

Abstract
BACKGROUND AND PURPOSE: Combined methylmalonic aciduria and homocystinuria (MMA-HC) is caused by impaired hepatic conversion of dietary cobalamin to methylcobalamin and adenosylcobalamin, resulting in decreased activity of methylmalonyl-CoA mutase and methionine synthase. Patients with the early-onset variety present within 12 months of age with severe neurologic, hematologic, and gastrointestinal abnormalities. We describe the neuroradiologic features of early-onset MMA-HC and discuss related pathophysiological mechanisms.
METHODS: Twelve infants with hypotonia, failure to thrive, poor feeding, and hematologic abnormalities were diagnosed with MMA-HC on the basis of a typical plasmatic and urinary metabolic profile and enzyme activity in fibroblastic cultures. Complementation studies were performed in two cases, and yielded a CblC result. MR imaging was performed at presentation in four cases and later in the others. All patients showed prompt biochemical improvement with intramuscular hydroxocobalamin administration, and most had moderate neurologic improvement.
RESULTS: Diffuse supratentorial white matter edema and dysmyelination was the typical MR picture at presentation, whereas white matter bulk loss characterized later stages of the disease. Nucleocapsular areas of gliosis were an additional finding in one case. One patient had tetraventricular hydrocephalus at presentation.
CONCLUSION: White matter damage is probably caused by reduced methyl group availability and nonphysiological fatty acids toxicity, whereas focal gliosis results from homocysteine-induced toxicity to the endothelium. Hydrocephalus may result from diffuse intracranial extracerebral arterial stiffness, known as reduced arterial pulsation hydrocephalus. MR imaging features at presentation and at follow-up are nonspecific.
Combined methylmalonic aciduria and homocystinuria (MMA-HC) is a rare condition resulting from impaired conversion of dietary vitamin B12 or cobalamin (Cbl) to its two metabolically active forms, methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl), in the hepatic cell (1). MeCbl and AdoCbl are essential coenzymes to N5-methyltetrahydrofolate: homocysteine methyltransferase (methionine synthase) and methylmalonyl-CoA mutase, whose functional deficiency results in methylmalonic acidemia and aciduria, hyperhomocysteinemia, homocystinuria, and hypomethioninemia.
Two distinct clinical phenotypes of MMA-HC, differing in age of onset, type of neurologic and systemic impairment, and outcome, are known. Patients with the early-onset variety present within 12 months of age with a host of severe neurologic, hematologic, and gastrointestinal abnormalities. According to Rosenblatt et al (2), an unfavorable outcome is expectable, with a significant percentage of deaths (29.5%) and moderate to severe residual neurologic deficit in survivors. Conversely, patients with the late-onset variety present after 4 years of age with a slightly milder picture dominated by psychomotor regression and acute neurologic dysfunction, and have a more promising outcome (2). The diagnosis is based on plasmatic and urinary amino and organic acid determinations as well as on the measurement of enzyme activity in fibroblastic cultures (2). Genetic heterogeneity within the disease is revealed by complementation studies, which identify three groups (CblC, CblD, and CblF), among which CblC is by far the most common (2). Recently, prenatal diagnosis of this condition based on amino and organic acid determinations on amniotic fluid supernatant has become feasible (3).
To the best of our knowledge, neuroimaging findings of MMA-HC have not been clearly established, and only a single case report has described the results of serial brain MR studies (4). We report a series of 12 infants with early-onset MMA-HC, discuss the clinical and neuroradiologic findings at various stages of the disease, and attempt to provide insight into the related pathophysiological mechanisms.
Methods
From a total of 16 patients who satisfied the criteria for the biochemical diagnosis of MMA-HC, 12 were included in the present study on the basis of availability of MR imaging studies for retrospective evaluation (four patients who had only CT examinations were excluded). The study population comprised seven boys and five girls whose average age at presentation was 1.9 months (range, 3 days to 6 months). The available clinical and neuroradiologic records were evaluated retrospectively, and the relevant information is displayed in the Table 1. Medical attention was sought for poor feeding, failure to thrive, and hypotonia; neurologic examination revealed moderate to severe psychomotor delay in all cases. In one patient (case 3), raised intracranial pressure due to hydrocephalus developed at age 3 months; it is noteworthy that two additional patients from the CT group also had tetraventricular hydrocephalus that required shunting by age 6 months. Patient 2 was diagnosed with hemolytic-uremic syndrome at presentation.
Clinical and neuroradiologic findings in 12 patients with combined methylmalonic aciduria and homocystinuria
All patients were given intramuscular hydroxo-Cbl, and some also received carnitine and betaine orally; their clinical status was recorded during a mean follow-up period of 6.7 years (range, 20 months to 12 years). A variable degree of clinical improvement was recorded in all patients, although all the children exhibited variable residual neurologic impairment with psychomotor retardation, brisk reflexes, nystagmus, and mild to moderate hypotonia.
