

Abstract
Enantiomeric cyclopropavir phosphates (+)-9 and (−)-9 were synthesized and investigated as substrates for GMP kinase. N2-Isobutyryl-di-O-acetylcyclopropavir (11) was converted to (+)-monoacetate 12 using hydrolysis catalyzed by porcine liver esterase. Phosphorylation via phosphite 13 gave after deacylation, phosphate (+)-9. Acid-catalyzed tetrahydropyranylation of (+)-monoacetate 12 gave, after deacylation, tetrahydropyranyl derivative 14. Phosphorylation via phosphite 15 furnished, after deprotection, enantiomeric phosphate (−)-9. Racemic diphosphate 16 was also synthesized. The phosphate (+)-9 is a relatively good substrate for GMP kinase with a KM value of 57 μM that is similar to that of the natural substrates GMP (61 μM) and dGMP (82 μM). In contrast, the enantiomer (−)-9 is not a good substrate (KM 1200 μM) indicating a significant enantioselectivity for the GMP kinase catalyzed reaction of monophosphate to diphosphate.
Keywords: Methylenecyclopropanes, nucleoside analogues, cyclopropavir, phosphates, enantiomers, enantioselectivity, antivirals, GMP kinase
INTRODUCTION
Methylenecyclopropane analogues of nucleosides are established antiviral agents that are particularly effective against herpesviruses such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), and human herpes virus 6 and 8 (HHV-6 and HHV-8).[1] The most potent analogues against CMV are the purine Z-isomers. In the first generation group, the 2-aminopurines like synguanol (1) have gained particular prominence (Chart 1). The anti-CMV potency of synguanol and other 2-aminopurine methylenecyclopropanes is strictly enantioselective; the S-(+)-enantiomers are effective and R-(−)-enantiomers are inactive.[2,3] In the second generation series, cyclopropavir (2) has emerged as the most potent anti-CMV analogue presently in preclinical development as a therapeutic agent against human cytomegalovirus (HCMV) infections.[4–7] Mechanism of action of methylenecyclopropanes, such as 1 and 2, undoubtedly includes the phosphorylation cascade observed for other nucleoside analogues: monophosphate–diphosphate–triphosphate. In the first generation series, potent antiviral activity of lipophilic phosphorylated prodrugs[8,9] 3 as well as inhibition of HIV-1 reverse transcriptase with synadenol triphosphate[10] (4) provided strong evidence for the above activation pathway. Resistance studies have supported involvement of HCMV-encoded UL97 phosphotransferase in the first phosphorylation step of synguanol[11] (1) and cyclopropavir[12–15](2).
CHART 1.
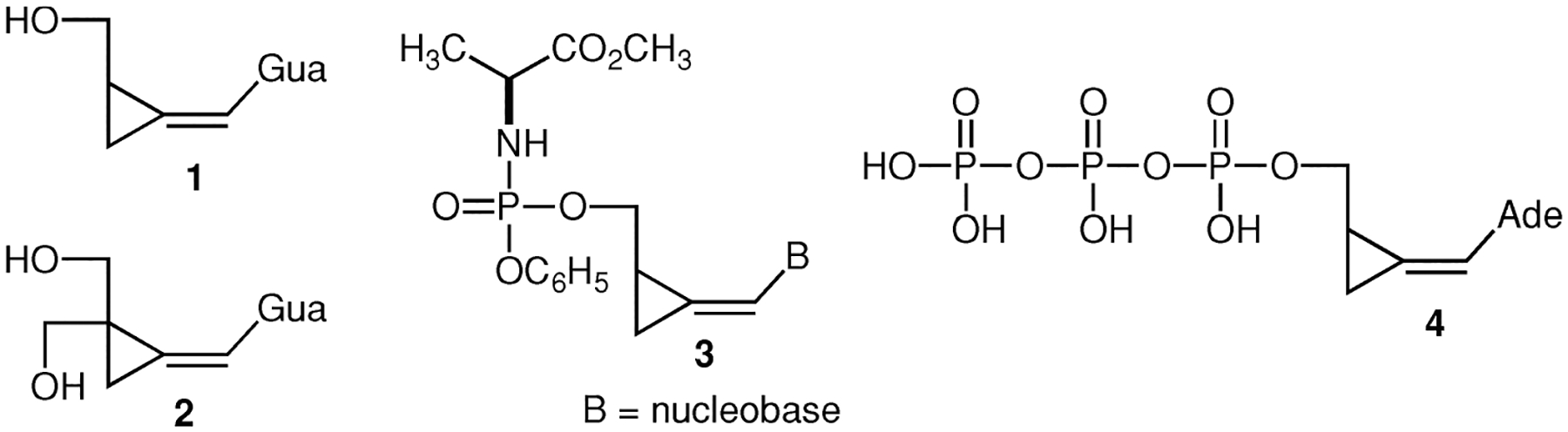
Cyclopropavir (2) can be considered a rigid analogue of ganciclovir (Cytovene, 5), an approved drug against HCMV infections (Scheme 1). Ganciclovir is also achiral but phosphorylation studies revealed that chirality is critically involved in its activation. Unlike methylenecyclopropane analogues, such as 1 and 2, ganciclovir (5) is also effective against herpes simplex 1 (HSV-1) infections. It was established that the first phosphorylation step is catalyzed by HSV-1 thymidine kinase to give the phosphates 6 in an enantioselective process.[16] It has to be stated that HCMV lacks thymidine kinase but it employs UL97 phosphotransferase[17,18] of unknown enantioselectivity in the first phosphorylation step of ganciclovir (5). It was also established that the formation of a diphosphate 7 is catalyzed by guanylate (GMP) kinase in an enantioselective process.[16] This enzyme is also involved in activation of other guanine nucleoside analogues.[19,20] In the last step, diphosphate 7 is converted to the respective triphosphate 8 by cellular kinases.
SCHEME 1.

