

Abstract
Background
The mechanism underlying endothelial dysfunction leading to cardiovascular disease in type 2 diabetes mellitus (T2DM) remains unclear. Here, we show that inhibition of histone deacetylase 3 (HDAC3) reduced inflammation and oxidative stress by regulating nuclear factor-E2-related factor 2 (Nrf2), which mediates the expression of anti-inflammatory- and pro-survival-related genes in the vascular endothelium, thereby improving endothelial function.
Methods
Nrf2 knockout (Nrf2 KO) C57BL/6 background mice, diabetic db/db mice, and control db/m mice were used to investigate the relationship between HDAC3 and Nrf2 in the endothelium in vivo. Human umbilical vein endothelial cells (HUVECs) cultured under high glucose-palmitic acid (HG-PA) conditions were used to explore the role of Kelch-like ECH-associated protein 1 (Keap1) –Nrf2–NAPDH oxidase 4 (Nox4) redox signaling in the vascular endothelium in vitro. Activity assays, immunofluorescence, western blotting, qRT-PCR, and immunoprecipitation assays were used to examine the effect of HDAC3 inhibition on inflammation, reactive oxygen species (ROS) production, and endothelial impairment, as well as the activity of Nrf2-related molecules.
Results
HDAC3 activity, but not its expression, was increased in db/db mice. This resulted in de-endothelialization and increased oxidative stress and pro-inflammatory marker expression in cells treated with the HDAC3 inhibitor RGFP966, which activated Nrf2 signaling. HDAC3 silencing decreased ROS production, inflammation, and damage-associated tube formation in HG-PA-treated HUVECs. The underlying mechanism involved the Keap1–Nrf2–Nox4 signaling pathway.
Conclusion
The results of this study suggest the potential of HDAC3 as a therapeutic target for the treatment of endothelial dysfunction in T2DM.

Video Abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s12964-020-00681-z.
Keywords: T2DM, Endothelial dysfunction, HDAC3, Nrf2
Background
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder characterized by reduced insulin action, increased hepatic glucose production, and the development of diabetic vascular complications [1]. Globally, approximately 425 million people are affected by T2DM [2]. Diabetic vasculopathy (DV) is a long-term complication of diabetes [3]. Endothelial dysfunction is the first step in the development of DV, and is associated with increased oxidative stress and inflammation [4, 5]. Developing effective methods to prevent or retard the progression of DV associated with oxidative stress is critical. Aberrant production of reactive oxygen species (ROS) activates nuclear factor-κB (NF-κB) [6], which plays a critical regulatory role in the release of pro-inflammatory cytokines, such as interleukin (IL)-1β, IL6, IL8, and tumor necrosis factor-alpha (TNF-α), as well as vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule-1 (ICAM-1) [7]. ROS are produced continuously as natural byproducts of the normal metabolism of oxygen and play important roles in redox signaling [8, 9]. Cells have evolved highly regulated endogenous antioxidant defense systems to counteract ROS overproduction, and the ability of endothelial cells to react to diabetic-like conditions by upregulating antioxidant responses has been investigated [10, 11].
Nuclear factor-E2-related factor 2 (Nrf2) is a redox-sensitive master regulatory transcription factor that plays a protective role in endothelial cells and resides in the cytoplasm bound to Kelch-like ECH-associated protein 1 (Keap1) under normal conditions [12]. Upon Keap1 destabilization or downregulation, the Keap1–Nrf2 complex is disrupted, resulting in the failure of Keap1–Nrf2 binding and nuclear translocation of Nrf2. In the nucleus, Nrf2 binds to antioxidant response elements and regulates the expression of genes encoding phase II detoxifying enzymes such as NAD(P)H dehydrogenase quinone 1, 2 (NQO1, NQO2), superoxide dismutase 2 (SOD2), heme oxygenase 1 (HO1), and catalase (CAT). This results in the production of antioxidants that act as scavengers for diabetes-induced free radicals and alleviate DV [13, 14].
A major defense against vascular injury is endothelial nitric oxide synthase (eNOS), which generates nitric oxide (NO) in the presence of optimal concentrations of the substrate L-arginine and the cofactor (6R)-5,6,7,8-tetrahydrobiopterin [15, 16]. Progressive vasculopathy is associated with NO deficiency caused by eNOS dysfunction [17, 18], a phenomenon referred to as “uncoupling”. Uncoupling of eNOS results in decreased bioavailability of NO as well as increased generation of ROS [19, 20]. However, the mechanisms by which eNOS is inactivated in DV remain unclear.
NADPH oxidase (Nox) is an enzyme complex that catalyzes the transfer of electrons across biological membranes, resulting in ROS production [21, 22]. The Nox complex consists of two membrane-bound catalytic subunits (p22phox and a Nox protein) and several cytosolic regulatory subunits (p47phox, p67phox, p40phox, and the small GTPase Rac) [23]. Nox4 is the predominant Nox isoform in endothelial cells, and its expression is 100‐fold higher than that of Nox1, Nox2, or Nox5 [24]. Nox family members generate superoxide, whereas Nox4 produces predominantly hydrogen peroxide [25]. Because the activity of Nox4 depends mainly on its expression level [26], abnormal expression of Nox4 affects cellular proliferation and apoptosis and is associated with the development of cardiovascular pathologies [27, 28].
Histone deacetylase (HDAC)-3 is a class I HDAC that is involved in tumor development, DM, inflammation, and cardiovascular and neurodegenerative diseases [29]. Small interfering RNA (siRNA)-mediated knockdown of HDAC3 prevents the binding of transcription factor(s) and polymerase(s) to the Nox4 promoter, thereby decreasing Nox4 transcription and Nox4-mediated ROS production in human endothelial cells [30]. In addition, Nox4–Nrf2 redox imbalance is associated with vascular endothelial cell dysfunction [21]. These findings led us to hypothesize that T2DM-induced inactivation of Nrf2 signaling caused by HDAC3 inhibition may contribute to the transcriptional regulation of Nox4.
Here, we show that siRNA-mediated inhibition of HDAC3 impaired the binding of Keap1 to Nrf2 by suppressing the synthesis of Keap1, thereby increasing Nrf2 levels by preventing Nrf2 ubiquitination and proteasomal degradation. The protective effect of HDAC3 inhibition in diabetes can be partly attributed to reduced eNOS uncoupling and activation of Nrf2 signaling through the modulation of Nox4–Nrf2 redox imbalance.
Materials and methods
Animals
Diabetic db/db mice and their control littermates, db/m, were obtained from Jackson Laboratories (Strain: BKS.Cg-Dock7m+/+Leprdb/J). 8 week old db/db mice were separated into 4 groups and subjected to the following treatment regimens: 1) ad libitum feeding of chow diets; 2) ad libitum feeding of chow diets and vehicle injection of dimethyl sulfoxide (DMSO; Sigma-Aldrich, D8418); 3) ad libitum feeding of chow diets and RGFP966 (MCE, HY-13909) 10 mg/kg was subcutaneously injected into the mice every other day for 10 weeks[31]. RGFP966′s formula is C21H19FN4O which is a highly selective HDAC3 inhibitor with an IC50 of 80 nM and shows no inhibition to other HDACs at concentrations up to 15 μM. RGFP966 was dissolved in DMSO and diluted in 30% hydroxypropyl beta-cyclodextrin and the final DMSO concentration was no more than 1%; 4) ad libitum feeding of chow diets and GKT137831 (Selleck, S7171) by daily gavage at a dose of 60 mg/kg/day for 10 weeks[32]. The GKT137831′s formula is C21H19ClN4O2, a member of the pyrazolopyridine dione family, which is an effective dual Nox1/4 inhibitor with an IC50 of 110/140 nM. It is worth pointing out that Nox4 was the predominant Nox isoform in endothelial cells, and its expression was 100‐fold higher than that of Nox1, Nox2, or Nox [24] and the GKT137831 was a widely used Nox4 inhibitor for animal administration [33, 34] as there is no specific Nox4 pharmacological inhibitor until now.
Additionally, mice with global Nrf2 knockout (Nrf2 KO) and their wild-type (WT) C57BL/6 control mice were purchased from Jackson laboratory. C57BL/6 mice were obtained from Model Animal Research Center of Nanjing University. Nrf2 KO mice with the T2DM background were established by feeding a high fat diet and by intraperitoneal streptozotocin injection (STZ) (Sigma-Aldrich, V900890). HFD (Medicience Ltd., Jiangsu, China, 21.9 kJ/g, 60% of energy as fat; MD12033), a month after exposure to the diets, the HFD-fed mice were administrated with 40 mg/kg/d STZ by intraperitoneal injection after overnight fasting for 5 consecutive days and normal diet mice were received the same volume of citrate buffer[35]. To assess the role of Nrf2 in T2DM, The randomized animal division results in 3 treatment groups in total. (1) HFD-STZ-induced mice were administrated with same volume of vehicle for 10 weeks. (2) HFD-STZ-induced T2DM mice were administrated with RGFP966 10 mg/kg subcutaneous injection for 10 weeks. (3) Nrf2 KO with HFD-STZ-induced T2DM mice were administrated with RGFP966 10 mg/kg subcutaneous injection for 10 weeks. After treatment, corresponding analyses were performed. All male mice were kept in a standard laboratory condition of temperature 21 ± 2℃, relative humidity 50 ± 15%, 12 h light-darkness cycles, with water and food available ad libitum. All animal experiments and methods performed in this study followed ethical guidelines for animal studies and were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University, China.
