

Abstract
Neurofibromatosis Type 1 (NF1) is an autosomal dominant tumor-predisposition disorder that is caused by a heterozygous loss of function variant in the NF1 gene, which encodes a protein called neurofibromin. The absence of neurofibromin causes increased activity in the Rat sarcoma protein (RAS) signalling pathway, which results in an increased growth and cell proliferation. As a result, both oncological and non-oncological comorbidities contribute to a high morbidity and mortality in these patients. Optic pathways gliomas, plexiform neurofibromas and malignant peripheral nerve sheath tumor (MPNST) are the most frequent NF1-associated tumors. The treatment of these complications is often challenging, since surgery may not be feasible due to the location, size, and infiltrative nature of these tumors, and standard chemotherapy or radiotherapy are burdened by significant toxicity and risk for secondary malignancies. For these reasons, following the novel discoveries of the pathophysiological mechanisms that lead to cell proliferation and tumorigenesis in NF1 patients, emerging drugs targeting specific signalling pathways (i.e. the MEK/ERK cascade), have been developed with promising results. (www.actabiomedica.it)
Keywords: NF1, Malignant Peripheral Nerve Sheath Tumor, MPNST, Optic Pathway Glioma, Plexiform Neurofibroma, Selumetinib, Mtor Inhibitors
Background
Neurofibromatosis Type 1 (NF1) is an autosomal dominant tumor-predisposition disorder that is caused by a heterozygous loss of function variant in the tumor suppressor gene NF1. The average global prevalence is 33/100,000 individuals, varying among different countries from 12.8/100,000 in Russia to 104/100,000 in Israel (1–3).
NF1 was first described as a multisystemic disease by Friedrich Von Recklinghausen, in 1882. Nearly one century later, the National Institution of Health (NIH) Consensus Development Conference identified the diagnostic criteria (1987) (Table 1), which are still in use nowadays (4,5). The clinical hallmarks of NF1 are highly heterogeneous, and encompass non-malignant and malignant features. The former comprise pigmentary abnormalities (multiple café-au-lait macules, axillary and inguinal freckling, Lisch nodules), neurofibromas, skeletal deformities, hypertension and neurocognitive deficits. Risk of cancer in NF1 patients is 2 to 5 times higher than in the general population (6,7). Malignancies can develop within or without the nervous system. Nervous system tumours include: optic pathway and brainstem glioma, glioblastoma, and malignant peripheral nerve sheath tumor (MPNST). Patients with NF1 also show an increased risk of tumours developing outside the nervous system, like gastrointestinal stromal tumor (GIST), breast cancer, leukaemia, phaeochromocytoma, duodenal carcinoid and rhabdomyosarcoma (8). Altogether, these clinical manifestations heavily affect life expectancy, which is in average 8-21 years shorter compared to the general population (6,9,10).
Table 1.
International Diagnostic criteria for Neurofibromatosis type 1 (4)
| NIH Consensus Development Conference Diagnostic Criteria for NF1 | |
| The diagnostic criteria for NF1 are met in an individual if two or more of the following are found: | 1. 6 cafe au lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals |
| 2. 2 neurofibromas of any type or one plexiform neurofibromas | |
| 3. Freckling in the axillary or inguinal regions | |
| 4. Optic pathways glioma | |
| 5. ≥ 2 Lisch nodules | |
| 6. A distinctive osseous lesion such as sphenoid dysplasia or thinning of long bone cortex, with or without pseudarthrosis | |
| 7. A first-degree relative (parent, sibling, or offspring) with NF-1 by the above criteria | |
Resective surgery is the first line therapeutic option for most of the NF1-associated oncological complications. However, satisfactory results are not always achieved due to local extension and invasion of vital areas, tumor size, and risk of postoperative regrowth. On the other hand, the use of chemotherapy and radiotherapy is limited by high toxicity rates in NF1 patients. Furthermore, chemo- and radiotherapy should be strictly reserved to highly selected patients, when other therapeutic options (including watchful waiting) are not possible, and discussed with both patients and caregivers for the significant risk of developing secondary dysplasias and tumors later in life, due to the intrinsic tumor-predisposition of this syndrome. For these reasons, in the era of precision medicine, novel targeted therapies are highly demanded. In this review, we will briefly summarize the recent advances in the pathophysiological understanding of NF1-associated tumors and the available evidence for new emerging drugs.
Genetics and pathophysiology of NF1
The NF1 gene is located on chromosome 17q11.2 and encodes a 250 kDa cytoplasmatic protein called neurofibromin. About half of the cases are sporadic and due to de novo mutations. The germline mutation rate of NF1 is some 10-fold higher than that observed for most other inherited disease genes. Currently, over 2600 different inherited mutations in NF1 have been reported in the Human Gene Mutation Database (HGMD®) as a cause of NF1 (11–16).
Neurofibromin is a large multi-domain protein that acts as tumor suppressor. Neurofibromin includes a guanosine triphosphatase (GTPase)–activating protein (GAP) domain. GAP stimulates a GTPase activity intrinsic to RAS to inactivate the signal transduction pathway by converting RAS–guanosine triphosphate (GTP) to RAS–guanosine diphosphate (GDP) (17). This negative regulation of RAS reduces cell proliferation and differentiation by forestalling activation of the downstream signalling pathways phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) and rapidly accelerated fibrosarcoma/mitogen activated protein kinase /extracellular signal regulated kinase (RAF/MEK/ERK) (7,18–21). Neurofibromin also regulates adenylyl cyclase and lowers the levels of intracellular cyclic adenosine monophosphate (cAMP) via RAS-dependent activation of atypical protein kinase C zeta (22). This protein is widely expressed in different organs and tissues, with high levels in the nervous system, and especially in neurons, astrocytes, oligodendrocytes, microglia, and Schwann cells (23,24).
From mutational analysis, the majority of germline NF1 mutations are predicted to be inactivating, resulting in almost complete absence of transcript or protein (25). Pathogenic mutations have been identified in most of its 61 exons, and include complete gene deletions, gene-disrupting chromosome rearrangements, smaller deletion or insertions, nonsense mutations, amino acid substitutions and splicing mutations (26). As a result, loss of neurofibromin expression leads to increased RAS activity and cell growth (27,28).
Some manifestations associated with NF1, such as cognitive problems, result from haploinsufficiency of NF1. Other clinical features require an additional somatic mutation, resulting in biallelic NF1 inactivation, as seen in the development of café-au-lait macules (CALMs), neurofibromas, GIST, glomus tumors, juvenile myelomonocytic leukemia (JMML), bone dysplasia and pheochromocytoma (25). Furthermore, mouse models of MPNST have shown that biallelic inactivation of the NF1 gene may not be sufficient for tumour formation and that additional genetic alterations such as mutation of TP53, CDKN2A or SUZ12, are required for the progression of MPNST (7,29,30).
Clinical evolution of the oncological complications in NF1
Most of the signs and symptoms of NF1 develop progressively from childhood to adulthood, and are rarely seen at birth. About 46% of the patients with sporadic NF1 do not meet the diagnostic criteria by the age of 1-year. When NF1 is suspected, annual monitoring until late childhood is necessary because 97% of the children with at least one feature of NF1 will eventually meet the diagnostic criteria by the age of eight (31). Skeletal deformities are frequently detected during infancy, while CALMs and axillary/inguinal freckling usually appear in childhood, and other typical signs and symptoms, including neurofibromas and lish nodules, only develop after puberty (Figure 1). Of note, also cognitive impairment and learning, memory, or attention deficits, are diagnosed lately during childhood (32–37). Early diagnosis is thus crucial to appropriately manage the neurocognitive and psycho-social issues and to reduce morbidity and mortality with preventive and therapeutic strategies.
Figure 1.