Phosphorylation cascade of ganciclovir (5).
The purpose of this study was two-fold: (i) synthesis of enantiomeric phosphates of cyclopropavir (+)-9 and (−)-9; and (ii) to determine their substrate affinity for GMP kinase.
RESULTS AND DISCUSSION
Synthesis
Acetylation of N2-isobutyrylganciclovir catalyzed by porcine pancreatic lipase (PPL) was employed for synthesis of protected enantiomers of ganciclovir.[21] We then assumed that N2-isobutyrylcyclopropavir (10) could be used as a starting point for both enantiomeric phosphates (+)-9 and (−)-9. Therefore, cyclopropavir (2) was transformed to compound 10 in 85% yield by acylation-deacylation procedure[21] (Scheme 2). Nevertheless, under the conditions described for N2-isobutyrylganciclovir21/porcine pancreatic lipase (PPL) in pyridine- benzene/ no acetylation was observed. For this reason, N2-isobutyrylcyclopropavir (10) was transformed to diacetate 11 (84%). The PLE-catalyzed hydrolysis afforded (+)-acetate 12 in 64% yield and 95% ee. Unreacted diacetate 11 (18%) and diol 10 (16%) were also isolated. Phosphorylation of (+)-12 was performed in analogy to the corresponding racemic[22] compound rac-12 (i-But = H). Reaction of (+)-12 with diphenyl phosphite in pyridine followed by triethylamine gave phosphite 13 (83%). The latter was converted to phosphate (+)-9 by oxidation of trimethylsilyl ester intermediate with iodine in pyridine. Deprotection in 80% acetic acid followed by ammonolysis gave (+)-9 in 82% yield. Synthesis of enantiomeric phosphate (−)-9 made use of a simple interchange of the protecting groups (Scheme 3). Starting material, tetrahydropyranyl derivative 14, was synthesized in 97% yield from acetate (+)-12 by a procedure described[22] for compound 14 obtained from acetate rac-12 (i-But = H). Phosphorylation followed the procedure employed for enantiomeric phosphate (+)-9. Intermediary phosphite 15 obtained in 93% yield was oxidized and the respective intermediate was deprotected by a treatment with 80% acetic acid to give phosphate (−)-9 (69%). Tentatively, the S configuration can be proposed for phosphate (+)-9 as based on the antiviral activity of S-(+)-synguanol.[2] We also prepared racemic diphosphate 16 to confirm identity of the product of GMP kinase catalyzed phosphorylation (Scheme 4). Compound 11 was transformed to racemic acetate rac-12 (93%) by a procedure previously used[22] for unprotected racemic compound rac-12 (i-But = H). Compound rac-12 was converted to phosphite rac-13 in 80% yield as described above for enantiomeric compound 13. Oxidation followed by activation with N,N-carbonyldiimidazole[23] and reaction with inorganic phosphate followed by deacylation gave the racemic diphosphate 16 in 62% yield.
SCHEME 2.
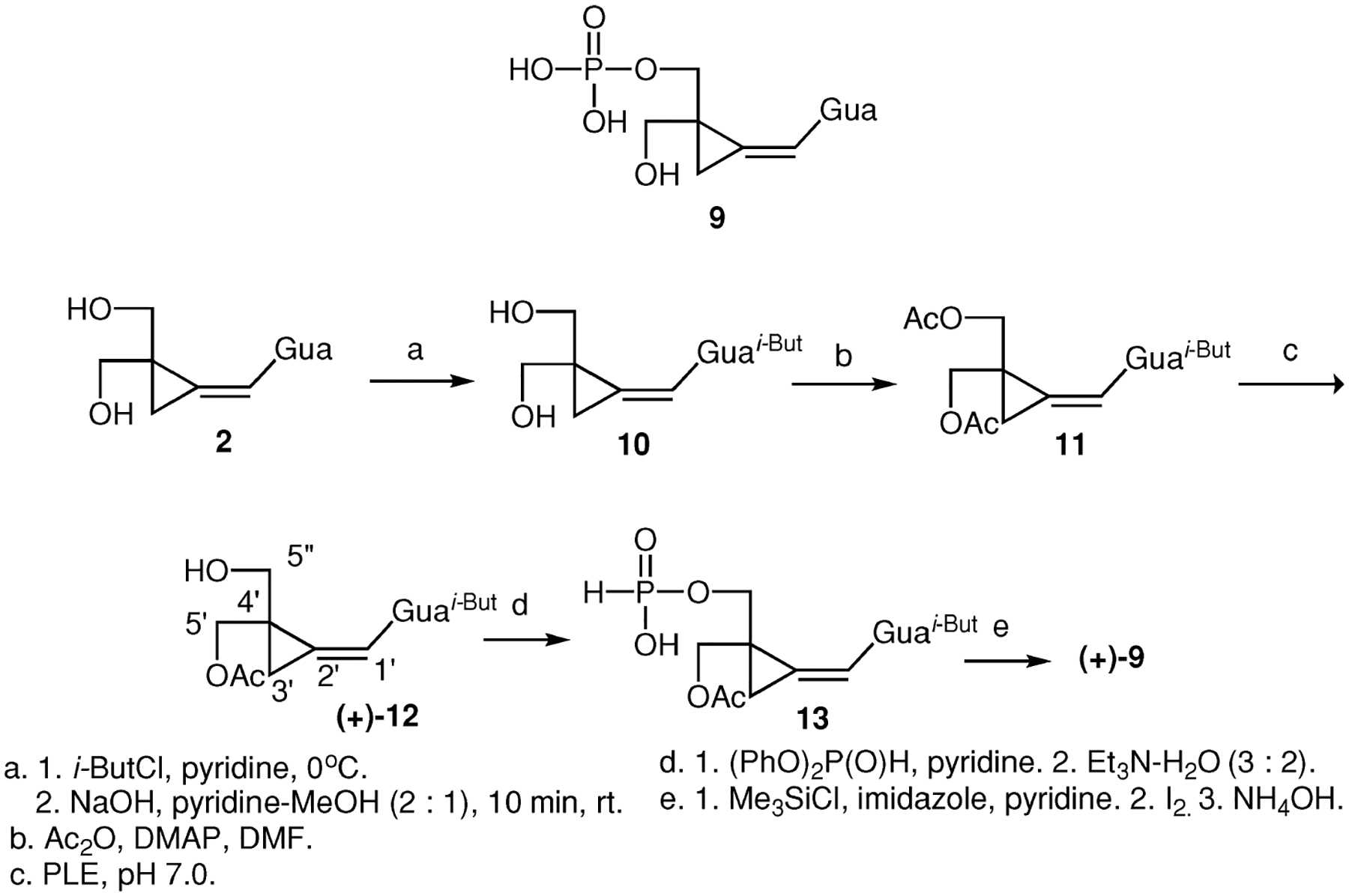
Synthesis of cyclopropavir phosphate (+)-9 (i-But, isobutyryl; THP, tetrahydropyranyl; COIm2, N,N′-carbonyldiimidazole).
