

Abstract
Photosensitivity is a sensitivity to ultraviolet radiation (UVR) commonly found in systemic lupus erythematosus (SLE) patients who have cutaneous disease (CLE). Upon even ambient UVR exposure, patients can develop inflammatory skin lesions that can reduce the quality of life. Additionally, UVR-exposed skin lesions can be associated with systemic disease flares marked by rising autoantibody titers and worsening kidney disease. Why SLE patients are photosensitive and how skin sensitivity leads to systemic disease flares are not well understood, and treatment options are limited. In recent years, the importance of immune cell-stromal interactions in tissue function and maintenance is being increasingly recognized. Here, we discuss SLE as an anatomic circuit and review recent findings in the pathogenesis of photosensitivity with a focus on immune cell-stromal circuitry in tissue health and disease.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that is strikingly associated with photosensitivity, a sensitivity to ultraviolet radiation (UVR) that results in the development of skin lesions. Notably, these lesions can be associated with triggering of systemic disease flares (Figure 1). Marked by circulating autoantibodies and inflammatory damage of the kidneys, brain, and heart among other organs, SLE is strongly associated with cutaneous lupus erythematosus (CLE). CLE can occur with and without systemic disease and can be divided into acute, subacute, and chronic forms. Acute CLE is most often associated with SLE, although SLE patients can have any type of CLE (1–4). Defined by the American College of Rheumatology (ACR) as “a skin rash as a result of an unusual reaction to sunlight” (5), photosensitivity is a common manifestation of SLE (1–4)). Current treatment options for photosensitivity are limited, with first-line treatment involving topical steroids, calcineurin inhibitors, and systemic anti-malarials such as hydroxychloroquine, which were fortuitously found to be effective during World War II (6). Reduced UVR exposure through sun avoidance, protective clothing, and sunscreen is effective and a mainstay in current therapy in the prevention of photosensitivity and its sequela (7–9). However, reduced UVR exposure can lead to reduced levels of UVR-dependent vitamin D synthesis seen in SLE patients(10), which is thought to contribute to poor bone health and osteoporosis (11). Furthermore, vitamin D is important in immune regulation; reduced Vitamin D can potentially exacerbate SLE and its symptoms (12). Photosensitivity has been shown to have a large negative impact on quality of life (13–16). Understanding the mechanisms of photosensitivity will provide insights into pathogenesis and treatment of both CLE and SLE.
Figure 1. Photosensitivity in lupus.

In patients with CLE or SLE, even ambient exposure to sunlight can trigger the development of skin lesions. In SLE patients, this photosensitivity can be associated with flares of systemic disease. Photos from American College of Rheumatology Image Library (c) 2020 American College of Rheumatology.
The histopathology of CLE lesions hints at some of the potential immune cell-stromal interactions. Although the lesions of acute, subacute, and chronic CLE have some distinct features, dermo–epidermal junction changes with basement membrane vacuolization and apoptotic keratinocytes are seen across the spectrum of changes (17, 18). There is often a perivascular and periadnexal mixed infiltrates comprised of lymphocytes and dendritic cells that can range from sparse to pronounced. Neutrophils can be found in early acute lesions and more densely in some of the rarer forms of CLE lesions (19). Monocytes, macrophages, dendritic cells, and plasmacytoid dendritic cells are also found in cutaneous lesions (20, 21). Langerhans cells have been noted to be less dendritic in morphology and to be present in fewer numbers (22). Linear immune deposits and complement deposited at the dermal epidermal junction form a “lupus band.” Non-lesional skin, despite the absence of overt inflammation, also shows abnormalities, and positive lupus bands, endothelial activation, and fewer Langerhans cells can be seen (18, 23, 24). Epidermal and dermal stromal cells such as keratinocytes and endothelial cells are involved then, as are resident immune cells such as Langerhans cells and infiltrating cells such as monocytes, neutrophils, plasmacytoid dendritic cells, and T cells.
UVR is a form of electromagnetic radiation that is subdivided into the three categories of UVA (320–400 nm), UVB (290– 320 nm), and UVC (200–290 nm) light. UVC emitted by the sun is filtered by the atmosphere and does not reach Earth’s surface. The majority, some 90 percent, of UVB is similarly filtered. The shorter UVB waves that do reach the skin do not penetrate deeper than the epidermis, while the longer UVA waves penetrate into the dermis (3, 25, 26). UVA contributes to ROS generation and photoaging while UVB is more effective at generating DNA breaks. Both UVA and UVB are considered to contribute to the proapoptotic and consequent immune suppressive effects and to photosensitive lesion development. However, UVA1 (340–400 nm) in the longer range of UVA waves can have therapeutic effects, ameliorating systemic disease in a murine lupus model and in limited clinical studies. Its efficacy is attributed to the induction of apoptosis specifically of T and B cells, thus targeting the disease-causing cells. The relative contributions of the different UVR components remain to be fully elucidated.
Work over the past several decades has led to a model involving skin-intrinsic dysfunction combined with immune cell dysfunction. Work by Golan et al and Furukawa et al in the late 1990’s showed that SLE keratinocytes were more sensitive to UVR-induced apoptosis (27, 28), a characteristic recently confirmed by Kahlenberg and colleagues (29), suggesting a keratinocyte-intrinsic contribution to the UVR-induced skin injury. These studies, combined with earlier findings, including those of Casciola-Rosen et al that SLE autoantibodies bind antigens expressed by UVR-exposed keratinocytes (30, 31), contribute to the prevailing model that UVR causes greater keratinocyte apoptosis in SLE and subsequent higher autoantigen levels, opportunity for the selection of autoantibodies, and autoantibody-mediated damage. The autoantigen levels are further increased by reduced clearance of apoptotic cells in SLE (32, 33). More recent work has focused on the mediators of skin inflammation, whereby innate immune cell accumulation is followed by lymphocytic infiltration, leading to the clinical lupus cutaneous lesions (34), and the role and regulation of IFN-I (35). In addition to contributing to tissue injury, immune cells can help to promote normal tissue function, as the complex circuitry between immune cells and the resident epithelial, mesenchymal, and endothelial cells of the “stromal” compartment has been increasingly appreciated across multiple systems (36). Here, we review recent findings in the pathogenesis of lupus photosensitivity from the framework of circuitry. First, we discuss SLE in terms of anatomic circuitry to identify potential loci for pathogenesis. Second, we discuss immune cell-stromal circuitry that has recently been shown to contribute to pathogenesis at some of these loci. We propose this framework to help us all think about tissue injury and autoimmunity in disease.
Photosensitivity and SLE pathogenesis as an anatomic circuit
Viewing SLE pathogenesis through the lens of anatomic immune circuitry (37–39) leads to the identification of several potential loci where dysfunction could contribute to disease (Figure 2). Assuming that response to UVR exposure in part stimulates lymph nodes and are not solely dependent on tissue activities of resident lymphocytes such as skin-resident memory T cells (40), the sensitive keratinocytes will apoptose and release autoantigens. These autoantigens then are brought from the skin via lymphatic vessels in association with dendritic cells or as soluble molecules to the draining lymph node, where autoimmune T and B cells become activated and develop into effector cells. The effector T cells and antibodies secreted by plasma cells will leave via the efferent lymphatics, pool into the blood circulation by way of the thoracic duct and then home from the blood into affected tissues. In a healthy host, inflammation at the site of injury likely leads to tissue repair and subsequent regulation of the immune response. A potential scenario in SLE is that mechanisms that turn off the response fail, leading to continued inflammation and tissue damage. The autoantibodies, potentially in the form of immune complexes (ICs), may also circulate to other tissues such as the kidneys, where they can deposit and cause inflammation. Similarly, lymphocytes can enter these tissues, especially in chronic inflammation perhaps caused by IC deposition or systemic inflammatory cytokines.
Figure 2. SLE as an anatomic immune circuit.
