Db (top 0.01% of 40,000 peptides tested in NetMHC), cells that express CB are not rejected unless an immunoregulatory axis is perturbed, which likely masked the immunogenicity of CB in previous studies. As such, CB-specific T cells model T cells specific to neoantigens that respond following PD-L1 blockade in patients. A limitation of the present work is that the tumor-specific T cells are specific to a foreign antigen rather than a naturally acquired neoantigen. However, clinically relevant neoepitopes are likely more similar to foreign than self-epitopes (Balachandran et al., 2017).
During infection, the size of the naive precursor T cell frequency correlates with magnitude of effector T cell response in mice (Kotturi et al., 2008) and humans (Schmidt et al., 2011) (reviewed in Jenkins and Moon, 2012). Using tetramer pull-down, we identified a ~1 log increase in the number of naive Ova-specific CD8+ T cells versus the number of naive CB-specific T cells in B6 mice. These results suggest that for neoepitopes that bind MHC strongly, a high precursor frequency of neoepitope-specific T cells (modeled by Ova257–264:H-2Kb-specific T cells) may promote antigen-loss variants, whereas a low precursor frequency (modeled by CB101–109:H-2Db-specific T cells) may instead result in preservation of neoantigen and induction of T cell exhaustion in PDA. Future studies that either modify the peptide MHC binding affinity or naive T cell precursor frequency are necessary to support this premise. Applying this paradigm to enumerate T cell precursor frequency specific to epitopes in shared driver mutations, viral epitopes in virally induced cancer, and shared self and tumor antigens is feasible in humans (Pittet et al., 1999).
Our study suggests that PD-L1 blockade acts systemically in PDA. We show that PD-L1 blockade induces the systemic expansion of tumor-specific T cells and recruitment of circulating T cells was critical for efficacy. Thus, αPD-L1 may not only re-invigorate intratumoral T cells. Unexpectedly, we also show a progressive dysfunction of systemic antigen-specific T cells in tumor-bearing mice, which could be due to IL-10, IL-35 (Das and Bar-Sagi, 2019), and/or TGFβ (Bellone et al., 1999). An additional advantage of our model is the ability to track circulating tumor-specific T cells, which we show can serve as a biomarker for tumor burden, tumor immunogenicity, and immunological response. In dual-treated mice, the kinetics of circulating CB-specific T cells reflected continual cycles of tumor recurrence and control and acquired phenotypic features consistent with generating memory T cells.
Tumor escape is a frequent consequence of immunotherapy, and following PD-L1 blockade, we show that tumor escape variants with a defect in MHC class I expression following IFNγ emerged. Acquired resistance following PD-1 blockade in a single PDA patient was reported (Hu et al., 2018a). Mutant KRAS, which is the most common gene mutated in PDA, is associated with HLA expression defects (He et al., 2013), and HLA loss is a common mechanism of acquired resistance (reviewed in Garrido et al., 2016). A lack of cell surface HLA has been attributed to defects in β2 m (Gettinger et al., 2017; Restifo et al., 1996; Zaretsky et al., 2016), loss of heterozygosity at the HLA locus (Tran et al., 2016), a decrease in antigen presentation machinery (Donia et al., 2017), epigenetic silencing (Paulson et al., 2018), or defects in IFNγR signaling (Zaretsky et al., 2016). In our study, because IFNγ-inducible PD-L1 expression is maintained, and PD-L1 and HLA operate via a similar IFNγ signaling axis converging on IRF1 (Garcia-Diaz et al., 2017), IFNγ signaling appears intact. Instead, we show a specific defect in Tap1 following IFNγ. Because hypermethylation of Tap1 occurs in colorectal cancer (Ling et al., 2017), this is one potential mechanism to assess in future studies.
We show that PD-1 and PD-L1 are relevant immune checkpoints in PDA. We identify that dual PD-1 + PD-L1 blockade elicits qualitatively distinct T cells with greater potential to respond to tumor recurrence compared with monotherapy. Dual blockade qualitatively changed intratumoral T cell phenotype by altering KLRG1, PD-1, and Lag3 expression and enhanced TNFα production. These observations are consistent with reports that TNFα may be a linchpin cytokine mediating tumor-immune equilibrium (Park et al., 2019) and suggest that dual blockade may alter the program of intratumoral T cell exhaustion, which can be imprinted within the first 2 weeks of antigen exposure (Philip et al., 2017) and includes loss of TNFα (Williams et al., 2017). Delaying dual blockade by 1 week retained survival benefit, whereas delayed monotherapy did not prolong survival. Further investigation into whether dual blockade affects the proportion of terminally versus progenitor exhausted T cell subpopulations (Miller et al., 2019) is warranted. Although single-agent PD-1 or PD-L1 antibodies are being tested in combination with numerous other drugs, combination PD-1 + PD-L1 blockade has not been tried in humans as far as we can identify. A clinical trial using a PD-1-PD-L1 bi-specific antibody is recruiting (ClinicalTrials. gov identifier NCT03936959). Our study suggests that this combination could be potentially beneficial in which there is a pre-existing endogenous antitumor T cell response and warrants further investigation in less immunogenic settings.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents including novel cell lines generated in this report should be directed to and will be fulfilled by the Lead Contact, Ingunn Stromnes (ingunn@umn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
University of Minnesota Institutional Animal Care and Use Committee approved all animal studies. We previously backcrossed the KrasLSL-G12D+;Trp53LSL-R172H/+;p48Cre (KPC) mice to > 99.6% C57BL/6J background (Stromnes et al., 2015) and were generously provided by Drs. Philip Greenberg and Sunil Hingorani at the Fred Hutchinson Cancer Research Center (Seattle, WA). All orthotopic experiments used 6–11 week old female and male C57BL/6J mice (Jackson laboratories).