SCHEME 3.

Synthesis of cyclopropavir phosphate (−)-9 (i-But, isobutyryl; THP, tetrahydropyranyl; COIm2, N,N′-carbonyldiimidazole).
SCHEME 4.

Synthesis of racemic cyclopropavir diphosphate 16 (i-But, isobutyryl; THP, tetrahydropyranyl; COIm2, N,N′-carbonyldiimidazole).
Guanylate Kinase Assays
Experiments to determine whether or not phosphate (+)-9 and/or (−)-9 are substrates for GMP kinase were performed under the conditions described[21] for enantiomeric ganciclovir phosphates 6 and ent-6 at 34°C. A time course (Figure 1) demonstrated that cyclopropavir diphosphate 16 formed from phosphate (+)-9 reached equilibrium after 3 hours with approximately 80% of diphosphate 16, whereas a maximum of only 10% of enantiomeric diphosphate 16 was formed from phosphate (−)-9 during the course of the experiment. This established a significant enantioselectivity for the reaction of (−)-9 to diphosphate 16 with GMP kinase.
FIGURE 1.

Time course of phosphorylation of enantiomeric cyclopropavir phosphates (+)-9 and (−)-9 catalyzed by GMP kinase.
Kinetic values of KM and Vmax for (+)-9 and (−)-9 were determined using the Lineweaver-Burk double reciprocal plot methodology (Table 1). A KM value of 57 μM was found for (+)-9 compared to 1200 μM for the (−)-9 enantiomer. The value for the (+)-enantiomer was similar to that found for the natural substrates GMP (61 μM) and dGMP (82 μM). Although the Vmax for (−)-9 (0.23 nmol/min) was somewhat greater than that of (+)-9 (0.16 nmol/min), the difference was not enough to make up for the relatively high KM resulting in the inability of GMP kinase to effectively phosphorylate (−)-9 to diphosphate 16 (Figure 1).
TABLE 1.
Kinetic values for enantiomeric cyclopropavir phosphates and natural substrates
| Compound | KM (μM) | Vmax (nmol/min) | Vmax/KM |
|---|---|---|---|
| Phosphate (+)-9 | 57 | 0.16 | 2.8 × 10−3 |
| Phosphate (−)-9 | 1200 | 0.23 | 1.9 × 10−4 |
| GMP | 61 | 0.33 | 5.4 × 10−3 |
| dGMP | 82 | 0.54 | 6.6 × 10−3 |
EXPERIMENTAL SECTION
General Methods
The NMR spectra were determined at 300 or 400 MHz (1H), 75 or 100 MHz (13C) and 121 or 162 MHz (31P) in DMSO-d6, UV spectra were measured in ethanol and mass spectra were run in electrospray ionization (ESI) mode (methanol-NaOAc) unless stated otherwise. High performance liquid chromatography (HPLC) was performed on Hamilton PRP-1 column, 150 × 4.1 mm, 10 μ (Hamilton Co., Reno, NV, USA; column 1) and Brownlee Anion Aquapore AX-300 column, 250 × 4.6 mm, 10 μ (Anspec, Inc., Ann Arbor, MI, USA; column 2). Chiral HPLC was run on Chirobiotic T column, 250 × 4.6 mm, 5 μ (Advanced Separation Technologies, Inc., Whippany, NJ, USA) unless stated otherwise.
Starting Materials and Enzymes
Cyclopropavir (2) was obtained from Microbiotix, Inc. (Worcester, MA, USA). Porcine liver esterase (PLE), phosphoenol pyruvate, lactate dehydrogenase, pyruvate kinase, bovine serum albumin (BSA), and bovine guanylate (GMP) kinase were products of Sigma-Aldrich Corp. (St. Louis, MO, USA).
(Z)-9-{[2,2-Bis(hydroxymethyl)cyclopropylidene]methyl}-N2-isobutyrylguanine (10):
Isobutyryl chloride (0.5 mL, 4.5 mmol) was added dropwise to a suspension of cyclopropavir (2, 220 mg, 0.84 mmol) in pyridine (20 mL) at 0°C. The mixture was stirred at room temperature for 16 h. The reaction was quenched with methanol (5 mL) and the solvents were evaporated. The residue was dissolved in (pyridine-methanol (2: 1, 30 mL) and the pH was carefully adjusted to 12.5 with 2M NaOH and the mixture was kept at room temperature for 10 minutes. The pH was then adjusted to 7.0 with 2M HCl and the solvents were evaporated. The residue was chromatographed on a silica gel column using dichloromethane-methanol (10 : 1) to give compound 10 (236 mg, 85%) as a white solid, m.p. 214–216°C. UV λmax 294 nm (ε 11,900), 240 nm (ε 26,800). 1H NMR δ 12.08, 11.70 (2bs, 2H, NH), 8.68 (s, 1H, H8), 7.16 (s, 1H, H1′), 5.02 (2 overlapped t, 2H, OH), 3.67, 3.49 and 3.66, 3.48 (2AB, 4H, J = 11.2 and 11.0 Hz, H5′), 2.75 (m, 1H, CH of isobutyryl), 1.35 (s, 2H, H3′), 1.10 (d, 6H, J = 7.2 Hz, CH3). 13C NMR 180.8 (CO), 155.5, 149.0, 147.7, 137.3, 120.4, 120.2 (purine, C2′), 110.6 (C1′), 62.7 (C5′), 35.4 (CH of isobutyryl), 31.5 (C4′), 19.5 (CH3), 11.7 (C3′). ESI-MS 334 (M + H, 6.0), 356 (100.0, M + Na), 689 (42.0, 2M + Na). Anal. Calcd for C15H19N5O4 × 2 H2O: C, 48.78; H, H, 6.28; N, 18.96. Found: C, 48.84; H, 6.22; N, 18.75.