Cell culture
HUVECs was purchased from Lonza, Basel, Switzerland and cultured in endothelial cell growth medium-2 (Lonza, EGM-2, CC-3156 & CC-4176) supplemented with 10% FBS and 1% penicillin/streptomycin in an incubator containing 95% air and 5% CO2 at 37℃ until the start of experiment. Before starting the experimental procedures, the medium was removed and replaced with phenol red-free low-glucose DMEM (Gibco BRL, 11054020) supplemented with 1% calf serum (Gibco BRL, 16010159) for 12 h, and then HUVECs were treated with EGM-2 consisting of either NG (5.5 mM) or HG (33 mM)—PA (100 μM) in the presence or absence of si-HDAC3 for 72 h; palmitic acid (Sigma-Aldrich, PA, 100 μM; P5585), NG was used as control in this study; mannitol (Sigma-Aldrich, MAN, 33 mM: 5.5 mM of glucose + 27.5 mM of D-mannitol; M4125) was served as the osmotic control for the HG-PA, pharmacological antioxidant molecules N-Acetyl-L-cysteine (Sigma-Aldrich, NAC, 2 mM; V900429) was pretreated for 2 h every day to evaluate the effect of si-HDAC3 on the oxidative stress. Media were changed every 24 h. The NOS inhibitor L-NAME (MCE, 100 μM; HY-18729A) co-incubated with HG-PA for 72 h was used to evaluate ROS accumulation during T2DM progress in vitro. The Sulforaphane (Selleck, SFN, 10 μM; S5771) treatment for 72 h, was used to facilitate Nrf2 nuclear translocation which were used positive control to reconfirm si-HDAC3 cytoprotection through the similar mechanism. Ubiquitination of Nrf2 were detected using immunoprecipitation after treatment with MG132 (Selleck, 5 μM; S2619) pretreatment for 10 h before si-HDAC3 administration.
For RNA interference, cells were transfected with human HDAC3 siRNA (si-HDAC3) (Santa Cruz Biotechnology, sc-355380), human Nrf2 siRNA (si-Nrf2) (Santa Cruz Biotechnology, sc-37030), human Nox4 siRNA (si-Nox4) (Santa Cruz Biotechnology, sc-41586) or control scramble siRNA by Lipofectamine 3000 for 12 h in Opti-MEM. After the transfection, cells were removed to full-growth medium for another 12 h and then analyzed for further studies.
TUNEL staining
The cells were then stained with an In Situ Cell Death Detection Kit (Roche, 11684795910) according to the manufacturer’s protocol. The stained cells were imaged with a confocal laser scanning microscope (TCS SP8, Leica). One hundred cells per field were counted and the percentage of TUNEL-positive cells was calculated.
In vitro angiogenesis (Tube formation) assay [36]
The in vitro angiogenic activity of HUVECs was determined by Matrigel tube formation assay. After the experimental period described above, the HUVECs were stained with cell-permeable dye, calcein (Corning, 354216), for 30 min and replated in 24-well plates precoated with 150 μL/well growth factor-reduced Matrigel (Corning, 354234) and incubated at 37℃ in cell culture incubator. After 12 h of incubation, capillary-like tube formation was observed with a computer-assisted microscope (Thermo Fisher Scientific, EVOS). Tube formation was defined as a tube-like structure exhibiting a length four times its width. The tube length in duplicate wells was counted and averaged using ImageJ software.
Aortic ring assays
To establish a direct action of HDAC3 on vascular, thoracic aortae from 8-week-old C57/BL6 mice were surgically isolated, cleaned and dissected into 0.5 mm rings. Rings were embedded in 1 mg/mL of type I collagen (Millipore, 08–115) in a 96-well plate as described previously [37]. When embedded, the rings were cultured in serum-free endothelial basal medium (EBM) (Lonza, CC-3121) consisting of either NG or HG-PA in the presence or absence of si-HDAC3. Endothelial microvessel sprouts growing out from the rings were counted during the exponential growth phase to obtain angiogenic response data. Before the regression phase, rings were fixed with 4% (w/v) paraformaldehyde for 30 min. Pictures were taken on day 12 with a computer assisted microscope (Eclipse Ni, NIKON), and the total number of branches was counted using ImageJ (National Institutes of Health).
Isolation of mouse aortic endothelial cells
Following the instructions by the established method for aortic endothelial cells isolation in mouse [38], briefly, the thoracic aortae of mice were surgically isolated and carefully rinsed free of blood with PBS containing 1000 U/mL heparin (Selleck, S1346), and immersed in 20% FBS-DMEM (1 g/L) containing 1000 U/mL heparin. The fat and connecting tissue were quickly removed under a stereoscopic microscope. The inside of the lumen was washed with serum-free DMEM (1 g/L), filled with collagenase II (Sigma-Aldrich, 2 mg/mL; C6685), and incubated at 37℃, for 45 min. Endothelial cells were removed by flushing with 5 mL 20% FBS-DMEM (1 g/L), and collected by centrifuging at 1200 rpm, for 5 min. The cells were then suspended gently with 20% FBS-DMEM (1 g/L), and seeded in a type I collagen-coated dish. After 7 to 10 days, confluent endothelial cells were observable.
HDAC3 activity assay
HDAC3 activity was assessed using the HDAC3 activity assay kit (Sigma, EPI004) according to the manufactures’ protocols.
DHE staining
Dihydroethidium (DHE) (Thermo Fisher Scientific, D11347) staining was performed as previously described [39]. HUVECs cultured in different medium stained with DHE (25 μΜ) for 30 min at 37 °C, then medium was replaced by low-glucose DMEM to monitor the generation of ROS. Pictures were captured using a computer-assisted microscope.
Immunofluorescence staining
For quantification of CD31, 3-NT or Ki67 positive area, immunofluorescence staining was performed within the aortic wall. Eight micrometer paraffin sections were cut and incubated with anti-CD31 (Abcam, ab28364) or anti-3-NT (Abcam, ab61392) or anti-Ki67 (CST, 12,075). After washing, samples for CD31 were incubated with Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (Abcam, ab150077) at a dilution of 1:200 for 60 min at room temperature, and samples for 3-NT were incubated with Alexa Fluor 647-conjugated anti-mouse IgG secondary antibody (Abcam, ab150115) at a dilution of 1:200 for 60 min at room temperature. The anti-Ki67 and TUNEL staining have their own fluorescent label, and aortic rings were conducted the Ki67 and TUNEL staining according to protocol. Cell nuclei were labeled by DAPI. The stained aortic walls were imaged with a confocal laser scanning microscope (Leica, TCS SP8). The total tissue area and the CD31, 3-NT or Ki67-stained positive area was measured with ImageJ software. Data were expressed as percentage of positive staining area per analyzed area.
For quantification of nuclear localization of Nrf2 in HUVECs, briefly, cultured cells in six-well plates were fixed with 4% (w/v) paraformaldehyde for 30 min and permeabilized with 0.5% (v/v) Triton-X 100 (Solarbio, Beijing, China) for 10 min at 37℃. After wash, the cells were blocked in 5% (v/v) BSA for 2 h and incubated with primary rabbit antibody against Nrf2 (Abcam, ab31163) at a dilution of 1:200 at 4 °C overnight. After incubation and wash, cells were blocked with Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibody (Abcam, ab150077) for 1 h. After washes with PBS, the cell nuclei were stained with DAPI for 19 min. Photo capture was performed by a confocal laser scanning microscope. Randomly selected the perspective nuclear localization of Nrf2 and DAPI cells were under the microscope.
Western blotting and antibodies
Equal amounts of protein lysates from HUVECs was harvested and lysed by SDS-PAGE and transferred to PVDF membrane (Merck Millipore, IPVH00010), then subjected to western blotting analysis: Membranes were blocked with 5% (v/v) bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% (v/v) Tween 20 (TBST) and incubated with primary antibodies overnight at 4℃. The proteins were visualized using an Image Quant LAS 4000 (GE Healthcare) system, and the secondary antibodies are: goat anti-rabbit HRP (Bio-Rad, 1706515), goat anti-mouse HRP (Bio-Rad, 1706516).