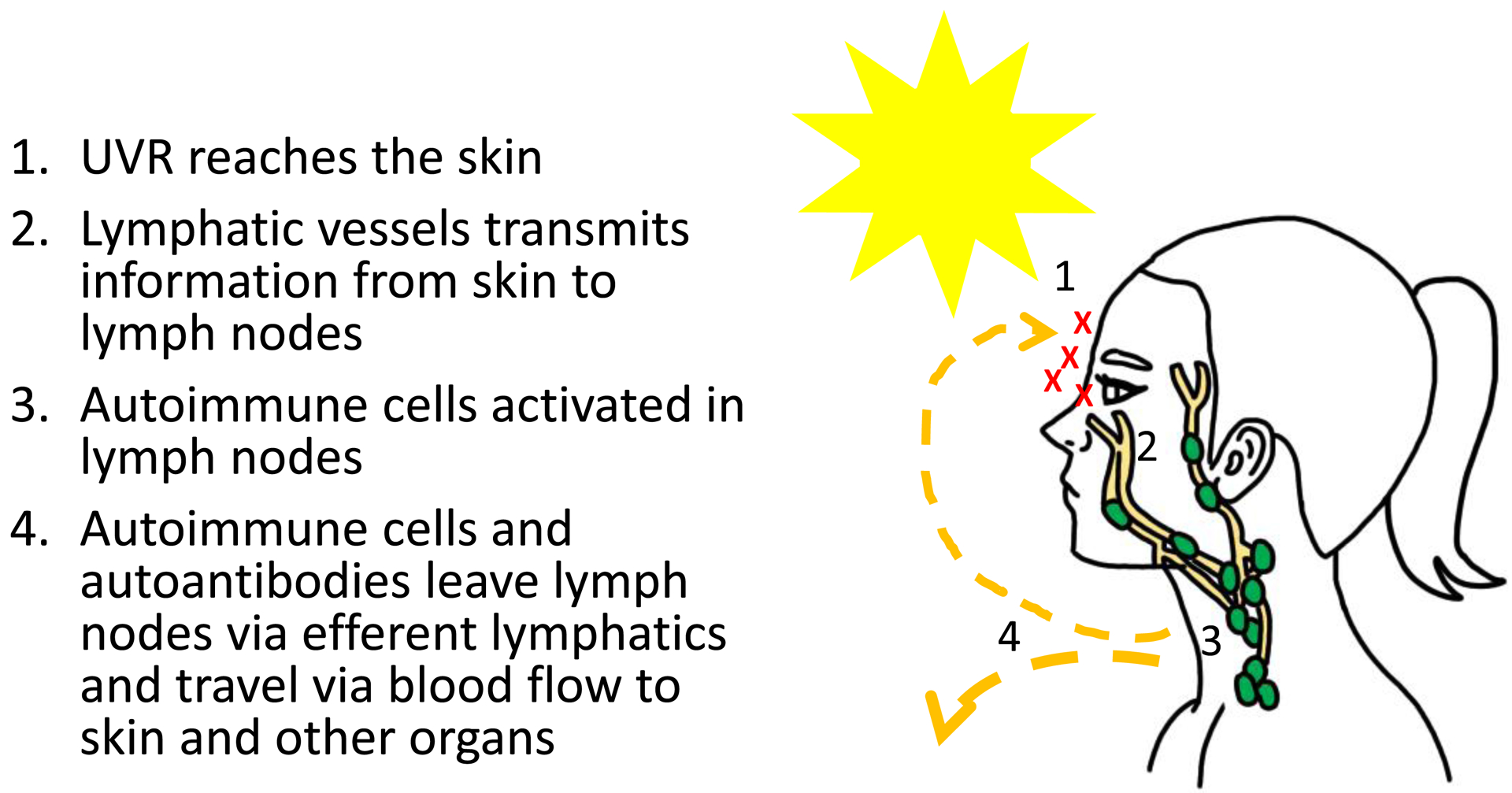
The circuitry that may contribute to propagating information from the skin to draining nodes and systemically, based on the principles of immune circuitry.
While autoimmune lymphocytes are a prerequisite, they may be insufficient for disease manifestations. Pathophysiologic function of tissues in the immune circuit may additionally contribute. Below we focus on the skin and the connection from events in the skin to systemic disease. Because SLE is fundamentally an immune disorder and the importance of immune cells in tissue function is increasingly appreciated, we discuss recent findings of immune cell-stromal circuitry that can contribute to disease pathophysiology.
Immune cell-stromal mechanisms in photosensitivity
Dysfunctional Langerhans cell-keratinocyte circuit:
We have recently shown that Langerhans cells (LCs) protect UVR-induced skin injury by limiting keratinocyte apoptosis via epidermal growth factor receptor (EGFR) ligands. The key mediator in this axis is LC-expressed a disintegrin and metalloproteinase 17 (ADAM17) (24), a requisite sheddase for the activation of many EGFR ligands among other substrates (41). UVR activates LC ADAM17 to cleave EGFR ligands in a cis-dependent manner providing these ligands to keratinocyte EGFR to limit UVR-induced keratinocyte apoptosis (24). Remarkably, LCs in photosensitive murine lupus models showed reduced ADAM17 mRNA expression and enzyme activity, suggesting that intrinsic LC dysfunction could contribute to photosensitivity. Non-lesional human SLE skin showed reduced epidermal EGFR phosphorylation and LC numbers, suggesting that SLE skin has an inadequate source of EGFR ligands perhaps from reduced LC numbers if not LC ADAM17 function. Together, our study suggested a mechanism whereby LC ADAM17 provides EGFR ligands that maintain skin barrier integrity (Figure 3A), and a dysfunctional LC-keratinocyte axis leads to a propensity to photosensitivity. Topical EGFR ligand supplementation ameliorated photosensitivity in lupus models, pointing to the potential therapeutic use of EGFR ligands in photosensitivity.
Figure 3. Immune-stromal circuits in the skin that may contribute to photosensitivity.
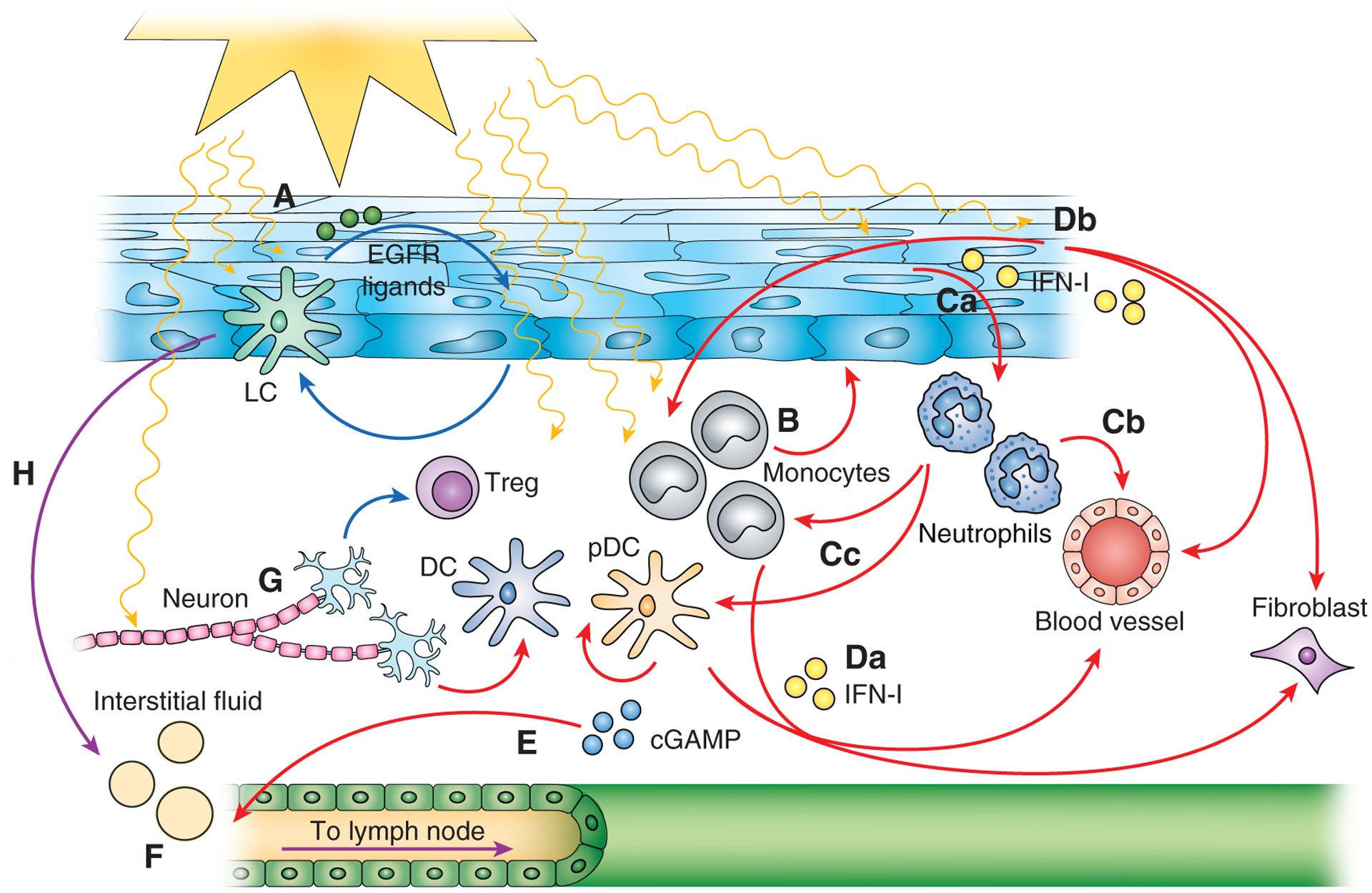
Protective circuits are denoted by blue arrows, pathogenic circuits by red arrows, and circuits that can have dual roles by purple arrows. (A) Langerhans cell-keratinocyte circuit. (B) Monocyte-epidermal circuit. (C) a. keratinocyte-neutrophil circuit; b. neutrophil-endothelial cell circuit; c. neutrophil-immune cell circuit. (D) IFN-I circuits involving IFN-I originating from a. immune cells and b. stromal cells. (E) cGAMP originating from skin for systemic signal transmission. (F) Lymphatic flow and function that connects skin to draining lymph nodes and systemic circulation. (G) Neuronal control of immunity. (H) Migration of skin-derived Langerhans cells and dendritic cells connects skin events to draining lymph nodes. See text for details.