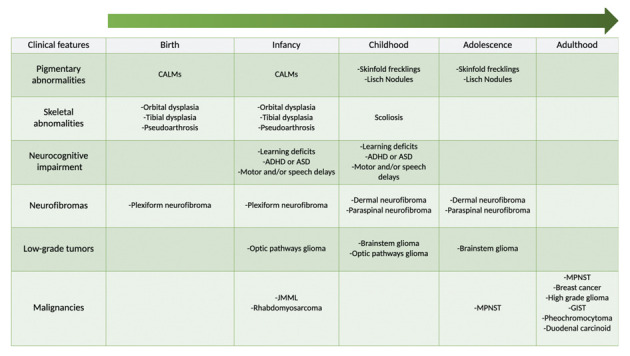
Clinical evolution in patients with neurofibromatosis type 1 (NF1).
ADHD: attention deficit/hyperactivity disorder; ASD: autism spectrum disorders; CALMS: café-au-lait macules; GIST: gastroIntestinal stromal tumors; JMML: juvenile myelomonocytic leukemia; MPNST: malignant peripheral nerve sheath tumour.
Development and severity of clinical features of NF1 can vary between individuals, but usually follow a common timeline. Café-au-lait spots can be detected early during infancy, while skinfold freckling develops later in childhood. Cognitive dysfunction has a high impact on NF1 children, since school-age children with NF1 have higher rates of developmental delay and cognitive impairment than their pairs, and many of them carry a concurrent diagnosis of attention deficit/hyperactivity disorder. Moreover, one-third of all children with NF1 have a mild to severe autism spectrum disorder. Typical signs and symptoms of NF1 as neurofibromas and Lisch nodules usually develop only after puberty (5). Plexiform neurofibromas are detected on clinical examination in approximately 27% of individuals with NF1. However, these tumors do not always cause symptoms and may be clinically silent, especially when they reside deep within the body (5). About 15-20% of the patients will develop a glioma. Patients with NF1 are also at risk to develop other malignancies in adulthood, like gastrointestinal stromal tumors, pheochromocytoma, duodenal carcinoid, high grade glioma and breast cancer.
Low grade tumors
1. Glioma
About 15-20% of children with NF1 will develop a glioma, with a median age at diagnosis of 4.9 years. Optic pathways gliomas (OPGs) are pylocitic astrocitomas arising from the optic nerve, they can be unilateral or bilateral, and are the most frequent form (66%) of NF1-related gliomas (38,39).
OPGs can involve every part of the optic nerve from the papilla to the optic radiations, with different symptoms according to the location. NF1-related OPGs are usually asymptomatic, slowly growing and non-aggressive. However, symptoms of tumor progression may include decreased visual acuity, abnormal pupillary function, decreased colour vision, optic nerve atrophy, proptosis or other complications due to compression of the surrounding structures (i.e. between 12 and 40% children with chiasmal OPG develop precocious puberty) (39,40). Postchiasmatic OPGs presenting before the age of 2 years or after the age of 8 years tend to be more aggressive and should therefore be carefully followed up. Although the 5-year overall survival for patients with low grade glioma is 85%, progression-free survival for those with unresectable/residual disease requiring treatment is significantly lower (40%) (41).
The second most frequent CNS tumor in NF1 patients is brainstem glioma, which represent about 17% of all tumors in children with NF1 (42). Other gliomas are rarer, typically develop later in adulthood, and can involve all areas of the brain (37,43–48).
2. Neurofibroma
Neurofibromas are benign peripheral nerve sheath tumors composed of neoplastic Schwann cells, fibroblast, blood vessels and mast cells. According to their location, they can be divided in four types: cutaneous, subcutaneous, spinal and plexiform. Cutaneous and subcutaneous neurofibromas develop during childhood or early adolescence. They are benign tumors, and have no malignant potential (8). Spinal neurofibromas develop from the spinal foramina and can cause nerve roots compression or spinal deformities (i.e. scoliosis, kyphoscoliosis and vertebral body anomalies). When symptomatic, they can cause both motor and sensitive neuropathy and should be surgically treated (8,49). Plexiform neurofibromas (PNF) are benign nerve sheath tumors that can be found in 30-50% of NF1 patients (50). They are mostly congenital, and arise from the deep peripheral nervous plexuses. As all neurofibromas, they are slowly growing, but can often become large and bulky, developing in complex and infiltrative shapes (51). PNF can occur anywhere throughout the body, but particularly in extremities, thoracic and pelvic region, and tend to surround and invade nearby tissues and structures (i.e. bones) causing pain, disfigurement, neurologic impairment and motor dysfunction (51,52). Furthermore, PNF have a 8-15% lifetime risk of malignant transformation in MPNST (53–55). Therefore, surgery should be considered early and all patients should undergo a careful presurgical evaluations, especially when PNF become symptomatic. Unfortunately surgical outcomes are often dissatisfactory, especially when only partial resection is attainable (56), with post-operative re-growth rates that can reach 44% (57).
Malignancies
1. Malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumors (MPNST) are rare, biologically aggressive soft tissue sarcomas derived from Schwann cells or pluripotent cells of the neural crest. About 22-50% of all cases are associated with NF1. The median age at diagnosis is between 20 and 40 years (10-20 years earlier compared to the sporadic cases). MPNST usually arise from a pre-existing PNF. MPNST most commonly develop in the limbs (45%), the trunk (34%) and the head or neck (19%) (51,58). The clinical presentation is usually characterized by a rapid enlargement causing mass effect and neuropathic symptoms, such as paraesthesia, motor weakness or radicular pain. The prognosis for NF1-related MPNST is poor, with a 50% of early metastatic involvement at diagnosis (mainly in the lungs) and a 5-year overall survival of 35-50% (59–61). As for other soft-tissue sarcomas, the best curative option is complete surgical resection, which is often not feasible due to location, size, and presence of metastasis. Furthermore, relapse rate is high and there is a lack of alternative therapeutic options (59). Adjuvant radiotherapy might be used to reduce local recurrence but needs a thorough risk-benefit evaluation for the heightened risk of secondary malignancies (62). Standard chemotherapy remains a treatment option in locally advanced or metastatic MPNST patients. It usually includes a combination of doxorubicin, ifosfamide, and etoposide, but response rate is usually poor compared to sporadic MPNST (17.9% vs 44.4%) (63).
2. Gastrointestinal Stromal tumor
Gastrointestinal stromal tumour (GIST) is a mesenchymal tumour that primarily arises in the gut mucosal wall. Unlike sporadic GISTs, those associated with NF1 usually lack somatic mutations of CD117 (c-KIT) or PDGFR-A (platelet-derived growth factor receptor A) (64,65). Instead, biallelic inactivation of the NF1 gene results in constitutive RAS activation, increasing the downstream mitogenic signalling through the MAP kinase cascade. Interestingly, gain-of-function mutations of c-KIT also activate many downstream signalling pathways including the RAS–MAP kinase cascade, suggesting a common pathogenetic mechanism in both sporadic and NF1-associated GISTs. As other tumors, NF1-associated GISTs have unique clinical features, compared to sporadic forms: they occur in younger patients (mean age at presentation 52.8 years), are multiple (60%) or develop in multiples sites, are smaller in size and with low mitotic activity, and occur mostly in the duodenum or small bowel. They are usually asymptomatic and incidentally detected during routine investigations. Surgery is the only modality that can offer a permanent cure of GIST, and complete surgical resection avoiding tumor rupture and injuries to the pseudocapsule is the initial treatment for primary and localized GISTs when the risk of morbidity and death from surgery is acceptable. The aims of surgery include complete resection with macroscopic and microscopic negative margins and functional preservation by wedge resection, when applicable. Unfortunately NF1 related GISTs show a variable but generally incomplete response to the tyrosine kinase inhibitor Imatinib treatment (64–69).