The diagnosis of MMA-HC was established within 1 month of the clinical presentation in seven cases, and at an average age of 2.7 years (range, 9 months to 6 years) in the remainder. The diagnostic examinations included biochemical measurements from plasma and urine (all cases), measurements of enzyme activity in fibroblastic cultures obtained from skin biopsy specimens (all cases), and complementation studies (two cases).
Plasmatic and urinary biochemical measurements included serum amino acid determinations by ion-exchange chromatography, bidimensional urine amino acid chromatography, and organic acid determinations by gas chromatography/mass spectrometry. The typical metabolite profile was represented by increased urinary excretion of methylmalonic acid and homocystine, increased plasmatic titers of methylmalonic acid and homocysteine, reduced plasmatic concentration of methionine, and normal serum values of Cbl and folic acid.
Enzyme studies were performed on cultured skin fibroblasts, and revealed decreased activity of both methylmalonyl-CoA mutase and N5-methyltetrahydrofolate: homocysteine methyltransferase (methionine synthase) that was restored by addition of hydroxo-Cbl to the culture medium.
Genetic complementation studies were carried out by polyethylene glycol-mediated fusion of patient fibroblast lines with previously classified fibroblast lines of each of the known complementation groups. These studies were performed in two patients (cases 1 and 3), who subsequently were classified into the CblC group, whereas they were not performed in the remainder; however, the measurements obtained from enzyme activity studies were consistent with those typical of the CblC and CblD groups for all patients.
All patients underwent MR imaging studies, which yielded an apparently heterogeneous picture of brain damage, mainly because these examinations were performed at different ages, thereby introducing bias due to the different stages of the disease and the time elapsed since treatment was started. The MR sequence parameters, static magnetic field power, and gradient field strength of the MR scanners varied among the individual cases.
Four patients (cases 1–4) had their initial neuroradiologic studies (MR imaging in three cases and CT in one) performed within 1 month of clinical presentation and before treatment was started; follow-up MR studies over a period of 20 months to 7 years were available for three of these patients, whereas one (case 3) was lost to follow-up. The remaining eight patients (cases 5–12) were first examined by MR imaging at an average age of 6.4 years (range, 3 to 10 years); one of these patients still had no clinical diagnosis at age 5 years (case 12).
Two experienced neuroradiologists independently analyzed the available radiologic material. The readers were blinded to the patient's name, age, and clinical status at the time of each investigation, and the various imaging studies were randomized before being presented to avoid the potential effect of a learning curve. Both interpreters were asked to assess the supratentorial white matter in terms of volume and presence of signal abnormalities. White matter volume was grouped into three categories: increased (swelling due to edema), normal, and reduced (bulk loss). The investigators were also asked to evaluate the presence of focal signal abnormalities in the gray and white matter that could be consistent with cerebrovascular ischemic damage (ie, foci of gliosis) and to assess the size of the ventricular system for the presence of hydrocephalus. Interobserver agreement was evaluated by calculating κ values (5) to determine how often the readers agreed on the above-mentioned points.
Results
Owing to the retrospective nature of this study, the available MR examinations were heterogeneous in nature, making it difficult to evaluate them objectively. However, neuroradiologic studies at presentation (cases 1–4) showed a picture of diffuse supratentorial white matter hypodensity on CT scans and hyperintensity on T2-weighted MR images (Figs 1 and 2). Both interpreters agreed that the affected white matter was moderately swollen with involvement of the U fibers in all cases. In one of the patients (case 3), raised intracranial pressure developed by age 6 months, and the patient was found to have tetraventricular hydrocephalus (Fig 3) whose cause could not be ascertained by means of imaging alone; no follow-up studies were available for this patient.
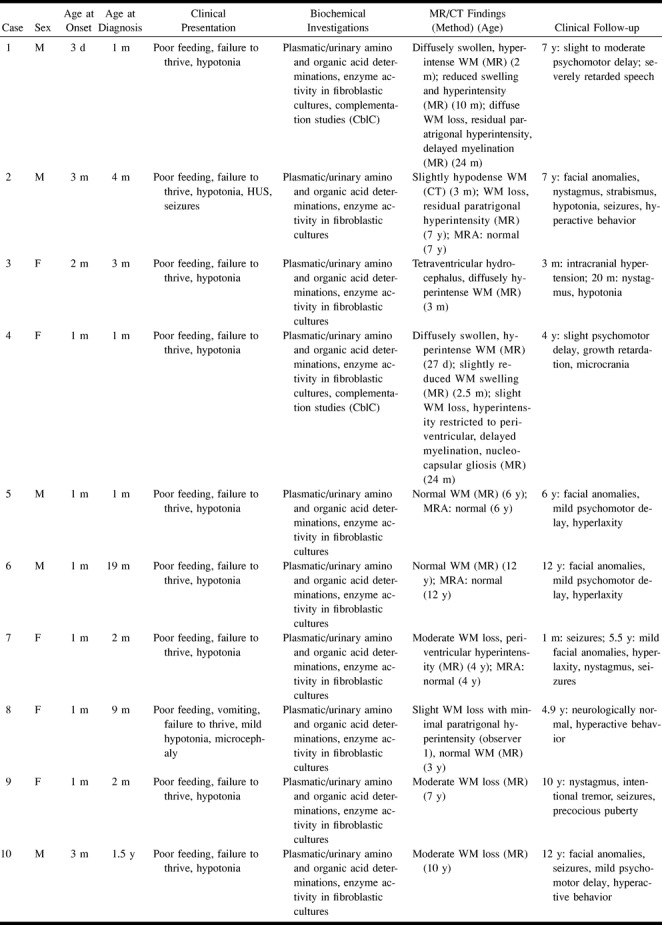
fig 1.