The LC-keratinocyte axis is consistent with the idea that immune cells, especially at barrier surfaces, can be tissue-protective. Keratinocyte ADAM17 expression and generation of EGFR ligands are critical for skin barrier maintenance during development but seem to play a minor role at homeostasis in adults (42). We also showed that LCs do not seem to play a major role in homeostatic adults, suggesting that LCs act as a dominant EGFR ligand source in times of stress including UVR exposure. These data echo the role that DCs have in promoting the survival of mesenchymal cells in inflamed lymph nodes and fibrotic dermis (43, 44) and the role the EGFR ligand amphiregulin plays in protecting the epithelial barrier in inflamed lung and gut by regulatory T cells and ILC2, respectively (45, 46). Immune cells can serve as guardians of tissue function, shoring up stromal cells in times of stress by providing extra resources. Dysfunction of this protective immune cell-stromal circuitry, then, can lead to tissue injury and damage.
Keratinocytes can also regulate LC function. Keratinocytes are known to provide IL-34, which is required for LC differentiation and continued self-renewal at homeostasis (47). IL-34 has also been shown to activate TGFβ to retain LCs in the skin(48). Additionally, keratinocyte MyD88, interleukin-1β (IL-1β), and tumor necrosis factor-α (TNF-α) have been implicated in the regulation of LC migration in murine models of atopic dermatitis (AD) and aged human skin, respectively (49) (50). While the causative factors that drive LC dysfunction in SLE models are not fully understood, it is possible that altered keratinocyte phenotypes contribute to LC dysfunction, fueling a pathogenic feed-forward circuitry that contributes to photosensitivity.
Monocyte and neutrophils-mediated tissue injury
Monocytes have been implicated to play a pathogenic role in photosensitivity. In humans exposed to UVR, monocytes are among the first cells that accumulate in the skin (51), although the cellular infiltrate in established CLE lesions largely consists of T-lymphocytes (52). In the MRL/lpr lupus model, UVR caused keratinocytes and dermal fibroblasts to secrete CSF-1, and experimental deletion of CSF-1 and the associated reduction in myeloid cell accumulation prevented UVR-induced lesion development (53). These results suggested that myeloid cells are important contributors to lesion development. In wild-type mice on a B6 background, UVR-induced monocyte accumulation corresponded with an upregulation of interferon-stimulated genes (ISGs), and monocyte depletion in CCR2-DTR mice prevented ISG expression. These data are consistent with the idea that monocytes can be major producers of pathogenic IFN-I (54, 55). Monocyte depletion also prevented UVR-induced increases in epidermal permeability (24), suggesting that monocytes contribute to skin damage in part by disrupting barrier integrity (Figure 3B). In tissues, monocyte phenotype and function are in part modulated by stromal cells. For example, fibroblast-derived CCL2, in addition to recruiting monocytes to the tissue, can modulate monocyte reactive oxygen species production (56). Additionally, notch ligands, potentially from endothelial cells, can act with TLR ligands to modulate monocyte phenotype and differentiation (57, 58). The fate of monocytes in skin—as monocytes, monocyte-derived dendritic cells, macrophages, or Langerhans cells (59, 60) – and how these cells interact with epidermal and dermal stromal cells in photosensitivity remain to be better understood.
Neutrophils are also recruited to skin early after UVR exposure. There, they phagocytose antigens released by dying keratinocytes and are stimulated by UVR along with ICs of autoantibodies with nuclear autoantigens from dead cells to release neutrophil extracellular traps (NETs) (Figure 3Ca) (61). Netting neutrophils can directly damage endothelial cells (Figure 3Cb) and the NETs, comprised in part by oxidized nucleic acids known to be more resistant to degradation, can induce plasmacytoid dendritic and other cells to upregulate IFN-I production (Figure 3Cc) (62, 63).
IFN-I in immune cell-stromal circuitry
IFN-I can be key mediators in interactions between immune cells and stromal cells in many settings and seems to play a role in photosensitivity. An IFN-I signature is observed in both lesional and non-lesional SLE skin (64–66), and UVR can upregulate IFN-I in skin in humans and mice (51, 54, 67). Recently, clinical trials in SLE patients have shown that anti-IFNAR1, anifrolumab, improves CLE (68, 69). While IFN-I appears to play a pathogenic role in humans, the data in mice are less clear. Topical application of TLR7 agonist imiquimod that drives IFN-I to wild-type B6 mice was sufficient to cause autoimmunity and photosensitivity (70), but global IFNAR deficiency exacerbated UVR-induced skin lesions in otherwise wildtype (i.e. non-lupus) mice (54). Whether this phenotype of global IFNAR deficiency reflects in part the protective role of IFNAR in epithelial maintenance and wound healing (71) remains to be seen. However, recent efforts delineating the exact roles, sources, and regulation of IFN-I at different time points after UVR exposure are starting to paint a picture as discussed below.
Nucleic acids, likely from cell injury or death, seem to be a key driver of the IFN-I response to UVR exposure. Elkon and colleagues have shown an important role for cyclic GMP-AMP synthase (cGAS), which detects cytosolic double-stranded DNA and produces cGAMP that binds to the stimulator of IFN genes (STING). STING then activates, via TBK1, IFN regulatory factor 3 (IRF3) along with NF-κB, which function together to turn on and amplify the transcription of IFN-I and other proinflammatory cytokines (72, 73). In mice, global cGAS and STING deficiency were both associated with reduced UVR-induced skin IFN-I signatures (54, 67). cGAS activity was especially important at the earliest time point, 6 hours, after a single dose of UVR (67). Whether STING is important during the same period of time was not tested, but STING was required for both IFN-I signature and inflammatory cytokine expression with a subacute multi-day regimen. This suggested that other DNA and RNA sensors that can also activate STING (74, 75) may be involved at later time points. In addition to potential involvement of different pathways over time, the key cellular sources of cGAS and STING still need to be worked out.