For nuclear Nrf2 accumulation assays, HUVECs were harvested and lysed to obtain cytoplasmic and nuclear lysates using the Keygen Nuclear-Cytosol Protein Extraction Kit from Nanjing KeyGen Biotech. Co., Ltd. (Nanjing, China).
The primary antibodies used to probe the membranes included Nrf2 (Santa Cruze Biotechnology, sc365949), Bax (Abcam, ab32503) and Bcl-2 (Abcam, ab59348) and GAPDH (CST, 5174), Lamin B (CST, 12,586), Keap1 (CST, 4678), c-Caspase-3 (CST, 9661), HDAC3 (CST, 85057), eNOS (CST, 32027), acetylated-lysine (CST, 9441), 3-NT (Millipore, 05–233), Nox4 (Origene, TA349083).
Assay of eNOS dimer/monomer
SDS-resistant eNOS dimers and monomers were assayed by using low-temperature SDS-PAGE as described previously [40]. Briefly, after being washed twice with ice-cold phosphate-buffered saline, D-glucose-treated HUVECs were lysed as described above, and protein lysates were mixed with loading buffer and loaded on gels without boiling. Proteins were separated with low-temperature SDS-PAGE under reducing conditions (with -mercaptoethanol). Gels and buffers were kept at 4℃ during the whole procedure.
Co-immunoprecipitation
Cell lysates with 500 μg proteins were incubated with 20 μL PureProteome™ Protein A/G Mix Magnetic Beads (Merck Millipore, LSKMAGAG10) and appropriate primary antibodies at 4℃ overnight according to the manufacturer’s protocols. The supernatants of the immunoprecipitates were subjected to immunoblotting for the detection of protein–protein interaction.
RNA extraction and qRT-PCR
Total RNA from HUVECs were isolated by Trizol reagent (BioTeke, RP1001) and reversely transcribed to cDNA using SuperScriptIII (Vazyme, R223-01). qRT-PCR was performed with products from Thermo fisher scientific Inc. (A31667) on a QuantStudio™ 3 Real-Time PCR Detection System. Glyceraldehyde-3- phosphate dehydrogenase (GAPDH) was used as a reference gene. The relative expression levels of mRNA were quantified using the ΔΔCt method. The specific primers are showed in Additional file 1: table 1.
Statistical analysis
Data were analyzed by GraphPad Prism 5.0 and results were expressed as mean ± standard error of the mean (S.E.M.). Differences of each sample were evaluated using the unpaired Student's two-tailed t-test or analysis of variance (ANOVA). A value of *P < 0.05 was considered significant. All experiments were repeated at least three times.
Results
HDAC3 inhibition rescues endothelial injury caused by T2DM in vivo and in vitro
The role of HDAC3 in the endothelium was investigated using endothelial cells isolated from aorta of diabetic db/db and nondiabetic db/m mice. HDAC3 activity, but not protein level, was significantly higher in the db/db group than in the db/m group (Fig. 1a and c). The relationship between HDAC3 and endothelial dysfunction was examined by treating mice with the specific HDAC3 inhibitor RGFP966 by subcutaneous injection at a dose of 10 mg/kg. HDAC3 inhibition significantly restored T2DM-induced de-endothelialization in diabetic mice compared with that in the vehicle-treated group, demonstrating the protective effect of HDAC3 inhibition against T2DM-induced endothelial impairment in vivo (Fig. 1e and i).
Fig. 1.

HDAC3 activity and its inhibition are related to endothelial function in T2DM. a, b The immunoblotting and quantitative analysis of HDAC3 protein level relative to GAPDH protein levels in the endothelial cells isolated from db/m mice or db/db mice. Values displayed are means ± SEM (n = 5). c Measurements of deacetylase activity of HDAC3 in different groups of endothelial cells isolated from db/m mice or db/db mice. d Levels of the oxidative damage marker 3-NT in HUVECs were detected by Western blot. e Representative immunofluorescence with CD31 from db/m mice, db/db mice, and RGFP966 or vehicle treated (subcutaneously, 10 mg/kg) db/db mice aorta tissue sections. The red area represented endothelium and the nucleus was blue. Scale bars: 200 μm. f Representative confocal images of oxidative damage marker 3-NT in aortal vascular endothelium. The red area represented endothelium, the green area represents 3-NT positive staining and the nucleus was blue. Scale bars: 40 μm. g Representative confocal images of apoptosis in aortal vascular endothelium. The red area represented endothelium, the green area represents TUNEL positive staining and the nucleus was blue. Scale bars: 40 μm. h The presence of immunofluorescence with CD31 and Ki67 of aortal vascular endothelium, scale bars: 20 μm, the green area represents Ki67 positive staining and the nucleus was blue. i–l Quantification of the number of CD31 positive area staining (e), the number of 3-NT staining (f), TUNEL + cells (g), the proportion of Ki67 positive staining (h). m The quantitative analysis of 3-NT protein immunoblotting, values displayed are means ± SEM (n = 4). Significance (c, i, j, k, l): *P < 0.05 vs. db/m mice; #P < 0.05 vs. db/db mice or vehicle treated db/db mice. Significance (m): *P < 0.05 vs. Con or MAN in scrambled HUVECs; #P < 0.05 vs. HG-PA in scrambled HUVECs. All above results in graphs from western blot were normalized to the first group
T2DM-induced endothelial dysfunction is mediated by several mechanisms, of which oxidative stress and pro-inflammatory responses originate in the endothelium. We used 3-nitrotyrosine (3-NT) as an oxidative stress marker and showed that si-HDAC3 treatment alleviated high glucose-palmitic acid (HG-PA)-induced oxidative stress from HUVECs (Fig. 1d and m). Immunofluorescence analysis showed that 3-NT staining intensity in the aortic vascular endothelium was higher in diabetic mice than in the db/m group, and this was decreased by HDAC3 inhibition (Fig. 1f and j). Next, we analyzed the effect of RGFP966 on T2DM-induced vascular apoptosis in diabetic aortic endothelial cells. The results showed that the accumulation of apoptosis puncta in the aortic vascular endothelium (labeled with CD31) was greater in db/db mice than in the corresponding control littermates, whereas treatment with RGFP966 significantly reduced apoptosis signals compared with those in the vehicle-treated group (Fig. 1g and k). RGFP966 treatment increased the Ki67 and CD31 positive area, indicating that HDAC3 inhibition promoted vascular endothelial proliferation during diabetic vascular impairment (Fig. 1h and l).
Similar results were obtained in the dihydroethidium (DHE) assay, which indicated that si-HDAC3 alleviated HG-PA-induced superoxide production (Fig. 2a and e). The HG-PA-induced increase in the number of TUNEL-positive cells (Fig. 2c and g), and the increased c-caspase 3 levels and Bax/Bcl-2 ratio (Fig. 2i–k), indicated that T2DM increased endothelial cell apoptosis, and these effects were reversed by si-HDAC3 treatment. In the aortic ring assay, aortic rings from C57BL/6 mice were cultured in different media. Aortic rings cultured in normal medium showed a well-structured microvessel network with clearly defined tubules and regular branching. HG-PA treatment dramatically impaired sprouting, whereas HDAC3 knockdown reversed this effect (Fig. 2b and f). Tube-forming activity was also significantly impaired in HUVECs exposed to HG-PA compared with the controls, whereas treatment with si-HDAC3 restored tube formation (Fig. 2d and h).
Fig. 2.