3. Pheochromocytoma
Pheochromocytomas are neuroendocrine catecholamine-secreting tumors, and occur in 2-2.9% of patients with NF1. Median age at presentation is 43 years (range 14–61 years) (70). This tumor is usually solitary, benign and localized in the adrenal glands, bilateral in 17% of the cases and metastatic or recurrent in 7.3%. Adrenalectomy remains the primary treatment of pheochromocytoma, with the entire gland being surgically removed in order to achieve cure. No differences have been described in the treatment and outcome of NF1-related pheochromocytoma compared to sporadic or other genetically determined forms of phaeochromocytoma (i.e. Multiple Endocrine Neoplasia type 2, Von Hippel Lindau syndrome, Hereditary paraganglioma–pheochromocytoma syndrome, Carney’s triad) (70–75).
4. Breast Cancer
Although rare in patients with NF-1, few studies have shown that women with NF1 are at a higher risk of developing early onset breast cancer with aggressive behaviour and a poorer prognosis, compared to the general population. Cancer management is not well defined in this population, these lesions are usually treated with a combination of surgery, chemotherapy, and radiation in relation to the stage at diagnosis, although risks of secondary fibrosarcomas may be increased by radiotherapy in this vulnerable population group (76–79).
5. Duodenal carcinoid
Carcinoid tumors of the gastrointestinal tract are neuroendocrine tumors (NETs). Most of the cases of carcinoids are sporadic, but approximately 26% of all carcinoid tumors occur in patients with NF1, with the most common site being the periampullary region. Mean age at presentation is 47.9 years, with a 59% female preponderance (80). Clinical symptoms are multiple, and vary depending on the tumor size, compression and dissemination. The most common presenting symptoms are jaundice (65%) and abdominal pain (31%). Biologically, the most common type of peri-ampullary NET in NF1 patients is somatostatinoma (40%). Surgical treatment is recommended: pancreaticoduodenectomy is the first choice approach for well-differentiated ampullary carcinoid >2 cm and for ampullary neuroendocrine carcinomas, while local tumor excision can be considered for carcinoids <2 cm. In patients who are not eligible for surgery, chemotherapy may be considered. Options for management of grade I and II tumors include octreotide, lanreotide, mTOR inhibitors (everolimus), and peptide-receptor radiotherapy (80–84).
6. Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is the most frequent soft-tissue sarcoma in children, and can be distinguished in alveolar and embryonal subtypes. Less than 1% of patient with NF1 develop RMS, and all have a embryonal histology (due to the known role of RAS activation in the pathogenesis of embryonal-type RMS). The median age at diagnosis is 2.9 years, significantly earlier compared to sporadic RMS (5 years). Frequent locations are pelvic and orbital. These patients tend to develop early non-metastatic RMS, most often in the pelvic sites, that appear to be genetically similar to sporadic cases. Complete resection is the best curative option and treatment does not differ from sporadic cases (85–90).
7. Juvenile myelomonocytic leukaemia
Juvenile myelomonocytic leukaemia (JMML) is a unique, aggressive hematopoietic disorder of infancy/early childhood caused by excessive proliferation of cells of monocytic and granulocytic lineages. Although JMML is an uncommon complication of NF1, it is estimated that patient with NF1 have a 200-350 fold increased risk of developing JMML, compared to the general population. Moreover, this association may be underestimated because patients with JMML may die at an age at which children do not manifest sufficient clinical signs to make the diagnosis of NF1. Allogeneic hematopoietic stem cell transplantation remains the therapy of choice for most patients with JMML, and should be recommended to any child with NF1-mutated JMML (91–93).
Emerging treatments for NF1-related tumors
Standard chemotherapy regimens are weighed by the toll of toxic effect that sometimes may lead to a discontinuation of therapy. Precision medicine is an approach that takes account for the characteristics of NF1 related tumors. Below an analysis of the current standard therapy and new, emerging drug for glioma, plexiform neurofibroma and malignant peripheral nerve sheath tumors. Table 2 illustrates novel target therapies that has been used or are currently under investigation.
Figure 2.

Signaling pathways and drug targets.
AC: Adenyl cyclase; cAMP: Cyclic adenosine monophosphate; AKT: Protein kinase B; GPCR: G-protein coupled receptor; GTP: Guanosine Triphosphate; MEK: Mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; mTOR: Mammalian target of rapamycine; PI3K: Phosphoinositide 3-kinase; RAF: Rapidly Accelerated Fibrosarcoma; RAS: Rat Sarcoma protein; R-TK: Receptor Tirosyne Kinase
1. Glioma
Despite the behaviour of this tumor is usually not aggressive, specific treatment might be necessary in case of tumor progression and clinical symptoms. The mainstay treatment is chemotherapy. Indications for radiation therapy and surgery are less frequent in NF1-associated gliomas. On one hand, radiotherapy it’s not recommended because of the heightened risk of secondary tumors and moyamoya syndrome (94-95). On the other hand, most of the times this tumors are not surgically approachable for a complete resection, although a palliative debulking might be needed under specific circumstances (e.g. vision loss, corneal exposure due to proptosis, or pituitary localization) (95,96). Carboplatin and vincristine are the recommended first line chemotherapy for OPG (97-98), and the treatment protocol should always be handled by a specialist oncologist. Second line drugs include vinblastine, vinorelbine and temozolomide (99-100). Other options combine TPCV (thioguanine, procarbazine, lomustine, and vincristine) and weekly vinblastine (98). Recently, a phase II study of bevacizumab plus irinotecan was conducted in children with recurrent low-grade glioma, NF-1 related or not, to measure sustained response and/or stable disease lasting ≥6 months and progression-free survival, the results of that study show that this therapeutic strategy could be useful (101).
All cited regimens seem to be effective but classic chemotherapy exposes children to toxic effects such as myelosuppression, allergic reactions, peripheral neuropathy, constipation, secondary malignancies, and infertility. Although effective, radiotherapy increases the risk of secondary malignancy, ototoxicity, endocrinopathies, and neurocognitive decline (102,103).
Among new emerging drugs, Selumetinib has shown promising results in the treatment of NF1-associated OPG. Selumetinib is an oral selective inhibitor of MEK 1 and 2. This inhibitor locks MEK1/2 into an inactive conformation that enables the binding of ATP and substrate but disrupts both the molecular interactions required for catalysis and the proper access to the ERK activation loop (104). First evidences of efficacy for selective MEK inhibition came from mouse models of NF1-deficient acute myeloid leukaemia, where it induced tumor regression (105-106). In 2017, the Pediatric Brain Tumor Consortium completed a phase I trial of Selumetinib in 38 children with recurrent, refractory, or progressive paediatric low-grade glioma, establishing the recommended phase II dose as 25 mg/m2 twice daily. Five of 25 patients treated at the recommended phase 2 dose achieved a partial response (41). Simultaneously, in a phase I trial, 17/24 (71%) patients with NF1-associated PNF showed partial response after treatment with Selumetinib (107). Both trials showed tolerable toxicities and equal recommended treatment doses. A recent phase II multicentre trial (108) with Selumetinib has shown at least a partial response (≥50% tumour reduction on MRI) in 40% of the patients with NF1-associated low grade glioma. These preliminary results suggest a comparable efficacy to conventional chemotherapy, with a higher tolerability, manageability and safety profile (109).
2. Plexiform neurofibroma
At present, the only curative option for PNF is resective surgery, and it should therefore be considered as soon as possible, whenever applicable. However, due to their infiltrative nature, eventually involving vital structures, and tendency for regrowth, surgery might not always be performed. Unfortunately, as a matter of fact, the medical treatment of PNF hasn’t found its keystone yet. As for many NF1-associated malignancies, radiotherapy is not recommended because of the risk of secondary malignancies (including radiation-induced MPNST, which typically have an even worse prognosis).Similarly, chemotherapeutic agents are not used because of their mutagenic nature and all drugs that have been used until now have shown little evidence of efficacy (56,62,110).
Among alternative treatments, interferon (INF) therapy has been reported in various studies (111,112) as an effective tumor-stabilizer. Jakacki and colleagues (111) eventually reported a 15-20% volume decrease in 29% of the patient. INF is safe and tolerable, and may be useful to reduce neuropathic pain. For this reason, a therapeutic trial of at least 6 months might be recommended, even if it will rarely be resolutive. The efficacy of Thalidomide (113) is less clear, as in a single study on 12 patients it showed a minor response in only 33%.
