

Resilient silica-nanoparticle lattices organized by DNA withstand harsh temperature, pressure and radiation conditions.
Abstract
Rapid developments of DNA-based assembly methods provide versatile capabilities in organizing nanoparticles (NPs) in three-dimensional (3D) organized nanomaterials, which is important for optics, catalysis, mechanics, and beyond. However, the use of these nanomaterials is often limited by the narrow range of conditions in which DNA lattices are stable. We demonstrate here an approach to creating an inorganic, silica-based replica of 3D periodic DNA-NP structures with different lattice symmetries. The created ordered nanomaterials, through the precise 3D mineralization, maintain the spatial topology of connections between NPs by DNA struts and exhibit a controllable degree of the porosity. The formed silicated DNA-NP lattices exhibit excellent resiliency. They are stable when exposed to extreme temperatures (>1000°C), pressures (8 GPa), and harsh radiation conditions and can be processed by the conventional nanolithography methods. The presented approach allows the use of a DNA assembly strategy to create organized nanomaterials for a broad range of operational conditions.
INTRODUCTION
Starting from the visionary idea of using DNA for organizing nanoscale matter (1) and from the early demonstration of using DNA motifs for linking inorganic particles (2, 3), DNA-based assembly methods have become a powerful approach to creating nanoscale structures, including planar (4–7) and three-dimensional (3D) architectures (8, 9) from DNA (6, 10–14), nanoparticle (NP) clusters (15–17), and 3D NP lattices guided by DNA grafted to particle surfaces (18–21). The usefulness of DNA-based nanomaterial assembly approaches is particularly attractive due to the intrinsic limitations of lithographic methods for fabrication of 3D arrays and incorporation of wet chemistry–derived nanoscale objects. Enabling 3D fabrication of rationally organized nanomaterials is critical for emerging technologies, including quantum information systems, neuromorphic computing, and optical and mechanical metamaterials (22–25), and these functionally desired structures are typically not attainable by the established nanofabrication approaches. From the self-assembly perspective, the challenge of creating structurally robust and rationally organized complex 3D nanomaterials is well recognized. For example, a comprehensive strategy for the assembly of designed NP lattices using polyhedral DNA origami constructs of different shapes (26–29) and particles of different geometries (30, 31) for the formation of diverse types of lattice symmetries has been demonstrated. Given the exquisite structural control offered by DNA in creating complex 3D structures with integrated functional nano-objects (13, 16, 32–35), it is increasingly important to have the ability to translate the assembly methodology into a generation of materials that are not limited by the environmental requirements of DNA. For instance, stabilization of DNA structures using polymers and peptoids for a range of buffer conditions was demonstrated (36, 37) as a method for preserving DNA structures in a broader range of liquid environments. Moreover, the future photonic, catalytic, and mechanical applications of DNA-assembled NP-based materials might operate at high temperatures, in vacuum or reactive environments, at elevated pressures or under radiation conditions. This challenge also requires an understanding of the behavior of nanostructures at these extreme conditions (38–42). Thus, it is important and timely to develop methods that can take advantage of the DNA-enabled assembly methodology for the creation of complexly designed and resilient 3D NP-based materials stable under a wide range of temperatures and environments.
Several approaches that use DNA for templating inorganic materials have been investigated. For example, DNA-metallization processes were actively developed for low-dimensional DNA (43, 44) structures using metals, oxides, and sulfides. However, due to uncontrolled nucleation, it has been difficult to achieve continuity of structures and thus replication of these architectures. The molecular intercalation (45) allows the modification of a double helix more precisely, but it does not permit stabilization over a broad range of conditions. The use of silica for templating organic structures has attracted much attention (46, 47). Silica offers excellent chemical and temperature stability and access to a broad range of functionalization chemistries and allows subsequent processing, such as atomic layer deposition. In this study, we demonstrate a method for converting 3D DNA-NP lattices into silica replicas while maintaining the topology of interparticle connections by DNA struts and the integrity of NP organization. We also show that such ordered 3D structures are resilient to high temperature, high pressure, and radiation. Our approach preserves the 3D architecture of the lattice, NP placement, and the connectivity of DNA origami struts while allowing one to tune the porosity of the lattices via strut thickness. Using in situ x-ray scattering and electron microscopy methods, we show that these materials are stable at extreme conditions (>1000°C) and pressures (8 GPa), and their architecture can be tuned through the selection of origami shape and NP, and through the silication process. The approach offers a plethora of opportunities for creating resilient 3D NP-integrated structured materials with control of nanometer-scale architecture, which can find applications in optics, catalysis, and nanomechanics.
The sol-gel approach (48) has been extensively used for creating structural materials using silica templating of lyotropic surfactant phases (46, 49) and block copolymers (50), but structural diversity is limited by the most common cubic, lamellar, and columnar morphologies of the underlying phase. Sol-gel methods were also used to silicate more complex objects, such as proteins or even mammalian cells (51, 52), to tailor morphology and surface chemistry of shaped NPs (53), as well as to embed NP assemblies into a silica matrix (54, 55). Recently, an existence of ultrasmall symmetric silica cages formed from micelles was reported (56) and such cages were used to form 2D lattices (57). Various methods of obtaining DNA-templated silicas, including the formation of hollow tubules and circular replicas (58), ordered nanochannel arrays, and porous DNA-silica xerogels (59), have been reported. More recently, sol-gel methods have been used to transition NP lattices assembled by DNA shells grafted to NP surfaces into a solid phase (55); however, in this approach, the entire lattice is fully encapsulated in silica matrix. The precise control over the silica architecture and lattice porosity is not possible. Nanoscale templated growth using DNA origami has been demonstrated for origami constructs fixed to a surface in native magnesium buffer (60) and DNA lattice in low magnesium buffer (61), and these approaches allow templating DNA nanostructure but do not incorporate nanocomponents. In the approach presented here, we show that silication can be used for creating 3D silica-NP architectures in the bulk and native magnesium buffer. These inorganic replicas of engineered DNA origami–AuNP superlattices (26, 27) exhibit remarkable resilience under harsh environmental conditions. Because of the practically unlimited opportunities in creating superlattices using designed origami motifs, the presented approach leads to the creation of by-design fabrication of 3D inorganic architectures that can be loaded with functional components.