The effect of HDAC3 inhibition in T2DM–induced endothelial dysfunction in vitro. a Fluorescent images of superoxide levels in HUVECs cultured either in Con (5.5 mM of glucose), MAN (33 mM: 5.5 mM of glucose + 27.5 mM of D-mannitol) or HG (33 mM)-PA (100 μM) medium with or without si-HDAC3 for 72 h, MAN was served as the osmotic control for the HG-PA. Superoxide was determined with the fluorescent indicator DHE, and the fluorescent intensity of DHE was observed with a computer-assisted microscope. Scale bars: 100 μm. b Representative images of aortic rings from db/m mice were cultured in medium. Scale bars: 200 μm. c TUNEL assay of HUVECs was determined. The apoptotic cells were labeled with green, and nuclei were stained with DAPI (blue). Scale bars: 100 μm. d Capillary-like tube formation was assessed by Matrigel angiogenesis assay in HUVECs. Scale bars: 300 μm. e–h Quantification of the Fluorescence intensity in (a), number of sprouts in (b), TUNEL + cells in (c), the tube length in (d). i Cell lysates of HUVECs were used to detect Bax, Bcl-2 as well as c-Caspase 3 protein levels by immunoblotting. j, k The quantitative analysis of each immunoblotting. The data are represented as the means ± SEM (n = 5). Significance: *P < 0.05 vs. Con or MAN in scrambled HUVECs. #P < 0.05 vs. HG-PA in scrambled HUVECs. All above results in graphs from western blot were normalized to the first group
Next, we examined the effect of HDAC3 inhibition on eNOS uncoupling, which leads to endothelial dysfunction [41]. For this purpose, the proportion of eNOS existing as dimers or monomers in HUVECs treated with HG-PA in the presence or absence of si-HDAC3 was determined. eNOS dimer formation and stability are critical for the production of NO by eNOS, and a decrease in the dimer-to-monomer ratio indicates eNOS uncoupling [42]. Low-temperature SDS-PAGE (without heat denaturation) of HUVECs lysates detected bands approximately twice the size of the eNOS monomer, probably corresponding to eNOS dimers. Exposure of the cells to HG-PA for 72 h significantly decreased the dimer-to-monomer ratio compared with that in the control, and this effect was markedly reversed in response to si-HDAC3 (Additional file 1: Fig. 1E and F). Under pathological conditions, eNOS uncoupling results in increased generation of superoxide anion; therefore, we examined eNOS uncoupling-induced intracellular superoxide generation using the DHE assay and confocal microscopy. Incubation of HUVECs with HG-PA in the presence of si-HDAC3 significantly decreased ROS production (Fig. 1a and e). The contribution of uncoupled eNOS-to-ROS production was determined by measuring superoxide generation in the presence and absence of the eNOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME). Inhibition of HG-PA-induced superoxide generation by L-NAME indicates that eNOS is a source of ROS and, therefore, that the enzyme is uncoupled (Additional file 1: Fig. 1a and 1b). Collectively, these findings indicate that endothelial dysfunction caused by uncoupled eNOS was decreased by si-HDAC3 in HUVECs exposed to HG-PA. These data provide a rationale for studying the mechanism(s) by which the enzyme is uncoupled in these cells. In addition, the results suggest HDAC3 inhibition as a potential novel approach to the treatment of T2DM-induced endothelial impairment.
HDAC3 inhibition activates Nrf2 signaling by decreasing Keap1–Nrf2 interaction
Because decreased nuclear Nrf2 signaling is associated with oxidative stress [43], we hypothesized that HDAC3 inhibition could protect against HG-PA-induced endothelial dysfunction by regulating Nrf2-modulated antioxidant gene expression. Western blot analysis of Nrf2 expression showed that HG-PA significantly downregulated nuclear and total Nrf2 in endothelial cells, whereas co-treatment with si-HDAC3 restored Nrf2 levels (Fig. 3a–d). Assessment of the ratio of nuclear Nrf2 (n-Nrf2)/lamin B to cytoplasmic Nrf2 (c-Nrf2)/GAPDH (Fig. 3e) confirmed the results of western blotting and immunofluorescence, showing that si-HDAC3 decreased Nrf2 nuclear translocation in endothelial cells (Fig. 4g).
Fig. 3.

HDAC3 inhibition activates Nrf2 signaling in vitro. a and c Expression of total (T-Nrf2), cytosolic (c-Nrf2) and nuclear (n-Nrf2) protein levels of Nrf2 were detected by Western blot. b, d and e The quantitative analysis of each immunoblots. f The mRNA level and the quantification of Keap1 were evaluated by qRT-PCR. g the protein level of Keap1 were detected by western blot. h The quantitative analysis of (g). i The mRNA level and the quantification of the Nrf2 target gene were evaluated by qRT-PCR. j The mRNA level and the quantification of the NF-κB target gene were evaluated by qRT-PCR. The data are represented as the means ± SEM (n = 4). Significance: *P < 0.05 vs. Con or MAN in scrambled HUVECs; #P < 0.05 vs. HG-PA in scrambled HUVECs. All above results in graphs were normalized to the first group
Fig. 4.

HDAC3 inhibition activates Nrf2 signaling by decreasing Keap1–Nrf2 interaction. a HUVECs were cultured in different condition, and the cell lysates were subjected to immunoprecipitation with Nrf2 antibody, followed by immunoblotting with indicated antibody. b and c The quantitative analysis of (a). d The cell lysates of HUVECs were subjected to immunoprecipitation with Nrf2 antibody, followed by immunoblotting with indicated antibody. e and f The quantitative analysis of (d). g Nrf2 nuclear translocation was determined in fixed cells by immunofluorescent staining in HUVECs. The red area represented Nrf2 and the nucleus was blue. Scale bars: 15 μm. The data are represented as the means ± SEM (n = 4). Significance: *P < 0.05 vs. MAN in scrambled HUVECs, #P < 0.05 vs. HG-PA in scrambled HUVECs, & P < 0.05 vs. HG-PA with si-HDAC3. All above results in graphs were normalized to first group
qRT-PCR analysis of the mRNA levels of Nrf2 target genes showed that HG-PA downregulated NQO1, NQO2, CAT, SOD2, and HO1, and this effect was partly rescued by si-HDAC3 treatment (Fig. 3i). T2DM-induced oxidative stress activates NF-κB, which regulates the release of pro-inflammatory cytokines such as IL1β, IL6, IL8, TNFα, VCAM-1, and ICAM-1. Consistently, the mRNA expression of NF-κB target genes was significantly higher in endothelial cells exposed to HG-PA than in those cultured in NG, and this effect was attenuated by si-HDAC3 or the pharmacological antioxidant molecule NAC (Fig. 3j).
Next, we explored the molecular mechanism underlying the effect of HDAC3 inhibition on activating the Nrf2 pathway. Keap1 sequesters Nrf2 in the cytoplasm, restricting Nrf2 from nuclear translocation and facilitating Nrf2 degradation [44]. Western blotting and qRT-PCR results showed that si-HDAC3 co-treatment suppressed the effect of HG-PA on upregulating Keap1 expression (Fig. 3f–h). Sulforaphane (SFN), which inactivates Keap1 and promotes Nrf2 nuclear translocation, was introduced as positive control to confirm the effect of si-HDAC3 (Fig. 3g and h). The results indicated that HDAC3 inhibition activated Nrf2 signaling by downregulating Keap1 and facilitating Nrf2 nuclear translocation.
Nrf2 is rapidly degraded by the ubiquitin–proteasome system under homeostatic conditions [45]. To determine whether the increase in Nrf2 caused by HDAC3 inhibition was associated with the suppression of Nrf2 ubiquitination, Nrf2 ubiquitination was analyzed by immunoprecipitation after treatment with MG132, a proteasome and protease inhibitor, in the presence or absence of si-HDAC3. As shown in Fig. 4a–c, si-HDAC3 alone or in combination with MG132 upregulated Nrf2 expression compared with that in response to HG-PA. Immunoprecipitation with anti-Nrf2 showed that HG-PA increased Nrf2 ubiquitination (lane 2) reduced by co-treatment with si-HDAC3 (lane 3), and this effect was reversed by MG132 administration (lane 4).
Next, cell lysates were immunoprecipitated using an anti-Nrf2 antibody and immunoblotted for Keap1 to detect the Nrf2–Keap1 interaction. Total lysates (input) were evaluated by western blotting using anti-Nrf2 antibodies. The results showed that si-HDAC3 co-treatment inhibited the Nrf2–Keap1 interaction compared with that in HG-PA-treated endothelial cells, and this effect was similar to that of SFN co-treatment in the HG-PA medium (Fig. 4d–f).
Thus, we speculate that HDAC3 inhibition activated Nrf2 signaling by inhibiting Keap1 synthesis rather than promoting Keap1 ubiquitination, which suppressed the Nrf2–Keap1 interaction-induced ubiquitination of Nrf2, facilitating Nrf2 nuclear translocation.
Nox4 inhibition counteracts T2DM-induced endothelial dysfunction partially through the Nrf2–Nox4 loop
The Nrf2 response is associated with the induction of Nox4, namely, activated Nrf2 signaling inhibits the transcription of Nox4 in the endothelium [46]. Therefore, we examined whether Nox4–Nrf2 imbalance plays a role in the diabetic endothelium. The results showed that HG-PA treatment upregulated Nox4 transcription in HUVECs (Fig. 5a, d, and e), and this was reversed by si-HDAC3 treatment. Although the link between HDAC3 and Nrf2 was demonstrated, the relationship between Nox4 and Nrf2 remained unclear. To examine this, Nrf2 was silenced by siRNA (Fig. 5f and g). Nox4 mRNA and protein levels were higher in cells treated with si-Nrf2 in combination with HG-PA than in those treated with HG-PA alone (Fig. 5f, h, and i). HDAC3 inhibition downregulated Nox4 to a similar level than SFN treatment, which inactivates Keap1 (Fig. 5n and o). These results indicate that HDAC3 inhibition activated Nrf2 signaling by decreasing Keap1 synthesis (Result 2), thereby downregulating Nox4. SiRNA-mediated Nox4 silencing increased Nrf2 levels (Fig. 5j and m), which was associated with an increase in the transcription of genes encoding antioxidant enzymes (Fig. 5p), defining a Nrf2–Nox4 negative feedback loop.