Cell lines
Primary tumor epithelial cells were isolated from C57BL/6 KPC mice as described (Stromnes et al., 2015) and maintained below passage 15. Primary tumor cells were transduced with retroviral vectors CBR-GFP (Stromnes et al., 2015), or similar Ovalbumin-GFP or GFP only. Following transduction, GFP+ cells were single cell sorted to establish clonal cells and cells were grown in basic media: 500 mL DMEM (GIBCO) +10% FBS (GIBCO) + 2.5 μg/ml Amphotericin B (GIBCO) + 100 μg/ml pen/strep (GIBCO) + 2.5 mg dextrose (Fisher Chemical) at 37°C and 5% CO2. Media was sterile filtered and stored in the dark at 4°C.
METHOD DETAILS
Production of retrovirus and transduction
Briefly, 2.2 × 106 Platinum-E (Plat-E, ATCC) retroviral packaging cells were plated on 10 cm tissue culture-treated plates in Plat-E media with antibiotic selection (DMEM + 10%FBS + 20mM L-glutamine + 100 U/ml Pen/strep + 10 μg/ml Blasticidin + 1 μg/ml Puromycin) for 24 h at 37°C, 5% CO2. On day 2, Plat-E cells were transfected with Retroviral vectors containing CBR-GFP (Stromnes et al., 2015), Ovalbumin-GFP, or GFP using Effectene (QIAGEN). On day 3, Plat-E media was replaced with basic media (DMEM + 10% FBS + 20 mM L-glutamine+ 100 U/ml Pen/strep) and cells incubated at 32°C + 5% CO2. On days 4 and 5, viral supernatants were harvested and passed through a 0.45 μM filter for immediate use.
MHC class I and PD-L1 staining following incubation with IFNγ in vitro
3×105 KPC tumor epithelial cells per well were cultured in 6-well plates in basic media ± 50 ng/ml recombinant mouse IFNγ (R&D Systems). After ~48 h, adherent tumor cells were lifted in 10 mM EDTA (Invitrogen), washed and stained with antibodies directly conjugated to PD-L1 (10F.9G2, PE/Cy7, BioLegend) and MHC class I alleles H-2Db (KH95, PerCP/Cy5.5, BioLegend) and H-2Kb (AF6–88.5, PE, BD PharMingen) as well as primary antibody against mesothelin (clone B35, MBL) followed by a polyclonal secondary antibody goat anti-Rat conjugated to Alexa Fluor 488 (Thermo Fisher). Data were acquired on Fortessa flow cytometer (BD) using FACSDiva software (BD) and analyzed using FlowJo version 10.
Orthotopic surgery
Briefly, after reaching surgical plane anesthesia via isoflurane, a small incision was made in the right flank to access the pancreas. Next, 5 ×104 KPC cells were injected into the pancreas in 20 μl of 60% matrigel (Discovery Labware) using an insulin syringe (Covidien) as described (Chai et al., 2013). Separate sets of sutures were used to close the peritoneum and skin (Ethicon).
In vivo monoclonal antibody and FTY720 administration
200 μg of αPD-1 (RMP1.14, BioXCell) or αPD-L1 (10F.9G2, BioXcell) diluted in saline were injected IP on days 7, 10, and 12 following orthotopic tumor implantation. For prolonged treatments, mice received 2 injections per week for 3 weeks also starting at day 7 post orthotopic tumor implantation. For FTY720 experiments, mice were injected with 0.5 mg/kg FTY720 (Cayman Chemical) diluted and administered according to manufacturer’s instructions in 1:1 solution of ethanol:PBS immediately prior to injection.
Tumor measurements by IVIS or high-resolution ultrasound
Abdominal hair was removed using Nair. For bioluminescence, mice were injected with 100 μg of D-Luciferin (Promega) i.p. followed by image acquisition 11 minutes later using 0.5 s exposure time. Biodistribution and tumor radiance was quantified in photons per second using IVIS 100 and Living Image software (Xenogen). High-resolution ultrasound imaging was conducted using Vevo 2100 (Visual Sonics). Identification of a tumor mass was by location within the pancreas, anatomic landmarks, hypoechoic features, and with clear tumor boundaries (Stromnes et al., 2015). Tumor volume based on ultrasound measurements were calculated using a modified ellipsoidal formula (Jensen et al., 2008): Tumor volume = 1/2(length × width2).