RESULTS
Our process was specifically devised to yield a conformal coating for the 3D nanoscale structure, thus preserving the entire 3D architecture, which is determined by the position of NPs in a lattice and interparticle connectivity by DNA struts of DNA frames (27). In our approach, the reaction conditions are optimized to achieve uniform silication within the 3D DNA-NP frameworks, thus templating results in an inorganic replica of the entire architecture with the ability to control its structurally defined porosity through the degree of silica coating. To form inorganic replicas, we balance mineralization of DNA origami and AuNP preserving the overall lattice architecture, including the topology of the connections between particles provided by the struts of the polyhedra origami that connect NPs. In Fig. 1A, we show a two-step sol-gel coating process with organic silica precursors, (trimethoxysilyl)propyl-N,N,N-trimethylammonium chloride (TMAPS) and tetraethoxysilane (TEOS). We structured our approach to yield conformal coating of the 3D nanostructure, which has both well-defined DNA origami objects and functionalized NPs with single-stranded DNA stands grafted to particles’ surfaces. We stress that, unlike DNA origami, in which its molecular structure is well defined, NPs are intrinsically polydisperse. This can result in a distribution of a number of strands in NP shells and, consequently, a larger degree of coating heterogeneity of NPs compared to origami.
Fig. 1. Mineralization of 3D DNA origami-gold nanoparticle superlattices into all-inorganic 3D silica-Au replicas with preserved architecture.
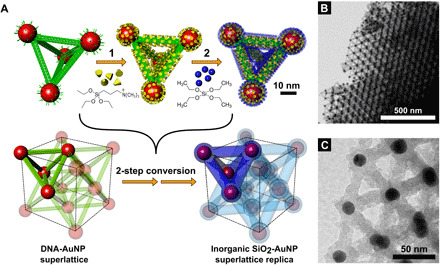
(A) Face-centered cubic (FCC) superlattice formed by programmable water-based self-assembly by DNA-functionalized AuNPs (red), 15-nm core, and DNA origami tetrahedra frames (green) is converted into an inorganic replica in a two-step sol-gel process . In the first step (1), positively charged ligand molecules (TMAPS) are added to coordinate phosphate groups in DNA strands acting as a heterogeneous nucleation site for SiO2 condensation after silica precursor (TEOS) is added in the second step (2). Low-magnification (B) and high-magnification (C) transmission electron microscopy (TEM) images reveal intricate details of air-dried silicated FCC AuNP superlattice that maintains an architecture of the assembled DNA-NP superlattice.
Thus, to control the uniformity of silica coating, we balanced the rate of precursor diffusion into the superlattice and the reaction rate of the precursor hydrolysis at the surface of the DNA struts of DNA frames that connect DNA-functionalized NP. In particular, the hydrolysis of SiO2 precursor (TEOS) and its deposition rate onto the DNA bundles must be much slower than the diffusive transport of the precursor into the superlattices. This provides an even coating on the struts that, in turn, avoids excessive silica deposition on the surface of superlattice grains, which would restrict subsequent diffusion of the additional precursor into the lattice interior. An additional constraint is the selectivity of the coating process, which limits the growth of the SiO2 shell to the DNA surface of the bundles and prevents the formation of silica elsewhere. The latter demand is met in the first step of the process. This involves the incubation of DNA in the diluted tris-acetate-EDTA (TAE) buffer with 10 mM MgCl2 in the presence of the DNA ligand, TMAPS, which, due to its positively charged headgroups, forms an anchoring layer between the negatively charged phosphate residues in the DNA backbone and the subsequently used silica precursor, TEOS (Fig. 1A, top). The use of the TMAPS linker is critical for the stability of SiO2-coated superlattices produced; a short incubation at 4°C of superlattices with the linker is required to facilitate the subsequent coating. This differs from previous 2D (60) and 3D (61) templating approaches that operate at room temperature. After addition of TEOS, the mixture is gently stirred for another 4 hours and temperature is gradually raised to 25°C over the course of 16 hours, thus allowing to coat an entire lattice (Fig. 1A, bottom). After the coating reactions, the stability of superlattices is greatly enhanced; they are stable against the removal of magnesium ions stabilizing the DNA bundles in origami, substitution of water to ethanol, and even complete removal of the solvent.
Figure 1B shows a low-magnification transmission electron microscopy (TEM) micrograph of a face-centered cubic (FCC) DNA origami–AuNP superlattice coated with 8 nm of silica. This FCC lattice was assembled using DNA tetrahedron frames and 15-nm AuNP (refer to the Supplementary Materials for details of lattice composition and synthesis). The intricate details of the lattice composed of DNA origami tetrahedral building blocks are revealed in the bright-field (BF) high-magnification TEM image (Fig. 1C). The silica coating is visible both on the surface of DNA bundles (17 nm diameter after silication) and as a ~5-nm layer on the 15-nm AuNP, resulting from the presence of thiol-functionalized single-stranded DNA on the surface of the particles. Silica coating is visible throughout the whole coated specimen, including small grains visualized via TEM and larger grains visible by scanning electron microscopy (SEM) (Fig. 2, A and B). Global preservation of the superlattice structure in all grains is further indicated by small-angle x-ray scattering (SAXS) experiments performed on the samples before (Fig. 2C) and after the silica coating (Fig. 2D). The same set of diffraction peaks, indicative of the FCC symmetry, is present both for the DNA-NP lattice in aqueous solution and for the same silicate lattice, where a slight shrinkage (about 5%) in lattice constant is observed after the coating. By comparison, unmodified DNA-based lattices will collapse upon drying, completely disrupting the 3D periodic order (figs. S5 and S6). The silication process does not destroy the lattice organization; in fact, it appears to improve structural order, as evidenced by peak sharpening and the presence of a larger number of higher-order peaks. This improvement in quality might be attributed to the suppression of fluctuations within the lattice and merging of domains during silication.
Fig. 2. Structural and elemental characterization of silica-AuNP replica lattice by electron microcopy.
(A) Scanning electron microscopy (SEM) images of grains of SiO2-coated FCC DNA-AuNP superlattice. (B) High-resolution SEM reveals that each grain is a monodomain with sharply terminated grain boundaries and dihedral angles corresponding to the symmetry of the lattice. Small-angle x-ray scattering (SAXS) patterns of the superlattice before coating (C) and (D) after the coating with SiO2. (E) High-angle annular dark-field (DF) scanning TEM micrographs of ethanol-washed SiO2-coated FCC DNA-AuNP superlattice after air drying. Energy-dispersive x-ray (EDX) analysis reveals the atomic composition of the sample confirming highly controlled localization of elements delivered to the lattice in the coating procedure. (F) An EDX spectrum of the image with indicated spectral positions of elements, (G) phosphorus (DNA strands in the origami and on the surface on AuNPs), (H) gold (AuNPs) mapping, (I) SiO2 (silica), and (J) overlaid Au, P, and Si elemental maps.