Fig. 5.

Nox4 inhibition counteracts T2DM-induced endothelial dysfunction partially through the Nrf2–Nox4 loop. a Cell lysates of HUVECs were used to detect indicated protein levels by immunoblotting. b–d The quantitative analysis of each immunoblots. e The mRNA level and the quantification of the Nox4 were evaluated by qRT-PCR. f Cell lysates of HUVECs were used to detect indicated protein levels by immunoblotting. g and h The quantitative analysis of each immunoblots. i The mRNA level and the quantification of the Nox4 were evaluated by qRT-PCR, which assay of HUVECs treated as indicated in (f). j Cell lysates of HUVECs were used to detect indicated protein levels by immunoblotting. k, l and m The quantitative analysis of each immunoblots in (j). n Cell lysates of HUVECs were subjected to immunoblot with Nox4 antibody. o The quantitative analysis of (n). p The mRNA level and the quantification of the Nrf2 target gene were evaluated by qRT-PCR. Data are represented as means ± SEM (n = 5). Significance: *P < 0.05 and **P < 0.01 vs. MAN in scrambled HUVECs, #P < 0.05 and ##P < 0.01 vs. HG-PA in scrambled HUVECs. All above results in graphs were normalized to first group
The results of TUNEL (Fig. 6a and e), DHE (Fig. 6b and f), tube formation (Fig. 6c and g), and aortic ring assays (Fig. 6d and h) demonstrated that downregulation of Nox4 alleviated T2DM-induced endothelial dysfunction. In vivo, db/db mice treated with the Nox4 inhibitor GKT137831 showed significantly reduced apoptosis in the aortic vascular endothelium (Fig. 6i and k) and attenuated T2DM-induced de-endothelialization compared with the vehicle-treated group. The results of the Ki67 assay (Fig. 6j and l) showed that Nox4 knockdown significantly improved vascular endothelial proliferation during diabetic vascular impairment. Taken together, the results indicate that downregulation of Nox4 by HDAC3 inhibition protected against T2DM-induced endothelial dysfunction through a mechanism involving Nrf2 and the activation of the Nrf2–Nox4 negative feedback loop.
Fig. 6.

Nox4 inhibition counteracted T2DM-induced endothelial dysfunction. a TUNEL assay of HUVECs cultured with HG-PA alone or with si-Nox4 for 72 h, Scale bars: 100 μm. b The fluorescent intensity of DHE was observed. Scale bars: 100 μm. c Capillary-like tube formation was assessed in HUVECs. Scale bars: 300 μm. d Representative images of aortic rings from db/m mice. Scale bars: 200 μm. e–h The quantitative analysis of TUNEL + cells in (a), fluorescent intensity in (b), the tube length in (c), the number of sprouts in (d). i Representative confocal images of apoptosis in aortal vascular endothelium from db/db mice or db/db mice gavaged at a dose of 60 mg/kg/day GKT137831. Scale bars: 40 μm. J The presence of immunofluorescence with CD31 and Ki67 of aortal vascular endothelium. Scale bars: 20 μm. k and l The quantitative analysis of TUNEL + cells and Ki67 of aortal vascular endothelium from db/db mice and GKT137831 treated db/db mice aorta tissue sections. Data are represented as means ± SEM (n = 5). Significance (e–h): *P < 0.05 vs. HG-PA in scrambled HUVECs. Significance (k and l): *P < 0.05 vs. vehicle-treated db/db mice. j Representative confocal images of oxidative damage marker 3-NT in aortal vascular endothelium. Scale bars: 40 μm. k The presence of immunofluorescence with CD31 and Ki67 of aortal vascular endothelium. Scale bars: 20 μm. l The quantitative analysis of TUNEL + cells from db/db mice and GKT137831 treated db/db mice aorta tissue sections. Data are represented as means ± SEM (n = 5). Significance (e–h): *P < 0.05 vs. HG-PA in scrambled HUVECs. Significance (k and l): *P < 0.05 vs. vehicle-treated db/db mice
Knockdown of Nrf2 abolishes the HDAC3 inhibition-induced protective effect in T2DM
To confirm the role of Nrf2 in the HDAC3 inhibition-mediated protective effect on diabetes-induced endothelial injury, si-HDAC3-treated HUVECs were incubated with HG-PA in the presence or absence of si-Nrf2. The results showed that si-Nrf2 abolished the si-HDAC3-induced nuclear accumulation of Nrf2 (Fig. 7a and b) and aggravated the si-HDAC3-induced the expression of Nox4 (Fig. 7e and g) compared with the scrambled HUVECs with the same treatment. Furthermore, HG-PA-induced oxidative damage detected by 3-NT was attenuated by HDAC3 inhibition, and this effect was reversed by si-Nrf2 co-treatment (Fig. 7c and d). si-Nrf2 eliminated the protective effect of HDAC3 inhibition on T2DM, as determined by the expression of antioxidant genes (Fig. 7i), increased apoptosis (Fig. 8j and h), increased oxidative stress (Fig. 8k and n), and aberrant tube formation (Fig. 8l and o) and vascular sprouting (Fig. 8m and p).
Fig. 7.

Knockdown of Nrf2 abolishes HDAC3 inhibition-induced protective effect in vitro. a HUVECs were transduced with si-Nrf2 and a scrambled sequence (scramble) respectively. Protein expression of nuclear accumulation of Nrf2 in HUVECs after transfecting with siRNA assayed by Western blot. b, c The quantitative analysis of nuclear accumulation of Nrf2 and 3-NT immunoblots. d Levels of the oxidative damage marker 3-NT in HUVECs was detected by Western blot. e HUVECs were transduced with si-Nrf2, scrambled sequence combined with si-HDAC3 or not. f, g The quantitative analysis of Nrf2 and Nox4 were evaluated. h Quantification of the TUNEL + cells in (j). i The mRNA level and the quantification of the Nrf2 target gene were evaluated by qRT-PCR. Data are represented as mean ± SEM (n = 5). Significance: *P < 0.05 vs. respective MAN in scrambled HUVECs, #P < 0.05 vs. respective HG-PA in scrambled HUVECs, % P < 0.05 vs. respective HG-PA with si-HDAC3 cultured HUVECs, & P < 0.05 vs. scrambled HUVECs with the same treatment. All above results in graphs (b), (c), (f), (g), (i) were normalized to first group. j TUNEL assay of HUVECs. Scale bars: 100 μm. k Superoxide was determined with DHE. Scale bars: 100 μm. l Capillary-like tube formation was assessed. Scale bars: 300 μm. m Representative confocal images of aortic rings. Scale bars: 400 μm. Values displayed are means ± SEM (n = 5). Significance (h), (n), (o), (p): *P < 0.05 vs. db/db mice, #P < 0./05 vs. RGFP966 treated db/db mice
Fig. 8.
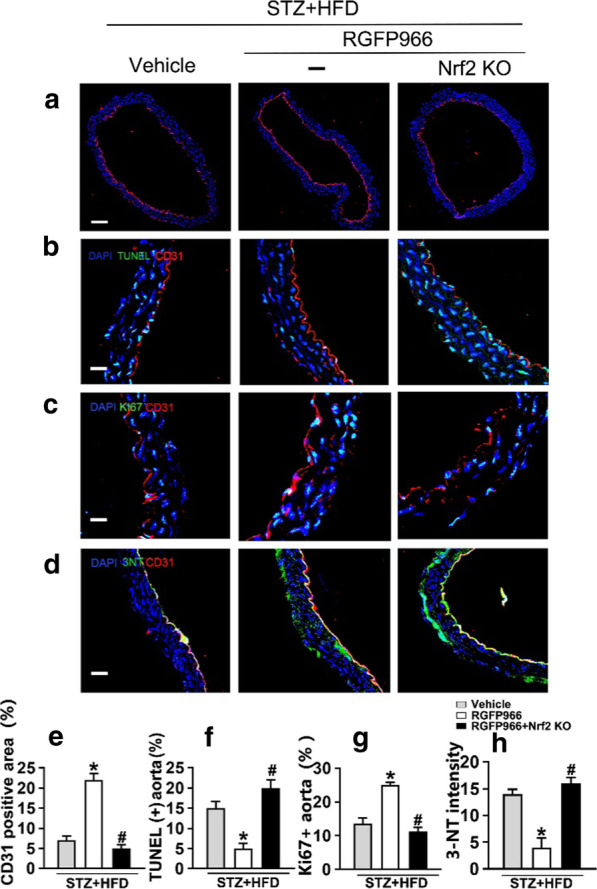
Knockdown of Nrf2 abolishes the HDAC3 inhibition-induced protective effect in vivo. a Representative immunofluorescence with CD31 from vehicle treated T2DM mice (fed by STZ and HFD), RGFP966 treated (10 mg/kg) T2DM mice or Nrf2 knockout mice with RGFP966 treated T2DM mice aorta tissue sections. Scale bars: 200 μm. b Representative confocal images of apoptosis in aortal vascular endothelium. Scale bars: 40 μm. c The presence of immunofluorescence with CD31 and Ki67 of aortal vascular endothelium. Scale bars: 20 μm. d Representative confocal images of oxidative damage marker 3-NT in aortal vascular endothelium. Scale bars: 40 μm. e–h Quantification of the CD31 positive area in (a), TUNEL + cells in (b), the proportion of Ki67 positive area in (c), the number of 3-NT staining in (d) respectively. Values displayed are means ± SEM (n = 5). Significance (e–h): *P < 0.05 vs. db/db mice, #P < 0.05 vs. RGFP966 treated db/db mice
Nrf2 KO mice with the T2DM background were established by feeding a high fat diet and by intraperitoneal streptozotocin injection (Additional file 1: Fig. 1c and d). The results of thoracic aortic sprouting, CD31 immunofluorescence staining (Fig. 8a and e), TUNEL assay (Fig. 8b and f), Ki67 detection (Fig. 8c and g), and 3-NT staining (Fig. 8d and h) indicated that Nrf2 knockout abrogated the RGFP966-induced protective effect on T2DM-induced aortic endothelial injury. Consistent with the observations in vitro, Nrf2 in the aortic endothelium was required for HDAC3 inhibition-mediated attenuation of T2DM-induced oxidative stress, inflammation, and vasculopathy.