Immune cells may be important expressors of IFN-I (Figure 3Da) and plasmacytoid DCs (pDCs) in lesional skin have been implicated as a major source (65). Consistent with this idea, depleting pDCs in SLE patients with anti-BDCA2 led to reduced skin IFN-I signatures and improved skin scores (76). In mice, pDCs were necessary for tape stripping-induced lesions in the NZBxNZW lupus model (77). However, other studies have emphasized the importance of inflammatory monocytes over pDCs in producing IFN-I (Figure 3Da). In UVR-exposed lupus patients, an increase in skin ISG expression correlated with T cell and monocyte infiltration but not pDC accumulation (51). Furthermore, a monocytic signature was observed in lesions of CLE patients (78), and inflammatory monocytes, but not pDCs, were also shown to be necessary for a UVR-induced IFN-I signature in wildtype mice (54). Together, the data point to monocytes and pDCs as the likely immune cell that are key sources of or are necessary for IFN-I, although work remains to better understand disease type and stage-specific contributions.
Stromal cells may also be critical sources of IFN-I (Figure 3Db). UVR induced keratinocytes cultured from non-lesional LE skin to upregulate IFN-κ (79), the only IFN-I besides IFNA10 that was detected to be upregulated in CLE lesions (29). In situ, IFN-κ was expressed in both the epidermis and dermis. Keratinocytes were a major source in healthy skin and expressed IFN-κ at higher levels in non-lesional SLE skin. Michelle Kahlenberg group recently showed that the keratinocyte IFN-κ overexpression contributed to their increased sensitivity to UVR-induced apoptosis, amplified responses to other IFN-I, and could stimulate dendritic cell activation (29). Keratinocytes are among the first cells to sense UVR exposure, and their subsequent upregulation of IFN-I may be one of the critical early events in photosensitive responses. While TLR stimulation will upregulate keratinocyte IFN-I expression (79), cGAS and STING are also functional in keratinocytes (80), and the early cGAS-dependent upregulation of IFN-I after UVR (67) may reflect keratinocyte rather than immune cell activity.
IFN-I can act on multiple immune and stromal cell types. DCs, macrophages, B cells, and T cells can all respond to IFN-I, stimulating downstream programs that can promote autoimmunity when effects are unbalanced (81–83). Stromal cells respond as well, with IFN-I presumably acting on keratinocytes to promote epithelial integrity during wound healing (71), but, likely at higher levels, promoting sensitivity to UVR-induced apoptosis and IL-6 expression (29, 79). Similar to the epithelium, endothelial cell barrier integrity is mediated by IFN-I, but high IFN-I levels can induce vascular activation and vasculopathy (84, 85). Fibroblasts can be activated by IFN-I to adopt a pro-inflammatory phenotype (81). Successful targeting of IFN-I in photosensitivity may have to be titrated, leaving enough tonic IFN-I signaling to maintain proper homeostatic tissue function, and consideration of IFN-λ, which can have similar effects (83, 86), may be needed.
From UVR exposure at the skin to systemic flare
A longstanding enigma has been how UVR exposure triggers not only skin lesions but also flares of systemic disease. Supporting the idea that the link between skin lesions and systemic disease reflects similar pathogenic mechanisms in all affected tissues is that the IFN-I gene expression signature found in SLE blood cells (87–89) is also found in lesional skin and kidney (64, 66, 90, 91). Remarkably, vascular activation in even non-lesional skin of SLE patients with kidney disease paralleled vascular activation in kidneys (23), suggesting non-lesional skin was not only abnormal but also a potential barometer of and accessible window into the systemic state. This concept was further reinforced recently by the Accelerated Medicines Partnership (AMP) consortium that showed via single cell RNA sequencing that epithelial cells from both non-lesional skin and diseased kidneys in SLE patients had interferon signatures (66). While these studies showed a clear connection between skin and systemic disease, the results do not establish that skin lesions beget systemic disease.
In mice, there is evidence that UVR will induce systemic effects that are relevant for SLE. BXSB male mice are a model of severe SLE that developed high autoantibody levels, nephritis, and eventually death after UVR exposure (92). The NZM2328 lupus model also showed immune activation with UVR, developing lymphadenopathy, increased IFN-β and IFN-κ in skin, and IFNAR-dependent activated T cell accumulation and Treg suppression in lymph nodes (93). Keith Elkon and colleagues recently showed that UVR treatment can lead to a rapid IFN-I response in skin that is propagated to blood and kidneys of wild type mice (94). The systemic IFN-I response, like the early skin response, was dependent on cGAS, and manipulation of extracellular cGAMP levels modulated the systemic IFN-I response. These results suggest a scenario whereby cGAMP generated in the skin after UVR exposure could enter the circulation and induce a systemic IFN-I response (Figure 3E). It will be interesting to further understand how the Treg effect in NZM2328 mice is disseminated beyond the draining lymph nodes and how the cGAMP circuit may operate differently in lupus models to contribute to pathology.
Circuitry to be explored in photosensitivity
Lymphatics
The lymphatic system functions to reduce inflammation in part by removing fluid from inflamed tissues and propagating tissue-specific information to the draining lymph node via antigens, antigen presenting cells, and cytokines. Increased vessel permeability and/or reduced lymphatic flow increases the magnitude and duration of UVR-induced skin inflammation as seen with inhibition of lymphatic-dependent VEGFR3 or VEGF-A overexpression-mediated vascular permeability (95, 96). Supplementation of VEGFR3 ligands, VEGF-C or VEGF-D, partially reduced UVR-induced damage (97, 98). Interestingly, lymphatic function can also be modulated by immune cells, with inflammatory cells including T cells contributing to lymphatic dysfunction (99, 100) and IRF4-dependent DCs maintaining lymphatic vessel integrity (101). While lymphatic vessels are not known to be dysfunctional in SLE (102), the association of lymphatic dysfunction with inflammation suggest the possibility of lymphatic-specific contributions to lupus photosensitivity.
While moving interstitial fluid out of the skin may reduce inflammation, reducing lymphatic flow may serve protective purposes. Experimentally, reduction of lymphatic flow with viral infection helped to limit systemic dissemination (103). Lymphatic flow may play a critical role in connecting skin pathology to systemic disease flares by transmitting pathogenic signals such as IFN-I to the draining lymph node, where it could help to activate responses or inhibit regulatory responses (93, 104). Or, taking the example of cGAMP discussed above, lymphatic vessels would bring cGAMP from the interstitial fluid to the draining lymph node to be sent via the efferent lymphatics to the blood circulation. Additionally, once brought to the lymph node, soluble molecules can be sent into the conduit system comprised by fibroblastic reticular cells (FRCs) to the basement membrane of postcapillary venules. From the basement membrane, molecules can traverse the endothelium to the vessel lumen and be delivered to the blood stream (105). Lymphatic vessels that drain the skin, then, critically bridge skin signals to the systemic circulation and may contribute to the induction of systemic disease flares after UVR exposure (Figure 3F).
In addition to transmitting proinflammatory signals, the lymphatics transmit regulatory signals that act directly on immune cells. Regulatory signals such as IL-10, Treg cells, and DCs with regulatory functions are all carried by lymphatic vessels to the draining nodes (106–108). Interestingly, fluid transport and DC migration to the lymph node are differentially affected by lymphatic flow disruption (109), suggesting that altered flow can lead to unbalanced information reaching the lymph node. Lymphatic endothelial cells can also directly promote T cell tolerance (110) (111) and induce DCs to adopt a regulatory phenotype (112). Lymphatic flow or phenotypic alterations, then, can modulate the information that reaches the lymph node, which could potentially result in immune dysregulation.
Neuronal-immune cell circuit
The skin is rich in sensory nerves that innervate both the epidermis and dermis. Sensory nerves with nociceptors that sense noxious stimuli can release neuropeptides and neurotransmitters to modulate skin resident and immune cells (113). For example, α-melanocyte-stimulating hormone (α-MSH) induces expansion of tolerogenic dendritic cells and regulatory T cells and can dampen skin inflammation in a psoriasis-like model. Ex vivo, α-MSH can reduce the activity of pathogenic Th17 cells from psoriasis patients (114). Similarly, the neurotransmitter dopamine can activate Tregs (115). In contrast, nociceptor neurons activated by imiquimod can induce dermal DCs to express IL-23, subsequent skin-resident γδ T cell expression of IL-17, and psoriasis-like inflammation (116). This effect may reflect neuronal expression of CGRP, which mediated a similar IL-17 response to Candida albicans infection (117). Interestingly, UVR activates nociceptor neurons that can then mediate vasodilation and other features of inflammation (118). Whether there is a role for nociceptor neurons in lupus photosensitivity and UVR-induced systemic disease flares remains to be determined (Figure 3G).
