

Zinc finger proteins targeted to MAPT safely repress tau mRNA in the adult mouse brain and protect against Aβ toxicity.
Abstract
Neuronal tau reduction confers resilience against β-amyloid and tau-related neurotoxicity in vitro and in vivo. Here, we introduce a novel translational approach to lower expression of the tau gene MAPT at the transcriptional level using gene-silencing zinc finger protein transcription factors (ZFP-TFs). Following a single administration of adeno-associated virus (AAV), either locally into the hippocampus or intravenously to enable whole-brain transduction, we selectively reduced tau messenger RNA and protein by 50 to 80% out to 11 months, the longest time point studied. Sustained tau lowering was achieved without detectable off-target effects, overt histopathological changes, or molecular alterations. Tau reduction with AAV ZFP-TFs was able to rescue neuronal damage around amyloid plaques in a mouse model of Alzheimer’s disease (APP/PS1 line). The highly specific, durable, and controlled knockdown of endogenous tau makes AAV-delivered ZFP-TFs a promising approach for the treatment of tau-related human brain diseases.
INTRODUCTION
The microtubule-binding protein tau is a key player in Alzheimer’s disease (AD) and frontotemporal dementia. The accumulation and aggregation of tau in the brain correlate with synaptic loss, neuronal loss, and cognitive decline (1–4). In patients with frontotemporal dementia, mutations in the tau gene, MAPT, lead to tau aggregation and cause widespread neurodegeneration (5), emphasizing the neurotoxic role of tau in these diseases. In addition to the neurotoxicity exerted by aggregated tau, soluble oligomeric forms of tau appear to be especially synaptotoxic (6). Mice engineered to lack expression of MAPT have been shown to be protected against β-amyloid (Aβ)–induced synaptotoxicity (7), as well as against stress-induced (8) and seizure-induced (9, 10) neuronal damage, and against learning and memory deficits resulting from traumatic brain injury (11). Moreover, reducing transgenic tau expression, even after tau has accumulated in mouse models of tauopathy, reverses the pathological effects of tau (12, 13). These findings support the idea that the reduction of tau protein could be used as a therapeutic approach in AD or other tauopathies.
Translation of the neuroprotective effect of tau repression into a therapeutic approach for neurodegenerative diseases requires a treatment that reduces endogenous tau in the adult brain. Previously, in vivo tau knockdown has been achieved through administration of antisense oligonucleotides (ASOs) that bind tau mRNA and prevent its translation (13, 14) or by immunotherapeutic intravenous injections of anti-tau antibodies (15–18). Although both approaches may facilitate tau protein reduction in the brain, they require chronic administrations to the patient and have limited ability to provide widespread knockdown.
Since recombinant adeno-associated viruses (rAAVs) can be used as viral vectors to efficiently transduce cells in the adult central nervous system (CNS) (19), we created a way to generate efficient, specific, and long-lasting down-regulation of the expression of endogenous tau by using a single viral administration: AAVs encoding engineered zinc finger protein (ZFP) arrays that precisely target a short region of the genomic mouse MAPT sequence and, through fusion to the KRAB repression domain of the human KOX1 transcription factor (TF), down-regulate MAPT gene expression. Using different AAV serotypes, we were able to reduce tau locally in the hippocampus—a brain region that is specifically affected by tau pathology in neurodegenerative diseases—through intracranial injections of AAV9 (adeno-associated virus serotype 9) or brain-wide through intravenous delivery of blood-brain barrier–crossing AAV-PHP.B (20). In both cases, a single AAV administration was sufficient to repress tau mRNA and all isoforms of the protein by 50 to 80% in the brain and for as long as we carried out the study—nearly 1 year—following the treatment.
Furthermore, we performed proof-of-principle experiments for the use of tau-targeted ZFP-TFs to treat neurodegeneration in a mouse model in vivo: The repression of endogenous tau appeared to protect neurons from toxicity in mice with AD-like Aβ pathology (APP/PS1 mice). Tau repression by ZFP-TFs reduced amyloid plaque–associated neuritic dystrophies, which are a tau-dependent pathological hallmark in these mice (21).
In summary, we present a novel translational approach to reduce tau in the adult brain, in which a single AAV ZFP-TF administration can produce potent, specific, and well-tolerated knockdown of endogenous neuronal tau. Our findings support the continued development of ZFP-TFs as a therapeutic platform for the treatment of tau protein–related disorders in the human brain.
RESULTS
Tau-targeted ZFP-TFs reduce endogenous tau expression in cultured primary mouse neurons
Tau reduction in the brain has been shown to be protective against pathological changes in different rodent models of neurodegeneration (7, 10, 22). To reduce all isoforms of tau in the adult mouse brain, we designed ZFPs—based on the backbone of human Zif268/EGR1—that target sequences within 500 base pairs of the transcription start site (TSS) of the mouse tau gene MAPT. The ZFPs efficiently binding the genomic MAPT sequence were then fused to a KRAB repression domain (TF). In each such ZFP-TF construct, the ZFP mediates the site-specific binding to the DNA, and the KRAB domain represses the endogenous expression of MAPT (Fig. 1A). When screening a panel of ~50 ZFP-TFs (out of 185 initial designs; fig. S1) for their dose-response activities on tau mRNA reduction in mouse neuroblastoma (N2a) cells, we identified candidates that robustly repressed tau mRNA and evaluated their off-target impact on global gene expression by RNA microarray. One candidate, ZFP-TF.89, targeting intron-1 of mouse MAPT (Fig. 1B), showed no detectable off-target transcriptome changes in N2a cells (fig. S1B and tables S1 and S2); we selected ZFP-TF.89 for further studies.