To investigate the distribution of individual elements in the formed AuNP-DNA-SiO2 superlattice and to correlate their locations, we conducted a detailed study using energy-dispersive x-ray spectroscopy (EDS); see the Supplementary Materials for details. The bottom panels in Fig. 2 show the EDS spectrum for the silicated lattice (Fig. 2F) elemental mapping for phosphorus (Fig. 2G), gold (Fig. 2H), and silicon (Fig. 2I). While the presence of gold atoms, as expected, is sharply concentrated at the location of nodes in the lattice (gold NPs), the remaining atoms tend to be distributed more evenly over the whole lattice. In particular, the phosphorus signal originating from the phosphodiester DNA backbone collocates with the middle region of the silicated struts and at the surface of gold NPs. Nitrogen atoms, present in DNA nucleotides and in the positively charged quaternary amine group of the ligands, are distributed in a similar fashion, indicating inclusion of the ligands into the growing silica shell. Figure 2J shows three elemental signals (Au, P, and Si) superimposed on high-angle annular dark-field (HAADF) micrograph, indicating an excellent colocalization of elements, as one would expect from a well-formed lattice replica.
DISCUSSION
Our silication approach allows us to generate a variety of silica lattices using an underlying 3D scaffold provided by DNA-NP lattices of different types, as determined by the DNA frame geometry and NP placement (26, 27). By varying the amount of silica precursor, a control over the thickness of deposited silica can be achieved. Thus, both the type of lattice and its filling can be regulated, which provides control over the design of 3D silica-NP ordered materials. Specifically, the amount of TEOS used in the templating step directly controls the thickness of the SiO2 shell grown around DNA bundles forming origami struts and DNA-functionalized NPs. Furthermore, we have noticed a threshold value at the molar ratio of TEOS:DNA of about 10:1, which is a minimum necessary to stabilize the superlattices during subsequent exchange of solvent and drying process. Below this value, sub-1-nm layers of SiO2 are formed and the lattices tend to collapse when the stabilizing Mg2+ ions are removed during the solvent exchange to ethanol.
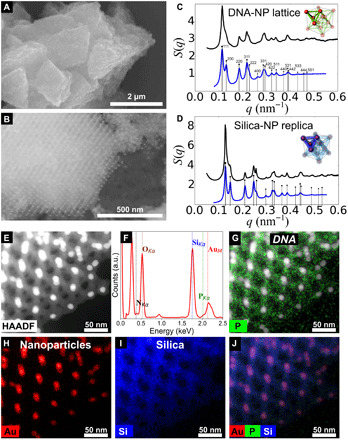
The formation of prescribed 3D NP lattices can be controlled through DNA polyhedral frames, in which vertices are encoded to bind NPs, and NPs play the role of nodes for connecting frames (27). The final symmetry of the lattice is determined by the topology of interparticle connections, as provided by frame linkages. In this study, we investigated NP assemblies formed directly by tetrahedral, cubic, and octahedral DNA frames, which induces an assembly of NP into FCC, simple cubic (SC), and FCC lattices, respectively. Figure 3 presents TEM images of three distinct coated superlattices oriented by adjusting the tilt angle of the sample holder in the optimal way to reveal the symmetry of the lattice and the degree of coating. The FCC lattice formed by DNA 15-nm AuNPs tetrahedra oriented in the (110) plane coated by using a 20:10:1 molar ratio of TEOS:linker:DNA nucleotide is shown in the DF image in Fig. 3A. This structure provides an example of a relatively thin, 5-nm silica-coating structure with large porosity. An SC lattice containing 10-nm AuNPs imaged in dark field in (100) orientation was coated with silica shell of intermediate thickness (15 nm) obtained at 90:15:1 TEOS:linker:DNA ratio. Conversely, an FCC superlattice made of octahedral frames, shown in Fig. 3C, was coated with a saturating ratio of TEOS:linker:DNA (50:10:1), which results in closing the voids and formation of space-filling silica matrix with preserved order of NPs.
Fig. 3. Superlattices formed with different polyhedra DNA origami and AuNP are mineralized with controllable coating thicknesses.
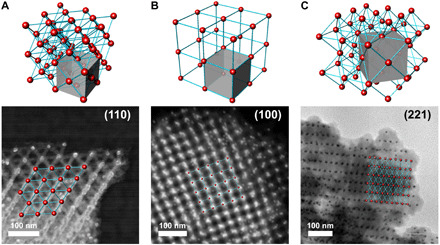
(A) FCC superlattice formed by 15-nm AuNPs and DNA origami tetrahedra with the unit cell shown in gray and a corresponding DF STEM image of a thinly coated grain imaged in the (110) plane. (B) Simple cubic superlattice built by DNA cubic cages and 10-nm AuNPs oriented in the (100) plane. Intermediate silica precursor amount was used, yielding 15-nm-thick coating around DNA struts. (C) Model of 10-nm AuNPs in octahedral DNA units forming FCC lattice and BF TEM image of the coated lattice imaged in the (221) plane. The amount of precursor used renders complete coating and filling of space between the particles and DNA.
The 3D silica architectures, formed from the AuNP-DNA-SiO2 superlattices, display a remarkable chemical and thermal stability. The buffer containing Mg2+ ions, which stabilizes DNA origami in the uncoated samples, is no longer necessary to maintain a DNA origami integrity. Thus, silicated lattices can be readily suspended in a broad range of polar solvents (water, ethanol, and methanol). To date, DNA-NP lattices have demonstrated enormous control over structure but have been quite limited in application due to the limited range of conditions under which they are stable, as compared to that required for real-life applications. To examine temperature stability of the lattices, we heated silicated FCC lattices up to 1200°C in a dry state. We observe neither lattice collapse nor change of the lattice symmetry via SAXS (Fig. 4H). This thermomechanical stability, which persists even during rapid thermal ramps (>100°C/min), is remarkable considering the fine structure and open nanoarchitecture. It is a manifestation of the chemical stability of SiO2 as well as the ability of open strut-like nanoarchitectures to redistribute large stresses throughout the lattice. The observed uniform shrinkage, approximately 5% of lattice constant, upon the high-temperature treatment, is related to the initial dehydration upon residual water evaporation. A subsequent solidification of sol-derived silica results in the isotropic compacting of the superlattice by a further 12%.
Fig. 4. Thermal stability of 3D silicated DNA-NP superlattice.