Langerhans cell and other APC migration
While LCs and dermal DCs migrate constitutively to draining lymph nodes to modulate T cell responses, our findings that LCs can modulate keratinocytes directly to limit UVR-mediated skin injury raise the possibility that LCs may directly modulate lymph node stromal cells (Figure 3H). Classical DCs can modulate vascular-stromal proliferation, growth, and survival with lymph node stimulation and expansion (44, 119–121) as well as FRC contractility to accommodate the expanding lymph node (122, 123). Footpad injection of DCs can stimulate lymph node endothelial and FRC proliferation, and depletion of migratory DCs reduces OVA/CFA-induced vascular-stromal expansion (120, 124, 125), supporting the idea that migratory APCs contribute to this modulation. As such, LCs could have contributed to studies that implicated migratory APCs which used CD11c-DTR, CD11c-cre, or CCR7-deficient mice (120, 122, 123, 125). How LC ADAM17 dysfunction, in addition to its contributions in photosensitivity, can affect lymph node function through the vascular-stromal or lymphoid compartments remains to be determined.
Conclusions
Here, we have reviewed and speculated on how immune cell-stromal circuits in the skin can contribute to photosensitivity and how UVR-induced effects on skin can be associated with flares of systemic disease. The state of the immune system and the tissues are inextricably linked, and there is much to be better understood about the extent to which tissue-modulating functions of immune cells and their interactions with stromal cells contribute to disease.
Support:
This work was supported by NIH R01AI079178, Lupus Research Alliance, and the St. Giles Foundation.
References
- 1.Okon LG, and Werth VP. 2013. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol 27: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsokos GC, Lo MS, Costa Reis P, and Sullivan KE. 2016. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 12: 716–730. [DOI] [PubMed] [Google Scholar]
- 3.Kim A, and Chong BF. 2013. Photosensitivity in cutaneous lupus erythematosus. Photodermatol Photoimmunol Photomed 29: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stannard JN, and Kahlenberg JM. 2016. Cutaneous lupus erythematosus: updates on pathogenesis and associations with systemic lupus. Current opinion in rheumatology 28: 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, Smolen JS, Wofsy D, Boumpas DT, Kamen DL, Jayne D, Cervera R, Costedoat-Chalumeau N, Diamond B, Gladman DD, Hahn B, Hiepe F, Jacobsen S, Khanna D, Lerstrøm K, Massarotti E, McCune J, Ruiz-Irastorza G, Sanchez-Guerrero J, Schneider M, Urowitz M, Bertsias G, Hoyer BF, Leuchten N, Tani C, Tedeschi SK, Touma Z, Schmajuk G, Anic B, Assan F, Chan TM, Clarke AE, Crow MK, Czirják L, Doria A, Graninger W, Halda-Kiss B, Hasni S, Izmirly PM, Jung M, Kumánovics G, Mariette X, Padjen I, Pego-Reigosa JM, Romero-Diaz J, Rúa-Figueroa Fernández Í, Seror R, Stummvoll GH, Tanaka Y, Tektonidou MG, Vasconcelos C, Vital EM, Wallace DJ, Yavuz S, Meroni PL, Fritzler MJ, Naden R, Dörner T, and Johnson SR. 2019. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis 78: 1151–1159. [DOI] [PubMed] [Google Scholar]
- 6.Tanenbaum L, and Tuffanelli DL. 1980. Antimalarial Agents: Chloroquine, Hydroxychloroquine, and Quinacrine. Archives of Dermatology 116: 587–591. [DOI] [PubMed] [Google Scholar]
- 7.Ahluwalia J, and Marsch A. 2019. Photosensitivity and photoprotection in patients with lupus erythematosus. Lupus 28: 697–702. [DOI] [PubMed] [Google Scholar]
- 8.Kuhn A, Gensch K, Haust M, Meuth AM, Boyer F, Dupuy P, Lehmann P, Metze D, and Ruzicka T. 2011. Photoprotective effects of a broad-spectrum sunscreen in ultraviolet-induced cutaneous lupus erythematosus: a randomized, vehicle-controlled, double-blind study. J Am Acad Dermatol 64: 37–48. [DOI] [PubMed] [Google Scholar]
- 9.Vilá LM, Mayor AM, Valentín AH, Rodríguez SI, Reyes ML, Acosta E, and Vilá S. 1999. Association of sunlight exposure and photoprotection measures with clinical outcome in systemic lupus erythematosus. P R Health Sci J 18: 89–94. [PubMed] [Google Scholar]
- 10.Sangüesa Gómez C, Flores Robles BJ, and Andréu JL. 2015. Bone health, vitamin D and lupus. Reumatol Clin 11: 232–236. [DOI] [PubMed] [Google Scholar]
- 11.Edens C, and Robinson AB. 2015. Systemic lupus erythematosus, bone health, and osteoporosis. Curr Opin Endocrinol Diabetes Obes 22: 422–431. [DOI] [PubMed] [Google Scholar]
- 12.Pludowski P, Holick MF, Pilz S, Wagner CL, Hollis BW, Grant WB, Shoenfeld Y, Lerchbaum E, Llewellyn DJ, Kienreich K, and Soni M. 2013. Vitamin D effects on musculoskeletal health, immunity, autoimmunity, cardiovascular disease, cancer, fertility, pregnancy, dementia and mortality-a review of recent evidence. Autoimmun Rev 12: 976–989. [DOI] [PubMed] [Google Scholar]
- 13.Mikita N, Ikeda T, Ishiguro M, and Furukawa F. 2011. Recent advances in cytokines in cutaneous and systemic lupus erythematosus. J Dermatol 38: 839–849. [DOI] [PubMed] [Google Scholar]
- 14.Ogunsanya ME, Kalb SJ, Kabaria A, and Chen S. 2017. A systematic review of patient-reported outcomes in patients with cutaneous lupus erythematosus. Br J Dermatol 176: 52–61. [DOI] [PubMed] [Google Scholar]
- 15.Tebbe B, and Orfanos CE. 1997. Epidemiology and socioeconomic impact of skin disease in lupus erythematosus. Lupus 6: 96–104. [DOI] [PubMed] [Google Scholar]
- 16.Foering K, Goreshi R, Klein R, Okawa J, Rose M, Cucchiara A, and Werth VP. 2012. Prevalence of self-report photosensitivity in cutaneous lupus erythematosus. J Am Acad Dermatol 66: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obermoser G, Sontheimer RD, and Zelger B. 2010. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus 19: 1050–1070. [DOI] [PubMed] [Google Scholar]
- 18.Crowson AN, and Magro C. 2001. The cutaneous pathology of lupus erythematosus: a review. J Cutan Pathol 28: 1–23. [DOI] [PubMed] [Google Scholar]
- 19.Johnson-Huang LM, McNutt NS, Krueger JG, and Lowes MA. 2009. Cytokine-producing dendritic cells in the pathogenesis of inflammatory skin diseases. Journal of clinical immunology 29: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reefman E, de Jong MC, Kuiper H, Jonkman MF, Limburg PC, Kallenberg CG, and Bijl M. 2006. Is disturbed clearance of apoptotic keratinocytes responsible for UVB-induced inflammatory skin lesions in systemic lupus erythematosus? Arthritis Res Ther 8: R156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, and Jahnsen FL. 2001. Plasmacytoid Dendritic Cells (Natural Interferon- α/β-Producing Cells) Accumulate in Cutaneous Lupus Erythematosus Lesions. The American journal of pathology 159: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sontheimer RD, and Pr B. 1982. Epidermal Langerhans cell involvement in cutaneous lupus erythematosus. J Invest Dermatol 79: 237–243. [DOI] [PubMed] [Google Scholar]
- 23.Izmirly PM, Shvartsbeyn M, Meehan S, Franks A, Braun A, Ginzler E, Xu SX, Yee H, Rivera TL, Esmon C, Barisoni L, Merrill JT, Buyon JP, and Clancy RM. 2012. Dysregulation of the microvasculature in nonlesional non-sun-exposed skin of patients with lupus nephritis. J Rheumatol 39: 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shipman WD, Chyou S, Ramanathan A, Izmirly PM, Sharma S, Pannellini T, Dasoveanu DC, Qing X, Magro CM, Granstein RD, Lowes MA, Pamer EG, Kaplan DH, Salmon JE, Mehrara BJ, Young JW, Clancy RM, Blobel CP, and Lu TT. 2018. A protective Langerhans cell–keratinocyte axis that is dysfunctional in photosensitivity. Science Translational Medicine 10: eaap9527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.York NR, and Jacobe HT. 2010. UVA1 phototherapy: a review of mechanism and therapeutic application. International Journal of Dermatology 49: 623–630. [DOI] [PubMed] [Google Scholar]
