

Abstract
BACKGROUND AND PURPOSE: The most abundant metabolite visible by proton magnetic resonance spectroscopy (MRS) in human brain is N-acetylaspartate (NAA), which is often used as a marker of viable neurons. NAA is anecdotally reported to be elevated in children with sickle cell disease (SCD), even though patients can have brain injury or atrophy. We measured NAA levels rigorously in SCD patients to test the hypothesis that NAA is elevated in this patient population.
METHODS: We evaluated 26 children with SCD and 25 age-similar healthy control subjects using a double spin-echo MRS technique to interrogate a 16 cc volume of interest in the basal ganglia. We acquired MRS spectra with an echo time (TE) of 30 ms to evaluate lipids, and with TE = 144 to show abundant metabolites against a flat baseline. We characterized metabolite relaxation properties and measured the water peak as an internal standard, to calculate the absolute quantity of metabolites.
RESULTS: The ratio of NAA:Choline was significantly elevated in basal ganglia of patients at both echo times (P <.016), and the absolute quantity of NAA was also elevated, with [NAA] 7–12% higher in patients than in control subjects. The measured increase in [NAA] cannot be explained by metabolite relaxation properties or by differences in tissue water content.
CONCLUSION: Brain NAA is greater in children with SCD than in healthy control subjects and appears not to be a reliable marker of viable neurons in SCD patients.
Proton MR spectroscopy (MRS) of the human brain makes it possible to measure metabolically important cellular components such as amino acids (including N-acetylaspartate [NAA]), amines, sugars, and bioenergetic metabolites in vivo (1). In healthy human brain, the most abundant metabolite visible on MRS is typically NAA, which is present almost exclusively in the nervous system (2). NAA is hypothesized to be a marker of the number of viable neurons (3). Consistent with this hypothesis, levels of NAA in the brain are reduced in a range of pathologic states, with dramatic reductions in patients with tumor or stroke (4, 5). A competing hypothesis proposes that NAA is an osmolyte, since microdialysis has shown that stepped changes in cellular hydration of brain sections can induce a redistribution of NAA from intracellular compartments to extracellular compartments (6). Thus, NAA is hypothesized to be a molecular pump in which translocation creates an osmotic environment that passively removes water from the intracellular milieu of neurons (7).
Brain NAA levels are anecdotally reported to be elevated in patients with sickle cell disease (SCD) (4). Because robust evidence suggests that children with SCD have brain injury (8, 9) and because no evidence suggests that children with SCD have enhanced neuronal proliferation, elevation of NAA values in SCD seems to be inconsistent with its hypothesized role as a marker of viable neurons. To better understand the role of NAA in the brain, we measured its levels in children with SCD, comparing patients with healthy age-similar control subjects.
Methods
Patients
We examined 26 patients (average age ± SD, 12.8 ± 2.7 years; range, 8–17 years; 16 boys, 10 girls), excluding those with clinical findings suggestive of a recent change in brain function that would require thorough diagnostic examination. Of the 26 patients, three were receiving transfusion, and two were receiving hydroxyurea treatment. The remaining 21 patients were not receiving any treatment at the time of evaluation. No patients were sedated for the examination. A total of 25 control subjects (12.0 ± 3.8 years; range, 8–22 years; 11 boys, 14 girls) were also evaluated. Parents or guardians of all subjects provided written informed consent after they received a brief description of the protocol and after subjects assented to their participation. Two additional patients with SCD were enrolled but retrospectively excluded: Patient motion had degraded the quality of the spectra for one patient, and the other patient had an oral spacer that caused unacceptable magnetic field inhomogeneity.