Identification of a click beetle epitope in C57BL/6 mice
To determine peptides with strong affinity to H-2Db, click beetle red amino acid sequence was analyzed using epitope prediction algorithms: syfpeithi (http://www.syfpeithi.de/bin/MHCServer.dll/EpitopePrediction.htm), BIMAS (https://www-bimas.cit.nih.gov/), and netMHC (version 4.0, http://www.cbs.dtu.dk/services/NetMHC/). Five peptides with ranking scores from the 3 algorithms were chosen for an in vivo competition priming assay. B6 mice were vaccinated with 100 μg of peptides along with 50 μg agonistic αCD40 (FGK145, Bioxcell) and 50 μg poly I:C (Sigma). Seven days post vaccination, splenocytes from primed and naive mice were re-stimulated in vitro with individual peptides for 4–5 h in the presence of Brefeldin A (BD Biosciences) followed by intracellular cytokine staining and flow cytometric analysis.
Peptide:MHC tetramers and magnetic bead-based detection from naive mice
H-2Db-restricted biotinylated monomer was produced by incubating CB101–109 peptide with purified H-2Db and β2 m followed by purification via Fast Protein Liquid Chromatography system (Aktaprime plus, GE health care) as described (Altman et al., 1996; Murali-Krishna et al., 1998). Biotinylated monomer was conjugated to streptavidin R-phycoerythrin (Invitrogen) to produce CB101–109/H-2Db tetramer. SIINFEKL-H-Kb tetramer was provided by the NIH tetramer core facility at Emory University conjugated to fluorochrome (http://tetramer.yerkes.emory.edu/reagents/class-I-mhc). To detect naive precursor CD8 T cells binding either peptide:MHC reagent, single cell suspensions of splenocytes were stained with tetramer for 60 minutes at room temperature, followed by 30 minute incubation at 4°C with magnetic beads conjugated to PE or APC, followed by enrichment using LS columns (Miltenyi), as described (Moon et al., 2007)
Preparation of mononuclear cells from tissues
Spleens were mechanically dissociated to single cells and red blood cells (RBCs) were lysed by incubation in ACK lysis buffer (GIBCO) for 1–2 minutes at room temperature in 15 mL conical tubes. 9 mL of T cell media (500 mL DMEM (GIBCO) + 10% FBS (GIBCO), 100 μg/ml pen/strep (GIBCO), 20 mM L-glutamine (GIBCO), 1x NEAA (GIBCO), and 50 uM betamercaptoethanol (Sigma)) was added to quench lysis. Cells were then spun at 1400 rpm for 5 minutes, resuspended in FACS buffer, and stained. Tumors were digested using 1 mg/ml collagenase IV (Sigma Aldrich) for 30 min in a 37°C shaker followed by mechanical digestion to single cells.
Flow cytometry of circulating tetramer+ T cells
Peripheral blood mononuclear cells (PBMCs) were collected in 20 mM EDTA in Eppendorf tubes. PBMCs were spun for 8 minutes at 10,000 rpm at 20°C and serum was removed. RBCs were lysed with ACK Lysis buffer (GIBCO) for 8 minutes at room temperature (rt). If incomplete lysis occurred, cells were spun down for a second time at 12,000 rpm for 30 s and the ACK lysis step was repeated. Mononuclear cells were stained 1:100 with CB101–109/H-2Db-PE tetramer in the presence of Fc block 1:100 (αCD16/32, Tonbo), and monoclonal antibodies at 1:100 specific to CD45 (30-F11, Biolegend), CD8α (53–6.7, ebioscience), LFA-1 (clone H155–78, Biolegend), CD44 (clone IM7, BD), CD62L (MEL-14, Biolegend), PD-1 (J43, Invitrogen), KLRG1 (2F1, Biolegend), Tim-3 (RMT3–23, Biolegend), Lag-3 (C9B7W, Biolegend). CD19 (1D3, BD), F4/80 (T45–2342, BD), and CD11c (N418, BD) were used for a dump channel along with ghost dye BV510 at 1:500 (Tonbo). Antibodies were diluted in FACs buffer (PBS + 2.5% FBS) and cells were stained at room temperature in the dark for 60 minutes and then added to 100 μl of cell counting beads (Sigma Aldrich). Stained cells were fixed with 0.4% paraformaldehyde and stored overnight in the dark at 4°C prior to FACs acquisition using a Fortessa 1770 flow cytometer and Facs Diva software (BD Biosciences) and analyzed using FlowJo software (version 10).