- 26.Vieyra-Garcia PA, and Wolf P. 2018. From Early Immunomodulatory Triggers to Immunosuppressive Outcome: Therapeutic Implications of the Complex Interplay Between the Wavebands of Sunlight and the Skin. Front Med (Lausanne) 5: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mevorach D, Zhou JL, Song X, and Elkon KB. 1998. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med 188: 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furukawa F, Itoh T, Wakita H, Yagi H, Tokura Y, Norris DA, and Takigawa M. 1999. Keratinocytes from patients with lupus erythematosus show enhanced cytotoxicity to ultraviolet radiation and to antibody-mediated cytotoxicity. Clin Exp Immunol 118: 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, Berthier CC, Swindell WR, Patrick MT, Shao S, Tsou P-S, Uppala R, Beamer MA, Srivastava A, Bielas SL, Harms PW, Getsios S, Elder JT, Voorhees JJ, Gudjonsson JE, and Kahlenberg JM. 2018. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Annals of the Rheumatic Diseases 77: 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golan TD, Elkon KB, Gharavi AE, and Krueger JG. 1992. Enhanced membrane binding of autoantibodies to cultured keratinocytes of systemic lupus erythematosus patients after ultraviolet B/ultraviolet A irradiation. Journal of Clinical Investigation 90: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casciola-Rosen LA, Anhalt G, and Rosen A. 1994. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 179: 1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung JH, Kwon OS, Eun HC, Youn JI, Song YW, Kim JG, and Cho KH. 1998. Apoptosis in the pathogenesis of cutaneous lupus erythematosus. Am J Dermatopathol 20: 233–241. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn A, Herrmann M, Kleber S, Beckmann-Welle M, Fehsel K, Martin-Villalba A, Lehmann P, Ruzicka T, Krammer PH, and Kolb-Bachofen V. 2006. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum 54: 939–950. [DOI] [PubMed] [Google Scholar]
- 34.Lehmann P, and Homey B. 2009. Clinic and pathophysiology of photosensitivity in lupus erythematosus. Autoimmun Rev 8: 456–461. [DOI] [PubMed] [Google Scholar]
- 35.Wolf SJ, Estadt SN, Gudjonsson JE, and Kahlenberg JM. 2018. Human and Murine Evidence for Mechanisms Driving Autoimmune Photosensitivity. Front Immunol 9: 2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rankin LC, and Artis D. 2018. Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell 173: 554–567. [DOI] [PubMed] [Google Scholar]
- 37.Baeyens AAL, and Schwab SR. 2020. Finding a Way Out: S1P Signaling and Immune Cell Migration. Annu Rev Immunol 38: 759–784. [DOI] [PubMed] [Google Scholar]
- 38.Hampton HR, and Chtanova T. 2019. Lymphatic Migration of Immune Cells. Front Immunol 10: 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Permanyer M, Bošnjak B, and Förster R. 2018. Dendritic cells, T cells and lymphatics: dialogues in migration and beyond. Current opinion in immunology 53: 173–179. [DOI] [PubMed] [Google Scholar]
- 40.Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, Scott MC, Fares-Frederickson NJ, Wijeyesinghe S, Thompson EA, Borges da Silva H, Vezys V, Youngblood B, and Masopust D. 2020. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nature Immunology 21: 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zunke F, and Rose-John S. 2017. The shedding protease ADAM17: Physiology and pathophysiology. BBA - Molec Cell Res. [DOI] [PubMed] [Google Scholar]
- 42.Franzke C-W, Cobzaru C, Triantafyllopoulou A, Löffek S, Horiuchi K, Threadgill DW, Kurz T, van Rooijen N, Bruckner-Tuderman L, and Blobel CP. 2012. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. The Journal of experimental medicine 209: 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chia JJ, Zhu T, Chyou S, Dasoveanu DC, Carballo C, Tian S, Magro CM, Rodeo S, Spiera RF, Ruddle NH, McGraw TE, Browning JL, Lafyatis R, Gordon JK, and Lu TT. 2016. Dendritic cells maintain dermal adipose-derived stromal cells in skin fibrosis. J Clin Invest 126: 4331–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar V, Dragos C Dasoveanu S Chyou T-C Tzeng C Rozo Y Liang W Stohl Y.-X. Fu, Nancy H. Ruddle, and Lu TT. 2015. A Dendritic-Cell-Stromal Axis Maintains Immune Responses in Lymph Nodes. Immunity 42: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, and Rudensky AY. 2015. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162: 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, and Artis D. 2015. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 112: 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, Kündig TM, Frei K, Ginhoux F, Merad M, and Becher B. 2012. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37: 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, Welty NE, Igyarto BZ, Wijeyesinghe S, Thompson EA, Matte C, Bartholin L, Kaplan A, Sheppard D, Bridges AG, Shlomchik WD, Masopust D, and Kaplan DH. 2016. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat Immunol 17: 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Didovic S, Opitz FV, Holzmann B, Förster I, and Weighardt H. 2016. Requirement of MyD88 signaling in keratinocytes for Langerhans cell migration and initiation of atopic dermatitis-like symptoms in mice. Eur J Immunol. [DOI] [PubMed] [Google Scholar]
- 50.Pilkington SM, Ogden S, Eaton LH, Dearman RJ, Kimber I, and Griffiths CEM. 2018. Lower levels of interleukin-1β gene expression are associated with impaired Langerhans’ cell migration in aged human skin. Immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reefman E, Kuiper H, Limburg PC, Kallenberg CG, and Bijl M. 2008. Type I interferons are involved in the development of ultraviolet B-induced inflammatory skin lesions in systemic lupus erythaematosus patients. Ann Rheum Dis 67: 11–18. [DOI] [PubMed] [Google Scholar]
- 52.Baltaci M, and Fritsch P. 2009. Histologic features of cutaneous lupus erythematosus. Autoimmun Rev 8: 467–473. [DOI] [PubMed] [Google Scholar]
- 53.Menke J, Hsu MY, Byrne KT, Lucas JA, Rabacal WA, Croker BP, Zong XH, Stanley ER, and Kelley VR. 2008. Sunlight triggers cutaneous lupus through a CSF-1-dependent mechanism in MRL-Fas(lpr) mice. J Immunol 181: 7367–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sontheimer C, Liggitt D, and Elkon KB. 2017. Ultraviolet B Irradiation Causes Stimulator of Interferon Genes–Dependent Production of Protective Type I Interferon in Mouse Skin by Recruited Inflammatory Monocytes. Arthritis & Rheumatology 69: 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee PY, Weinstein JS, Nacionales DC, Scumpia PO, Li Y, Butfiloski E, van Rooijen N, Moldawer L, Satoh M, and Reeves WH. 2008. A Novel Type I IFN-Producing Cell Subset in Murine Lupus. J Immunol 180: 5101–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dasoveanu DC, Park HJ, Ly CL, Shipman WD, Chyou S, Kumar V, Tarlinton D, Ludewig B, Mehrara BJ, and Lu TT. 2020. Lymph node stromal CCL2 limits antibody responses. Sci Immunol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gamrekelashvili J, Giagnorio R, Jussofie J, Soehnlein O, Duchene J, Briseño CG, Ramasamy SK, Krishnasamy K, Limbourg A, Häger C, Kapanadze T, Ishifune C, Hinkel R, Radtke F, Strobl LJ, Zimber-Strobl U, Napp LC, Bauersachs J, Haller H, Yasutomo K, Kupatt C, Murphy KM, Adams RH, Weber C, and Limbourg FP. 2016. Regulation of monocyte cell fate by blood vessels mediated by Notch signalling. Nature Communications 7: 12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gamrekelashvili J, Kapanadze T, Sablotny S, Ratiu C, Dastagir K, Lochner M, Karbach S, Wenzel P, Sitnow A, Fleig S, Sparwasser T, Kalinke U, Holzmann B, Haller H, and Limbourg FP. 2020. Notch and TLR signaling coordinate monocyte cell fate and inflammation. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferrer IR, West HC, Henderson S, Ushakov DS, Santos e Sousa P, Strid J, Chakraverty R, Yates AJ, and Bennett CL. 2019. A wave of monocytes is recruited to replenish the long-term Langerhans cell network after immune injury. Science Immunology 4: eaax8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh TP, Zhang HH, Borek I, Wolf P, Hedrick MN, Singh SP, Kelsall BL, Clausen BE, and Farber JM. 2016. Monocyte-derived inflammatory Langerhans cells and dermal dendritic cells mediate psoriasis-like inflammation. Nature Communications 7: 13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, and Kaplan MJ. 2016. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22: 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, and Kaplan MJ. 2011. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187: 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, Tüting T, Hartmann G, and Barchet W. 2013. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39: 482–495. [DOI] [PubMed] [Google Scholar]