Since neurofibromin controls cell growth by negatively regulating the mTOR pathway activity, it seems reasonable to use mTOR inhibitors to manage NF1-related tumors (18). Sirolimus is a safe and well tolerated mTOR inhibitor that has been used to lengthen time to progression with fair success (mean increased time to progression: 4 months), but unfortunately failed in achieving a significant response in tumor shrinking or pain relief (114).
Sunitinib malate is a powerful, highly selective Tyrosine Kinase receptor inhibitor with activity against c-Kit, PDGFR, and vascular endothelial growth factor receptor (VEGFR), which are all implicated in the pathogenesis of MPNSTs. Preclinical studies showed that Sunitinib can induce reduction in PNF number and size, decreased mast cell infiltration, diminished fibroblast collagen deposition, and reduced metabolic activity (115). A phase II trial with Sunitinib was prematurely terminated because one patient died for uncertain (but possibly drug-related) causes (NCT01402817).
Meanwhile, other protein kinase inhibitors have undergone clinical trials for the treatment of PNF. A phase I trial (116) with Sorafenib, a protein kinase inhibitor with activity against RAF, PDGFRb, c-KIT and VEGFR-2, showed scarce tolerability at substantially lower doses than the MTD, in children with refractory PNF. On the contrary, Pirfenidone, an oral anti-fibrotic and anti-inflammatory agent, demonstrated good tolerability in a phase II study (117), although it did not demonstrate clinical effectiveness and was not warranted further evaluation in children with progressive PN. Similarly, Tipirfanib, which selectively inhibits HRAS, did not offer significant efficacy compared to placebo (118,119). Imatinib Mesylate, a tyrosine kinase inhibitor with antineoplastic activity, targets c-KIT ligands secreted by biallelic NF1-inacrivated Schwann cells and is able to decrease the volume of PNF in mouse models (120). A phase II trial with Imatinib reported a 17% response with a ≥20% tumor reduction, although a few study limitations (i.e. relatively small sample size and significant heterogeneity of the selected population) may have underestimated its therapeutic effect (120).
Among emerging drugs for NF1, so far Selumetinib seems the most promising for the treatment of PNF. In a recent clinical trial on 24 patients with PNF (107), 71% showed partial tumor regression after a median follow up of 18 months, which is significantly high if compared to the response rates of imatinib (17%) (120) and interferon-alpha-2b (29%) (111). Moreover, all patients showed evidence of some degree of tumor reduction, with a response that remained stable without disease progression in 15/17. The most frequent toxic effects involved mainly the skin and the gastrointestinal tract, with a side-effect profile similar to adults (121), or an asymptomatic increase of the creatin kinase (107). Very recently, a phase II trial with Selumetinib in 50 children with inoperable PNF evidenced a 74% rate of partial response (defined as a ≥20% volume decrease), with a stable response in 56% after approximately one year (12 therapy cycles). In this study only a few children showed disease progression, and most of them (5/6) had experienced a dose reduction before progression. Notably, in addition to tumor shrinkage, 68% experienced improvements in neurofibroma-related complications such as pain or functional limitations. Toxic effects were similar to those evidenced in phase I, and always reversible. Taken together, these results identify Selumetinib as the most promising drug for the treatment of PNF, since its high tolerability and low toxicity profile may allow early prolonged treatments (122).
Finally, there are several ongoing trials with selective tyrosine kinase inhibitors like Nilotinib (NCT01275586), Trametinib (NCT03363217) (123), or Cabozantinib (NCT02101736), and mTOR pathway inhibitors like Everolimus (NCT01365468).
3. MPNST
The recent understandings in the pathogenesis of MPNST have led to the development of preclinical mouse models for the study of targeted agents and precision medicine. Unfortunately, most of these trials have been inconclusive, but several other are still ongoing. In a recent phase I/II study (124) Sirolimus was used in combination with Ganetespib, a novel injectable small molecule inhibitor of Hsp90, to treat MPNST. Despite the promising preclinical rationale and tolerability of the combination therapy, no significant responses were observed. Alike, several other mechanisms of actions are currently under investigation. These include the use of small molecules, like PLX3397 (an inhibitor of CSF1 and KIT) used in combination with mTor pathway inhibitors (NCT02584647) (125), or modified BET inhibitors to overcome resistance in MPNST (126). Knowing that many MPNST arise from previous PNF, however, the best approach would be to prevent malignant degeneration in high risk patients. In the future, the identification of risk factors, early biomarkers and eventually disease modifying drugs (like the promising Selumetinib) may radically change the natural history of these aggressive tumors.
Neurofibromin 1 (NF1) accelerates the conversion from active Guanosine Triphosphate bound RAS to inactive Guanosine Diphosphate bound RAS. RAS signalling transduces extracellular signals from ligand-activated receptors (Receptor Tirosyne Kinase and G-protein coupled receptor). Loss of neurofibromin results in elevated RAS signalling. GTP-RAS activates a multitude of effectors protein, including the RAF and the MEK/ERK signalling cascades, which promote proliferation, and the PI3K/mTOR pathway, which promotes growth and cell survival.
Conflict of Interest:
Each author declares that he or she has no commercial associations (e.g. consultancies, stock ownership, equity interest, patent/licensing arrangement etc.) that might pose a conflict of interest in connection with the submitted article
References
- 1.Williams VC, Lucas J, Babcock MA, Gutmann DH, Bruce B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123(1):124–133. doi: 10.1542/peds.2007-3204. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- 2.Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141(1):71–74. doi: 10.1001/archderm.141.1.71. doi: 10.1001/archderm.141.1.71. [DOI] [PubMed] [Google Scholar]
- 3.Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: A total population study in Finland. J Invest Dermatol. 2015;135(3):904–906. doi: 10.1038/jid.2014.465. doi: 10.1038/jid.2014.465. [DOI] [PubMed] [Google Scholar]
- 4.NIH National Institutes of Health. Consensus Development Conference Neurofibromatosis: Conference Statement. Arch Neurol. 1988;45:575–78. [PubMed] [Google Scholar]
- 5.Cimino PJ, Gutmann DH. Neurofibromatosis type 1. Handb Clin Neurol. 2018;148:799–811. doi: 10.1016/B978-0-444-64076-5.00051-X. doi: 10.1016/B978-0-444-64076-5.00051-X. [DOI] [PubMed] [Google Scholar]
- 6.Duong TA, Sbidian E, Valeyrie-Allanore L, et al. Mortality associated with neurofibromatosis 1: A cohort study of 1895 patients in 1980-2006 in France. Orphanet J Rare Dis. 2011;6(1) doi: 10.1186/1750-1172-6-18. doi: 10.1186/1750-1172-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Prim. 2017;3:1–18. doi: 10.1038/nrdp.2017.4. doi: 10.1038/nrdp.2017.4. [DOI] [PubMed] [Google Scholar]
- 8.Hirbe AC, Gutmann DH. Neurofibromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014;13(8):834–843. doi: 10.1016/S1474-4422(14)70063-8. doi: 10.1016/S1474-4422(14)70063-8. [DOI] [PubMed] [Google Scholar]