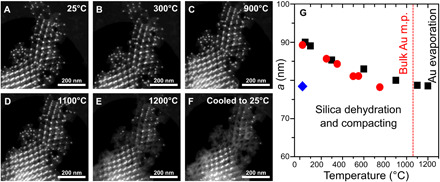
Silica-coated superlattice is heated in situ under STEM observation from 25° to 1200°C (A to E) and cooled back to room temperature (F). Once heated beyond the melting point of gold (1064°C), some of the NPs disappear, leaving behind a hollow SiO2 replica of the superlattice (F). The process is accompanied by the gradual shrinkage of the superlattice, (G) as tracked by the analysis of scattering peak shifts in the SAXS data (red dots) and Fourier-transformed TEM images (black squares). The lattice constant after the heating (blue diamond) is ~13% smaller when compared to the starting material.
The thermal behavior of superlattices can be directly tracked by in situ observation upon heating the formed silica replica lattice in the transmission electron microscope. Figure 4 shows a time-resolved image series (TEM mode) of a silica FCC superlattice composed of DNA origami tetrahedra heated from room temperature to 900°C. The first observed feature is the thermochemical stability of the structure even at temperatures where the DNA structures had already undergone a decomposition/pyrolysis process. On the other hand, the organic removal and additional mechanical stress relief during the experiment lead to shrinking of the structure, as seen in the fast Fourier transform of each image, presenting a reduction of 11 to 13% of the superlattice parameters (Fig. 4 and fig. S16), corroborating the conclusions of temperature-resolved SAXS analysis. Thus, this in situ heating TEM experiment provides a direct evidence that this phenomenon occurs without structural degradation on the local scale, i.e., the FCC structure remains intact.
Interesting phenomena are observed for NPs when the lattice is heated to yet higher temperatures (to 1200°C): The gold NPs shrink and some of them eventually disappear at a temperature above 1100°C, while a fraction of NP is still preserved. The shrinkage is presumably related to increased mobility of gold as the temperature reaches the melting point, whereupon the metal may diffuse out of the initial lattice positions and throughout the 3D silica scaffold (movie S1). A fraction of silica scaffolded sites seal NPs, hindering the diffusion even at 1200°C. We have examined the possibility of gold diffusion by tracking through the use of EDS, in the presence of NP and after their disappearance. Figure S16 shows the EDS mapping for the structure at room temperature (fig. S2, A to D) and the same area after a heating cycle (fig. S2, E and F), which indicates the shrinking of gold NP, but there is not any relevant trace of gold signal over the rest of the main SiO2 scaffold. To eliminate the effect of prolonged irradiation of gold particles with the electron beam, known to promote sample changes in microscopy experiments (e.g., sputtering, melting, and recrystallization), we have studied and compared the high-temperature sample areas located away from the electron beam. We observed that the superlattice in this experiment (movie S1 and fig. S2) and other structures located away of the electron beam irradiation area, after the heating cycle (fig. S17) exhibited the same pattern of phenomena with gold particles shrinking and vanishing, indicating purely thermal NP evaporation mechanism. Thus, we conclude that substantial thermally induced migration is the main path for the vanishing of particles at high temperatures. These experiments also reveal a remarkable resilience of the lattice, which remains intact despite the mobility and redistribution of gold metal at vertices of the lattice strut positions.
Besides the observation of the gold NP’s disappearance, their melting process inside silica encapsulation was also possible to follow by an in situ observation. During the heating experiment at ~1150°C, the annular DF signal [scanning TEM (STEM)] (movie S1) showed quick oscillations of contrast over time, inferring changes in the crystal order. Although signals from the larger angle of collection of electrons (>80 mrad, as in HAADF-STEM) have almost exclusively Rutherford scattered electrons (i.e., no diffraction information), lower angles of collection of electrons [as in medium/low angle annular dark field (MAADF-STEM/LAADF-STEM) or BF-STEM] convey the contribution of coherent elastic scattering (i.e., diffraction contribution). Because any phase transition (as melting) will interfere with the diffraction signal contribution, it will be noticed only in signals with lower angles of collection of electrons, but unnoticed in higher angles, which is exactly observed in this experiment (see movie S2 of the same experiment). This supports the conclusion that this is a melting process.
To further probe the mechanical properties of formed silica-NP lattices, we have performed high-pressure compression experiments. By controlling a silica growth, the thickness of struts was changed, thus resulting in porous (about 20% silica) and fully silica filled FCC lattices. Samples of silica-coated 3D DNA-gold NP superlattice were placed in a diamond anvil cell and studied by in situ high-pressure SAXS at a pressure up to 8 GPa (Fig. 5). The scattering pattern indicates that the lattice is preserved under isotropic compression up to the highest probed pressure, 8 GPa. No significant changes in the lattice quality are observed at the intermediate-high pressure (3.9 GPa), as shown by the broadness and number of scattering peaks. However, at 8 GPa, some degradation of order becomes apparent, although the lattice still persists because the scattering pattern that corresponds to the FCC lattice is present. Using the second-order Birch-Murnaghan equation of state, we obtained bulk modules of measured lattices from SAXS data (see the Supplementary Materials). The fitted bulk modulus for the 20% silica-coated FCC superlattice is 30.1 ± 5.5 GPa, and the value for the 100% silica-coated FCC superlattice is 50.4 ± 9.9 GPa, which is compared favorably with the reported bulk modulus (36 and 50 GPa) of silica (62). We note that these silicated lattices show a mechanical response that differs drastically from DNA-NP lattices (63). It has been shown that distribution of bulk modulus can arise due to different conditions of pH and concentration in solution, affecting grain structure at the different scales. However, in our study, by creating a nanoporous lattice, we observe that such a lattice with 20% filling nevertheless offers about 60% of the bulk modulus. Several effects can contribute to this observation: (i) hydrostatic pressure of oil penetrating lattice in our high-pressure experiment can offset the measurement of bulk modulus; (ii) a microscale structure of struts, analogously to effects in nanoporous gold (64); and (iii) larger-scale heterogeneity in a sample. Thus, future studies with a direct mechanical indentation of lattices are required to elucidate these observations.
Fig. 5. Mechanical properties of the silicated FCC DNA-NP superlattice under compression stress.