Case 1.
A and B, Axial T2-weighted MR images at 2 months show the supratentorial white matter is markedly edematous and hyperintense, the U fibers are diffusely involved, the basal ganglia are spared, and the ventricular size is normal. Periencephalic CSF collections are still consistent with immaturity of CSF absorption at this age.
C and D, Axial T2-weighted MR images at 10 months show the supratentorial white matter is still abnormally hyperintense, but edema has resolved and a certain degree of white matter loss is becoming apparent, especially around the trigones of the lateral ventricles and in the parietal lobes. The basal ganglia are normal. An arachnoid cyst (asterisk) has developed adjacent to the right frontal lobe.
E and F, Axial T2-weighted MR images at 24 months show the white matter loss is now particularly severe in the paratrigonal areas, where the cortex nearly abuts on the ventricular surface, and in the parietal lobes; however, there is bulk loss throughout the whole supratentorial white matter. There is also delayed myelination, as shown by absent hypointensity in the anterior limbs of both internal capsules. The right frontal arachnoid cyst (asterisk) is essentially unchanged in size.
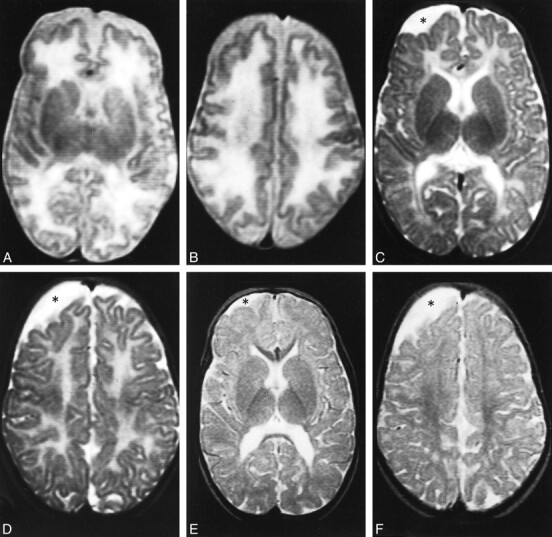
fig 3.

Case 3. Axial T2-weighted MR image at age 3 months shows marked dilatation of the lateral ventricles. The white matter is markedly and diffusely hyperintense whereas the basal ganglia are spared.
fig 4. Case 4.
A and B, Axial proton density–weighted (A) and T2-weighted (B) MR images at 24 months show slight supratentorial white matter loss and delayed myelination with hyperintense areas in the posterior paraventricular regions (arrows). A focal area of gliosis in the nucleocapsular regions is depicted as hyperintense focus both on proton density—and T2-weighted images (arrowhead).
MR studies performed later in the course of the disease (11 patients overall) displayed in most cases a variable degree of supratentorial white matter loss with preferential posterior involvement (Figs 1 and 2); both observers identified white matter bulk loss in eight patients and a normal picture in two, whereas in one case the white matter was judged as mildly atrophic by observer 1 and normal by observer 2. Thus, the interobserver agreement as to white matter size was good (κ = .74). Abnormal signal intensity of the residual supratentorial white matter was considered to be present in six cases and absent in four cases by both observers, whereas there was disagreement between the two readers regarding the presence of slight peritrigonal signal abnormalities in one case. Therefore, agreement on the presence of abnormal hyperintensity of residual periventricular white matter on T2-weighted images was excellent (κ = .81). Delayed myelination was seen on MR examinations performed at age 24 months in two patients (Figs 1 and 4), whereas focal areas of gliosis were detected in the nucleocapsular regions in patient 4 on a follow-up MR study performed at age 24 months (Fig 4).