- 9.Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: An analysis using U.S. death certificates. Am J Hum Genet. 2001;68(5):1110–1118. doi: 10.1086/320121. doi: 10.1086/320121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frances Hannan, Ivan Ho, James Jiayuan Tong, Yinghua Zhu PN and YZ. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum Mol Genet 2006. 2006;1(15):1087–1098. doi: 10.1093/hmg/ddl023. doi: 10.1038/jid.2014.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics. 2017;11(1) doi: 10.1186/s40246-017-0109-3. doi: 10.1186/s40246-017-0109-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am J Med Genet Part A. 2010;152(2):327–332. doi: 10.1002/ajmg.a.33139. doi: 10.1002/ajmg.a.33139. [DOI] [PubMed] [Google Scholar]
- 13.Rad E, Tee AR. Neurofibromatosis type 1: Fundamental insights into cell signalling and cancer. Semin Cell Dev Biol. 2016;52:39–46. doi: 10.1016/j.semcdb.2016.02.007. doi: 10.1016/j.semcdb.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Ballester R, Marchuk D, Boguski M, et al. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63(4):851–859. doi: 10.1016/0092-8674(90)90151-4. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 15.Martin GA, Viskoohil D, Bollag G, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63(4):843–849. doi: 10.1016/0092-8674(90)90150-d. doi: 10.1016/0092-8674(90)90150-D. [DOI] [PubMed] [Google Scholar]
- 16.Ars E, Kruyer H, Morell M, et al. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet. 2003;40(6) doi: 10.1136/jmg.40.6.e82. doi: 10.1136/jmg.40.6.e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu G, O’Connell P, Viskochil D, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62(3):599–608. doi: 10.1016/0092-8674(90)90024-9. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 18.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 Tumor Suppressor Critically Regulates TSC2 and MTOR. 2005 doi: 10.1073/pnas.0503224102. www.pnas.orgcgidoi10.1073pnas.0503224102 . Accessed March 23, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic Analysis Reveals Hyperactivation of the Mammalian Target of Rapamycin Pathway in Neurofibromatosis 1-Associated Human and Mouse Brain Tumors. 2005 doi: 10.1158/0008-5472.CAN-04-4058. http://prospector.ucsf.edu/ucsfhtml4.0/msfit.htm . Accessed March 23, 2020. [DOI] [PubMed] [Google Scholar]
- 20.Karajannis MA, Ferner RE. Neurofibromatosis-related tumors: Emerging biology and therapies. Curr Opin Pediatr. 2015;27(1):26–33. doi: 10.1097/MOP.0000000000000169. doi: 10.1097/MOP.0000000000000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulte A, Ewald F, Spyra M, et al. Combined targeting of AKT and mTOR inhibits proliferation of human NF1-Associated malignant peripheral nerve sheath tumour cells in vitro but not in a xenograft mouse model in vivo. Int J Mol Sci. 2020;21(4) doi: 10.3390/ijms21041548. doi: 10.3390/ijms21041548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anastasaki C, Gutmann DH. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum Mol Genet. 2014;23(25):6712–6721. doi: 10.1093/hmg/ddu389. doi: 10.1093/hmg/ddu389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Declue JE, Cohen BD, Lowy DR, Lerner AB. Identification and Characterization of the Neurofibromatosis Type 1 Protein Product Communicated By. 1991;88 doi: 10.1073/pnas.88.22.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutmann DH, Wood DL, Collins FS. Identification of the Neurofibromatosis Type 1 Gene Product (Protein/Antibodies/GTPase-Activating Protein. 1991;88 doi: 10.1073/pnas.88.21.9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James A, Walkera MU. Emerging therapeutic targets for neurofibromatosis type 1 (NF1) Expert Opin Ther Targets. 2018;22(5):419–437. doi: 10.1080/14728222.2018.1465931. doi: 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amy Theos, MD, Bruce R, Korf MD P. Clinical. Pathophysiology of Neurofibromatosis Type 1. Physiol Med A Ser Artic Link Med WITH Sci. 2006:842–849. doi: 10.7326/0003-4819-144-11-200606060-00010. [DOI] [PubMed] [Google Scholar]
- 27.Tanya N. Basu, David H, Gutmannt Jonathan A, Fletcer Thomas W. Glover FSC & JD. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:715–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 28.Gideon Bollag, D, Wade Clapp, Shane Shih, Felix Adler, You Yan Zhang, Patricia Thompson, Beverly J, Lange Melvin H Freedman Frank McCormick TJ & KS. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nature. 1996 doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 29.Verdijk RM, Den Bakker MA, Dubbink HJ, Hop WCJ, Dinjens WNM, Kros JM. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J Neuropathol Exp Neurol. 2010;69(1):16–26. doi: 10.1097/NEN.0b013e3181c55d55. doi: 10.1097/NEN.0b013e3181c55d55. [DOI] [PubMed] [Google Scholar]
- 30.Korfhage J, Lombard DB. Malignant peripheral nerve sheath tumors: From epigenome to bedside. Mol Cancer Res. 2019;17(7):1417–1428. doi: 10.1158/1541-7786.MCR-19-0147. doi: 10.1158/1541-7786.MCR-19-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3):608–614. doi: 10.1542/peds.105.3.608. doi: 10.1542/peds.105.3.608. [DOI] [PubMed] [Google Scholar]
- 32.Parisi P, Vanacore N, Belcastro V, et al. Clinical guidelines in pediatric headache: evaluation of quality using the AGREE II instrument. J Headache Pain. 2014;15(1):57. doi: 10.1186/1129-2377-15-57. doi: 10.1186/1129-2377-15-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foiadelli T, Piccorossi A, Sacchi L, et al. Clinical characteristics of headache in Italian adolescents aged 11-16 years: a cross-sectional questionnaire school-based study. doi: 10.1186/s13052-018-0486-9. doi: 10.1186/s13052-018-0486-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garone G, Reale A, Vanacore N, et al. Acute ataxia in paediatric emergency departments: A multicentre Italian study. Arch Dis Child. 2019;104(8):768–774. doi: 10.1136/archdischild-2018-315487. doi: 10.1136/archdischild-2018-315487. [DOI] [PubMed] [Google Scholar]
- 35.Obringer AC, Meadows AT, Zackai EH. The Diagnosis of Neurofibromatosis-1 in the Child Under the Age of 6 Years. Am J Dis Child. 1989;143(6):717–719. doi: 10.1001/archpedi.1989.02150180099028. doi: 10.1001/archpedi.1989.02150180099028. [DOI] [PubMed] [Google Scholar]
- 36.D.H G, A A, J.C C, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. J Am Med Assoc. 1997;278(1):51–57. http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=emed4&NEWS=N&AN=1997195390 . [PubMed] [Google Scholar]
- 37.Gutmann DH, Parada LF, Silva AJ, Ratner N. Neurofibromatosis type 1: Modeling CNS dysfunction. J Neurosci. 2012;32(41):14087–14093. doi: 10.1523/JNEUROSCI.3242-12.2012. doi: 10.1523/JNEUROSCI.3242-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bornhorst M, Frappaz D, Packer RJ. Pilocytic astrocytomas. Handb Clin Neurol. 2016;134:329–344. doi: 10.1016/B978-0-12-802997-8.00020-7. doi: 10.1016/B978-0-12-802997-8.00020-7. [DOI] [PubMed] [Google Scholar]
- 39.Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: The effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr. 1995;127(5):718–722. doi: 10.1016/s0022-3476(95)70159-1. doi: 10.1016/S0022-3476(95)70159-1. [DOI] [PubMed] [Google Scholar]