MR Imaging
Patients and control subjects underwent an examination that included MR imaging and MRS at 1.5T, with MR imaging used to screen for brain injury and to characterize tissue type in the MRS volume of interest (VOI). A T1-weighted gradient-echo image set was acquired in the transverse plane with the following parameters: TR/TE = 175/ 3.77, section thickness = 5 mm, FOV = 21 cm, matrix = 256 × 256, sections = 19, distance factor = 0.2, and time for one acquisition = 1 minute 42 seconds. A T2-weighted turbo spin-echo image set was acquired in the same orientation by using the following parameters: TR/TE1/TE2 = 3500/16/109, section thickness = 5 mm, FOV = 21 cm, matrix = 256 × 256, sections = 19, distance factor = 0.2, and time for one acquisition = 4 minutes 7 seconds. Screening MR imaging showed evidence of disease-related brain injury in six patients and normal findings in all control subjects. This screening was done using previously established criteria (9) to identify patients with lacunae, loss of white matter volume, encephalomalacia, or leukoencephalopathy.
An inversion-recovery sequence (10) was done at the same section level and section orientation as those of the MRS voxel to measure gray matter, white matter, and CSF volumes in the spectroscopic volume. The inversion-recovery sequence has been used to measure tissue T1 (10), but here it was segmented according to established methods (10) to estimate the proportion of gray matter in the spectroscopic VOI. Parameters were as follows: TR/TE/TI = 2500/20/100, 500, 900, and 2389; section thickness = 5 mm; FOV = 21 cm; and time four acquisitions = approximately 4 minutes.
MR Spectroscopy
During MRS, a single, 16-mL VOI in the basal ganglia (Fig 1A) was examined by using a double spin-echo pulse sequence (11) with water suppression (12). Several MRS sequences were included: 1) a sequence with a TR/TE/NEX of 1500/30/128 to show lipids and other metabolites with a short spin-spin relaxation (T2) time; 2) a long-echo sequence with a TR/TE/NEX of 1500/144/128 to show major metabolites against a flat baseline; and 3) in some subjects (18 patients and 12 control subjects), a sequence with a TR/TE/NEX of 5000/144/40 to show most metabolites in the fully-relaxed state with minimal metabolite saturation. This sequence was repeated again without water suppression (NEX = 1) using the water signal intensity as an internal concentration standard for metabolites (13).
Fig 1.
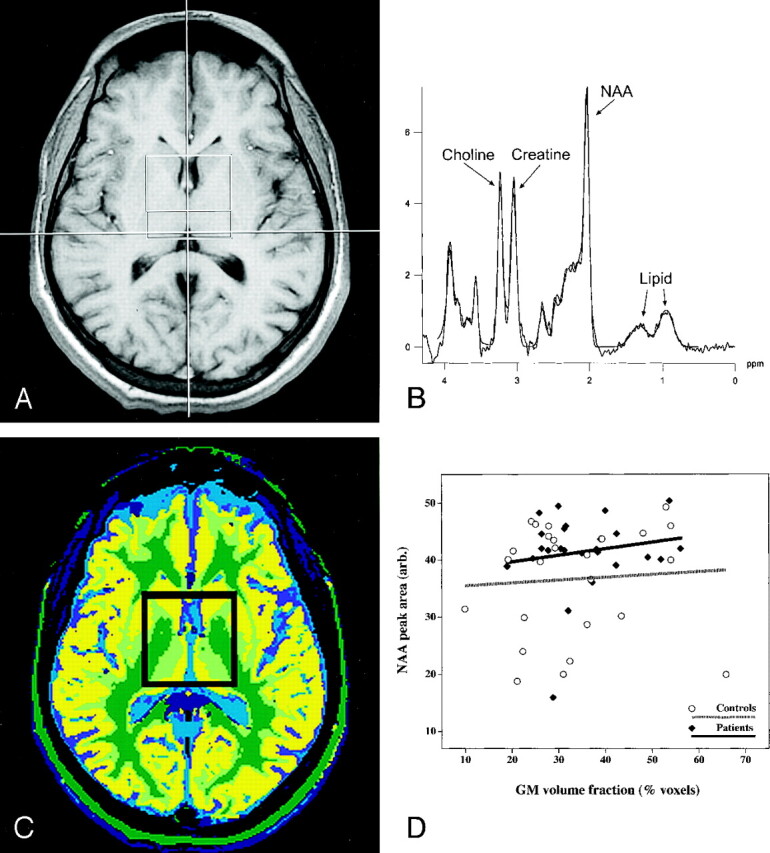
MR spectroscopic findings obtained in a patient with SCD.