The severity of white matter loss grossly correlated with the degree of residual psychomotor retardation. In fact, two patients (cases 5 and 6) with particularly mild residual neurologic deficit had essentially normal MR studies, whereas one patient (case 12) who was diagnosed with MMA-HC as late as 6 years of age displayed a particularly severe degree of white matter atrophy as compared with other patients from the same group whose diagnosis and initiation of treatment came earlier. However, we were unable to perform a statistical analysis of such correlation owing to inherent difficulties in objectively quantifying the severity of white matter disease and the different techniques used in the various studies. Therefore, neuroradiologic findings from both patient groups, albeit heterogeneous, appeared to respond to a common pattern; namely, supratentorial white matter involvement with slight to moderate posterior prevalence and involvement of the U fibers. A picture of diffuse white matter edema and abnormal myelination was typical of the initial stages of the disease (100% of cases studied at presentation), whereas tetraventricular hydrocephalus was detected in one patient. A variably severe white matter bulk loss with signal abnormalities affecting the residual paraventricular white matter was found in later stages in the majority of cases. Specifically, white matter bulk loss and residual periventricular hyperintensities were identified in 54.5% of cases by observer 1 and in 63% of cases by observer 2. Nucleocapsular foci of gliosis were detected in one case (8.3%).
Discussion
Disorders of Cbl metabolism can be divided into those associated with deficiency of this essential cofactor, which generally occur in adulthood, and those due to inborn metabolic errors, which usually elicit medical attention in infancy and childhood (6). Cbl is an essential vitamin synthesized in microorganisms, ingested from diverse dietary sources, absorbed from the intestinal mucosa, and transported to the liver along the portal circulation. Once entered in the hepatic cell, Cbl is converted to either MeCbl in the cytosol or to AdoCbl in the mitochondria (6) (Fig 5). MeCbl is an essential cofactor for N5-methyltetrahydrofolate: homocysteine methyltransferase (methionine synthase), an enzyme catalyzing the cytosolic remethylation of homocysteine to methionine, whereas AdoCbl is a cofactor for methylmalonyl-CoA mutase, which catalyzes the intramitochondrial isomerization of methylmalonyl-CoA to succinyl-CoA in the organic and amino acid oxidative process (Fig 5). MMA-HC results from combined impaired hepatic synthesis of MeCbl and AdoCbl, resulting in secondary decreased activity of both methionine synthase (producing increased plasmatic concentration of homocysteine with subsequent homocystinuria) and methylmalonyl-CoA mutase (which results in methylmalonic acidemia and aciduria) (2). Complementation studies are used to further categorize patients with inborn Cbl metabolic disorders. These studies show that patients with MMA-HC belong to groups CblC, CblD, and CblF, among which CblC is by far the most common (2); other complementation groups comprise patients who have isolated methylmalonic acidemia (CblA, CblB) or hyperhomocysteinemia (CblE, CblG) (7). As a consequence, MMA-HC should be considered as an autonomous, well-defined entity that is different from isolated forms of methylmalonic acidemia and hyperhomocysteinemia both on biochemical and genetic grounds. This is reflected in different neuroradiologic features, as will be discussed later.
fig 5.

The metabolism of MMA-HC. The disease results from impaired hepatic conversion of dietary cobalamin (Cbl) to both methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl). Complementation studies individuate three genetic subsets (CblC, CblD, and CblF) within MMA-HC, whereas other complementation groups represent biosynthetic defects that are restricted to either AdoCbl (CblA, CblB) or MeCbl (Cbl E, CblG) and that, as a consequence, represent different diseases. Absence of MeCbl and AdoCbl results in defective activity of methionine synthase (MS) and methylmalonyl-CoA mutase, respectively; the eventual biochemical picture is represented by hyperhomocysteinemia, homocystinuria, hypomethioninemia, methylmalonic acidemia, and methylmalonic aciduria. CBS indicates cystathionine β-synthetase; MTHFR, 5,10-methylenetetrahydrofolate reductase. Modified from (7) and reproduced with permission from Springer, Berlin
Infants with early-onset MMA-HC present with gastrointestinal, hematologic, and neurologic problems; minor facial anomalies (such as long face, high forehead, large, flappy, and low-set ears, and a flat philtrum) often develop as the patients grow (8). Gastrointestinal involvement manifests with feeding difficulties, vomiting, atrophic stomatitis, glossitis, alternating diarrhea and constipation, and failure to thrive (7). Hematologic disorders include megaloblastic anemia, thrombocytopenia, and the hemolytic uremic syndrome (HUS), a pathophysiologically unclear condition characterized by acute renal failure, microangiopathic hemolytic anemia, and thrombocytopenia (9). Neurologic manifestations of early-onset MMA-HC include hypotonia, seizures, psychomotor delay, lethargy, ataxia, brisk reflexes, and optic atrophy; moreover, HUS may produce neurologic complications, such as cerebral ischemic insults with hemiparesis, cortical blindness, seizures, and reduced level of consciousness, that are known to represent significant mortality risk factors in these patients (10, 11). Regardless of the possible association with HUS, the prognosis of early-onset MMA-HC is usually dismal, with a high percentage of deaths and significant neurologic deficits in survivors (2). MMA-HC is treated with intramuscular supplementation of hydroxo-Cbl; betaine and carnitine may be used as additives. Although the biochemical and hematologic response to treatment is usually rapid (7), uncertainty about prognosis and long-term response to treatment complicates both patient care and assessment of the effectiveness of treatment (2).