- 40.Guillamo JS, Créange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain. 2003;126(1):152–160. doi: 10.1093/brain/awg016. doi: 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- 41.Banerjee A, Jakacki RI, Onar-Thomas A, et al. A phase i trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 2017;19(8):1135–1144. doi: 10.1093/neuonc/now282. doi: 10.1093/neuonc/now282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peltonen S, Kallionpää RA, Rantanen M, et al. Pediatric malignancies in neurofibromatosis type 1: A population-based cohort study. Int J Cancer. 2019;145(11):2926–2932. doi: 10.1002/ijc.32187. doi: 10.1002/ijc.32187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cynthia J, Campen MD1, David H, Gutmann MD P. Optic Pathway Gliomas in Neurofibromatosis Type 1. J Child Neurol. 2018;33(1):73–81. doi: 10.1177/0883073817739509. doi: 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeid JL, Charrow J, Sandu M, Goldman S, Listernick R. Orbital optic nerve gliomas in children with neurofibromatosis type 1. J AAPOS. 2006;10(6):534–539. doi: 10.1016/j.jaapos.2006.03.014. doi: 10.1016/j.jaapos.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 45.Balcer LJ, Liu GT, Heller G, et al. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: Relation to tumor location by magnetic resonance imaging. Am J Ophthalmol. 2001;131(4):442–445. doi: 10.1016/s0002-9394(00)00852-7. doi: 10.1016/S0002-9394(00)00852-7. [DOI] [PubMed] [Google Scholar]
- 46.Palumbo P, Lombardi F, Siragusa G, et al. Involvement of NOS2 activity on human glioma cell growth, clonogenic potential, and neurosphere generation. Int J Mol Sci. 2018;19(9) doi: 10.3390/ijms19092801. doi: 10.3390/ijms19092801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palumbo P, Lombardi F, Augello FR, et al. NOS2 inhibitor 1400W induces autophagic flux and influences extracellular vesicle profile in human glioblastoma U87MG cell line. Int J Mol Sci. 2019;20(12) doi: 10.3390/ijms20123010. doi: 10.3390/ijms20123010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raysi Dehcordi S, Ricci A, Di Vitantonio H, et al. Stemness Marker Detection in the Periphery of Glioblastoma and Ability of Glioblastoma to Generate Glioma Stem Cells: Clinical Correlations. World Neurosurg. 2017;105:895–905. doi: 10.1016/j.wneu.2017.05.099. doi: 10.1016/j.wneu.2017.05.099. [DOI] [PubMed] [Google Scholar]
- 49.Taleb FS, Guha A, Arnold PM, Fehlings MG, Massicotte EM. Surgical management of cervical spine manifestations of neurofibromatosis Type 1: Long-term clinical and radiological follow-up in 22 cases. J Neurosurg Spine. 2011;14(3):356–366. doi: 10.3171/2010.9.SPINE09242. doi: 10.3171/2010.9.SPINE09242. [DOI] [PubMed] [Google Scholar]
- 50.Mautner V-F, Asuagbor FA, Dombi E, et al. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro Oncol. 2008;10(4):593–598. doi: 10.1215/15228517-2008-011. doi: 10.1215/15228517-2008-011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen R, Kluwe L, Fuensterer C, Kentsch M, Friedrich RE, Mautner VF. Plexiform neurofibromas in children with neurofibromatosis type 1: Frequency and associated clinical deficits. J Pediatr. 2011;159(4) doi: 10.1016/j.jpeds.2011.04.008. doi: 10.1016/j.jpeds.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 52.Pascual-Castroviejo I, Lopez-Pereira P, Savasta S, Lopez-Gutierrez JC, Lago CM, Cisternino M. Neurofibromatosis type 1 with external genitalia involvement. Presentation of 4 patients. J Pediatr Surg. 2008;43(11):1998–2003. doi: 10.1016/j.jpedsurg.2008.01.074. doi: 10.1016/j.jpedsurg.2008.01.074. [DOI] [PubMed] [Google Scholar]
- 53.Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis. J Med Genet. 2002;39(5):311–314. doi: 10.1136/jmg.39.5.311. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uusitalo E, Rantanen M, Kallionpää RA, et al. Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol. 2016;34(17):1978–1986. doi: 10.1200/JCO.2015.65.3576. doi: 10.1200/JCO.2015.65.3576. [DOI] [PubMed] [Google Scholar]
- 55.Higham CS, Dombi E, Rogiers A, et al. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro Oncol. 2018;20(6):818–825. doi: 10.1093/neuonc/noy013. doi: 10.1093/neuonc/noy013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Packer RJ, Rosser T. Therapy for plexiform neurofibromas in children with neurofibromatosis 1: An overview. J Child Neurol. 2002;17(8):638–641. doi: 10.1177/088307380201700816. doi: 10.1177/088307380201700816. [DOI] [PubMed] [Google Scholar]
- 57.Needle MN, Cnaan A, Dattilo J, et al. Prognostic signs in the surgical management of plexiform neurofibroma: The children’s hospital of Philadelphia experience, 1974-1994. J Pediatr. 1997;131(5):678–682. doi: 10.1016/s0022-3476(97)70092-1. doi: 10.1016/S0022-3476(97)70092-1. [DOI] [PubMed] [Google Scholar]
- 58.Stucky CCH, Johnson KN, Gray RJ, et al. Malignant Peripheral Nerve Sheath Tumors (MPNST): The Mayo Clinic experience. Ann Surg Oncol. 2012;19(3):878–885. doi: 10.1245/s10434-011-1978-7. doi: 10.1245/s10434-011-1978-7. [DOI] [PubMed] [Google Scholar]
- 59.Khu KJ, Midha R. Malignant Peripheral Nerve Sheath Tumors. World Neurosurg. 2016;94:566–567. doi: 10.1016/j.wneu.2016.07.054. doi: 10.1016/j.wneu.2016.07.054. [DOI] [PubMed] [Google Scholar]
- 60.Valentin T, Le Cesne A, Ray-Coquard I, et al. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO) Eur J Cancer. 2016;56:77–84. doi: 10.1016/j.ejca.2015.12.015. doi: 10.1016/j.ejca.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 61.Widemann BC. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr Oncol Rep. 2009;11(4):322–328. doi: 10.1007/s11912-009-0045-z. doi: 10.1007/s11912-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamanaka R, Hayano A. Radiation-Induced Malignant Peripheral Nerve Sheath Tumors: A Systematic Review. World Neurosurg. 2017;105:961–970.e8. doi: 10.1016/j.wneu.2017.06.010. doi: 10.1016/j.wneu.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 63.Higham CS, Steinberg SM, Dombi E, et al. Clinical Study SARC006: Phase II Trial of Chemotherapy in Sporadic and Neurofibromatosis Type 1 Associated Chemotherapy-Naive Malignant Peripheral Nerve Sheath Tumors. Artic ID. 2017. 2017 doi: 10.1155/2017/8685638. doi: 10.1155/2017/8685638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agaimy A, Vassos N, Croner RS. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): Clinicopathological spectrum with pathogenetic considerations. Int J Clin Exp Pathol. 2012;5(9):852–862. [PMC free article] [PubMed] [Google Scholar]
- 65.Salvi PF, Lorenzon L, Caterino S, Antolino L, Antonelli MS, Balducci G. Gastrointestinal stromal tumors associated with neurofibromatosis 1: A single centre experience and systematic review of the literature including 252 cases. Int J Surg Oncol. 2013;2013 doi: 10.1155/2013/398570. doi: 10.1155/2013/398570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Popescu I, Andrei S. Gastrointestinal stromal tumors. Chir. 2008;103(2):155–170. doi: 10.33748/jradidn.v1i3.25. [PubMed] [Google Scholar]
- 67.Lee JL, Kim JY, Ryu MH, et al. Response to imatinib in KIT-and PDGFRA-wild type gastrointestinal stromal associated with neurofibromatosis type 1. Dig Dis Sci. 2006;51(6):1043–1046. doi: 10.1007/s10620-006-8003-1. doi: 10.1007/s10620-006-8003-1. [DOI] [PubMed] [Google Scholar]