A, Axial image shows a 4 × 4 × 1-cm VOI centered in the basal ganglia.
B, Short-echo spectrum acquired with a TR/TE of 1500/30 shows NAA, lipid, and other metabolites with a short spin-spin relaxation time (T2).
C, Spectroscopic VOI was segmented and classified by using a set of inversion recovery images acquired at the same section position to calculate the proportion of gray matter in the VOI.
D, Proportion of gray matter in the VOI was regressed against tissue NAA. Amount of NAA in the VOI did not change as a function of the gray matter volume in either patients or control subjects.
Data Analysis
Resonance areas were integrated by using standard software provided with the machine (Symphony; Siemens, Erlangen, Germany). We modified the integration parameters to include two lipid resonances that were visible in both patient (Fig 1B) and control spectra. This approach also compensated for the possibility of an artifactual elevation of NAA produced by a broad metabolite peak underlying the upfield region of the spectra.
Signal intensity processing included line broadening (Hanning filter, 700-ms width at half-height), zero-filling (to 2048 time points), fast Fourier transformation, baseline correction (over the spectral range of 0–4.3 ppm with a sixth-order polynomial), and phase correction (zero- and first-order). Resonance areas proportional to metabolite concentration were determined by fitting gaussian peaks to the spectral data. Use of ratios for metabolite quantitation can compensate for differences in coil loading or magnetic field homogeneity (5, 14).
Possible Experimental Artifacts
The spin-lattice relaxation time (T1) and the spin-spin relaxation time (T2) of metabolites can affect their visibility in proton spectra. Tissue NAA levels could be artifactually elevated if disease alters either the T1 (15) or the T2 (16) of NAA. Therefore, we calculated the T1 and T2 of major metabolites in the spectra of both patients and control subjects. Metabolite T1 was inferred by comparing metabolite resonance areas at TR = 1500 ms to the resonance areas of these metabolites at TR = 5000 ms (holding TE constant at 144 ms). We assumed that NAA was fully relaxed at the long TR; therefore, the resonance area decrement at TR = 1500 ms should correlate with the degree of T1 saturation of the metabolite. Similarly, metabolite T2 was estimated from the metabolite peak areas at TE = 30 and 144 ms (both at TR = 1500 ms) by assuming monoexponential relaxation of metabolites. We used the following equation: metabolite T2 = ΔTE/(−ln [α144/α30]), where ΔTE = 114 (the difference between TE = 144 and TE = 30 in milliseconds), and α is the peak area of a given metabolite at the TE indicated by the subscript (17).
We then used the calculated T1 and T2 relaxation times, along with the unsuppressed water signal intensity, to estimate the absolute concentration of NAA, choline (Cho), and creatine (Cr) (13, 18), assuming the T2 of water to be 78.5 ms (17).
If the proportion of gray matter or white matter in the brain differed between patients and control subjects, a difference in tissue type could potentially explain any excess of NAA in patient spectra. To address this issue, we segmented and classified tissue in the VOI as gray matter, white matter, or a partial volume admixture of these two tissues by using the inversion-recovery images segmented according to established methods (10). In this analysis, we ignored CSF because it was no more than a small percentage of the volume of the VOI, and CSF usually has a negligible content of NAA (5).
Another possibility to explain the apparent increase in NAA levels in patients is that the absolute quantity of NAA in brain is unchanged but the total tissue water content is reduced; therefore, the tissue NAA concentration is increased. To assess this possibility, we measured the unsuppressed water peak at TE = 144 ms by fitting a gaussian line shape and evaluating the peak area as an indication of water proton abundance.
Results
Metabolite Levels
Patients had a proportion of NAA in the basal ganglia higher than that of healthy control subjects (Table 1). Analysis of the short-echo spectrum (Fig 1B) showed a higher ratio of NAA to Cho in patients than in control subjects (P = .012), and replicated long-echo spectra confirmed this finding (P = .016). Not surprisingly, the ratio of NAA/Cho differed somewhat with the two TEs (Table 1), as the contribution of macromolecules was likely increased at short TE. Furthermore, each of the ratios reported (Table 1) is an estimate of the population mean based on a sample that included only 25 patients and 25 control subjects.