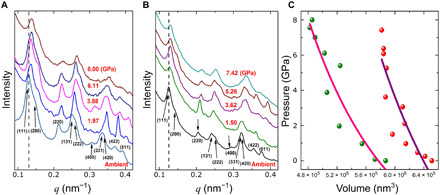
SAXS patterns of the coated superlattice composed of DNA origami tetrahedra and 15-nm gold NPs under high hydrostatic pressure generated using Almax plate diamond anvil cell. (A) Scattering data of the sample containing 20% silica by volume compressed to different pressures up to 8.00 GPa and (B) completely filled with silica. The vertical dashed line is used to guide the eye to track the 111-peak shift under increasing pressure. (C) Evolution of the unit cell volume of the superlattice as a function of pressure, coated by a varied amount of SiO2. Green markers, 20% silica; red markers, 100% interstitial volume filled by silica. The solid lines indicate fitting results to the second-order Birch-Murnaghan equation of state and the calculated bulk moduli (B0) of the superlattice coated to a different thickness of silica (see the Supplementary Materials).
The strength and durability afforded to the structure were allowed for processing materials using typical semiconductor industry methods that are nearly impossible to apply to soft matter–based structures. Silicated lattices can be postprocessed using both e-beam and focused ion beam (FIB) milling. By using the unique capabilities of the FIB/SEM to section nanometer-scale slices from a structure, the entire superlattice can be sliced, imaged, and reconstructed to reveal the full 3D nature of the lattice (see Fig. 6 and figs. S20 and S21). With this technique, we visualize the growth of an ordered grain evolving from a disordered aggregate state (Fig. 6). The lattice facets along (111) planes with a truncation on top of the pyramid correspond to the (100) plane. The ability to slice through the sample allows imaging the interior of assemblies. Through this method, it will thus be possible to obtain information about defects and dislocations of nanoscale superlattices unlocking untold amounts of information pertaining to the nucleation and defect formation mechanisms in self-assembled lattices.
Fig. 6. 3D imaging of the silicated nanoparticle lattice using a focused ion beam processing and serial reconstruction.
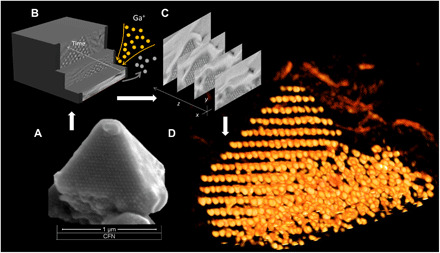
(A) SEM image of a DNA-NP superlattice structure. (B) Representation of the FIB/SEM collection process wherein a 10-nm layer of sample was removed sequentially by ion milling. (C) Slices of SEM images showing the evolution of pyramid domain. (D) 3D representation of AuNP lattice showing ordered and disordered domains within the assembled volume (see also supplementary movies and the Supplementary Materials).
The radiation robustness of the silica encapsulated lattices is further demonstrated by its stability during bombardment by x-ray photons using high-flux synchrotron source, such as National Synchrotron Light Source II (NSLS II) (BNL). DNA-NP lattices typically degrade after exposure to x-ray beam during SAXS measurements. The damage onset is indicated by changes in a scattering pattern, such as a decrease of intensity of diffraction peaks and a disappearance of high-order peaks (fig. S22). In comparison with native DNA-NP lattices, silica encapsulated lattices could withstand a significantly higher radiation exposure, as probed by x-ray imaging using irradiation by nanofocused x-ray beam (see the Supplementary Materials). The damage is pronounced by appearance of the hole in the exposed area (fig. S23). We estimate that silication allows to sustain the delivered radiation dose about six orders of magnitude higher than a native DNA-NP lattice (table S1). The high tolerance to the radiation of processed DNA-NP lattices opens opportunities to apply flux-intensive x-ray methods, such as x-ray tomography and ptychography, to nondestructive real-space 3D probing of DNA-assembled lattices. The greater penetration depth of x-rays would supersede electron microscopies’ limited range and allow in-depth analysis of local structure, defects, and dislocation not otherwise possible with scattering techniques alone. The presented method for creating highly structured NP-based materials might also be potentially applicable for the fabrication of engineered radiation-shielding nanocomposite materials (65) and nuclear nanotechnology applications requiring highly radiation-tolerable and highly architectured nanomaterials (66).
We present a facile synthetic approach to a new class of periodic supramolecular nanostructure obtained by templated growth of amorphous silica on gold NP-DNA superlattices and subsequent calcination of the latter. The process results in all-inorganic replica preserving the arrangement of gold NPs with DNA strands converted to SiO2. Such structures, given their fine structure, are amazingly stable, even in solvent-free environments. The in situ heating TEM observations of superlattices demonstrated their great thermomechanical stability, with no loss or changing of the main SiO2 structure order, and only modest shrinkage. The possibility of removing the organic part and the gold NPs, prediction of superlattice parameter shrinkage without structure order changing, provides a promising avenue toward the construction of a new class of nanostructured porous materials.
MATERIALS AND METHODS
Silica-coating process
All chemicals used in the conversion of AuNP-DNA superlattices were purchased from Sigma-Aldrich. AuNP-DNA superlattices were gently centrifuged to remove unbound DNA and redispersed in 10 times diluted standard TAE buffer containing 10 mM MgCl2 to obtain a final concentration of the lattice-bound DNA of 91 mmol/μl (single-nucleotide basis is used throughout this protocol). The linker, TMAPS, was added at the 5:1 molar ratio with respect to the bound DNA concentration and incubated at 4°C for 30 min upon gentle stirring, followed by the addition of silica precursor, TEOS. Silica precursor diluted in absolute ethanol is added at the end of the initial incubation with the linker. The dilution facilitates handling small amounts of liquid and also improves the miscibility of the precursor with the coating buffer and thus the homogeneity of the coating mixture. The molar ratio of the TEOS–to–lattice-bound DNA was varied depending on the desired coating thickness from 5:1 to 1000:1. Following the 4-hour incubation period at 4°C, the temperature of the reaction mixture was gradually increased to room temperature over a course of 16 hours with gentle agitation throughout the process. Silica-coated superlattices were harvested by centrifugation and washed with ethanol-water for several times and air-dried directly on TEM grids or Si wafers for electron microscopy studies.
Conversion to all-inorganic replicas
The conversion of hybrid inorganic-inorganic superlattices to all-inorganic AuNP-SiO2 replicas was performed by the oxidative calcination in a high-temperature furnace at 500°C for 1 hour in the O2 atmosphere. The calcined samples were subsequently studied by SAXS.
Small-angle X-ray scattering
SAXS experiments were carried out at the NSLS II coherent hard x-ray scattering (CHX) and complex materials scattering (CMS) beamlines. The scattering data were collected with a Dectris photon-counting area detectors and converted to 1D scattering intensity versus wave vector transfer, q = (4π/λ) sin (θ/2), where λ = 0.8551 Å and θ are the wavelength of incident x-ray and the scattering angle, respectively. The scattering angle was calibrated using silver behenate as a standard. The structure factor S(q) was calculated as Ia(q)/Ip(q), where Ia(q) and Ip(q) are background-corrected 1D scattering intensities extracted by angular averaging of detector images for assembled systems and dissociated particles, respectively.