(Z)-9-{[2,2-Bis(acetoxymethyl)cyclopropylidene]methyl}-N2-isobutyrylguanine (11):
A mixture of isobutyryl derivative 10 (200 mg, 0.6 mmol), 4-(N-dimethylamino)pyridine (DMAP, 10 mg, 0.08 mmol) in DMF (20 mL) and acetic anhydride (0.4 mL, 4.2 mmol) was stirred for 1 hour at room temperature. Solvent was evaporated and the crude product was chromatographed on a silica gel column using dichloromethane-methanol (50 : 1 to 25 : 1) to give diacetate 11 (202 mg, 84%) as a white solid, m.p. 124–126°C. UV λmax 292 nm (ε 12,600), 236 nm (ε 26,100). 1H NMR δ 12.10, 11.69 (2bs, 2H, NH), 8.22 (s, 1H, H8), 7.25 (s, 1H, H1′), 4.34, 4.07 (AB, 4H, J = 11.6 Hz, H5′), 2.75 (m, 1H, CH of isobutyryl), 1.95 (s, 6H, CH3 of acetate), 1.69 (s, 2H, H3′), 1.10 (d, J = 6.5 Hz, 6H, CH3 of isobutyryl). 13C NMR 180.9, 170.7 (CO), 155.4, 149.2, 148.1, 137.0, 120.5 (purine), 118.6 (C2′), 112.6 (C1′), 66.0 (C5′), 35.5 (CH of isobutyryl), 25.3 (C4′), 21.1 (CH3 of acetate), 19.5 (CH3 of isobutyryl), 13.4 (C3′). ESI-MS 418 (M + H, 3.6), 440 (100.0, M + Na). Anal. Calcd for C19H23N5O6 × 1.2 H2O: C, 51.98; H, 5.79; N, 15.95. Found: C, 51.91; H, 5.74; N, 15.82.
(+)-(R,Z)-9-{[2-(Acetoxymethyl)-2-(hydroxymethyl)cyclopropylidene]methyl}-N2-isobutyrylguanine (12):
A mixture of diacetate 11 (100 mg, 0.25 mmol) and porcine liver esterase (PLE, 210 mg, 8,610 units) in DMF (15 mL) and 0.02 M Na2HPO4 (pH 7.0, 100 mL) was stirred for 45 minutes at room temperature, then lyophilized. The residue was sonicated with dichloromethane-methanol (3 : 1, 3 × 100 mL) and the insoluble portion was filtered off. The solvents were evaporated and the crude product was chromatographed on a silica gel column using dichloromethane-methanol (10: 1) to give monoacetate 12 (60 mg, 64%) as a white solid. Starting diacetate 11 (18 mg, 18%) and diol 10 (13 mg, 16%) were also obtained.
(+)-Enantiomer 12: m.p. 192–195°C, [α]D25 21.7° (c 1.0, DMSO), chiral HPLC in methanol as an eluent, flow rate 1 mL/min, detection at 290 nm, retention time (RT) 5.5 minutes, 97.6%, ee 95.2%)/ and 6.6 minutes /(−)-enantiomer 12, 2.4%/. UV λmax 291 nm (ε 12,800), 236 nm (ε 27,400). 1H NMR δ 12.07, 11.71 (2bs, 2H, NH), 8.47 (s, 1H, H8), 7.20 (s, 1H, H1′), 5.29 (bs, 1H, OH), 4.21, 4.10 (AB, 2H, J = 11.6 Hz, CH2OAc), 3.76, 3.44 (AB, 2H, J = 10.8 Hz, CH2OH), 2.75 (m, 1H, CH of isobutyryl), 1.92 (s, 3H, CH3 of acetate) 1.52, 1.50 (AB, 2H, J = 9.8 Hz, H3′), 1.10 (d, J = 7.3 Hz, 6H, CH3 of isobutyryl). 13C NMR 180.9, 170.7 (CO), 155.4, 149.1, 147.9, 137.1, 120.5, 119.2 (), 111.6 (purine, C2′, C1v), 65.5, 63.2 (C5′), 35.5 (CH of isobutyryl), 28.2 (C4′), 21.2 (CH3 of acetate), 19.5 (CH3 of isobutyryl), 12.5 (C3′). ESI-M 376 (M + H, 7.2), 398 (100.0, M + Na). Anal. Calcd for C17H21N5O5: C, 54.38; H, 5.64; N, 18.66. Found: C, 54.18; H, 5.71; N, 18.37.
The racemic compound (rac-12) was prepared using a modified procedure for unprotected acetate (rac-12, i-But = H).[22] A mixture of compound 10 (27 mg, 0.08 mmol), trimethyl orthoacetate (16 μl, 0.12 mmol) and p-toluenesulfonic acid (trace) in CH2Cl2 (2 mL) was stirred at room temperature for 2 hours. Triethylamine (0.1 mL) was added, the solvent was evaporated, and the solution of residue in acetic acid (80%, 2 mL) was stirred at room temperature for 30 minutes. The solvent was removed in vacuo and the crude product was chromatographed on a silica gel column in CH2Cl2-MeOH (15 : 1) to give rac-12 (27 mg, 93%).