We found that diffuse supratentorial white matter swelling with prominent hyperintensity on T2-weighted MR images is the most common feature of the early, untreated stages of the disease; later on, variably severe white matter bulk loss, which is more pronounced posteriorly, and T2 hyperintensities of the residual white matter become prominent features, sometimes associated with delayed myelination. The pathophysiological mechanisms underlying the white matter abnormalities detected by MR imaging have been associated with edema and abnormal myelination (7). In fact, myelinopathy is recognized as a dominant pathologic feature in the hyperhomocysteinemias together with vascular abnormalities; however, the relative contribution to the clinical picture varies according to the specific disease (7). In MMA-HC, myelinopathy appears to predominate on vascular damage, as opposed to isolated homocystinuria caused by cystathionine β-synthase (7). The cause of myelin derangement is poorly understood. Evidence suggests that deficiency of S-adenosylmethionine, a crucial methyl donor in the human brain, is strongly related to white matter demyelination; in fact, a relationship has been established experimentally between the presence of demyelination and deficiency of S-adenosylmethionine in the CSF, whereas remyelination under treatment is associated with a return of CSF S-adenosylmethionine levels to normal (7). In MMA-HC, decreased activity of methionine synthase reduces the availability of methionine for conversion to S-adenosylmethionine, possibly providing an explanation for the picture of severe myelinopathy and subsequent white matter loss. Additionally, deficient activity of methylmalonyl-CoA mutase increases the tissue levels of methylmalonyl-CoA and its precursor, propionyl-CoA. As a consequence, nonphysiological fatty acids containing an odd number of carbon atoms are synthesized and incorporated into neuronal lipids; such biochemical abnormality may contribute to the neurologic picture.
The basal ganglia, and particularly the globi pallidi, were free of diffuse signal alterations in all the cases we observed. This represents a distinctive feature with respect to isolated methylmalonic aciduria resulting from methylmalonyl-CoA mutase deficiency, in which T2 prolongation in the globi pallidi due to cytotoxic edema and necrosis represents a characteristic feature (12, 13). The case of MMA-HC reported by Enns et al (4) also displayed pallidal T2 hyperintensity both at 15 and 27 months of age. The involvement of the globi pallidi in methylmalonic aciduria has no definite explanation, although it is probably related to elevated metabolic levels and high energy demands, resulting in greater vulnerability during metabolic insults (12, 13). According to our experience, the involvement of these nuclei is usually negligible in MMA-HC as compared with isolated methylmalonic aciduria, and progression to pallidal necrosis does not ensue.
As was stated previously, cerebrovascular disease is a well-known feature of hyperhomocysteinemias, although its contribution to the overall clinical and radiologic picture is variable, depending on the specific underlying metabolic derangement. Experimental studies (14) have shown that excess homocysteine generates superoxide and hydrogen peroxide (which have been linked to damage to arterial endothelium), changes coagulation factor levels so as to encourage clot formation, prevents small arteries from dilating so they are more vulnerable to obstruction, and causes smooth muscle cells in the arterial wall to multiply. To our knowledge, cerebral infarcts and vascular thrombosis have not been reported in patients with inborn errors of Cbl metabolism, as opposed to hyperhomocysteinemia due to cystathionine β-synthase deficiency, in which vascular disease dominates. This is probably related to lower plasmatic concentration of homocysteine (7). We only found one case in which focal areas of gliosis in the nucleocapsular regions were associated with the typical picture of supratentorial white matter involvement; however, neither stroke nor transient ischemic attacks were recorded in the patient's clinical history. We surmise that minor ischemic episodes contribute only rarely to the pathologic and neuroradiologic picture of MMA-HC, and that the severity of such episodes is unlikely to significantly affect the clinical course of the disease.
Although one patient in our series had HUS, neither clinical nor radiologic evidence of brain ischemia was found. HUS is defined as a triad of azotemia, thrombocytopenia, and microangiopathic hemolytic anemia, often preceded by a prodrome of hematic diarrhea (15). It is known that HUS is part of the phenotypic spectrum of MMA-HC (2, 16), but the underlying pathophysiological mechanism remains obscure, although endothelial damage of glomerular capillaries is likely to play a critical role (9). Brain complications of HUS include cortical and basal ganglia infarctions that are often hemorrhagic, and reversible abnormal T2 prolongation in the callosal splenium, representing local edema (17); however, these brain complications occur only in 20% of children with HUS (10). As a consequence, it is not surprising that we found no evidence of brain ischemia in our patient with HUS, although HUS may potentially cause brain ischemic disease.