TABLE 1:
Ratios of metabolite peak areas
| Ratio | Patients | Control Subjects | Difference (%) | Mann-Whitney U* |
|---|---|---|---|---|
| TR/TE = 1500/30 | ||||
| NAA/Cr | 1.59 ± 0.60 | 1.20 ± 0.27 | 32.5 | 0.002 |
| NAA/Cho | 1.90 ± 0.33 | 1.60 ± 0.37 | 18.8 | 0.012 |
| Cho/Cr | 0.85 ± 0.37 | 0.76 ± 0.10 | 11.8 | NS |
| Tr/TE = 1500/144 | ||||
| NAA/Cr | 1.65 ± 0.27 | 1.63 ± 0.23 | 1.2 | NS |
| NAA/Cho | 1.62 ± 0.24 | 1.50 ± 0.17 | 8.0 | 0.016 |
| Cho/Cr | 1.04 ± 0.20 | 1.09 ± 0.15 | −4.6 | NS |
Ties included in the analysis.
We noted substantial upfield resonances on the short-echo spectra of patients (Fig 1B) and control subjects (data not shown) that were absent from long-echo spectra; these findings can be present on spectra from pediatric patients with brain tumor (5). Because the resonances were absent at long-echoes, they were attributed to lipid rather than to lactate. Lipid resonances did not differ between groups, but the central gray matter of all subjects had lipid levels higher than those reported in spectra from adults. Whether lipid resonances are related to the subject’s age or to voxel position is not known. However, if such resonances were present but not fitted by the model, this could potentially lead to an artifact in quantitation of the NAA resonance.
The ratio of Cho to Cr was similar in patients and control subjects, implying that the Cho concentration was not reduced in patients. Comparable brain Cho values between patients and control subjects would confirm that the brain tissue of patients with SCD have more NAA (8). If NAA is a neuronal marker, patients would have been expected to have stable or reduced NAA levels, as children with SCD often have both brain injury and cognitive impairment (9).
Possible Experimental Artifacts
We tested whether the elevation of NAA in patients was artifactual by measuring metabolite relaxation properties. We found no differences in metabolite T1 or T2 that could explain the increased levels of NAA in patients. Among patients, the NAA peak was 66% ± 4% relaxed at short TR, whereas in control subjects, the NAA peak was 65% ± 5% relaxed (not significant). We also noted no difference in the saturation of Cho or Cr and no systematic change in the ratios measured. The calculated T1 of NAA in patients was normal (1388 ± 129 ms), and not significantly different from the T1 of NAA in control subjects (1450 ± 238 ms) (13). Similarly, the calculated T2 of NAA in patients with SCD was normal (318 ± 234 ms) and not significantly different from the T2 of NAA in control subjects (284 ± 139 ms) (13). Differences between patients and control subjects in the T2 of other metabolites were not significant. Therefore, we could not explain the elevated NAA levels in patients as a function of disease-related differences in either metabolite T1 or T2.
We calculated the absolute concentration of metabolites and found that the concentration of NAA was 12% higher in patients (12.2 mmol/L) than in control subjects (10.9 mmol/L), the concentration of Cr was 4% lower in patients (7.8 mmol/L) than in control subjects (8.1 mmol/L), and the concentration of Cho was equivalent (1.6 mmol/L). All metabolite concentrations were well within the range of reported values for the human brain (13, 14, 18), suggesting that the increase in NAA/Cho that we found represents an actual increase in concentration of NAA in patients.
We segmented tissue in the VOI (Fig 1C) to determine whether high NAA levels in patients might be a function of differing tissue composition in the VOI. In patients, amounts of gray matter in the VOI were similar to those of control subjects (Table 2), but patients also had more white matter (P = .001) and less partial-volume of gray matter and white matter (P = .01). When tissue NAA was regressed against the proportion of gray matter in the VOI, we noted no significant relationship between NAA and gray matter in patients or in control subjects (Fig 1D), but patients had 7% more NAA in the VOI.