(Z)-9-{[2-(Acetoxymethyl)-2-(hydroxymethyl)cyclopropylidene]methyl}-N2-isobutyrylguanine Phosphite (13):
Acetate (+)-12 (110 mg, 0.3 mmol) was added dropwise to diphenyl phosphite (1 mL, 5 mmol) in pyridine (5 mL). The mixture was stirred for 16 hours at room temperature, triethylamine-water (3 : 2, 5 mL) was added and the stirring was continued for 2 hours. Volatile components were evaporated and the residue was chromatographed on a silica gel column using chloroform-methanol-triethylamine (75 : 4 : 2). The crude product was passed through Dowex-50W (H+) column (elution with water) to give 112 mg (83%) of phosphite 13 as a white solid. 1H and 31P NMR indicated the presence of about 15% of deacetylated compound. UV λmax 274 nm, 230 nm. 1H NMR δ 12.11, 11.71 (2s, 2H, NH), 8.18 (s, 1H, H8), 7.27 (s, 1H, H1′), 6.71 (d, J = 667.3 Hz, 1H, P-H), 4.35, 4.02 (AB, 2H, J = 11.4 Hz, CH2OAc), 4.18–4.05 (m, 2H, CH2OP), 2.76 (m, 1H, CH of isobutyryl), 1.91 (s, 3H, acetate), 1.68 (collapsed AB, 2H, J = 11.0 Hz, H3′), 1.10 (d, 6H, J = 7.3 Hz, CH3 of isobutyryl). 13C NMR 180.9, 170.6 (CO), 155.4, 149.1, 148.1, 137.0, 120.5, 118.2, 112.7 (purine, C2′, C1′), 65.9 (d, J = 4.5 Hz, CH2OP), 65.5 (CH2OAc), 35.4 (CH of isobutyryl), 26.4 (d, J = 8.1 Hz, C4′), 21.1 (CH3 of OAc), 19.5 (CH3 of isobutyryl), 13.1 (C3′). 31P NMR 5.96, 5.81. Negative ESI-MS 438 (M – H, 100.0).
The racemic compound rac-13 was obtained from racemic acetate rac-12 (25 mg, 0.7 mmol), diphenyl phosphite (0.2 mL, 1 mmol), in pyridine (1 mL), reaction time 23 hours following the procedure described above to give rac-13 (33 mg, 80%).
(+)-(Z-9-{[2,2-Bis(hydroxymethyl)cyclopropylidene]methyl}guanine Phosphate/(+)-9:
A mixture of phosphite 13 (87.8 mg, 0.2 mmol) and imidazole (67 mg, 8 mmol) in pyridine (5 mL) was sonicated for 20 minutes. Trimethylsilyl chloride (0.5 mL, 4.0 mmol) was then added dropwise with stirring at room temperature. After 20 minutes, iodine (103 mg, 0.4 mmol) in pyridine (1 mL) was added and the stirring was continued for 16 hours. The solvent was evaporated and the residue was partitioned between water (30 mL) and dichloromethane (3 × 30 mL). Aqueous ammonia (15 mL) was added to the aqueous portion and the mixture was heated at 50–60°C for 16 hours. The solution was lyophilized after removing ammonia in vacuo (aspirator). The product was passed through the Dowex-50W X2–200 (H+) column with water as an eluent. The appropriate fractions were combined and they were lyophilized to give phosphate (+)-9 (51 mg, 82%), which was converted to ammonium salt by adding aqueous ammonia (30%) and lyophilization. HPLC (column 1, 150 × 4.1 mm, 10 μ, 0.05 M KH2PO4 in 5% methanol as eluent, flow rate 1 mL/min, retention time 6.38 minutes, purity >99%). UV (H2O) λmax 269 nm (ε 11,400), 230 nm (ε 27,800). [α]D27 27.6° (c 0.5, H2O). 1H and 13C NMR (D2O) corresponded to those of racemic sodium salt.22 31P NMR 1.36. Negative ESI-MS (MeOH) 342 (M – H, 100.0).
(−)-(Z)-9-{[2-(Hydroxymethyl)-2-(2-tetrahydropyranyloxymethyl) cyclopropylidene]-methyl}guanine (14):
A mixture of (+)-acetate 12 (21.2 mg, 0.06 mmol), 3,4-dihydro-2H-pyran (86 μL, 0.95 mmol) and methanesulfonic acid (4 μL, 0.06 mmol) was stirred for 16 hours at room temperature. The reaction was quenched with triethylamine (0.1 mL) and the volatile components were evaporated. The residue was dissolved in aqueous ammonia (30%, 10 mL) and the solution was heated at 50–60°C for 4 hours. Ammonia was evaporated in vacuo (aspirator) and the solution was lyophilized. The crude product was chromatographed on a silica gel column using dichloromethane-methanol (10 : 1) to give 19.9 mg (97%) of compound 14 as a white solid, m.p. 268–271°C, [α]D27 −3.2° (c 1.0, DMSO). UV, 1H, 13C NMR and mass spectra corresponded to the known racemic compound[22] and differing only by a diastereoisomeric composition.