Flow cytometric analysis of T cells from tissues
To determine peptide-specific cytokine production, single cell suspension of spleen and tumor were cultured in complete T cell media in the presence of Golgiplug (1:500 BD Biosciences) and CB101–109 peptide (1 μg/ml Genscript) for 5 hours at 37°C. Next, cells were stained for Ghost live-dead (Tonbo), CD45, CD8α, CD44, PD-1 as described above, then fixed and permeabilized (BD Fixation Kit) and stained for intracellular expression of IFNγ (XMG1.2, Biolegend, 1:100) and TNFα (MP6-Xt22, Biolegend, 1:100) overnight in the dark at 4°C. To profile the phenotype of splenic and intratumoral T cells, mononuclear cells were stained with CB101–109/H-2Db-PE tetramer (1:100) in the presence of 1:500 Fc block (anti-CD16/32, Tonbo), and monoclonal antibodies to the following antibodies were used for surface stain (same clones as stated above): CD45, CD8, CD44, PD-1, Tim-3, Lag-3, and TIGIT (1G9, Biolegend). Following fixation with the intracellular fixation and permeabilization Foxp3 kit (Tonbo), Ki67 (B56, Biolegend) was used to stain the cells for 30 minutes in the dark on ice. Cells were washed and analyzed using a Fortessa 1770 flow cytometer and Facs Diva software (BD Biosciences) and analyzed using FlowJo software (version 10).
Calculation of cell numbers per gram of milliliter of tissue
Spleen and tumors were weighed ex vivo to calculate total grams of tissue used for single cell suspensions for flow cytometry. The number of cell counting beads and number of live CD45+ cells collected per tube were determined using FlowJo software and the equation: #CD45+ cells per tube (n) = (#Beads/#Cells) × (Concentration of beads × Volume of beads added). Total number of cells collected from the entire single cell suspension was determined by multiplying n by total number of stains. Cell numbers were normalized to gram of tissue by dividing cell numbers by gram (spleen or tumor) or milliliters (blood samples) collected.
Immunofluorescence
Tissues were embedded in OCT (Tissue-Tek) and stored at −80°C. 7 μm sections were cut using a Cryostat and fixed in acetone at −20°C for 10 min. Sections were rehydrated with PBS + 1% bovine serum albumin (BSA) and incubated for 1 hr at rt with primary antibodies to goat anti-mouse PD-L1 (R&D Systems, AF1019 1:500), rat anti-mouse CD31 (BD, 1:100), rabbit anti-mouse Lyve1 (Novus, 1:500), CD8α (BD, 53–6.7, 1:100), panCK-FITC (Sigma-Aldrich, F3418, 1:200) diluted in PBS + 1% BSA. Slides were washed 3X in PBS + 1% BSA and incubated with anti-goat AF647 (Jackson ImmunoResearch, 1:500), anti-rat AF546 (Invitrogen, 1:500), and anti-rabbit AF647 (Life Technologies, 1:500) for 1 hr at rt in the dark. Additional panels included F4/80 in PE (Tonbo, BM8.1, 1:100), alpha-SMA (Invitrogen, 1A4, 1:00), rabbit anti-mouse Ly6G (BD, 1:100). Following staining slides were then washed 3X with PBS + 1% BSA, washed 3X with PBS, and mounted in DAPI Prolong Gold (Life Technologies). Images were acquired on a Leica DM6000 epifluorescent microscope at the University of Minnesota Center for Immunology using Imaris 9.1.0 (Bitplane). To determine CD8 T cell number, individual cells and DAPI-stained nuclei from n = 3 mice per group and a minimum of n = 3 fields per mouse were manually counted and recorded by an investigator blinded to the experimental conditions with the assistance of Cell Counter plug-in in Fiji2.0. PD-L1 staining intensity was measured by pixel intensity from n = 3 mice per group and a minimum of n = 3 fields per mouse using Fiji2.0.
Gene expression of antigen presentation genes in PDA cell lines
KPC2a tumors from 2 mice treated with αPD-L1 (harvested at day 45), 1 mouse treated with αPD-1 (harvested at day 45), and one mouse treated with dual blockade (harvested at day 63) were expanded in vitro using culture conditions described above. Parental, KPC2a, and re-derived cell lines were plated at equal numbers and cultured in vitro ± recombinant mouse IFNγ (100ng/mL, R&D Systems) for 24 hours in 6-well plates. Cells were lifted in 10mM EDTA (Invitrogen), RNA was extracted (QIAGEN RNeasy Mini Kit) and concentration/purity was assessed by Nanodrop. cDNA was generated using RT Buffer Mix and RT Enzyme Mix (Thermo Fisher). Real time PCR was performed in triplicate on a BioRad CFX96 Touch Real-Time PCR Detection System by measuring SYBR Green (BioRad) fluorescence for 40 cycles. MSLN was used as a negative control and ATP5b as a reference gene because they are unlikely to be influenced by IFNγ. Calculations were performed according to the geNorm method to calculate fold change and standard error (Vandesompele et al., 2002).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad software. All mouse experiments reflect n = 3–8 mice per group per time point. Student’s t test was used to compare 2-group data. One-way ANOVA and Tukey post-test were used for multiple comparisons. Log-Rank Mantel Cox test was used to test for statistical differences in mouse survival. Data are expressed as mean ± SEM p < 0.05 was considered significant. *, p < 0.05; ** p < 0.005; ***, p < 0.0005.