TABLE 2:
Segmented brain volumes in the VOI used for MRS
| Brain volume in VOI | Mean ± SD (%) |
Mann-Whitney U | |
|---|---|---|---|
| Patients | Control Subjects | ||
| White matter | 22.9 ± 9.2 | 15.6 ± 4.7 | 0.0012 |
| Partial volume | 41.6 ± 12.4 | 51.0 ± 13.1 | 0.0106 |
| White manner + partial volume | 64.4 ± 9.6 | 66.6 ± 13.4 | NS |
| Gray matter | 35.6 ± 9.6 | 33.4 ± 13.4 | NS |
Finally, we calculated the concentration of tissue water in the VOI by using the unsuppressed water peak and found no significant difference between patients and control subjects. The water resonance area in patients was 25,778 ± 1680, which was not significantly different from the water resonance area in control subjects (25,382 ± 1174). Therefore, we detected no difference between patients and control subjects in the degree of brain tissue hydration.
Discussion
We have shown that the ratio of NAA/Cho is significantly elevated at long and short TEs in patients with SCD (Table 1). In addition, the NAA/Cr ratio is significantly elevated at a short TE. This apparent elevation in NAA is unexplained by artifacts such as a change in the T1 or T2 relaxation properties of NAA or by an overall reduction of tissue water content.
Metabolite T1 and T2 relaxation were both determined by using two-point methods, which are subject to inaccuracy, but the T1 and T2 values that we report are consistent with the literature (15–18). In addition, our metabolite concentrations are within the range of reported values for the human brain (13, 14, 18), suggesting that the increase in NAA/Cho that we found represents an actual increase in concentration of NAA in patients.
A limitation of our study was that all spectra were analyzed by using software available at the console (Siemens). This software specifically fits the following resonances at short echoes: Cho, Cr, the NAA singlet and doublet, the glutamate/glutamine singlets and the doublet, and the inositol doublet. Any other major contributions to the spectra could create artifacts in spectral quantification. However, findings on visual inspection of the spectra did not suggest that macromolecular contributions were an important source of error in our subjects, even on short-echo spectra (Fig 1B), and we found no systematic differences between patients and control subjects. Another problem with the software is that incomplete outer-volume suppression can cause artifactual lipid contamination of the spectra. However, the major source of unsuppressed lipids is generally subcutaneous fat (5), and VOI placement in this study avoided subcutaneous fat (Fig 1A). Although we recognize that MRS studies have many potential sources of inherent error, we believe our data are as free of such problems as any other dataset.
Children with SCD are known to have brain injury as a consequence of their disease (9). At a mean age of only 10 years, the prevalence of infarction, ischemia, or atrophy in patients with SCD is estimated to be 44%, while the prevalence of arterial vasculopathy is estimated to be 55%. The prevalence of brain injury in the patients evaluated in this study was roughly consistent with the published prevalence of brain injury (9), given that we specifically excluded patients with clinical findings suggestive of recent brain injury. The prevalence of brain injury is known to increase with age (9), suggesting an ongoing disease process associated with injury. In this context, it seems somewhat unlikely that children with SCD had neuronal proliferation. Nevertheless, our study was limited in that we are unable to exclude the possibility of neuronal proliferation in patients of SCD.
Our findings call into question the role of NAA as a marker of the number of viable neurons in brain tissue. Many other observations may also be problematic if NAA is to be interpreted as a marker of neuronal viability. The key issue is that if NAA is a neuronal marker, it should be present whenever neurons are present, and it should be stable under conditions in which neuronal number is stable. Yet, a case report of a child with only mild developmental disability described no NAA resonance at all in the brain (1). The NAA peak is also known to transiently decrease after acute brain injury (19), and NAA can show stable increases after therapy for moyamoya disease (20), amyotrophic lateral sclerosis (21), or Wernicke encephalopathy (22). The finding that NAA deficits can be reversed over a relatively short period argues strongly that NAA is not a reliable marker of neuronal loss. Furthermore, NAA levels can be chronically elevated in disease states not associated with neuronal proliferation, including Canavan disease (23), Pelizaeus-Merzbacher disease (24), and familial bipolar I disorder (25). In aggregate, these results suggest that NAA is not a reliable marker of viable neurons (19, 26).