(Z)-9-{[2-(2-Tetrahydropyranyloxymethyl)-2-(hydroxymethyl) cyclopropylidene]-methyl}guanine phosphite (15):
A solution of compound 14 (16.6 mg, 0.05 mmol) in pyridine (1 mL) was added dropwise to diphenyl phosphite (85–90%, 0.2 mL, 0.89 mmol) in pyridine (1 mL) with stirring at room temperature. The stirring was continued for 16 hours, triethylamine-water (3 : 2, 1 mL) was added and the mixture was stirred for 2 hours. The volatile components were evaporated and the crude product was chromatographed on a silica gel column using dichloromethane-methanol (4 : 1 to 1.5 : 1) to give phosphite 15 (19 mg, 93%) as a white solid. UV λmax 274 nm (ε 10,100), 230 nm (ε 24,300). 1H NMR δ 11.17 (bs, 1H, NH), 8.25, 8.18 (2s, 1H, H8), 7.13 (s, 1H, H1′), 6.83 (bs, 2H, NH2), 6.59 (d, 1H, J = 600.5 Hz, P-H), 4.58, 4.48 (2s, 1H, CHO of THP), 4.00 (bs, 1H, OH), 3.76–3.24 (2AB, overlapped with H2O, H5′, H5″, CH2O of THP), 1.10–1.70 (m, 8H, CH2 of THP, H3′). 13C NMR 157.6, 154.9, 150.4, 134.6, 116.8, 111.9 (purine, C2′, C1′), 98.5, 98.3 (CHO of THP), 69.2, 68.7 (CH2O of THP), 64.3, 61.7, 61.3 (C5′, C5″), 30.5, 27.6, 25.6. 25.5, 19.5, 19.2 (3xCH2 of THP, C4′), 12.5 (C3′). Negative ESI-MS (MeOH) 410 (M – H, 100.0).
(−) - (Z) - 9 - {[2,2-Bis(hydroxymethyl)cyclopropylidene]methyl}guanine phosphate/(−)-9/:
A mixture of phosphite 15 (19 mg, 0.045 mmol) and of imidazole (12.1 mg, 0.18 mmol) in pyridine (2 mL) was sonicated for 20 minutes at room temperature. Trimethylsilyl chloride (0.15 mL, 1.2 mmol) was then added dropwise and the mixture was stirred for 20 minutes. Iodine (30 mg, 0.09 mmol) in pyridine (1 mL) was added dropwise and the mixture was stirred for 16 hours. The solvent was removed in vacuo and the residue was stirred in acetic acid (80%, 10 mL) at room temperature for 24 hours. The solution was lyophilized and the residue was stirred in aqueous ammonia (30%, 50 mL) at room temperature for 3 hours. The volatile components were evaporated and the crude product was chromatographed on Dowex-50 WX2–200 (H+) column using water as an eluent. The appropriate fractions were collected and they were lyophilized to give phosphate (−)-9 (10.5 mg, 69%) as a white powder. Ammonium salt was obtained by adding aqueous ammonia and subsequent lyophilization. HPLC /see enantiomer (+)-9/retention time 6.40 minutes, purity 99%, [α]27D −29.6° (c 0.5, H2O). UV (H2O) λmax 268 (ε 10,400), 229 (ε 24,600). 1H NMR corresponded to that of the racemic sodium salt.[22] 31P NMR 1.30. Negative ESI-MS (MeOH) 342 (M – H, 100.0).
(Z)-9-{[2,2-Bis(hydroxymethyl)cyclopropylidene]methyl}guanine Diphosphate (16):
A mixture of racemic phosphite rac-13 (23 mg, 0.052 mmol) and imidazole (18 mg, 0.26 mmol) was stirred at room temperature for 30 minutes. Me3SiCl (0.2 mL 1.6 mmol) was added and the stirring was continued for 20 minutes. After addition of iodine (27 mg, 0.08 mmol) and stirring for another 20 minutes the volatile components were evaporated. The residue was partitioned between water (20 mL) and CH2Cl2 (2 × 20 mL). The aqueous portion was lyophilized, crude product was put on the column of Dowex 50 /H(+)/, which was eluted with water. The appropriate fractions were lyophilized to give phosphate (18 mg, 75%). This product (16 mg, 0.035 mmol) and tributylamine (8.4 μL, 0.035 mmol) in DMF (0.5 mL) was stirred for 30 minutes at room temperature. N,N′-Carbonyldiimidazole (28.6 mg, 0.175 mmol) was added and the stirring was continued for 3 hours. The reaction was quenched with methanol (11.4 μL, 0.28 mmol) and, after 30 minutes, bis(tributylammonium) phosphate (200 mg, 0.71 mmol) was added and the mixture was stirred for 16 h. The solvents were removed in vacuo, the residue was dissolved in aqueous ammonia (30%, 20 mL) and the solution was stirred for 60 hours at room temperature. The volatiles were evaporated and the crude product was chromatographed on DEAE Sephadex A-25 /HCO3(−), 20 × 1 cm/ column using a discontinuous gradient of NH4HCO3: 0.1, 0.15, 0.2, 0.25, and 0.3 M NH4HCO3 (20 mL each). Appropriate fractions were combined and lyophilized to give product 16 (10 mg, 62%) as an ammonium salt. HPLC (see phosphate 13) retention time 3.20 minutes, purity 87.5%. 1H NMR (D2O) δ 8.29 (s, 1H, H8), 7.08 (s, 1H, H1′), 4.12 (m, 1H), 4.12, 3.89 (2m, 2H, CH2OP), 3.71, 3.58 (AB, 2H, J = 11.9 Hz, CH2OH), 1.46 (m, 2H, H3′). 31P NMR −7.49 (d, J = 21.4 Hz, Pβ), −9.97 (d, J = 21.4 Hz, Pα). Negative ESI-MS 422 (100.0, M – H).