One of the patients included in our study (case 3) presented with raised intracranial pressure due to hydrocephalus; moreover, it is noteworthy that two other children who were not included in the study because of the unavailability of MR examinations had tetraventricular hydrocephalus at or shortly after presentation. Hydrocephalus was previously described as the presenting sign of MMA-HC in two cases in the literature, both of whom had concurrent HUS (9, 18); however, HUS was not present in our patient. It is difficult to explain why hydrocephalus occurs in this disease if one takes into consideration only the traditional theories of CSF production, circulation, and absorption; it is noteworthy that previous authors have not attempted any such explanation (9, 18). Recently, however, Greitz and Greitz (19) published a new theory of CSF dynamics and the development of hydrocephalus (Fig 6). According to their theory, CSF is produced by the choroid plexus, flows in bulk through the aqueduct, and exits the ventricles through the fourth ventricular foramina; however, once in the subarachnoid space, CSF is mixed and dispersed evenly by pulsations of the intracranial extracerebral arteries and is subsequently absorbed throughout the brain and spinal cord via their capillaries, rather than through the arachnoid villi as was previously believed. Additionally, rhythmic pulsations of the intracranial extracerebral arteries occurring at each heartbeat result in the arterial pulse being damped, so that the intraarterial pressure transmitted downstream to the brain is reduced. The normally damped intracranial arterial pressure that is propagated downstream produces an intracerebral pulse pressure and a pulsatile transcerebral mantle pressure gradient, which is responsible for keeping the ventricles patent and of normal size. According to these authors, communicating hydrocephalus may be produced by whatever disorder results in increased arterial stiffness, and may therefore be termed reduced arterial pulsation hydrocephalus (19). In fact, pathologic processes, such as arteriopathy or leptomeningitis, that result in increased stiffness of the intracranial extracerebral arteries cause the arterial pulse pressure to be propagated downstream undamped, elevating the intracerebral pressure and the transcerebral mantle pressure gradient, thus leading to ventricular dilatation and higher intraventricular pulse pressure. We speculate that the toxic effect of homocysteine to the arterial wall, albeit insufficient to produce significant focal ischemic damage, could slightly reduce the compliance of extracerebral intracranial arteries, thereby producing reduced arterial pulsation hydrocephalus through the mechanisms explained above. Obviously, further experimental studies are needed to prove whether such a hypothesis is tenable.
fig 6.

Greitz and Greitz's theory on the pathogenesis of hydrocephalus.
A, Normal systole. The systolic pulse wave causes a large expansion of the arteries with a concomitant and successful dampening of the arterial pressure. The pressure is immediately transmitted to the entire subarachnoid space. After some delay, a small pressure wave is transmitted to the brain via the intracerebral arteries. This causes a slight brain expansion and a transcerebral mantle pressure gradient of normal magnitude, which keeps the ventricles patent and of normal size.
B, Hydrocephalus systole. The arterial pulsations are restricted, so little or no dampening of the arterial pressure occurs. The undamped pulse pressure is therefore transmitted into the brain, giving rise to an increased transcerebral mantle pressure gradient. Ventricular dilatation results. Reproduced from (19) with permission from Lippincott Williams & Wilkins.
We are aware of several important limitations to our study. First, as this was a retrospective review, the type and number of imaging examinations available for review were very heterogeneous. Although we saw a total of 16 patients with MMA-HC, we decided to exclude four children who only underwent CT in an effort to maximally homogenize our neuroradiologic series. However, the stage of the disease at which MR examinations were performed and the time elapsed since the initiation of treatment varied significantly among patients, and serial studies ranging from very early to late in the disease were available in only a small fraction; therefore, we were unable to conclusively establish that MR imaging is the method of choice and that it should be used to follow the results of therapy. Moreover, because many of the MR studies were performed in late stages of the disease, we were unable to determine what exactly is the natural history of disease progression and how treatment affects it.
Second, proton MR spectroscopy was not performed. To our knowledge, only one study in the literature reported on MR spectroscopic findings in patients with MMA-HC (20); in one patient from that study, MR imaging showed abnormal signal in both basal ganglia, and the proton MR spectrum obtained from those regions showed decreases in N-acetyl aspartate to 3.8 mmol/L, whereas both MR imaging and MR spectroscopy were normal in the other patient. To date, there is not sufficient evidence to suggest that MR spectroscopy may play a significant role in the diagnosis of this disease; however, we surmise that it could prove useful, especially in the earlier stages when abnormal T2 prolongation becomes apparent in the supratentorial white matter. Further studies are needed to identify a precise role for MR spectroscopy in the diagnostic work-up of MMA-HC patients.
Third, MR studies of the spinal cord were not performed. Spongiform white matter degeneration and demyelination of the dorsal and lateral columns of the cord (so-called subacute combined degeneration of the spinal cord) is a known complication of severe Cbl deficiency (21), which, however, has become exceedingly rare and is unlikely to occur if treatment is initiated early. Nevertheless, we acknowledge that patients with MMA-HC should also undergo spinal cord MR imaging to obtain a complete neuroradiologic picture of their disease at presentation.