Conclusion
Our findings suggest that NAA in brain spectra does not reflect neuronal viability in children with SCD. Instead, we hypothesize that changes in the central gray matter in patients with SCD—including the relative increase in NAA (Table 1), the relative reduction in partial volume admixture of gray matter and white matter (Table 2), and the known reduction in gray matter T1 (27)—are interrelated. These observations suggest that gray matter is abnormal in patients with SCD. Our results do not seem to be consistent with the commonly accepted role of NAA as a marker of neuronal number or neuronal viability (3–5). Redistribution of NAA from the intracellular space to the extracellular space has been proposed as a way to maintain osmotic balance in tissue (6, 7). Therefore, the hypothesis that NAA is an osmolyte may be more consistent with our results. We note that if NAA were primarily an osmolyte, NAA levels could still indirectly reflect neuronal viability. Nevertheless, we can be certain that NAA serves some purpose in the brain other than as a useful marker of neuronal viability. Further research is needed to evaluate the possibility that changes in NAA indicate osmotic stress in patients with SCD. This issue could be important in the management of SCD, as dehydration is a known risk factor for stroke (4).
Acknowledgments
We thank the patients and families of St Jude, who dedicated their time and effort to this research; the MR Technologists who did the imaging; and Gisele Hankins, RN, who coordinated this study.
Footnotes
Support by the National Heart, Lung, and Blood Institute (RO1 HL60022, to R.G.S.), Washington, DC, and the American Lebanese Syrian Associated Charities, Memphis, TN.
References
- 1.Martin E, Capone A, Schneider J, Hennig J, Thiel T. Absence of N-acetylaspartate in the human brain: impact on neurospectroscopy? Ann Neurol 2001;49:518–521 [PubMed] [Google Scholar]
- 2.Birken DL, Oldendorf WH. N-acetyl-L-aspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. Neurosci Biobehav Rev 1989;13:23–31 [DOI] [PubMed] [Google Scholar]
- 3.Meyerhoff DJ, MacKay S, Bachman L, et al. Reduced brain N-acetyl-aspartate suggests neuronal loss in cognitively impaired human immuno-deficiency virus-seropositive individuals: in vivo 1H magnetic resonance spectroscopic imaging. Neurology 1993;43:509–515 [DOI] [PubMed] [Google Scholar]
- 4.Zimmerman RA, Wang ZJ. The value of proton MR spectroscopy in pediatric metabolic brain disease. AJNR Am J Neuroradiol 1997;18:1872–1879 [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor JS, Ogg RJ, Langston JW. Proton MR spectroscopy of pediatric brain tumors. Neuroimag Clin N Am 1998;8:753–779 [PubMed] [Google Scholar]
- 6.Taylor DL, Davies SEC, Obrenovitch TP, et al. Investigation into the role of N-acetylaspartate in cerebral osmoregulation. J Neurochem 1995;65:275–281 [DOI] [PubMed] [Google Scholar]
- 7.Baslow MH. Evidence supporting a role for N-acetyl-L-aspartate as a molecular water pump in myelinated neurons in the central nervous system: an analytical review. Neurochem. Int 2002;40:295–300 [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Bogdan AR, Zimmerman R, Gusnard DA, Leigh JS, Ohene-Frempong K. Investigation of stroke in sickle cell disease by 1H nuclear magnetic resonance spectroscopy. Neuroradiology 1992;35:57–65 [DOI] [PubMed] [Google Scholar]
- 9.Steen RG, Emudianughe T, Hankins G, et al. Brain imaging findings in pediatric sickle cell disease patients. Radiology 2003;228:216–225 [DOI] [PubMed] [Google Scholar]