High-pressure experiments
The high pressure is generated by Almax plate diamond anvil cell; the culet size of Boehler-Almax–designed diamond is 450 μm with bevel. Tungsten is chosen as a gasket material and is preindented to 90 μm. A hole with a diameter of 300 μm is drilled in the center of the preindented tungsten gasket to serve as a sample chamber. Then, ruby balls together with the silica-coated superlattice sample dispersed in the high-pressure liquid medium (a mixture of methanol, ethanol, and water, 16:3:1 volume ratio) are loaded into the sample hole. The pressure is calibrated by the fluorescence shift of the ruby R1 line; the quasi-hydrostatic condition can be monitored by the separation and width of ruby R1 and R2 lines.
The high-pressure SAXS was carried out at the CMS beamline of NSLS II, Brookhaven National Laboratory. The wavelength of x-ray is 0.918 Å. The SAXS data were collected by MAR charge-coupled device detector. The sample to detector distance and geometric parameters were calibrated by using silver behenate. The 2D x-ray patterns were converted to 1D intensity by Fit2d software.
In situ heating (S)TEM experiments
TEM samples were prepared by dropping the suspension of DNA-AuNP superlattice in a disposable 3-mm-diameter microelectromechanical system (MEMS) device (which serves as both the heating element and the specimen support grid) or in an amorphous carbon film supported on nickel grids. The isothermal in situ heating experiments were performed in two microscopes. STEM imaging was carried out by using a MEMS-based heating holder (FEI NanoEx-i/v) in the FEI Talos F200X TEM equipped with X-FEG electron source and a four-quadrant 0.9-sr energy-dispersive x-ray spectrometer (windowless) operated at 200 kV. Additional in situ experiments (TEM imaging) were performed in a Gatan 628 single-tilt heating holder (with an Inconel-based resistive heater) in JEOL JEM2100F with Schottky field-emitter source. Both microscopes were operated at 200 kV. The thermal ramp was executed by heating the sample at 0.5°C/s from room temperature up to the desired temperature (900°C or 1200°C in the experiments herein). The sample was held at the maximum temperature for 10 min and then cooled down to room temperature at the rate of –0.5°C/s.
Serial sectioning experiments
DNA-NP superlattices (20 nm Au) were drop-cast onto a clean silicon wafer. Imaging and serial sectioning were performed using FEI Helios NanoLab 660 SEM/FIB; Auto Slice and View G3 software suites were used to automate collection. The particle cluster was imaged with an Elstar in-lens BSE detector (TLD-BSE) with a 3 μs dwell time and a 100 pA current. A sacrificial layer of Pt was deposited on top of the sample to help avoid damaging the sample before slicing, after which a trench was milled surrounding the sample and the fiducial marker was placed with Pt for Auto slice and view functionality. The FIB current was set to 0.43 nA; a 10 nm step size for layer removal was selected. A total of 151 slices were collected in this method.
Avizo/Amira software was used to perform corrections, filter, and view data.
Supplementary Material
Acknowledgments
We thank the Imaging Facility of CUNY Advanced Science Research Center for instrument use and technical assistance. Funding: This research used resources of the Center for Functional Nanomaterials and National Synchrotron Light Source II, which are U.S. DOE Office of Science Facilities, at Brookhaven National Laboratory under contract no. DE-SC0012704. The DNA design work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, grant DE-SC0008772. This work was supported by U.S. Department of Defense, Army Research Office, W911NF-19-1-0395. Author contributions: P.W.M., K.G.Y., and O.G. conceived and designed the experiments. W.L., Y.T., C.T., C.M., and A.M. performed the assembly experiments. P.W.M., K.G.Y., C.M., and O.G. conducted the SAXS study, and K.G.Y. contributed to the model fitting of SAXS data. P.W.M., C.M., and A.M. performed the silica growth experiments. M.A.L.C., K.K., P.W.M., and E.A.S. contributed to electron microscopy experiments including EDS and in situ thermal studies. C.M. performed in situ mechanical pressure studies. A.M. performed x-ray radiation and serial sectioning of the superlattice along with computation reconstruction of the structure. P.W.M., A.M., and O.G. wrote the paper. O.G. supervised the project. All authors discussed the results, helped assemble the figures, and edited or reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabf0617/DC1
REFERENCES AND NOTES
- 1.Seeman N. C., Nucleic acid junctions and lattices. J. Theor. Biol. 99, 237–247 (1982). [DOI] [PubMed] [Google Scholar]
- 2.Alivisatos A. P., Johnsson K. P., Peng X., Wilson T. E., Loweth C. J., Bruchez M. P. Jr., Schultz P. G., Organization of ‘nanocrystal molecules’ using DNA. Nature 382, 609–611 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Mirkin C. A., Letsinger R. L., Mucic R. C., Storhoff J. J., A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 382, 607–609 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Winfree E., Liu F., Wenzler L. A., Seeman N. C., Design and self-assembly of two-dimensional DNA crystals. Nature 394, 539–544 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Mao C., Sun W., Seeman N. C., Designed two-dimensional DNA holliday junction arrays visualized by atomic force microscopy. J. Am. Chem. Soc. 121, 5437–5443 (1999). [Google Scholar]
- 6.Rothemund P. W. K., Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Shekhirev M., Sutter E., Sutter P., In situ atomic force microscopy of the reconfiguration of on-surface self-assembled DNA-nanoparticle superlattices. Adv. Funct. Mater. 29, 1806924 (2019). [Google Scholar]
- 8.Douglas S. M., Dietz H., Liedl T., Högberg B., Graf F., Shih W. M., Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 459, 414–418 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seeman N. C., Gang O., Three-dimensional molecular and nanoparticle crystallization by DNA nanotechnology. MRS Bull. 42, 904–912 (2017). [Google Scholar]