- 64.Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, and Jahnsen FL. 2001. Plasmacytoid Dendritic Cells (Natural Interferon- {{alpha}}/{beta}-Producing Cells) Accumulate in Cutaneous Lupus Erythematosus Lesions. Am J Pathol 159: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meller S, Winterberg F, Gilliet M, Muller A, Lauceviciute I, Rieker J, Neumann NJ, Kubitza R, Gombert M, Bunemann E, Wiesner U, Franken-Kunkel P, Kanzler H, Dieu-Nosjean MC, Amara A, Ruzicka T, Lehmann P, Zlotnik A, and Homey B. 2005. Ultraviolet radiation-induced injury, chemokines, and leukocyte recruitment: An amplification cycle triggering cutaneous lupus erythematosus. Arthritis Rheum 52: 1504–1516. [DOI] [PubMed] [Google Scholar]
- 66.Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, Izmirly P, Clancy R, Belmont HM, Koenigsberg M, Mokrzycki M, Rominieki H, Graham JA, Rocca JP, Bornkamp N, Jordan N, Schulte E, Wu M, Pullman J, Slowikowski K, Raychaudhuri S, Guthridge J, James J, Buyon J, Tuschl T, and Putterman C. 2019. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol 20: 915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, Baum R, Kawasumi M, Green R, Gale M Jr., Kalus A, Werth VP, and Elkon KB. 2020. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Scientific reports 10: 7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, and Tummala R. 2020. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. The New England journal of medicine 382: 211–221. [DOI] [PubMed] [Google Scholar]
- 69.Merrill JT, Furie R, Werth VP, Khamashta M, Drappa J, Wang L, Illei G, and Tummala R. 2018. Anifrolumab effects on rash and arthritis: impact of the type I interferon gene signature in the phase IIb MUSE study in patients with systemic lupus erythematosus. Lupus science & medicine 5: e000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yokogawa M, Takaishi M, Nakajima K, Kamijima R, Fujimoto C, Kataoka S, Terada Y, and Sano S. 2014. Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: a new model of systemic Lupus erythematosus. Arthritis Rheumatol 66: 694–706. [DOI] [PubMed] [Google Scholar]
- 71.Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, Arai N, Gallo RL, Digiovanni J, and Gilliet M. 2010. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med 207: 2921–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tanaka Y, and Chen ZJ. 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5: ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, and Chen ZJ. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630. [DOI] [PubMed] [Google Scholar]
- 74.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, Jakobsen MR, Nevels MM, Bowie AG, and Unterholzner L. 2018. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol Cell 71: 745–760.e745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zevini A, Olagnier D, and Hiscott J. 2017. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol 38: 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, Wang W, Christmann R, Gardet A, Pellerin A, Hamann S, Auluck P, Barbey C, Gulati P, Rabah D, and Franchimont N. 2019. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest 129: 1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guiducci C, Tripodo C, Gong M, Sangaletti S, Colombo MP, Coffman RL, and Barrat FJ. 2010. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med 207: 2931–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berthier CC, Tsoi LC, Reed TJ, Stannard JN, Myers EM, Namas R, Xing X, Lazar S, Lowe L, Kretzler M, Gudjonsson JE, and Kahlenberg JM. 2019. Molecular Profiling of Cutaneous Lupus Lesions Identifies Subgroups Distinct from Clinical Phenotypes. Journal of clinical medicine 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stannard JN, Reed TJ, Myers E, Lowe L, Sarkar MK, Xing X, Gudjonsson JE, and Kahlenberg JM. 2017. Lupus Skin Is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. J Invest Dermatol 137: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Almine JF, O’Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, and Unterholzner L. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nature Communications 8: 14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ivashkiv LB, and Donlin LT. 2014. Regulation of type I interferon responses. Nat Rev Immunol 14: 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale AS, Glass DD, Lopez-Ocasio M, Baur R, Kanakabandi K, Porcella SF, and Shevach EM. 2018. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLOS Pathogens 14: e1006985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lazear HM, Schoggins JW, and Diamond MS. 2019. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 50: 907–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barrat FJ, and Lu TT. 2019. Role of type I interferons and innate immunity in systemic sclerosis: unbalanced activities on distinct cell types? Current opinion in rheumatology 31: 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin GR, Henare K, Salazar C, Scheidl-Yee T, Eggen LJ, Tailor PP, Kim JH, Podstawka J, Fritzler MJ, Kelly MM, Yipp BG, and Jirik FR. 2019. Expression of a constitutively active human STING mutant in hematopoietic cells produces an Ifnar1-dependent vasculopathy in mice. Life Science Alliance 2: e201800215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goel RR, Wang X, O’Neil LJ, Nakabo S, Hasneen K, Gupta S, Wigerblad G, Blanco LP, Kopp JB, Morasso MI, Kotenko SV, Yu ZX, Carmona-Rivera C, and Kaplan MJ. 2020. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc Natl Acad Sci U S A 117: 5409–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, and Pascual V. 2003. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J. Exp. Med 197: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, and Behrens TW. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. PNAS 100: 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crow MK, Kirou KA, and Wohlgemuth J. 2003. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity 36: 481–490. [DOI] [PubMed] [Google Scholar]
- 90.Blomberg S, Eloranta ML, Cederblad B, Nordlin K, Alm GV, and Ronnblom L. 2001. Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus 10: 484–490. [DOI] [PubMed] [Google Scholar]
- 91.Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, Chicoine A, Eisenhaure TM, Jonsson AH, Li S, Lieb DJ, Zhang F, Slowikowski K, Browne EP, Noma A, Sutherby D, Steelman S, Smilek DE, Tosta P, Apruzzese W, Massarotti E, Dall’Era M, Park M, Kamen DL, Furie RA, Payan-Schober F, Pendergraft WF 3rd, McInnis EA, Buyon JP, Petri MA, Putterman C, Kalunian KC, Woodle ES, Lederer JA, Hildeman DA, Nusbaum C, Raychaudhuri S, Kretzler M, Anolik JH, Brenner MB, Wofsy D, Hacohen N, and Diamond B. 2019. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol 20: 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.----Ansel JC, Mountz J, Steinberg AD, DeFabo E, and Green 1985. Effects of UV radiation on autoimmune strains of mice: increased mortality and accelerated autoimmunity in BXSB male mice. J Invest Dermatol 85: 181–186. [DOI] [PubMed] [Google Scholar]