- 10.Glass JO, Reddick WE, Goloubeva O, Yo V, Steen RG. Hybrid artificial neural network segmentation of precise and accurate inversion recovery (PAIR) images from normal human brain. Magn Reson Imaging 2000;18:1245–1253 [DOI] [PubMed] [Google Scholar]
- 11.Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann N Y Acad Sci 1987;508:333–348 [DOI] [PubMed] [Google Scholar]
- 12.Ogg RJ, Kingsley PB, Taylor JS. WET, a T1- and B1-insensitive water-suppression method for in vivo localized 1H NMR spectroscopy. J Magn Reson B 1994;104:1–10 [DOI] [PubMed] [Google Scholar]
- 13.Christiansen P, Henriksen O, Stubgaard M, Gideon P, Larsson HBW. In vivo quantification of brain metabolites by 1H-MRS using water as an internal standard. Magn Reson Imaging 1992;11:107–118 [DOI] [PubMed] [Google Scholar]
- 14.Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson. Med 1993;30:672–679 [DOI] [PubMed] [Google Scholar]
- 15.Olson DP, Hirashima F, Yurgelun-Todd DA, Renshaw PF. Relaxation effects in quantitative MRS in schizophrenia. In: Nasrallah HA, Delisi LE, eds. International Congress of Schizophrenia Research. Colorado Springs: Schizophrenia Research;2003. :244
- 16.Ke Y, Coyle JT, Simpson NS, Gruber SA, Renshaw PF, Yurgelun-Todd DA. Frontal brain NAA T2 values are significantly lower in schizophrenia. In: Nasrallah HA, Delisi LE, eds. International Conference on Schizophrenia Research. Colorado Springs: Schizophrenia Research;2003. :242
- 17.Whittall KP, MacKay AL, Graeb DA, Nugent RA, Li DKB, Paty DW. In vivo measurement of T2 distributions and water contents in normal human brain. Magn Reson Med 1997;37:34–43 [DOI] [PubMed] [Google Scholar]
- 18.Kreis R, Ernst T, Ross BD. Absolute quantitation of water and metabolites in the human brain, II: metabolite concentrations. J Magn Reson B 1993;102:9–19 [Google Scholar]
- 19.de Stefano N, Matthews PM, Arnold DL. Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med 1995;34:721–727 [DOI] [PubMed] [Google Scholar]
- 20.Shimizu H, Shirane R, Fujiwara S, Takahashi A, Yoshimoto T. Proton magnetic resonance spectroscopy in children with moya-moya disease. Clin Neurol Neurosurg 1997;99:S64–S67 [DOI] [PubMed] [Google Scholar]
- 21.Kalra S, Cashman NR, Genge A, Arnold DL. Recovery of N-acetylaspartate in corticomotor neurons of patients with ALS after riluzole therapy. Neuroreport 1998;9:1757–1761 [DOI] [PubMed] [Google Scholar]
- 22.Mascalchi M, Belli G, Guerrini L, Nistri M, Del Sappia I, Vallari N. Proton MR spectroscopy of Wernicke encephalopathy. AJNR Am J Neuroradiol 2002;23:1803–1806 [PMC free article] [PubMed] [Google Scholar]
- 23.Grodd W, Krageloh-Mann I, Petersen D, Trefz FK, Harzer K. In vivo assessment of N-acetylaspartate in brain in spongy degeneration (Canavan’s disease) by proton spectroscopy. Lancet 1990;336:437–438 [DOI] [PubMed] [Google Scholar]
- 24.Takanashi J, Inoue K, Tomita M, Kurihara A, Morita F, Ikehira H, et al. Brain N-acetylaspartate is elevated in Pelizaeus-Merzbacher disease with PLP1 duplication. Neurology 2002;58:237–241 [DOI] [PubMed] [Google Scholar]
- 25.Deicken RF, Eliaz Y, Feiwell R, Schuff N. Increased thalamic N-acetylaspartate in male patients with familial bipolar I disorder. Psychiatr Res Neuroimag 2001;106:35–45 [DOI] [PubMed] [Google Scholar]
- 26.Barker PB. N-acetyl aspartate-a neuronal maker? Ann Neurol 2001;49:423–424 [PubMed] [Google Scholar]
- 27.Steen RG, Xiong X, Mulhern RK, Langston JW, Wang WC. Subtle brain abnormalities in children with sickle cell disease: relationship to blood hematocrit. Ann Neurol 1999;45:279–286 [DOI] [PubMed] [Google Scholar]