DATA AND CODE AVAILABILITY
This study did not generate or analyze large datasets/code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-PD-L1 (clone 10F.9G2) | Biolegend | Cat No. 124313 |
| Anti-MHC class I H-2Db (clone KH95) | Biolegend | Cat No.111518 |
| Anti-MHC class I H-2Kb (clone AF6–88.5) | BD | Cat No. 561072 |
| Anti-mouse PD-1 (clone RMP1.14) | BioXcell | Cat No. BE0146 |
| Anti-mouse PD-L1 (clone 10F.9G2) | BioXcell | Cat No. BE0101 |
| Primary antibody against mouse mesothelin (clone B35) | MBL | Cat No. D233–3 |
| Goat anti-rat secondary antibody conjugated to AF488 | Thermo Fisher | Cat No. A-11006 |
| Agonistic anti-mouse anti-CD40 (clone FGK45) | BioXcell | BE0016–2 |
| Fc block (anti-CD16/32) | Tonbo | 70-1061-U100 |
| Anti-CD45 (clone 30-F11) | Biolegend | 103147 |
| Anti-CD8a (clone 53–6.7) | Thermo Fisher | 45-0081-82 |
| Anti-CD44 (clone IM7) | BD | 564392 |
| Anti-CD62L (clone MEL-14) | Biolegend | 104406 |
| Anti-PD-1 (CD274, clone J43) | Thermo Fisher | 25-9985-82 |
| Anti-KLRG1 (clone 2F1) | Biolegend | 138408 |
| Anti-Tim-3 (clone RMT3-23) | Biolegend | 119725 |
| Anti-Lag-3 (CD223, clone C9B7W) | Biolegend | 125227 |
| Anti-CD19 (clone 1D3) | BD | 563557 |
| Anti-F4/80 (clone T45-2342) | BD | 565614 |
| Anti-CD11c (clone N418) | BD | 744180 |
| Ghost dye live-dead in BV510 | Tonbo | 13-0870-T500 |
| Ghost dye live-dead in APCef780 | Tonbo | 13-0865-T500 |
| Anti-IFN-gamma (clone XMG1.2) | Biolegend | 505810 |
| Anti-TIGIT (clone 1G9) | Biolegend | 372706 |
| Anti-CD69 (clone H1.2F3) | Biolegend | 104506 |
| Anti-Ki67 (clone 16A8) | Biolegend | 652418 |
| Goat anti-mouse PD-L1 | R&D systems | AF1019-SP |
| Rat anti-mouse CD31 | BD | 550274 |
| Rabbit anti-mouse Lyve-1 | Novus | NB600–1008 |
| Anti-panCK-FITC (clone C-11) | Sigma-Aldrich | F3418 |
| Anti-goat AF647 | Jackson Immunoresearch | 705-605-147 |
| Anti-rat AF546 | Invitrogen | A-11081 |
| Anti-rabbit AF647 | Life Technologies | A27040 |
| Anti-F4/80 (clone BM8) | Tonbo | 4801-U100 |
| Anti-αSMA (clone 1A4) | Thermo FIsher | 14-9760-82 |
| Rabbit anti-mouse Ly6G | BD | 551459 |
| Anti-LFA-1 (clone H155–78) | Biolegend | 141008 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FTY720 | Cayman Chemical | Cat No. 10008639 |
| D-Luciferin | Gold Bio | LUCK-100 |
| Poly I:C | Sigma-Aldrich | P1530 |
| Recombinant mouse IFNγ | R&D systems | 485-MI-100 |
| Click beetle red luciferase derived peptides | Genscript | N/A |
| Streptavidin R-PE | Invitrogen | S866 |
| Red blood cell lysis buffer | GIBCO | A1049201 |
| DAPI Prolong Gold | Life Technologies | P36931 |
| Amphotericin B | GIBCO | 15290018 |
| Penicillin/streptomycin | GIBCO | 10378016 |
| Dextrose | Fisher Scientific | D15–500 |
| L-glutamine | Thermo Fisher | 25030081 |
| Blasticidin | Thermo Fisher | A1113903 |
| Puromycin | Thermo Fisher | A1113803 |
| EDTA | Thermo Fisher | 15575020 |
| Collagenase IV | Sigma Aldrich | V900893 |
| NEAA | GIBCO | 11-140-050 |
| Betamercaptoethanol | GIBCO | M6250 |
| D-PBS (no phenol red) | GIBCO | 14040133 |