- 93.Wolf SJ, Estadt SN, Theros J, Moore T, Ellis J, Liu J, Reed TJ, Jacob CO, Gudjonsson JE, and Kahlenberg JM. 2019. Ultraviolet light induces increased T cell activation in lupus-prone mice via type I IFN-dependent inhibition of T regulatory cells. J Autoimmun 103: 102291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, Kawasumi M, Green R, Gale M, Kalus A, Werth VP, and Elkon KB. 2019. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. bioRxiv: 835132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kajiya K, Hirakawa S, and Detmar M. 2006. Vascular endothelial growth factor-A mediates ultraviolet B-induced impairment of lymphatic vessel function. Am J Pathol 169: 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kajiya K, and Detmar M. 2006. An Important Role of Lymphatic Vessels in the Control of UVB-Induced Edema Formation and Inflammation. J Invest Dermatol 126: 920–922. [DOI] [PubMed] [Google Scholar]
- 97.Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M, Werner S, Alitalo K, and Detmar M. 2011. An important role of lymphatic vessel activation in limiting acute inflammation. Blood 117: 4667–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kajiya K, Sawane M, Huggenberger R, and Detmar M. 2009. Activation of the VEGFR-3 pathway by VEGF-C attenuates UVB-induced edema formation and skin inflammation by promoting lymphangiogenesis. J Invest Dermatol 129: 1292–1298. [DOI] [PubMed] [Google Scholar]
- 99.Fonseca DM, Hand TW, Han SJ, Gerner MY, Glatman Zaretsky A, Byrd AL, Harrison OJ, Ortiz AM, Quinones M, Trinchieri G, Brenchley JM, Brodsky IE, Germain RN, Randolph GJ, and Belkaid Y. 2015. Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell 163: 354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Torrisi JS, Hespe GE, Cuzzone DA, Savetsky IL, Nitti MD, Gardenier JC, Nores GDG, Jowhar D, Kataru RP, and Mehrara BJ. 2016. Inhibition of Inflammation and iNOS Improves Lymphatic Function in Obesity. Scientific reports 6: 19817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ivanov S, Scallan JP, Kim K-W, Werth K, Johnson MW, Saunders BT, Wang PL, Kuan EL, Straub AC, Ouhachi M, Weinstein EG, Williams JW, Briseño C, Colonna M, Isakson BE, Gautier EL, Förster R, Davis MJ, Zinselmeyer BH, and Randolph GJ. 2016. CCR7 and IRF4-dependent dendritic cells regulate lymphatic collecting vessel permeability. The Journal of Clinical Investigation 126: 1581–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schwartz N, Chalasani MLS, Li TM, Feng Z, Shipman WD, and Lu TT. 2019. Lymphatic Function in Autoimmune Diseases. Front Immunol 10: 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Loo CP, Nelson NA, Lane RS, Booth JL, Loprinzi Hardin SC, Thomas A, Slifka MK, Nolz JC, and Lund AW. 2017. Lymphatic Vessels Balance Viral Dissemination and Immune Activation following Cutaneous Viral Infection. Cell Reports 20: 3176–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barrat FJ, Crow MK, and Ivashkiv LB. 2019. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol 20: 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, and von Andrian UH. 2001. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med 194: 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ding W, Beissert S, Deng L, Miranda E, Cassetty C, Seiffert K, Campton KL, Yan Z, Murphy GF, Bluestone JA, and Granstein RD. 2003. Altered cutaneous immune parameters in transgenic mice overexpressing viral IL-10 in the epidermis. J Clin Invest 111: 1923–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tomura M, Honda T, Tanizaki H, Otsuka A, Egawa G, Tokura Y, Waldmann H, Hori S, Cyster JG, Watanabe T, Miyachi Y, Kanagawa O, and Kabashima K. 2010. Activated regulatory T cells are the major T cell type emigrating from the skin during a cutaneous immune response in mice. J Clin Invest 120: 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Clausen BE, and Stoitzner P. 2015. Functional Specialization of Skin Dendritic Cell Subsets in Regulating T Cell Responses. Frontiers in immunology 6: 534–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Platt AM, Rutkowski JM, Martel C, Kuan EL, Ivanov S, Swartz MA, and Randolph GJ. 2013. Normal dendritic cell mobilization to lymph nodes under conditions of severe lymphatic hypoplasia. J Immunol 190: 4608–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Card CM, Yu SS, and Swartz MA. 2014. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Invest 124: 943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krishnamurty AT, and Turley SJ. 2020. Lymph node stromal cells: cartographers of the immune system. Nat Immunol 21: 369–380. [DOI] [PubMed] [Google Scholar]
- 112.Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, and Skobe M. 2009. Inflamed Lymphatic Endothelium Suppresses Dendritic Cell Maturation and Function via Mac-1/ICAM-1-Dependent Mechanism. The Journal of Immunology 183: 1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Blake KJ, Jiang XR, and Chiu IM. 2019. Neuronal Regulation of Immunity in the Skin and Lungs. Trends Neurosci 42: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Auriemma M, Brzoska T, Klenner L, Kupas V, Goerge T, Voskort M, Zhao Z, Sparwasser T, Luger TA, and Loser K. 2012. alpha-MSH-stimulated tolerogenic dendritic cells induce functional regulatory T cells and ameliorate ongoing skin inflammation. J Invest Dermatol 132: 1814–1824. [DOI] [PubMed] [Google Scholar]
- 115.Kerage D, Sloan EK, Mattarollo SR, and McCombe PA. 2019. Interaction of neurotransmitters and neurochemicals with lymphocytes. J Neuroimmunol 332: 99–111. [DOI] [PubMed] [Google Scholar]
- 116.Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, Paust S, Wood JN, and von Andrian UH. 2014. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510: 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kashem Sakeen W., Riedl Maureen S., Yao C, Christopher N. Honda, Vulchanova L, and Kaplan Daniel H.. 2015. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 43: 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Scholzen TE, Brzoska T, Kalden DH, O’Reilly F, Armstrong CA, Luger TA, and Ansel JC. 1999. Effect of ultraviolet light on the release of neuropeptides and neuroendocrine hormones in the skin: mediators of photodermatitis and cutaneous inflammation. J Investig Dermatol Symp Proc 4: 55–60. [DOI] [PubMed] [Google Scholar]
- 119.Tzeng TC, Chyou S, Tian S, Webster B, Carpenter AC, Guaiquil VH, and Lu TT. 2010. CD11chi dendritic cells regulate the re-establishment of vascular quiescence and stabilization after immune stimulation of lymph nodes. J Immunol 184: 4247–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Benahmed F, Chyou S, Dasoveanu D, Chen J, Kumar V, Iwakura Y, and Lu TT. 2014. Multiple CD11c+ cells collaboratively express IL-1beta to modulate stromal vascular endothelial growth factor and lymph node vascular-stromal growth. J Immunol 192: 4153–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang C-Y, Vogt TK, Favre S. p., Scarpellino L, Huang H-Y, Tacchini-Cottier F, and Luther SA. 2014. Trapping of naive lymphocytes triggers rapid growth and remodeling of the fibroblast network in reactive murine lymph nodes. Proc Natl Acad Sci U S A 111: e109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Acton SE, Farrugia AJ, Astarita JL, Mourao-Sa D, Jenkins RP, Nye E, Hooper S, van Blijswijk J, Rogers NC, Snelgrove KJ, Rosewell I, Moita LF, Stamp G, Turley SJ, Sahai E, and Reis e Sousa C. 2014. Dendritic cells control fibroblastic reticular network tension and lymph node expansion. Nature 514: 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Astarita JL, Cremasco V, Fu J, Darnell MC, Peck JR, Nieves-Bonilla JM, Song K, Kondo Y, Woodruff MC, Gogineni A, Onder L, Ludewig B, Weimer RM, Carroll MC, Mooney DJ, Xia L, and Turley SJ. 2015. The CLEC-2-podoplanin axis controls the contractility of fibroblastic reticular cells and lymph node microarchitecture. Nat Immunol 16: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, and Lu TT. 2006. Regulation of lymph node vascular growth by dendritic cells. J Exp Med 203: 1903–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chyou S, Benahmed F, Chen J, Kumar V, Tian S, Lipp M, and Lu TT. 2011. Coordinated regulation of lymph node vascular-stromal growth first by CD11c+ cells and then by T and B cells. J Immunol 187: 5558–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]