Discussion
In vivo and in vitro studies have demonstrated that T2DM can progressively compromise endothelial function in association with excessive ROS production and inflammation, thereby contributing to T2DM-associated vascular complications. HDACs are epigenetic regulators that catalyze the removal of acetyl groups from lysine residues in a variety of proteins. HDACs deacetylate nucleosome histones and alter the electrostatic properties of chromatin to repress gene expression [47, 48]. HDAC inhibition improves diabetic complications [49]. Total inhibition of HDACs, particularly that of class I HDACs, is associated with endothelial dysfunction, which is an important factor in the pathogenesis of vascular changes [2, 50, 51]. This is accompanied by the production of ROS, which play a causative role in the development and progression of diabetic vascular complications. HDAC3, a class I HDAC that is present in the nucleus and cytoplasm of cells [52], is involved in tumor development, DM, inflammation, and cardiovascular and neurodegenerative diseases [29]. The mechanism of HDAC3 inhibition in anti-inflammation, involves the removal of inhibitory NF-κB p65 acetylation at K122, 123, 314, and 315 [53]. Moreover, HDAC3 deletion induces cardiac hypertrophy, and global deletion of HDAC3 results in embryonic lethality at E9.5 in the heart [52], whereas its inhibition prevents type 1 diabetic cardiomyopathy via epigenetic regulation of the DUSP5–ERK1/2 pathway [54]. Jung and Seong demonstrated that knockdown of endogenous HDAC3 in endothelial cells increases lysine acetylation of endogenous eNOS [55]. The increase in lysine acetylation of eNOS increases NO production by endothelial cells in which HDAC3 is knocked down without affecting eNOS expression. However, HDAC3 also plays a non-deacetylase role in the occurrence and development of diseases. Loss of HDAC3 function protects β-cells from cytokine-induced apoptosis and maintains proper glucose-stimulated insulin secretion [56–58]. Inhibition of HDAC3 in diabetic mice ameliorates diabetes-induced liver damage, and reduces oxidative stress and aortic injury by activating Nrf2 signaling [59]. Increased HDAC3 activity or levels in patients with T2DM is correlated with inflammatory marker expression, poor glycemic control, and insulin resistance [60]. HDAC3 expression is involved in the differentiation of embryonic stem cells into endothelial progenitors; it is critical for endothelial cell survival and as a pro-survival molecule [61–63]. Therefore, we hypothesized that HDAC3 might be a therapeutic target for the treatment of endothelial damage.
In this study, we investigated the effect of HDAC3 inhibition on vascular endothelial function using db/db mice and cultured HUVECs. The results can be summarized as follows: (1) HDAC3 activity was higher in the thoracic aortic vascular endothelium of db/db mice than in that of control mice; (2) HDAC3 inhibition may prevent T2DM-triggered endothelial dysfunction including excessive apoptosis, sprouting, and angiogenesis loss in vivo and in vitro; (3) the protective effect of HDAC3 inhibition on the endothelium is mediated by the activation of Nrf2 signaling, which involves decreasing the Keap1–Nrf2 interaction by reducing Keap1 level; 4) HDAC3 inhibition elicits Nrf2 signaling in association with Nox4 downregulation in the T2DM endothelium; siRNA-mediated Nox4 silencing prevents endothelial dysfunction and activates Nrf2 signaling, defining a Nrf2–Nox4 negative feedback loop. The present results provide insight into the role of HDAC3 in the development of diabetic endothelial dysfunction. However, because other HDAC inhibitors, especially HDAC class I inhibitors, have a protective effect on the cardiovascular system [64–66], the involvement of other HDACs cannot be excluded.
In the resting cell, Nrf2 is sequestered in the cytoplasm by Keap1 and continuously targeted for ubiquitination [67]. Modification of critical cysteine residues in Keap1 leads to the dissociation of the Keap1–Nrf2 complex or increased Nrf2 stability [68]. Civantos and Zhang investigated the association between diabetes-induced liver damage or nephropathy, and increased Nrf2 activity and miR-200a upregulation [59, 69]. However, the relationship between Keap1, Nrf2, and miR-200a in the diabetes-induced endothelium remains unclear and will be further explored in the future.
HDAC3 inhibition decreases Nox4 transcription despite the open chromatin structure in human endothelial cells, thereby improving endothelial function. This may be attributed to HDAC inhibition in endothelial cells associated with increased chromatin accessibility in the human Nox4 promoter region without significant changes in DNA methylation [30]. The present data on the role of HDAC3 in endothelial cells support the role of HDAC3 inhibition on promoting an anti-inflammatory phenotype by altering Nox4 transcriptional levels. Evidence supports the important role of Nox4 in redox signaling. Elsherbiny indicated that doxorubicin‐induced renal damage involves a redox imbalance caused by Nox4 upregulation and Nrf2 downregulation [70]. Hecker confirmed the effect of ROS‐induced Nox4–Nrf2 redox imbalance in persistent lung fibrosis [71]. In addition, inhibitor of NAPDH oxidase, DPI, downregulates Nox4 expression and promotes sliver nanoparticles-induced dysfunction by upregulating Nrf2 [21]. This is consistent with the present results showing that si-Nox4 upregulated Nrf2 and activated Nrf2 signaling (Fig. 5j and p). Nox4–Nrf2 redox imbalance is thus an important factor for maintaining endothelial function, and restoring balance could be a potential therapeutic strategy.
Conclusions
In this study, we showed that HDAC3 inhibition decreases T2DM-induced endothelial dysfunction, thereby improving T2DM-induced injury in the vascular endothelium. This protective effect can be partly attributed to the activation of Nrf2 signaling through the suppression of Keap1 level, modulation of the Nox4–Nrf2 redox imbalance, and inhibition of eNOS uncoupling. Keap1 expression and Nox4 transcription were increased in the T2DM endothelium, and HDAC3 inhibition reversed these effects, thereby protecting the endothelium in a Nrf2-dependent manner. The precise mechanism underlying the effect of HDAC3 inhibition on the modulation of Keap1 and Nox4–Nrf2 remains to be elucidated. Future studies should investigate the potential of HDAC3 as an epigenetic regulator in diabetes-associated vasculopathy.
Supplementary Information
Additional file 1: Supplementary Figure 1. HDAC3 inhibition decreases the HG-PA-induced eNOS uncoupling in vitro and the verification of Nrf2 KO in mice.
Acknowledgements
Not applicable.