- 10.Zheng J., Birktoft J. J., Chen Y., Wang T., Sha R., Constantinou P. E., Ginell S. L., Mao C., Seeman N. C., From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 461, 74–77 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han D., Pal S., Nangreave J., Deng Z., Liu Y., Yan H., DNA origami with complex curvatures in three-dimensional space. Science 332, 342–346 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Zhang F., Jiang S., Wu S., Li Y., Mao C., Liu Y., Yan H., Complex wireframe DNA origami nanostructures with multi-arm junction vertices. Nat. Nanotechnol. 10, 779–784 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Ma N., Minevich B., Liu J., Ji M., Tian Y., Gang O., Directional assembly of nanoparticles by DNA shapes: Towards designed architectures and functionality. Top. Curr. Chem. 378, 36 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Zhang F., Simmons C. R., Gates J., Liu Y., Yan H., Self-assembly of a 3D DNA crystal structure with rationally designed six-fold symmetry. Angew. Chem. Int. Ed. 57, 12504–12507 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Tian Y., Wang T., Liu W., Xin H. L., Li H., Ke Y., Shih W. M., Gang O., Prescribed nanoparticle cluster architectures and low-dimensional arrays built using octahedral DNA origami frames. Nat. Nanotechnol. 10, 637–644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H., Li M., Wang K., Tian Y., Chen J.-S., Fountaine K. T., DiMarzio D., Liu M., Cotlet M., Gang O., Polarized single-particle quantum dot emitters through programmable cluster assembly. ACS Nano 14, 1369–1378 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Sutter E., Zhang B., Sutter S., Sutter P., In situ electron microscopy of the self-assembly of single-stranded DNA-functionalized Au nanoparticles in aqueous solution. Nanoscale 11, 34–44 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Nykypanchuk D., Maye M. M., van der Lelie D., Gang O., DNA-guided crystallization of colloidal nanoparticles. Nature 451, 549–552 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Park S. Y., Lytton-Jean A. K. R., Lee B., Weigand S., Schatz G. C., Mirkin C. A., DNA-programmable nanoparticle crystallization. Nature 451, 553–556 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Xiong H., van der Lelie D., Gang O., Phase behavior of nanoparticles assembled by DNA linkers. Phys. Rev. Lett. 102, 015504 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Lewis D. J., Zornberg L. Z., Carter D. J. D., Macfarlane R. J., Single-crystal Winterbottom constructions of nanoparticle superlattices. Nat. Mater. 19, 719–724 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Kadic M., Milton G. W., van Hecke M., Wegener M., 3D metamaterials. Nat. Rev. Phys. 1, 198–210 (2019). [Google Scholar]
- 23.Roy K., Jaiswal A., Panda P., Towards spike-based machine intelligence with neuromorphic computing. Nature 575, 607–617 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Liddle J. A., Gallatin G. M., Nanomanufacturing: A perspective. ACS Nano 10, 2995–3014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kjaergaard M., Schwartz M. E., Braumüller J., Krantz P., Wang J. I.-J., Gustavsson S., Oliver W. D., Superconducting qubits: Current state of play. Annu. Rev. Condens. Matter Phys. 11, 369–395 (2020). [Google Scholar]
- 26.Liu W., Tagawa M., Xin H. L., Wang T., Emamy H., Li H., Yager K. G., Starr F. W., Tkachenko A. V., Gang O., Diamond family of nanoparticle superlattices. Science 351, 582–586 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Y., Zhang Y., Wang T., Xin H. L., Li H., Gang O., Lattice engineering through nanoparticle–DNA frameworks. Nat. Mater. 15, 654–661 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian Y., Lhermitte J. R., Bai L., Vo T., Xin H. L., Li H., Li R., Fukuto M., Yager K. G., Kahn J. S., Xiong Y., Minevich B., Kumar S. K., Gang O., Ordered three-dimensional nanomaterials using DNA-prescribed and valence-controlled material voxels. Nat. Mater. 19, 789–796 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Zhang T., Hartl C., Frank K., Heuer-Jungemann A., Fischer S., Nickels P. C., Nickel B., Liedl T., 3D DNA origami crystals. Adv. Mater. 30, 1800273 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Jones M. R., Macfarlane R. J., Lee B., Zhang J., Young K. L., Senesi A. J., Mirkin C. A., DNA-nanoparticle superlattices formed from anisotropic building blocks. Nat. Mater. 9, 913–917 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Lu F., Vo T., Zhang Y., Frenkel A., Yager K. G., Kumar S., Gang O., Unusual packing of soft-shelled nanocubes. Sci. Adv. 5, eaaw2399 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., Lu F., Yager K. G., van der Lelie D., Gang O., A general strategy for the DNA-mediated self-assembly of functional nanoparticles into heterogeneous systems. Nat. Nanotechnol. 8, 865–872 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Gang O., Tkachenko A. V., DNA-programmable particle superlattices: Assembly, phases, and dynamic control. MRS Bull. 41, 381–387 (2016). [Google Scholar]
- 34.Tian C., Cordeiro M. A. L., Lhermitte J., Xin H. L., Shani L., Liu M., Ma C., Yeshurun Y., DiMarzio D., Gang O., Supra-nanoparticle functional assemblies through programmable stacking. ACS Nano 11, 7036–7048 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Zhang C., Macfarlane R. J., Young K. L., Choi C. H. J., Hao L., Auyeung E., Liu G., Zhou X., Mirkin C. A., A general approach to DNA-programmable atom equivalents. Nat. Mater. 12, 741–746 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Agarwal N. P., Matthies M., Gür F. N., Osada K., Schmidt T. L., Block copolymer micellization as a protection strategy for DNA origami. Angew. Chem. Int. Ed. 56, 5460–5464 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Ponnuswamy N., Bastings M. M. C., Nathwani B., Ryu J. H., Chou L. Y. T., Vinther M., Li W. A., Anastassacos F. M., Mooney D. J., Shih W. M., Oligolysine-based coating protects DNA nanostructures from low-salt denaturation and nuclease degradation. Nat. Commun. 8, 15654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai F., Bian K., Li B., Wu H., Alarid L. J., Schunk H. C., Clem P. G., Fan H., Nanomaterials under stress: A new opportunity for nanomaterials synthesis and engineering. MRS Bull. 40, 961–970 (2015). [Google Scholar]