Conclusion
It is known that survivors with MMA-HC show a variable, often severe degree of residual neurologic dysfunction, mild to moderate mental retardation, and delayed development of motor skills; however, it has been questioned whether these findings result from delayed diagnosis and administration of proper treatment or, rather, from the inadequacy of current treatment strategies to correct the intracellular defect of Cbl metabolism, which may be critical in neurologic development (22). Because all patients in our series had similar clinical presentations and were given the same treatment, the fact that white matter bulk loss was not a consistent feature of late disease stages could reflect biochemical or genetic heterogeneity among affected patients. Although current therapeutic schemes probably fail to halt the pathologic progression of the disease, they succeed in at least reducing the severity and rapidity of such progression in most cases, as is suggested by the increased severity of the late-stage clinical and neuroradiologic picture among patients in whom diagnosis and initiation of treatment come late.
We found that MR imaging features of MMA-HC at presentation are nonspecific and do not permit differentiation from other metabolic leukoencephalopathies; therefore, the diagnosis relies heavily on clinical and laboratory investigations. Owing to the retrospective nature of the present study and the paucity of patients who were followed up serially with MR imaging, we were prevented from making definitive conclusions about the usefulness of MR imaging in the diagnosis and follow-up; prospective studies are awaited to prove our suggestion that serial MR imaging can be useful for monitoring the course of the disease and the response to treatment. Small infants with MMA-HC may rarely present with tetraventricular hydrocephalus; this disease should therefore be considered in the event of an otherwise unexplained hydrocephalus in the first 6 months of life. Finally, the determination of plasmatic methylmalonic acid and homocysteine titers should be part of the routine clinical investigation of failure to thrive, neurologic disorders, and unexplained anemia or cytopenia in children (2).
TABLE 1:
Continued
fig 2.
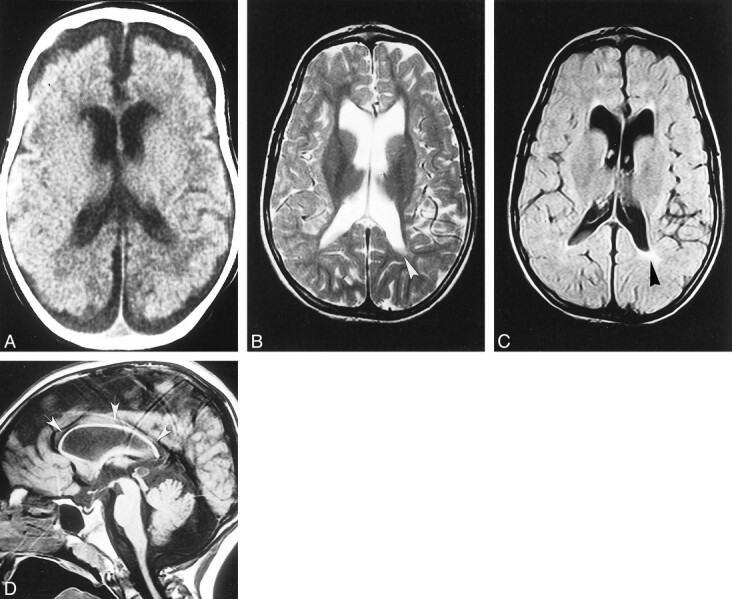
Case 2.
A, Unenhanced CT scan at age 3 months shows the supratentorial white matter is probably slightly hypodense, especially in the paraventricular and subcortical regions; however, swelling is not particularly pronounced in this case. Both the ventricles and the subarachnoid spaces are slightly dilated, consistent with immaturity of CSF absorption at this age.
B–D, Axial T2-weighted (B), axial fluid-attenuated inversion-recovery (C), and sagittal T1-weighted (D) MR images at 7 years show marked and diffuse bulk loss in the supratentorial white matter; the cortex nearly abuts on the ventricular surface. Slight residual hyperintensity is seen in the periventricular regions, especially posterior to the left trigone (arrowheads, B and C). The basal ganglia are spared, and the lateral ventricles are slightly enlarged ex-vacuo. Notice marked, diffuse thinning of the corpus callosum (arrows, D).
Acknowledgments
We thank B. Fowler, Head of Laboratories, Basler Kinderspital, Basel, Switzerland, for performing enzyme activity assays on cultured fibroblast lines from our patient cohort, and D. Greitz, MR Research Center, Karolinska Hospital, Stockholm, Sweden, for fruitful discussion about the pathogenesis of hydrocephalus and its possible implications in MMA-HC.
Footnotes
Presented at the annual meeting of the American Society of Neuroradiology, Atlanta, April 2000.
Address reprint requests to Andrea Rossi, MD, Department of Pediatric Neuroradiology, G. Gaslini Children's Research Hospital, I-16147 Genova, Italy.