| Golgiplug | BD | 555029 |
| OCT Tissue-Tek | Electron Microscopy Sciences | 6255001 |
| Acetone | Sigma Aldrich | V800023 |
| Critical Commercial Assays | ||
| BD fixation kit | BD | 554714 |
| Foxp3 fixation and permeabilization kit | Tonbo | TNB-0607-KIT |
| Effectene kit | QIAGEN | 301425 |
| QIAgen RNeasy kit | QIAGEN | 74104 |
| SYBR Green master mix | Thermo Fisher | 4344463 |
| High Capacity RNA to cDNA kit | Thermo Fisher | 438706 |
| Experimental Models: Cell Lines | ||
| KPC PDA cells and clones | This paper | N/A |
| Plat-E cells | CELL BIOLABS | RV-101 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J KPC mice | Fred Hutchinson Cancer Research Center | Stromnes et al., 2015 |
| C57BL/6J mice | Jackson laboratories | 000664 |
| Oligonucleotides (primers) | ||
| Msln R 5’ TGG ACC TTG TGA ACG AGA TTC 3’ | This paper | IDT |
| Msln F 5’ TGG ATC AGG GAC TCA GGA TAG 3’ | This paper | IDT |
| H2-Db R 5’ GGT GAC TTC ACC TTT AGA TCT GGG 3’ | Lv, D. et. al., 2015 | IDT |
| H2-Db F 5’ AGT GGT GCT GCA GAG CAT TAC AA 3’ | Lv, D. et. al., 2015 | IDT |
| H2-Kb R 5’ GGT GAC TTT ATC TTC AGG TCT GCT 3’ | This paper | IDT |
| H2-Kb F 5’ GCT GGT GAA GCA GAG AGA CTC AG 3’ | This paper | IDT |
| Tap1 R 5’ AGT TCA GGC AGA AGT TGG AAG 3’ | This paper | IDT |
| Tap1 F 5’ GCC CAG GTA CAG AAT TCC C 3’ | This paper | IDT |
| Lmp2 R 5’ GGT TAT GTG GAC GCA GCT TA 3’ | This paper | IDT |
| Lmp2 F 5’ GGT GAC CAG GTA GAT GAC AC 3’ | This paper | IDT |
| Lmp7 R 5’ GCT GCT TTC CAA CAT GAT GC 3’ | This paper | IDT |
| Lmp7 F 5’ CCG AGT CCC ATT GTC ATC TAC 3’ | This paper | IDT |
| ATP5b R 5’ CAA TGC AGG AAA GGA TCA CCA 3’ | This paper | IDT |
| ATP5b F 5’ CAT CCA AAT GGG CAA AGG TG 3’ | This paper | IDT |
| Recombinant DNA | ||
| CBR-GFP retroviral vector | Stromnes et al., 2015; Smith et al., 2017 | N/A |
| GFP-only retroviral vector | This report | N/A |
| Ovalbumin-GFP retroviral vector | This report | N/A |
| Software and Algorithms | ||
| NetMHC version 4.0 | http://cbs.dtu.dk/services/NetMHC/ | N/A |
| SYFPEITHI | http://www.syfpeithi.de/bin/MHCServer.dll/EdiptopePrediction.htm | N/A |
| BIMAS | https://bimas.cit.nih/gov/ | N/A |
| FlowJo version 10 | FlowJo | N/A |
| FACS Diva | BD | N/A |
| GraphPad | Prism | N/A |
| IVIS100 with Living Image Software | Xenogen | N/A |
| Imaris9.1.0 | Bitplane | N/A |
| Vevo 2100 | Visual Sonics Fujifilm | N/A |
| Real Touch CFX96 PCR detection system | BioRad | N/A |
| Other | ||
| DMEM | GIBCO | 11960051 |
| FCS | GIBCO | 26140079 |
| Cell counting beads | Sigma | P7458 |
| Magnetic beads conjugated to PE | Miltenyi | 130-048-801 |
| Matrigel | Discovery Labware | 08-774-552 |
| 4.0 Sutures | Ethicon | 1611G |
| 5.0 Sutures | Ethicon | 8580H |
| Magnetic beads conjugated to APC | Miltenyi | 130-090-855 |
| LS Columns | Miltenyi | 130-042-401 |
| QuadroMACS separator | Miltenyi | 130-090-076 |
| CB101–109-H-2Db tetramer | This report | N/A |
| SIINFEKL-H-2K-b tetramer - APC | http://tetramer.yerkes.emory.edu/reagents/class-I-mhc | N/A |
Highlights.