Abbreviations
- T2DM
Type 2 diabetes mellitus
- DV
Diabetic vasculopathy
- NG
Normal glucose
- HG-PA
High glucose-palmitic acid
- Nrf2
Nuclear factor (erythroid-derived 2)-like 2
- eNOS
Endothelial nitric oxide synthase
- NO
Nitric oxide
- L-NAME
NG-nitro-L-arginine methyl ester
- Keap1
Kelch-like ECH-associated protein 1
- NF-κB
Nuclear factor-κB
- Nox4
NAPDH oxidase 4
- SFN
Sulforaphane
- ARE
Antioxidant response element
- IL-1β, 6, 8
Interleukins-1β, 6, 8
- TNF-α
Tumor necrosis factor-alpha
- VCAM-1
Vascular cell adhesion molecular-1, ICAM-1: intracellular adhesion molecular-1
- NQO
NAD(P)H dehydrogenase quinone
- SOD2
Superoxide dismutase 2
- HO1
Heme oxygenase 1
- CAT
Catalase
- HDAC
Histone deacetylases
- HUVECs
Human umbilical vein endothelial cells
- DHE
Dihydroethidium
- EBM
Endothelial basal medium
- EGM-2
Endothelial cell growth medium-2
- NAC
N-Acetyl-L-cysteine
- si-HDAC3
HDAC3 siRNA
- si-Nrf2
Nrf2 siRNA
- si-Nox4
Nox4 siRNA
- MAN
Mannitol
- qRT-PCR
Quantitative real time-polymerase chain reaction
- BSA
Bovine serum albumin
- ROS
Reactive oxygen species
- HFD
High fat diet
- STZ
Streptozotocin
Authors’ contributions
Shuai Huang, Gen Chen, Litai Jin and Weitao Cong conceived, designed and supervised the study. Jia Sun, Yunjie Chen, Nan Wang researched the data. Gen Chen, Yunjie Chen, Zhicheng Hu and Yetong Dong contributed to the discussion and design of the project. Shuai Huang, Enzhao Shen and Wenjie Gong wrote the paper. Shuai Huang and Gen Chen are the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Funding
This work was supported by Grants from Natural Science Foundation of China (81770498, 81773346, 81900240).
Availability of data and materials
All data generated in this study are included in the manuscript.
Declarations
Ethics approval and consent to participate
Animal use and care protocol was approved by the Animals Use and Care Committee of the Wenzhou Medical University, Wenzhou, China.
Consent for publication
All the authors give their consent for publication.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shuai Huang and Gen Chen are first authors and also contributed equally to this work.
Contributor Information
Shuai Huang, Email: 15257713128@163.com.
Gen Chen, Email: chengen86374062@126.com.
Jia Sun, Email: annfightingaza@163.com.
Yunjie Chen, Email: cyj960621@163.com.
Nan Wang, Email: wn18815138519@163.com.
Yetong Dong, Email: soohixxx@163.com.
Enzhao Shen, Email: sez1239818559@163.com.
Zhicheng Hu, Email: chengcheng751@qq.com.
Wenjie Gong, Email: 694050970@qq.com.
Litai Jin, Email: jin_litai@126.com.
Weitao Cong, Email: cwt97126@126.com.
References
- 1.Olokoba AB, Obateru OA, Olokoba LB. Type 2 diabetes mellitus: a review of current trends. Oman Med J. 2012;27:269–273. doi: 10.5001/omj.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, Malanda B. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018;138:271–281. doi: 10.1016/j.diabres.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 3.Bhatt MP, Lim YC, Ha KS. C-peptide replacement therapy as an emerging strategy for preventing diabetic vasculopathy. Cardiovasc Res. 2014;104:234–244. doi: 10.1093/cvr/cvu211. [DOI] [PubMed] [Google Scholar]
- 4.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–592. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Vallee A, Lecarpentier Y. Crosstalk between peroxisome proliferator-activated receptor gamma and the canonical wnt/beta-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front Immunol. 2018;9:745. doi: 10.3389/fimmu.2018.00745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang SC, Liao PY, Hung SJ, Ge JS, Chen SM, Lai JC, Hsiao YP, Yang JH. Topical application of glycolic acid suppresses the UVB induced IL-6, IL-8, MCP-1 and COX-2 inflammation by modulating NF-kappaB signaling pathway in keratinocytes and mice skin. J Dermatol Sci. 2017;86:238–248. doi: 10.1016/j.jdermsci.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Gao L, Mann GE. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res. 2009;82:9–20. doi: 10.1093/cvr/cvp031. [DOI] [PubMed] [Google Scholar]
- 9.Cheng X, Siow RC, Mann GE. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal. 2011;14:469–487. doi: 10.1089/ars.2010.3283. [DOI] [PubMed] [Google Scholar]
- 10.Sykiotis GP, Habeos IG, Samuelson AV, Bohmann D. The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation. Curr Opin Clin Nutr Metab Care. 2011;14:41–48. doi: 10.1097/MCO.0b013e32834136f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaziri ND. Protective effect of Nrf2 and catalase in maternal diabetes-induced perinatal hypertension and kidney disease. Diabetes. 2012;61:2400–2402. doi: 10.2337/db12-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tkachev VO, Menshchikova EB, Zenkov NK. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Mosc) 2011;76:407–422. doi: 10.1134/s0006297911040031. [DOI] [PubMed] [Google Scholar]
- 13.Chapple SJ, Siow RC, Mann GE. Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int J Biochem Cell Biol. 2012;44:1315–1320. doi: 10.1016/j.biocel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 14.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 15.Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:413–420. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- 16.Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 17.Lee GH, Hoang TH, Jung ES, Jung SJ, Chae SW, Chae HJ: Mulberry extract attenuates endothelial dysfunction through the regulation of uncoupling endothelial nitric oxide synthase in high fat diet rats. Nutrients 2019, 11. [DOI] [PMC free article] [PubMed]
- 18.Badran M, Abuyassin B, Golbidi S, Ayas N, Laher I. Alpha lipoic acid improves endothelial function and oxidative stress in mice exposed to chronic intermittent hypoxia. Oxid Med Cell Longev. 2019;2019:4093018. doi: 10.1155/2019/4093018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faria AM, Papadimitriou A, Silva KC, Lopes de Faria JM, Lopes de Faria JB. Uncoupling endothelial nitric oxide synthase is ameliorated by green tea in experimental diabetes by re-establishing tetrahydrobiopterin levels. Diabetes. 2012;61:1838–1847. doi: 10.2337/db11-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komers R, Schutzer WE, Reed JF, Lindsley JN, Oyama TT, Buck DC, Mader SL, Anderson S. Altered endothelial nitric oxide synthase targeting and conformation and caveolin-1 expression in the diabetic kidney. Diabetes. 2006;55:1651–1659. doi: 10.2337/db05-1595. [DOI] [PubMed] [Google Scholar]
- 21.Sun X, Yang Y, Shi J, Wang C, Yu Z, Zhang H. NOX4- and Nrf2-mediated oxidative stress induced by silver nanoparticles in vascular endothelial cells. 2017, 37:1428–1437 [DOI] [PubMed]
- 22.Murdoch CE, Alom-Ruiz SP, Wang M, Zhang M, Walker S, Yu B, Brewer A, Shah AM. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res Cardiol. 2011;106:527–538. doi: 10.1007/s00395-011-0179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyano K, Ueno N, Takeya R, Sumimoto H. Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. J Biol Chem. 2006;281:21857–21868. doi: 10.1074/jbc.M513665200. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Goettsch C, Xia N, Horke S, Morawietz H, Forstermann U, Li H. Differential roles of PKCalpha and PKCepsilon in controlling the gene expression of Nox4 in human endothelial cells. Free Radic Biol Med. 2008;44:1656–1667. doi: 10.1016/j.freeradbiomed.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 25.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 27.Basuroy S, Bhattacharya S, Leffler CW, Parfenova H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am J Physiol Cell Physiol. 2009;296:C422–432. doi: 10.1152/ajpcell.00381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brandes RP, Schroder K. Differential vascular functions of Nox family NADPH oxidases. Curr Opin Lipidol. 2008;19:513–518. doi: 10.1097/MOL.0b013e32830c91e3. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Chen Y, Jiang Q, Song W, Zhang L. Therapeutic potential of selective histone deacetylase 3 inhibition. Eur J Med Chem. 2019;162:534–542. doi: 10.1016/j.ejmech.2018.10.072. [DOI] [PubMed] [Google Scholar]
- 30.Siuda D, Zechner U, El Hajj N, Prawitt D, Langer D, Xia N, Horke S, Pautz A, Kleinert H, Forstermann U, Li H. Transcriptional regulation of Nox4 by histone deacetylases in human endothelial cells. Basic Res Cardiol. 2012;107:283. doi: 10.1007/s00395-012-0283-3. [DOI] [PubMed] [Google Scholar]
- 31.Xu Z, Tong Q, Zhang Z, Wang S, Zheng Y, Liu Q, Qian LB, Chen SY, Sun J, Cai L: Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clinical science (London, England : 1979) 2017, 131:1841–1857. [DOI] [PMC free article] [PubMed]