Guanylate Kinase Assay
Method A
The procedure for phosphorylation of enantiomeric ganciclovir phosphates 6 and ent-6 was followed.[21] A stock GMP kinase assay buffer (0.1 M Tris-acetate, pH 7.6; 0.1 M KCl; 10 mM MgCl2; 4 mM ATP; 1.5 mM phosphoenol pyruvate; 0.1 mg/mL BSA; 11 units/mL lactate dehydrogenase; and 5 units/mL pyruvate kinase) was prepared. Phosphate (+)-9 or (−)-9 (0.14 mg, 0.4 μmol), NADH (1.5 mM, 4 μL) was preincubated in stock buffer (0.5 mL) at 34°C for 5 minutes. GMP kinase from bovine liver (6 μg, 0.08 unit) was then added and the reaction was allowed to proceed at 34°C. At specific time intervals, a 70 μL aliquot was removed, filtered through a 0.45 μ filter, and analyzed by HPLC for the conversion to diphosphate 16. The HPLC was run as described for phosphates (+)-9 and (−)-9. The reaction times are as follows: ATP and ADP 1.71 minutes, diphosphates 16 3.19 minutes, and phosphates 9 6.40 minutes. The mobility of diphosphates formed was identical with that of racemic compound 16. The results are shown in Figure 1.
In a control experiment, conversion of GMP to GDP was followed under the conditions described above. The HPLC was performed using column 2 and 0.6 M KH2PO4 (pH 4.5) as a mobile phase. The eluate was monitored at 265 nm and flow rate was 2.0 mL/min. The reaction times are as follows: GMP 3.28 minutes, ADP 5.04 minutes, GDP 8.80 minutes, ATP 18.50 minutes. After 0.5 hours, >95% conversion to GDP was observed.
Method B
The GMP kinase assay was adopted from Marshalko et al.[21] Briefly, GMP kinase buffer (0.05 M Tris, pH 7.6; 0.05 M KCl; 5.0 mM MgCl2), ATP (2.0 mM), BSA (0.1 mg/mL) (all final concentrations), and substrate (GMP, dGMP, phosphates (+)-9 and (−)-9; 5.0–2000 μM) were incubated at 37°C for 20 minutes prior to introduction of enzyme. At time zero, bovine GMP kinase was added to the solution to give a final concentration ranging from 0.01 to 0.2 units/mL (unit is defined as the amount of enzyme necessary to convert 1.0 μmol of GMP to GDP in 1.0 min at pH 7.5 at 30°C). At the designated times, an aliquot was removed, placed on ice, and proteins precipitated with 0.04 volumes of 10 M perchloric acid. The samples were centrifuged, the supernatants neutralized with KOH and the samples were stored at −20°C until analyzed by HPLC.
Phosphates (+)-9, (−)-9 and 16 were separated and quantified by reversed phase HPLC (Beckman Coulter, Fullerton, CA, USA; System Gold Programmable Solvent Module 125 and System Gold Programmable Detector Module 166 controlled by 32 Karat Software (ver. 7.0). Before injection, each sample was centrifuged at 14,000 rpm for 5 minutes to remove any remaining particulate matter. Samples were loaded onto a 10 μm Alphabond C18 300 × 3.9 mm reversed phase column (Alltech, Deerfield, IL, USA) at a flow rate of 2.0 mLs/min and phosphorylated cyclopropavir derivatives were eluted using 300 mM potassium phosphate (pH 3.0) and 100% methanol (5% methanol linear gradient over 30 minutes). Phosphorylated cyclopropavir derivatives were quantified by comparing their peak area with that of a known amount of the appropriate standard at a wavelength of 254 nm.
In experiments with GMP and dGMP as substrates, nucleotides were separated and quantified by strong anion-exchange HPLC using a Beckman Coulter (Fullerton, CA, USA) Proteome Lab PF 2D Protein Fractionation System controlled by 32 Karat Software (ver. 7.0). Before injection, each sample was centrifuged as above to remove any remaining particulate matter. Samples were then loaded onto a Partisil 5 SAX analytical 4.6 × 250 mm column (Whatman, Clifton, NJ, USA) at a flow rate of 2.0 mL/min and phosphorylated nucleotides were eluted using a 60 minute linear gradient of 10 mM ammonium phosphate (pH 3.0) to 500 mM ammonium phosphate (pH 3.0) followed by 30 minutes of 500 mM ammonium phosphate (pH 3.0). Nucleotides were quantified by comparing their peak area with that of a known amount of the appropriate standard at a wavelength of 254 nm.
Acknowledgments
We thank L. M. Hrihorczuk from the Central Instrumentation Facility (D. M. Coleman, Director) for mass spectra. The work described herein was supported by Research Grant 1 RO1 CA32779 from the National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
REFERENCES
- 1.Zemlicka J Methylenecyclopropane analogues of nucleosides as anti-herpes agents, in Advances in Antiviral Drug Design, ed. De Clercq E, Elsevier, Amsterdam, The Netherlands, 2007, pp. 113–165. [Google Scholar]
- 2.Qiu Y-L; Geiser F; Kira T; Gullen E; Cheng Y-C; Ptak RG; Breitenbach JM; Drach JC;Hartline CB; Kern ER; Zemlicka J Synthesis and enantioselectivity of the antiviral effects of (R,Z)-, (S,Z)-methylenecyclopropane analogues of purine nucleosides and phosphoralaninate prodrugs: influence of heterocyclic base, type of virus and host cells. Antiviral Chem. Chemother 2000, 11, 191–202. [DOI] [PubMed] [Google Scholar]
- 3.Chen X; Kern ER; Drach JC; Gullen E; Cheng Y-C; Zemlicka J Structure-activity relationshipsof (S,Z)-2-aminopurine methylenecyclopropane analogues of nucleosides. Variation of purine-6 substituents and activity against herpesviruses and hepatitis B virus. J. Med. Chem 2003, 46, 1531–1537. [DOI] [PubMed] [Google Scholar]