A peptide:MHC tetramer is created to study pancreas cancer-specific T cells
PD-L1 blockade expands peripheral T cells required for antitumor efficacy
Tumor escape variants defective in IFNγ-inducible Tap1 and MHC class I emerge
PD-1 + PD-L1 blockade enhances T cell functionality and longevity and promotes cure
ACKNOWLEDGMENTS
We thank Dr. Pamela Rosato for help generating the CB tetramer. We thank the NIH Tetramer Core for generously supplying us with fluorescently labeled Ova257-264 :H-2Kb tetramer. We thank Dr. Jason Mitchell at the Center for Immunology (CFI) for initial help with microscopy. We thank colleagues at the CFI at the University of Minnesota for helpful discussions. A.L.B. is supported by a computational training award from the American Association of Immunologists. E.J.S. is supported in part by an Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship. I.W. is part of the Wayzata High Schools Honors Mentor Connection Program in Minnesota. M.O. is supported by the University of Minnesota Undergraduate Research Opportunities Program (UROP). I.M.S. is supported by an American Association for Cancer Research (AACR) Pancreatic Cancer Action Network Career Development Award (17-20-25-STRO), an Institutional Research Grant (124166-IRG-58-001-55-IRG65) from the American Cancer Society, and support from the Masonic Cancer Center (University of Minnesota Medical School).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.059.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, and Kedl RM (2004). Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med 199, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, and Davis MM (1996). Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96. [DOI] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. ; Australian Pancreatic Cancer Genome Initiative (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. [DOI] [PubMed] [Google Scholar]
- Balachandran VP, quksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. ; Australian Pancreatic Cancer Genome Initiative; Garvan Institute of Medical Research; Prince of Wales Hospital; Royal North Shore Hospital; University of Glasgow; St Vincent’s Hospital; QIMR Berghofer Medical Research Institute; University of Melbourne, Centre for Cancer Research; University of Queensland, Institute for Molecular Bioscience; Bankstown Hospital; Liverpool Hospital; Royal Prince Alfred Hospital, Chris O’Brien Lifehouse; Westmead Hospital; Fremantle Hospital; St John of God Healthcare; Royal Adelaide Hospital; Flinders Medical Centre; Envoi Pathology; Princess Alexandria Hospital; Austin Hospital; Johns Hopkins Medical Institutes; ARC-Net Centre for Applied Research on Cancer (2017). Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone G, Turletti A, Artusio E, Mareschi K, Carbone A, Tibaudi D, Robecchi A, Emanuelli G, and Rodeck U (1999). Tumor-associated transforming growth factor-beta and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am. J. Pathol 155, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. ; Australian Pancreatic Cancer Genome Initiative (2012). Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchini BR, Southworth TL, Fontaine DM, Kohrt D, Welcome FS, Florentine CM, Henricks ER, DeBartolo DB, Michelini E, Cevenini L, et al. (2017). Red-emitting chimeric firefly luciferase for in vivo imaging in low ATP cellular environments. Anal. Biochem 534, 36–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai MG, Kim-Fuchs C, Angst E, and Sloan EK (2013). Bioluminescent orthotopic model of pancreatic cancer progression. J. Vis. Exp (76) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaravalli M, Reni M, and O’Reilly EM (2017). Pancreatic ductal adenocarcinoma: State-of-the-art 2017 and new therapeutic strategies. Cancer Treat. Rev 60, 32–43. [DOI] [PubMed] [Google Scholar]
- Chun J, and Hartung HP (2010). Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol 33, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, Drerup JM, Padron A, Conejo-Garcia J, Murthy K, et al. (2016). Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 76, 6964–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic bio-markers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, and Bar-Sagi D (2019). BTK signaling drives CD1dhiCD5+ regulatory B-cell differentiation to promote pancreatic carcinogenesis. Oncogene 38, 3316–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia M, Harbst K, van Buuren M, Kvistborg P, Lindberg MF, Andersen R, Idorn M, Munir Ahmad S, Ellebæk E, Mueller A, et al. (2017). Acquired immune resistance follows complete tumor regression without loss of target antigens or IFNγ signaling. Cancer Res. 77, 4562–4566. [DOI] [PubMed] [Google Scholar]
- Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, Bajor DL, Byrne KT, Stanger BZ, Riley JL, et al. (2016). Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 1, 88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley K, Kim V, Jaffee E, and Zheng L (2016). Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 381, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, and van Hall T (2016). The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol 39, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, et al. (2017). Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 7, 1420–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, et al. (2016). Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med 22, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MP, Woodroofe CC, Wood MG, Que I, Van’t Root M, Ridwan Y, Shi C, Kirkland TA, Encell LP, Wood KV, et al. (2018). Click beetle luciferase mutant and near infrared naphthyl-luciferins for improved bioluminescence imaging. Nat. Commun 9, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, Kanai M, Mori Y, Matsumoto S, Chikuma S, et al. (2015). Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol 33, 4015–4022. [DOI] [PubMed] [Google Scholar]
- Hartley GP, Chow L, Ammons DT, Wheat WH, and Dow SW (2018). Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol. Res 6, 1260–1273. [DOI] [PubMed] [Google Scholar]
- He XP, Song FJ, Liu XY, Wang Z, Li XX, Liu FY, Chen G, and Jiang WP (2013). The relationship between KRAS gene mutations and HLA class I antigen downregulation in the metastasis of non-small cell lung cancer. J. Int. Med. Res 41, 1473–1483. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Ho PC, and Kaech SM (2017). Reenergizing T cell anti-tumor immunity by harnessing immunometabolic checkpoints and machineries. Curr. Opin. Immunol 46, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZI, Hellmann MD, Wolchok JD, Vyas M, Shia J, Stadler ZK, Diaz LA Jr., and O’Reilly EM (2018a). Acquired resistance to immunotherapy in MMR-D pancreatic cancer. J. Immunother. Cancer 6, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZI, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, Lowery MA, Diaz LA Jr., Mandelker D, Yu KH, et al. (2018b). Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: challenges and recommendations. Clin. Cancer Res 24, 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lieskovan S, and Ribas A (2017). New combination strategies using programmed cell death 1/programmed cell death ligand 1 checkpoint inhibitors as a backbone. Cancer J. 23, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudrisier D, Mazarguil H, Laval F, Oldstone MB, and Gairin JE (1996). Binding of viral antigens to major histocompatibility complex class I H-2Db molecules is controlled by dominant negative elements at peptide non-anchor residues. Implications for peptide selection and presentation. J. Biol. Chem 271, 17829–17836. [DOI] [PubMed] [Google Scholar]