- 32.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 33.Mroueh FM, Noureldein M, Zeidan YH, Boutary S, Irani SAM, Eid S, Haddad M, Barakat R, Harb F, Costantine J, Kanj R, Sauleau EA, Ouhtit A, Azar ST, Eid AH, Eid AA. Unmasking the interplay between mTOR and Nox4: novel insights into the mechanism connecting diabetes and cancer. FASEB J. 2019;33:14051–14066. doi: 10.1096/fj.201900396RR. [DOI] [PubMed] [Google Scholar]
- 34.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, Abboud HE. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation. 2015;131:643–655. doi: 10.1161/CIRCULATIONAHA.114.011079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan J, Wang C, Jin Y, Meng Q, Liu Q, Liu Z, Liu K, Sun H. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol Res. 2018;130:466–480. doi: 10.1016/j.phrs.2017.12.026. [DOI] [PubMed] [Google Scholar]
- 36.Brill A, Elinav H, Varon D. Differential role of platelet granular mediators in angiogenesis. Cardiovasc Res. 2004;63:226–235. doi: 10.1016/j.cardiores.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Aplin AC, Nicosia RF. The rat aortic ring model of angiogenesis. Methods Mol Biol. 2015;1214:255–264. doi: 10.1007/978-1-4939-1462-3_16. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. J Atheroscler Thromb. 2005;12:138–142. doi: 10.5551/jat.12.138. [DOI] [PubMed] [Google Scholar]
- 39.Cong W, Niu C, Lv L, Ni M, Ruan D, Chi L, Wang Y, Yu Q, Zhan K, Xuan Y, et al. Metallothionein prevents age-associated cardiomyopathy via inhibiting NF-kappaB pathway activation and associated nitrative damage to 2-OGD. Antioxid Redox Signal. 2016;25:936–952. doi: 10.1089/ars.2016.6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Investig. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meza CA, La Favor JD, Kim DH, Hickner RC: Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int J Mol Sci 2019, 20. [DOI] [PMC free article] [PubMed]
- 42.Aa E, Dy L, Lj R. K K, Y G: Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol. 2013;33:3439–3460. doi: 10.1128/MCB.00217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jyrkkanen HK, Kansanen E, Inkala M, Kivela AM, Hurttila H, Heinonen SE, Goldsteins G, Jauhiainen S, Tiainen S, Makkonen H, et al. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ Res. 2008;103:e1–9. doi: 10.1161/CIRCRESAHA.108.176883. [DOI] [PubMed] [Google Scholar]
- 44.Miyata T, Suzuki N. van Ypersele de Strihou C: Diabetic nephropathy: are there new and potentially promising therapies targeting oxygen biology? Kidney Int. 2013;84:693–702. doi: 10.1038/ki.2013.74. [DOI] [PubMed] [Google Scholar]
- 45.Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem. 2003;278:2396–2402. doi: 10.1074/jbc.M209195200. [DOI] [PubMed] [Google Scholar]
- 46.Liang E, Ma M, Wang L, Liu X, Xu J, Zhang M, Yang R, Zhao Y. The BET/BRD inhibitor JQ1 attenuates diabetes-induced cognitive impairment in rats by targeting Nox4-Nrf2 redox imbalance. Biochem Biophys Res Commun. 2018;495:204–211. doi: 10.1016/j.bbrc.2017.11.020. [DOI] [PubMed] [Google Scholar]
- 47.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 48.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, Kelstrup CD, Dmytriyev A, Choudhary C, Lundby C, Olsen JV. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2:419–431. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan S, Komarya SK, Jena G. Phenylbutyrate and beta-cell function: contribution of histone deacetylases and ER stress inhibition. Epigenomics. 2017;9:711–720. doi: 10.2217/epi-2016-0160. [DOI] [PubMed] [Google Scholar]
- 50.Grundy SM, Howard B, Smith S, Jr, Eckel R, Redberg R, Bonow RO. Prevention Conference VI: diabetes and cardiovascular disease: executive summary: conference proceeding for healthcare professionals from a special writing group of the American Heart Association. Circulation. 2002;105:2231–2239. doi: 10.1161/01.cir.0000013952.86046.dd. [DOI] [PubMed] [Google Scholar]
- 51.Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol. 1985;2009(106):326–332. doi: 10.1152/japplphysiol.91353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ziesche E, Kettner-Buhrow D, Weber A, Wittwer T, Jurida L, Soelch J, Muller H, Newel D, Kronich P, Schneider H, et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res. 2013;41:90–109. doi: 10.1093/nar/gks916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu Z, Tong Q, Zhang Z, Wang S, Zheng Y, Liu Q, Qian LB, Chen SY, Sun J, Cai L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin Sci (Lond) 2017;131:1841–1857. doi: 10.1042/CS20170064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jung SB, Kim CS, Naqvi A, Yamamori T, Mattagajasingh I, Hoffman TA, Cole MP, Kumar A, Dericco JS, Jeon BH, Irani K. Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ Res. 2010;107:877–887. doi: 10.1161/CIRCRESAHA.110.222968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chou DH, Holson EB, Wagner FF, Tang AJ, Maglathlin RL, Lewis TA, Schreiber SL, Wagner BK. Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem Biol. 2012;19:669–673. doi: 10.1016/j.chembiol.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lundh M, Galbo T, Poulsen SS, Mandrup-Poulsen T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes Metab. 2015;17:703–707. doi: 10.1111/dom.12470. [DOI] [PubMed] [Google Scholar]
- 58.Wagner FF, Lundh M, Kaya T, McCarren P, Zhang YL, Chattopadhyay S, Gale JP, Galbo T, Fisher SL, Meier BC, et al. An Isochemogenic set of inhibitors to define the therapeutic potential of histone deacetylases in beta-cell protection. ACS Chem Biol. 2016;11:363–374. doi: 10.1021/acschembio.5b00640. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J, Xu Z, Gu J, Jiang S, Liu Q, Zheng Y, Freedman JH, Sun J, Cai L. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am J Physiol Endocrinol Metab. 2018;315:E150–e162. doi: 10.1152/ajpendo.00465.2017. [DOI] [PubMed] [Google Scholar]
- 60.Sathishkumar C, Prabu P, Balakumar M, Lenin R, Prabhu D, Anjana RM, Mohan V, Balasubramanyam M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin Epigenet. 2016;8:125. doi: 10.1186/s13148-016-0293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao Q, Zeng L, Zhang Z, Margariti A, Ali ZA, Channon KM, Xu Q, Hu Y. Sca-1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler Thromb Vasc Biol. 2006;26:2244–2251. doi: 10.1161/01.ATV.0000240251.50215.50. [DOI] [PubMed] [Google Scholar]
- 62.Zeng L, Xiao Q, Margariti A, Zhang Z, Zampetaki A, Patel S, Capogrossi MC, Hu Y, Xu Q. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J Cell Biol. 2006;174:1059–1069. doi: 10.1083/jcb.200605113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, Pepe AE, Wang G, Habi O, deFalco E, et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation. 2010;121:132–142. doi: 10.1161/CIRCULATIONAHA.109.890491. [DOI] [PubMed] [Google Scholar]
- 64.Bagchi RA, Weeks KL. Histone deacetylases in cardiovascular and metabolic diseases. J Mol Cell Cardiol. 2019;130:151–159. doi: 10.1016/j.yjmcc.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 65.Inoue K, Kobayashi M, Yano K, Miura M, Izumi A, Mataki C, Doi T, Hamakubo T, Reid PC, Hume DA, et al: Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler Thromb Vasc Biol 2006, 26:2652–2659. [DOI] [PubMed]
- 66.Wu J, Jiang Z, Zhang H, Liang W, Huang W, Zhang H, Li Y, Wang Z, Wang J, Jia Y, et al. Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic Biol Med. 2018;124:454–465. doi: 10.1016/j.freeradbiomed.2018.06.034. [DOI] [PubMed] [Google Scholar]
- 67.Juurlink BH. Dietary Nrf2 activators inhibit atherogenic processes. Atherosclerosis. 2012;225:29–33. doi: 10.1016/j.atherosclerosis.2012.08.032. [DOI] [PubMed] [Google Scholar]
- 68.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Civantos E, Bosch E, Ramirez E, Zhenyukh O, Egido J, Lorenzo O, Mas S. Sitagliptin ameliorates oxidative stress in experimental diabetic nephropathy by diminishing the miR-200a/Keap-1/Nrf2 antioxidant pathway. Diabetes Metab Syndr Obes. 2017;10:207–222. doi: 10.2147/DMSO.S132537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elsherbiny NM, El-Sherbiny M. Thymoquinone attenuates Doxorubicin-induced nephrotoxicity in rats: Role of Nrf2 and NOX4. Chem Biol Interact. 2014;223:102–108. doi: 10.1016/j.cbi.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 71.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, Thannickal VJ. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med 2014, 6:231–247. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary Figure 1. HDAC3 inhibition decreases the HG-PA-induced eNOS uncoupling in vitro and the verification of Nrf2 KO in mice.
Data Availability Statement
All data generated in this study are included in the manuscript.