- 68.Wang JH, Lasota J, Miettinen M. Succinate dehydrogenase subunit B (SDHB) is expressed in neurofibromatosis 1-associated gastrointestinal stromal tumors (Gists): Implications for the SDHB expression based classification of Gists. J Cancer. 2011;2(1):90–93. doi: 10.7150/jca.2.90. doi: 10.7150/jca.2.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishida T, Blay J-Y, Seiichi Hirota, Kitagawa Y, Yoon , Kang K. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer. 19 doi: 10.1007/s10120-015-0526-8. doi: 10.1007/s10120-015-0526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF, Bancos I. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin Endocrinol (Oxf) 2017;86(1):141–149. doi: 10.1111/cen.13163. doi: 10.1111/cen.13163. [DOI] [PubMed] [Google Scholar]
- 71.Santos P, Pimenta T, Taveira-Gomes A. Hereditary pheochromocytoma. Int J Surg Pathol. 2014;22(5):393–400. doi: 10.1177/1066896914537683. doi: 10.1177/1066896914537683. [DOI] [PubMed] [Google Scholar]
- 72.Lew JI, Jacome FJ, Solorzano CC. Neurofibromatosis-associated pheochromocytoma. J Am Coll Surg. 2006;202(3):550–551. doi: 10.1016/j.jamcollsurg.2005.09.028. doi: 10.1016/j.jamcollsurg.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 73.Lenders JWM, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. 2005:665–675. doi: 10.1016/S0140-6736(05)67139-5. [DOI] [PubMed] [Google Scholar]
- 74.Tsirlin A, Oo Y, Sharma R, Kansara A, Gliwa A, Banerji MA. Maturitas Pheochromocytoma: A review. 2014;77:229–238. doi: 10.1016/j.maturitas.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 75.Bausch B, Borozdin W, Neumann HPH. Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma [13] N Engl J Med. 2006;354(25):2729–2731. doi: 10.1056/NEJMc066006. doi: 10.1056/NEJMc066006. [DOI] [PubMed] [Google Scholar]
- 76.Kumar N, Pandey AN, Kumari S, Kishore S. Breast Cancer Associated with Von Recklinghausen’s Disease: Case Report and Review of Literature. Indian J Surg Oncol. 2014;5(3):205–207. doi: 10.1007/s13193-014-0327-2. doi: 10.1007/s13193-014-0327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khalil J, Afif M, Elkacemi H, Benoulaid M, Kebdani T, Benjaafar N. Breast cancer associated with neurofibromatosis type 1: A case series and review of the literature. J Med Case Rep. 2015;9(1):4–7. doi: 10.1186/s13256-015-0533-8. doi: 10.1186/s13256-015-0533-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Howell SJ, Hockenhull K, Salih Z. Evans DG. Increased risk of breast cancer in neurofibromatosis type 1: Current insights. Breast Cancer Targets Ther. 2017;9:531–536. doi: 10.2147/BCTT.S111397. doi: 10.2147/BCTT.S111397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chaudhry US, Yang L, Askeland RW, Fajardo LL. Metaplastic Breast Cancer in a Patient with Neurofibromatosis. J Clin Imaging Sci. 2015;5(1):5–8. doi: 10.4103/2156-7514.154102. doi: 10.4103/2156-7514.154102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Relles D, Baek J, Witkiewicz A, Yeo CJ. Periampullary and duodenal neoplasms in neurofibromatosis type 1: Two cases and an updated 20-year review of the literature yielding 76 cases. J Gastrointest Surg. 2010;14(6):1052–1061. doi: 10.1007/s11605-009-1123-0. doi: 10.1007/s11605-009-1123-0. [DOI] [PubMed] [Google Scholar]
- 81.Burke AP, Sobin LH, Shekitka KM, Federspiel BH, Helwig EB. Somatostatin-producing duodenal carcinoids in patients with von recklinghausen’s neurofibromatosis. A predilection for black patients. Cancer. 1990;65(7):1591–1595. doi: 10.1002/1097-0142(19900401)65:7<1591::aid-cncr2820650723>3.0.co;2-n. doi: 10.1002/1097-0142(19900401)65:7<1591::AID-CNCR2820650723>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 82.Abdessayed N, Gupta R, Mestiri S, Bdioui A, Trimech M, Mokni M. Rare triad of periampullary carcinoid, duodenal gastrointestinal stromal tumor and plexiform neurofibroma at hepatic hilum in neurofibromatosis type 1: A case report. BMC Cancer. 2017;17(1):4–8. doi: 10.1186/s12885-017-3567-z. doi: 10.1186/s12885-017-3567-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hartel M, Wente MN, Sido B, Friess H, Büchler MW. Carcinoid of the ampulla of Vater. J Gastroenterol Hepatol. 2005;20(5):676–681. doi: 10.1111/j.1440-1746.2005.03744.x. doi: 10.1111/j.1440-1746.2005.03744.x. [DOI] [PubMed] [Google Scholar]
- 84.Laird AM, Libutti SK. Management of Other Gastric and Duodenal Neuroendocrine Tumors. Surg Oncol Clin N Am. 2020;29(2):253–266. doi: 10.1016/j.soc.2019.11.009. doi: 10.1016/j.soc.2019.11.009. [DOI] [PubMed] [Google Scholar]
- 85.I M, M T, N K, T S, N N, J A. Neurofibromatosis Type 1 and Childhood Cancer. Cancer. 1993;72(9) doi: 10.1002/1097-0142(19931101)72:9<2746::aid-cncr2820720936>3.0.co;2-w. doi: 10.1002/1097-0142(19931101)72:9<2746::AID-CNCR2820720936>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 86.Sung L, Anderson JR, Arndt C, Raney RB, Meyer WH, Pappo AS. Neurofibromatosis in children with rhabdomyosarcoma: A report from the intergroup rhabdomyosarcoma study IV. J Pediatr. 2004;144(5):666–668. doi: 10.1016/j.jpeds.2004.02.026. doi: 10.1016/j.jpeds.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 87.Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4(2):216–231. doi: 10.1158/2159-8290.CD-13-0639. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Paulson V, Chandler G, Rakheja D, et al. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosom Cancer. 2011;50(6):397–408. doi: 10.1002/gcc.20864. doi: 10.1002/gcc.20864. [DOI] [PubMed] [Google Scholar]
- 89.Martinelli S, McDowell HP, Delle Vigne S, et al. RAS signaling dysregulation in human embryonal rhabdomyosarcoma. Genes Chromosom Cancer. 2009;48(11):975–982. doi: 10.1002/gcc.20702. doi: 10.1002/gcc.20702. [DOI] [PubMed] [Google Scholar]
- 90.Shukla N, Ameur N, Yilmaz I, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res. 2012;18(3):748–757. doi: 10.1158/1078-0432.CCR-11-2056. doi: 10.1158/1078-0432.CCR-11-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Paulus S, Koronowska S, Fölster-Holst R. Association Between Juvenile Myelomonocytic Leukemia, Juvenile Xanthogranulomas and Neurofibromatosis Type 1: Case Report and Review of the Literature. Pediatr Dermatol. 2017;34(2):114–118. doi: 10.1111/pde.13064. doi: 10.1111/pde.13064. [DOI] [PubMed] [Google Scholar]
- 92.Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015;125(7):1083–1090. doi: 10.1182/blood-2014-08-550483. doi: 10.1182/blood-2014-08-550483. [DOI] [PubMed] [Google Scholar]
- 93.Niemeyer CM, Flotho C. Juvenile myelomonocytic leukemia: Who’s the driver at the wheel? Blood. 2019;133(10):1060–1070. doi: 10.1182/blood-2018-11-844688. doi: 10.1182/blood-2018-11-844688. [DOI] [PubMed] [Google Scholar]
- 94.Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: Controversies and recommendations. Ann Neurol. 2007;61(3):189–198. doi: 10.1002/ana.21107. doi: 10.1002/ana.21107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nicolin G, Parkin P, Mabbott D, et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer. 2009;53(7):1231–1237. doi: 10.1002/pbc.22198. doi: 10.1002/pbc.22198. [DOI] [PubMed] [Google Scholar]
- 96.Luzzi S, Giotta Lucifero A, Del Maestro M, et al. Anterolateral Approach for Retrostyloid Superior Parapharyngeal Space Schwannomas Involving the Jugular Foramen Area: A 20-Year Experience. World Neurosurg. 2019;132:e40–e52. doi: 10.1016/j.wneu.2019.09.006. doi: 10.1016/j.wneu.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 97.Packer RJ, Lange B, Ater J, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol. 1993;11(5):850–856. doi: 10.1200/JCO.1993.11.5.850. doi: 10.1200/JCO.1993.11.5.850. [DOI] [PubMed] [Google Scholar]
- 98.Terashima K, Karger AG. Chemotherapy of Intracranial Gliomas in Children. In: Progress in Neurological Surgery. 2018;31:162–167. doi: 10.1159/000467377. doi: 10.1159/000467377. [DOI] [PubMed] [Google Scholar]