- Jenkins MK, and Moon JJ (2012). The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol 188, 4135–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MM, Jørgensen JT, Binderup T, and Kjaer A (2008). Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Med. Imaging 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. (2016). Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med 22, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA 3rd, Yarchoan M, Lee V, Laheru DA, and Jaffee EM (2017). Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res 23, 1656–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, Wool-man S, Vithayathil T, Glickman LH, Ndubaku CO, et al. (2018). Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 3, 122857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarquist J, Chitrakar A, Pennock ND, Kilgore AM, Blain T, Zheng C, Danhorn T, Walton K, Jiang L, Sun J, et al. (2018). Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci. Immunol 3, eaas9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotturi MF, Scott I, Wolfe T, Peters B, Sidney J, Cheroutre H, von Herrath MG, Buchmeier MJ, Grey H, and Sette A (2008). Naive precursor frequencies and MHC binding rather than the degree of epitope diversity shape CD8+ T cell immunodominance. J. Immunol 181, 2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. (2015). PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, et al. (2018). Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49, 178–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling A, Löfgren-Burström A, Larsson P, Li X, Wikberg ML,Öberg Å, Stenling R, Edin S, and Palmqvist R (2017). TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. OncoImmunology 6, e1356143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane LM, Abdel-Hakeem MS, and Wherry EJ (2019). CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, and Jenkins MK (2007). Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, and Ahmed R (1998). Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8, 177–187. [DOI] [PubMed] [Google Scholar]
- Obar JJ, Khanna KM, and Lefrançois L (2008). Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity 28, 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, et al. (2019). Tissue-resident memory CD8+ T cells promote melanoma-immune equilibrium in skin. Nature 565, 366–371. [DOI] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, Valente WJ, Koelle SJ, Church CD, Vandeven N, et al. (2018). Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat. Commun 9, 3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penaranda C, Tang Q, Ruddle NH, and Bluestone JA (2010). Prevention of diabetes by FTY720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes 59, 1461–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittet MJ, Valmori D, Dunbar PR, Speiser DE, Liénard D, Lejeune F, Fleischhauer K, Cerundolo V, Cerottini JC, and Romero P (1999). High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med 190, 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, and Rosenberg SA (1996). Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst 88, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, and Rosenberg SA (2010). Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother 33, 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, et al. (2016). Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J, Neumann-Haefelin C, Altay T, Gostick E, Price DA, Lohmann V, Blum HE, and Thimme R (2011). Immunodominance of HLA-A2-restricted hepatitis C virus-specific CD8+ T cell responses is linked to naive-precursor frequency. J. Virol 85, 5232–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et al. (2016). The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganova I, Moroz E, Cohen I, Moroz M, Mane M, Zurita J, Shenker L, Ponomarev V, and Blasberg R (2016). Enhancement of PSMA-directed CAR adoptive immunotherapy by PD-1/PD-L1 blockade. Mol. Ther. Oncolytics 4, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibaud V, Meyer N, Lamant L, Vigarios E, Mazieres J, and Delord JP (2016). Dermatologic complications of anti-PD-1/PD-L1 immune checkpoint antibodies. Curr. Opin. Oncol 28, 254–263. [DOI] [PubMed] [Google Scholar]
- Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, Pillarisetty VG, Pillai SPS, Wittrup KD, and Stephan MT (2017). Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest 127, 2176–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas C, Gombos A, Latifyan S, Piccart-Gebhart M, Kok M, and Buisseret L (2017). Targeting immune checkpoints in breast cancer: an update of early results. ESMO Open 2, e000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. (2016). CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 29, 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, and Masopust D (2015). Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, and Hingorani SR (2014). Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 63, 1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, Dotson AM, Tan X, Hotes JL, Greenberg PD, and Hingorani SR (2015). T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 28, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, and Hingorani SR (2017). T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol. Res 5, 978–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang GH, et al. (2018). Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 8, 1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ur Rahman S, Stanton M, Casey PG, Spagnuolo A, Bensi G, Hill C, Francis KP, Tangney M, and Gahan CGM (2017). Development of a click beetle luciferase reporter system for enhanced bioluminescence imaging of Listeria monocytogenes: analysis in cell culture and murine infection models. Front. Microbiol 8, 1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, and Speleman F (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide RH (2018). The immune revolution: a case for priming, not checkpoint. Cancer Cell 33, 563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, and Gajewski TF (2017). The EGR2 targets LAG-3 and 4–1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J. Exp. Med 214, 381–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, and Vonderheide RH (2015). Induction of T cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol. Res 3, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med 24, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze large datasets/code.