- 39.Bai F., Bian K., Huang X., Wang Z., Fan H., Pressure induced nanoparticle phase behavior, property, and applications. Chem. Rev. 119, 7673–7717 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Patra T. K., Chan H., Podsiadlo P., Shevchenko E. V., Sankaranarayanan S. K. R. S., Narayanan B., Ligand dynamics control structure, elasticity, and high-pressure behavior of nanoparticle superlattices. Nanoscale 11, 10655–10666 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Zhang X., Hattar K., Chen Y., Shao L., Li J., Sun C., Yu K., Li N., Taheri M. L., Wang H., Wang J., Nastasi M., Radiation damage in nanostructured materials. Prog. Mater. Sci. 96, 217–321 (2018). [Google Scholar]
- 42.Bringa E. M., Monk J. D., Caro A., Misra A., Zepeda-Ruiz L., Duchaineau M., Abraham F., Nastasi M., Picraux S. T., Wang Y. Q., Farkas D., Are nanoporous materials radiation resistant? Nano Lett. 12, 3351–3355 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Keren K., Krueger M., Gilad R., Ben-Yoseph G., Sivan U., Braun E., Sequence-specific molecular lithography on single DNA molecules. Science 297, 72–75 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Liu J., Geng Y., Pound E., Gyawali S., Ashton J. R., Hickey J., Woolley A. T., Harb J. N., Metallization of branched DNA origami for nanoelectronic circuit fabrication. ACS Nano 5, 2240–2247 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Pal S., Zhang Y., Kumar S. K., Gang O., Dynamic tuning of DNA-nanoparticle superlattices by molecular intercalation of double helix. J. Am. Chem. Soc. 137, 4030–4033 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Raman N. K., Anderson M. T., Brinker C. J., Template-based approaches to the preparation of amorphous, nanoporous silicas. Chem. Mater. 8, 1682–1701 (1996). [Google Scholar]
- 47.Kaehr B., Townson J. L., Kalinich R. M., Awad Y. H., Swartzentruber B. S., Dunphy D. R., Brinker C. J., Cellular complexity captured in durable silica biocomposites. Proc. Natl. Acad. Sci. U.S.A. 109, 17336–17341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brinker C. J., Hydrolysis and condensation of silicates: Effects on structure. J. Non-Cryst. Solids 100, 31–50 (1988). [Google Scholar]
- 49.Kresge C. T., Leonowicz M. E., Roth W. J., Vartuli J. C., Beck J. S., Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 359, 710–712 (1992). [Google Scholar]
- 50.Alberius P. C. A., Frindell K. L., Hayward R. C., Kramer E. J., Stucky G. D., Chmelka B. F., General predictive syntheses of cubic, hexagonal, and lamellar silica and titania mesostructured thin films. Chem. Mater. 14, 3284–3294 (2002). [Google Scholar]
- 51.Bhatia R. B., Brinker C. J., Gupta A. K., Singh A. K., Aqueous sol−gel process for protein encapsulation. Chem. Mater. 12, 2434–2441 (2000). [Google Scholar]
- 52.Lee J., Choi J., Park J. H., Kim M.-H., Hong D., Cho H., Yang S. H., Choi I. S., Cytoprotective silica coating of individual mammalian cells through bioinspired silicification. Angew. Chem. Int. Ed. 53, 8056–8059 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Désert A., Hubert C., Fu Z., Moulet L., Majimel J., Barboteau P., Thill A., Lansalot M., Bourgeat-Lami E., Duguet E., Ravaine S., Synthesis and site-specific functionalization of tetravalent, hexavalent, and dodecavalent silica particles. Angew. Chem. Int. Ed. 52, 11068–11072 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Fan H., Yang K., Boye D. M., Sigmon T., Malloy K. J., Xu H., López G. P., Brinker C. J., Self-assembly of ordered, robust, three-dimensional gold nanocrystal/silica arrays. Science 304, 567–571 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Auyeung E., Macfarlane R. J., Choi C. H. J., Cutler J. I., Mirkin C. A., Transitioning DNA-engineered nanoparticle superlattices from solution to the solid state. Adv. Mater. 24, 5181–5186 (2012). [DOI] [PubMed] [Google Scholar]
- 56.Ma K., Gong Y., Aubert T., Turker M. Z., Kao T., Doerschuk P. C., Wiesner U., Self-assembly of highly symmetrical, ultrasmall inorganic cages directed by surfactant micelles. Nature 558, 577–580 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Aubert T., Ma K., Tan K. W., Wiesner U., Two-dimensional superstructures of silica cages. Adv. Mater. 32, e1908362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Numata M., Sugiyasu K., Hasegawa T., Shinkai S., Sol–gel reaction using DNA as a template: An attempt toward transcription of DNA into inorganic materials. Angew. Chem. Int. Ed. 43, 3279–3283 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Satoh S., Fugetsu B., Nomizu M., Nishi N., Functional DNA–silica composite prepared by sol–gel method. Polym. J. 37, 94–101 (2005). [Google Scholar]
- 60.Liu X., Zhang F., Jing X., Pan M., Liu P., Li W., Zhu B., Li J., Chen H., Wang L., Lin J., Liu Y., Zhao D., Yan H., Fan C., Complex silica composite nanomaterials templated with DNA origami. Nature 559, 593–598 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Nguyen L., Döblinger M., Liedl T., Heuer-Jungemann A., DNA-origami-templated silica growth by sol–gel chemistry. Angew. Chem. Int. Ed. 58, 912–916 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Huang L., Kieffer J., Amorphous-amorphous transitions in silica glass. I. Reversible transitions and thermomechanical anomalies. Phys. Rev. B 69, 224203 (2004). [Google Scholar]
- 63.Srivastava S., Nykypanchuk D., Maye M. M., Tkachenko A. V., Gang O., Super-compressible DNA nanoparticle lattices. Soft Matter 9, 10452–10457 (2013). [Google Scholar]
- 64.Biener J., Hodge A. M., Hayes J. R., Volkert C. A., Zepeda-Ruiz L. A., Hamza A. V., Abraham F. F., Size effects on the mechanical behavior of nanoporous Au. Nano Lett. 6, 2379–2382 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Nambiar S., Yeow J. T. W., Polymer-composite materials for radiation protection. ACS Appl. Mater. Interfaces 4, 5717–5726 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Beyerlein I. J., Caro A., Demkowicz M. J., Mara N. A., Misra A., Uberuaga B. P., Radiation damage tolerant nanomaterials. Mater. Today 16, 443–449 (2013). [Google Scholar]
- 67.Seeman N. C., De novo design of sequences for nucleic acid structural engineering. J. Biomol. Struct. Dyn. 8, 573–581 (1990). [DOI] [PubMed] [Google Scholar]
- 68.Mao H. K., Xu J., Bell P. M., Calibration of the ruby pressure gauge to 800 kbar under quasi-hydrostatic conditions. J. Geophys. Res. Solid Earth 91, 4673–4676 (1986). [Google Scholar]
- 69.Podsiadlo P., Lee B., Prakapenka V. B., Krylova G. V., Schaller R. D., Demortière A., Shevchenko E. V., High-pressure structural stability and elasticity of supercrystals self-assembled from nanocrystals. Nano Lett. 11, 579–588 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabf0617/DC1