- 4.Zhou S; Breitenbach JM; Borysko KZ; Drach JC; Kern ER; Gullen E; Cheng Y-C; Zemlicka J Synthesis and antiviral activity of (Z)- and (E)-2,2-[bis(hydroxymethyl)cyclopropylidene]methylpurines and -pyrimidines: Second-generation methylenecyclopropane analogues of nucleosides. J. Med. Chem 2004, 47, 566–575. [DOI] [PubMed] [Google Scholar]
- 5.Kern ER; Kushner NL; Hartline CB; Williams-Azziz SL; Harden EA; Zhou S; Zemlicka J; Prichard MN In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother 2005, 49, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kern ER; Bidanset DJ; Hartline CB; Yan Z; Zemlicka J; Quenelle DC Oral activity of amethylenecyclopropane analog, cyclopropavir, in animal models for cytomegalovirus infections. Antimicrob. Agents Chemother 2004, 48, 4745–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowlin T; Brooks J; Zemlicka J Preclinical pharmacokinetic, toxicokinetic and toxicology resultsfor cyclopropavir, a promising new agent for the treatment of beta- and gamma-herpesviruses. 22nd International Conference on Antiviral Research, Miami Beach, Florida, May 3–7, 2009, Abstract 99, Antiviral Res 2009, 82, A46. [Google Scholar]
- 8.Qiu Y-L; Ptak RG; Breitenbach JM; Lin J-S; Cheng Y-C; Drach JC; Kern ER; Zemlicka J Synthesis and antiviral activity of phosphoralaninate derivatives of methylenecyclopropane analogues of nucleosides. Antiviral Res. 1999, 43, 37–53. [DOI] [PubMed] [Google Scholar]
- 9.Yoshimura K; Feldman R; Kodama E; Kavlick MF; Qiu Y-L; Zemlicka J; Mitsuya H Invitro reduction of human immunodeficiency virus type 1 variants resistant to phosphoralaninate prodrugs of Z-methylenecyclopropane nucleoside analogues. Antimicrob, Agents Chemother 1999, 43, 2479–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang R; Harada S; Mitsuya H; Zemlicka J Inhibition of human immunodeficiency virus reversetranscriptase by synadenol triphosphate and its E-Isomer. J. Med. Chem 2003, 46, 4799–4802. [DOI] [PubMed] [Google Scholar]
- 11.Baldanti F; Sarasini A; Drach JC; Zemlicka J; Gerna G Z-isomers of 2hydroxymethylcyclopropylidenemethyl adenine (synadenol) and guanine (synguanol) are active against ganciclovir- and foscarnet-resistant human cytomegalovirus UL97 mutants. Antiviral Res 2002, 56, 273–278. [DOI] [PubMed] [Google Scholar]
- 12.Kern ER; Kushner NL; Hartline CB; Williams-Azziz SL; Harden EA; Zhou S, Zemlicka J; Prichard MN in vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother 2005, 49, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breitenbach JM; Borysko KZ; Zemlicka J; Drach JC Resistance of human cytomegalovirus withsingle and double mutations in UL97 to first and second generation of methylenecyclopropane purines. 19th International Conference on Antiviral Research, San Juan, Puerto Rico, May 7–l1, 2006, Abstract 61, Antiviral Res. 2006, 70, A69. [Google Scholar]
- 14.Borysko KZ; Breitenbach JM; Gentry BG; Zemlicka J; Drach JC Selection of humancytomegalovirus resistant to a second generation methylenecyclopropane purine. 20th International Conference on Antiviral Research, Palm Springs, CA, USA, April 29–May 3, 2007, Abstract 139, Antiviral Res. 2007, 74, A83. [Google Scholar]
- 15.Borysko KZ; Gentry BG; Breitenbach JM; Zemlicka J; Drach JC Resistance of humancytomegalovirus to cyclopropavir involves a novel mutation in UL97. 21st International Conference on Antiviral Research, Montreal, Quebec, Canada, April 13–17, 2008, Abstract 98, Antiviral Res. 2008, 78, A54. [Google Scholar]
- 16.Karkas JD; Germershausen JG; Tolman RL; MacCoss M; Wagner AF; Liou R; Bostedor R Stereochemical considerations in the enzymatic phosphorylation and antiviral activity of acyclonucleosides. I. Phosphorylation of 2′ -nor-2′ -deoxyguanosine. Biochem. Biophys. Acta 1987, 911, 127–135. [DOI] [PubMed] [Google Scholar]
- 17.Littler E; Stuart AD; Chee MS Human cytomegalovirus UL97 open reading frame encodes aprotein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 1992, 358, 160–162. [DOI] [PubMed] [Google Scholar]
- 18.Sullivan V; Talarico CL; Stanat SC; Davis M; Coen DM; Biron KK A protein kinasehomologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 1992, 358, 162–164. [DOI] [PubMed] [Google Scholar]
- 19.Tolman RL Structural requirements for enzymatic activation of acyclonucleotide analogues, in Nucleotide Analogues as Antiviral Agents, ed. Martin JC, American Chemical Society, Washington, D.C., 1989, pp 35–50. [Google Scholar]
- 20.Miller WH; Daluge SM; Garvey EP; Hopkins S; Reardon JE; Boyd FL; Miller RL Phosphorylation of carbovir enantiomers by cellular enzymes determines the stereoselectivity of antiviral activity. J. Biol. Chem 1992, 267, 21220–21224. [PubMed] [Google Scholar]
- 21.Marshalko SJ; Schweitzer BI; Beardsley GP Chiral chemical synthesis of DNA containing (S)-9(1,3-dihydroxy-2-propoxymethyl)guanine (DHPG) and effects on thermal stability, duplex structure, and thermodynamics of duplex formation. Biochemistry 1995, 34, 9235–9248. [DOI] [PubMed] [Google Scholar]
- 22.Yan Z; Kern ER; Gullen E; Cheng Y-C; Drach JC; Zemlicka J Nucleotides and pronucleotidesof 2,2-bis(hydroxymethyl)methylenecyclopropane analogues of purine nucleosides: Synthesis and antiviral activity. J. Med. Chem 2005, 48, 91–99. [DOI] [PubMed] [Google Scholar]
- 23.Hoard DE; Ott DG Conversion off mono- and oligodeoxynucleotides to 5′-triphosphates. J. Am. Chem. Soc 1965, 87, 1785–1789. [DOI] [PubMed] [Google Scholar]