- 99.Cappellano AM, Petrilli AS, da Silva NS, et al. Single agent vinorelbine in pediatric patients with progressive optic pathway glioma. J Neurooncol. 2015;121(2):405–412. doi: 10.1007/s11060-014-1652-6. doi: 10.1007/s11060-014-1652-6. [DOI] [PubMed] [Google Scholar]
- 100.Gururangan S, Fisher MJ, Allen JC, et al. Temozolomide in Children with progressive low-grade glioma1. Neuro Oncol. 2007;9(2):161–168. doi: 10.1215/15228517-2006-030. doi: 10.1215/15228517-2006-030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gururangan S, Fangusaro J, Poussaint TY, et al. Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas-a Pediatric Brain Tumor Consortium study. doi: 10.1093/neuonc/not154. doi: 10.1093/neuonc/not154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ater JL, Zhou T, Holmes E, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: A report from the Children’s Oncology Group. J Clin Oncol. 2012;30(21):2641–2647. doi: 10.1200/JCO.2011.36.6054. doi: 10.1200/JCO.2011.36.6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chadderton RD, West CGH, Schulz S, et al. Radiotherapy in the Treatment of Low-Grade Astrocytomas I1. The Physical and Cognitive Sequelae. 1995;11 doi: 10.1007/BF00334961. Springer-Verlag. [DOI] [PubMed] [Google Scholar]
- 104.Yeh TC, Marsh V, Bernat BA, et al. Cancer Therapy: Preclinical Biological Characterization of ARRY-142886 (AZD6244), a Potent, Highly Selective Mitogen-Activated Protein Kinase Kinase 1 / 2 Inhibitor. 2007;142886(5):1576–1584. doi: 10.1158/1078-0432.CCR-06-1150. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 105.Lauchle JO, Kim D, Le DT, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461(7262):411–414. doi: 10.1038/nature08279. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jessen WJ, Miller SJ, Jousma E, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123(1):340–347. doi: 10.1172/JCI60578. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eva Dombi, M.D, Andrea Baldwin, C.P.N.P, Leigh J. Marcus, M.D, Michael J. Fisher, M.D, Brian Weiss, M.D, AeRang Kim, M.D., Ph.D, Patricia Whitcomb, R.N, Staci Martin, Ph.D, Lindsey E. Aschbacher-Smith, M.S, Tilat A. Rizvi, Ph.D, Jianqiang Wu, M. MD. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N Engl J Med. 2016;375(26):2550–2560. doi: 10.1056/NEJMoa1605943. doi: 10.1056/NEJMoa1605943.Activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fangusaro J, Onar-Thomas A, Young Poussaint T, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20(7):1011–1022. doi: 10.1016/S1470-2045(19)30277-3. doi: 10.1016/S1470-2045(19)30277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bouffet E, Jakacki R, Goldman S, et al. Phase II study of weekly vinblastine in recurrent or refractory pediatric low-grade glioma. J Clin Oncol. 2012;30(12):1358–1363. doi: 10.1200/JCO.2011.34.5843. doi: 10.1200/JCO.2011.34.5843. [DOI] [PubMed] [Google Scholar]
- 110.Avery RA, Katowitz JA, Fisher MJ, et al. Orbital/Peri-Orbital Plexiform Neurofibromas in Children with Neurofibromatosis type 1: Multi-disciplinary Recommendations for Care HHS Public Access. Ophthalmology. 2017;124(1):123–132. doi: 10.1016/j.ophtha.2016.09.020. doi: 10.1016/j.ophtha.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jakacki RI, Dombi E, Potter DM, et al. Phase i trial of pegylated interferon-α-2b in young patients with plexiform neurofibromas. Neurology. 2011;76(3):265–272. doi: 10.1212/WNL.0b013e318207b031. doi: 10.1212/WNL.0b013e318207b031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kebudi R, Cakir FB, Gorgun O. Interferon-α for unresectable progressive and symptomatic plexiform neurofibromas. J Pediatr Hematol Oncol. 2013;35(3) doi: 10.1097/MPH.0b013e318270cd24. doi: 10.1097/MPH.0b013e318270cd24. [DOI] [PubMed] [Google Scholar]
- 113.Gupta A, Cohen BH, Ruggieri P, Packer RJ, Phillips PC. Phase I study of thalidomide for the treatment of plexiform neurofibroma in neurofibromatosis 1. Neurology. 2003;60(1):130–132. doi: 10.1212/01.wnl.0000042321.94839.78. doi: 10.1212/01.WNL.0000042321.94839.78. [DOI] [PubMed] [Google Scholar]
- 114.Weiss B, Widemann BC, Wolters P, et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: a Neurofibromatosis Clinical Trials Consortium phase II study. doi: 10.1093/neuonc/nou235. doi: 10.1093/neuonc/nou235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ferguson MJ, Rhodes SD, Jiang L, et al. Preclinical Evidence for the Use of Sunitinib Malate in the Treatment of Plexiform Neurofibromas. Pediatr Blood Cancer. 2016;63(2):206–213. doi: 10.1002/pbc.25763. doi: 10.1002/pbc.25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim A, Dombi E, Tepas K, et al. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2013;60(3):396–401. doi: 10.1002/pbc.24281. doi: 10.1002/pbc.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Widemann BC, Babovic-Vuksanovic D, Dombi E, et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr Blood Cancer. 2014;61(9):1598–1602. doi: 10.1002/pbc.25041. doi: 10.1002/pbc.25041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Widemann BC, Dombi E, Gillespie A, et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. doi: 10.1093/neuonc/nou004. doi: 10.1093/neuonc/nou004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Untch BR, Dos Anjos V, Garcia-Rendueles MER, et al. Tipifarnib Inhibits HRAS-Driven dedifferentiated thyroid cancers. Cancer Res. 2018;78(16):4642–4657. doi: 10.1158/0008-5472.CAN-17-1925. doi: 10.1158/0008-5472.CAN-17-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Robertson KA, Nalepa G, Yang FC, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet Oncol. 2012;13(12):1218–1224. doi: 10.1016/S1470-2045(12)70414-X. doi: 10.1016/S1470-2045(12)70414-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Balagula Y, Huston KB, Busam KJ, Lacouture ME, Chapman PB, Myskowski PL. Dermatologic side effects associated with the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886) Invest New Drugs. 2011;29(5):1114–1121. doi: 10.1007/s10637-010-9567-3. doi: 10.1007/s10637-010-9567-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gross AM, Wolters PL, Dombi E, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382(15):1430–1442. doi: 10.1056/NEJMoa1912735. doi: 10.1056/NEJMoa1912735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Perreault S, Larouche V, Tabori U, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway: TRAM-01. BMC Cancer. 2019;19(1):1–9. doi: 10.1186/s12885-019-6442-2. doi: 10.1186/s12885-019-6442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim A, Lu Y, Okuno SH, et al. Targeting Refractory Sarcomas and Malignant Peripheral Nerve Sheath Tumors in a Phase I/II Study of Sirolimus in Combination with Ganetespib (SARC023) Sarcoma. 2020;2020 doi: 10.1155/2020/5784876. doi: 10.1155/2020/5784876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Patwardhan PP, Surriga O, Beckman MJ, et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin Cancer Res. 2014;20(12):3146–3158. doi: 10.1158/1078-0432.CCR-13-2576. doi: 10.1158/1078-0432.CCR-13-2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cooper JM, Patel AJ, Chen Z, et al. Overcoming BET inhibitor resistance in malignant peripheral nerve sheath tumors. Clin Cancer Res. 2019;25(11):3404–3416. doi: 10.1158/1078-0432.CCR-18-2437. doi: 10.1158/1078-0432.CCR-18-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]