References
- 1.Baumgartner ER, Wick H, Maurer R, Egli N, Steinmann B. Congenital defect in intracellular cobalamin metabolism resulting in homocystinuria and methylmalonic aciduria. Helv Paediat Acta 1979;34:465-482 [PubMed] [Google Scholar]
- 2.Rosenblatt DS, Aspler AL, Shevell MI, Pletcher BA, Fenton WA, Seashore MR. Clinical heterogeneity and prognosis in combined methylmalonic aciduria and homocystinuria (cblC). J Inherit Metab Dis 1997;20:528-538 [DOI] [PubMed] [Google Scholar]
- 3.Merinero B, Perez-Cerda C, Garcia MJ, et al. Reliability of biochemical parameters used in prenatal diagnosis of combined methylmalonic aciduria and homocystinuria. Prenat Diagn 1998;18:947-952 [PubMed] [Google Scholar]
- 4.Enns GM, Barkovich AJ, Rosenblatt DS, et al. Progressive neurological deterioration and MRI changes in cblC methylmalonic acidemia treated with hydroxocobalamin. J Inherit Metab Dis 1999;22:599-607 [DOI] [PubMed] [Google Scholar]
- 5.Landis R, Koch G. The measurement of observer agreement for categorical data. Biometrics 1977;33:159-174 [PubMed] [Google Scholar]
- 6.Shinnar S, Singer HS. Cobalamin C mutation (methylmalonic aciduria and homocystinuria) in adolescence: a treatable cause of dementia and myelopathy. New Engl J Med 1984;311:451-454 [DOI] [PubMed] [Google Scholar]
- 7.van der Knaap MS, Valk J. Magnetic Resonance of Myelin, Myelination, and Myelin Disorders. 2nd ed. Berlin: Springer 1995;223-230
- 8.Cerone R, Schiaffino MC, Caruso U, Lupino S, Gatti R. Minor facial anomalies in combined methylmalonic aciduria and homocystinuria due to defect in cobalamin metabolism. J Inherit Metab Dis 1999;22:247-250 [DOI] [PubMed] [Google Scholar]
- 9.Geraghty MT, Perlman EJ, Martin LS, et al. Cobalamin C defect associated with hemolytic-uremic syndrome. J Pediatr 1992;120:934-937 [DOI] [PubMed] [Google Scholar]
- 10.Hahn JS, Havens PL, Higgins JJ. Neurological complications of hemolytic-uremic syndrome. J Child Neurol 1989;4:108-113 [DOI] [PubMed] [Google Scholar]
- 11.Sheth KJ, Swick HM, Haworth N. Neurological involvement in hemolytic-uremic syndrome. Ann Neurol 1986;19:90-93 [DOI] [PubMed] [Google Scholar]
- 12.Andreula CF, De Blasi R, Carella A. CT and MRI studies of methylmalonic acidemia. AJNR Am J Neuroradiol 1991;12:410-412 [PMC free article] [PubMed] [Google Scholar]
- 13.Brismar J, Ozand PT. CT and MR of the brain in disorders of the propionate and methylmalonate metabolism. AJNR Am J Neuroradiol 1994;15:1459-1473 [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson JC, Spence JD. Vitamins and progression of atherosclerosis in hyper-homocyst(e)inaemia. Lancet 1998;24:263. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan BS, Thomson PD, de Chadarevian JP. The hemolytic uremic syndrome. Pediatr Clin North Am 1976;23:761-777 [DOI] [PubMed] [Google Scholar]
- 16.Cerone R, Barbano G, Maritano L, et al. Syndrome hemoltytique et uremique neonatal, acidurie methylmalonique et homocystinurie par deficit intracellulaire de la vitamine B12. Arch Pediatr 1994;1:762-763 [PubMed] [Google Scholar]
- 17.Ogura H, Takaoka M, Kishi M, et al. Reversible MR findings of hemolytic uremic syndrome with mild encephalopathy. AJNR Am J Neuroradiol 1998;19:1144-1145 [PMC free article] [PubMed] [Google Scholar]
- 18.Traboulsi EI, Silva JC, Geraghty MT, Maumenee IH, Valle D, Green WR. Ocular histopathologic characteristics of cobalamin C type vitamin B12 defect with methylmalonic aciduria and homocystinuria. Am J Ophthalmol 1992;113:269-280 [DOI] [PubMed] [Google Scholar]
- 19.Greitz D, Greitz T. The pathogenesis and hemodynamics of hydrocephalus: proposal for a new understanding. Int J Neuroradiol 1997;3:367-375 [Google Scholar]
- 20.Lam WWM, Wang ZJ, Zhao H, et al. 1H MR spectroscopy of the basal ganglia in childhood: a semiquantitative analysis. Neuroradiology 1998;40:315-323 [DOI] [PubMed] [Google Scholar]
- 21.Katsaros VK, Glocker FX, Hemmer B, Schumacher M. MRI of spinal cord and brain lesions in subacute combined degeneration. Neuroradiology 1998;40:716-719 [DOI] [PubMed] [Google Scholar]
- 22.Bellini C, Cerone R, Bonacci W, et al. Biochemical diagnosis and outcome of 2 years treatment in a patient with combined methylmalonic aciduria and homocystinuria. Eur J Pediatr 1992;151:818-820 [DOI] [PubMed] [Google Scholar]