Fig. 1. Tau-targeted ZFP-TFs reduce mouse tau mRNA and protein expression in vitro.
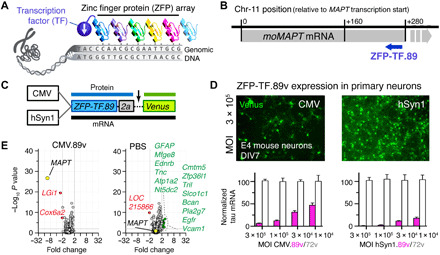
(A) Principle of ZFP-TF target DNA sequence recognition. Engineered arrays of ZFPs recognizing specific DNA triplets bind a specific genomic DNA sequence. Fusion to a TF enables repression of target gene transcription. (B) The binding locations of the lead ZFP-TF.89 downstream of the mouse MAPT gene TSS in the mouse chromosome 11 ZFP-TF.89. Gray bar indicates the location of the MAPT gene. Blue arrow indicates the coding direction of the ZFP-TF.89. (C) Adeno-associated virus serotype 9 (AAV9) constructs with CMV (ubiquitous) and hSyn1 (neuron-specific) promoters for expression of nuclear ZFP-TF.89v (N-terminal SV40 nuclear localization sequence on ZFPs) and cytoplasmic yellow/green fluorescent protein Venus as a transduction marker, separated by a self-cleaving 2a peptide. (D) Expression of AAV ZFP-TF.89v under CMV and hSyn1 promoter in primary cortical mouse neurons (DIV 7; 4 days p.i.). hSyn1-driven 89v expression shows higher tau mRNA repression (pink bars) at the same viral doses (MOI). ZFP-TF.72v, a control ZFP-TF with ZFP array without binding sequence in the mouse genome, shows no tau repression (white bars). Data are presented as means ± SD, n = 3 experiments, and data are normalized to tau mRNA in ZFP-TF.72v–expressing neurons at the highest dose (MOI = 3 × 105). (E) mRNA array data (volcano plots) show small changes in gene expression—other than MAPT—for CMV.89 or PBS compared to CMV.72v-treated primary neurons. Other significantly (>2-fold change, P < 0.01) down-regulated (red) and up-regulated (green) genes are listed. RNA array data are available in table S3. Data are presented as means ± SEM. One-way ANOVA with Sidak’s test, n = 5 to 6 biological replicates.
Tau is an abundant microtubule-binding protein in the brain that is predominantly expressed in neurons and, to a lesser extent, in oligodendrocytes (23, 24). Neurons, as well as astrocytes and oligodendrocytes at a lower frequency (25), can be transduced by AAV9 particles, and when regulated by ubiquitous promoters like cytomegalovirus (CMV), all three cell types could, in principle, produce the ZFP-TF. To target specifically neuronal tau, one can, for example, use the human synapsin-1 promoter (hSyn1), which drives neuron-specific strong transgene expression in all neuronal subtypes producing synapses (26).
For delivery of ZFP-TF.89 into neurons, we created and compared the vectors CMV.89.2a.Venus (abbreviated as CMV.89v) with the commonly used ubiquitous CMV promoter and hSyn1.89.2a.Venus (abbreviated as hSyn1.89v) with the human synapsin-1 promoter for neuron-specific tau reduction. In both vectors, the ZFP-TF is followed by a self-cleaving 2a peptide sequence (27, 28) and the yellow/green fluorescent protein Venus as a cytoplasmic transduction reporter (Fig. 1C). Notably, ZFP-TFs carry an SV40 nuclear localization sequence (PKKKRKV). To control for tau repression specificity in our study, we generated non–tau-targeting control ZFP-TFs (CMV.72v and hSyn1.72v) with no predicted target sites in the mouse genome. In addition, CMV.GFP was used as a control for effects of the KRAB domain expression. We packaged these vectors in the viral serotype AAV9 because of its previous successful use in other CNS gene therapy approaches (29).
Primary neurons transduced with CMV.89v and hSyn.89v produce one mRNA (ZFP-TF-89.2a.Venus) that is translated into separate proteins: nuclear ZFP-TF.89.2a and cytoplasmic Venus. Expression of CMV.89v and hSyn1.89v in primary cortical mouse neurons resulted in efficient and viral dose-dependent repression of tau mRNA after 10 days (Fig. 1D). CMV.72v and hSyn1.72v did not repress tau transcripts at the same viral doses. Expression of CMV.89 produced only minimal off-target gene expression changes in neurons as measured by microarray analysis (Fig. 1E and table S3), similar to our observations in N2a cells (fig. S1B).
ZFP-TF.89v reduces hippocampal tau by ~80% in vivo
To test the ability of ZFP-TF.89v to reduce tau in the adult brain, we stereotactically injected AAVs CMV.89v and hSyn.89 into the hippocampus of adult wild-type (WT) mice, a brain region that is substantially affected by tau-mediated damage in AD and other neurodegenerative diseases. Unilateral double injections of 1.5 μl of AAV each into the anterior and posterior left hippocampus [CMV.89v: 1.1 × 1010 vector genomes (vg) in 3 μl] achieved good coverage of the entire AAV-injected hippocampal formation (Fig. 2A). The contralateral right hippocampus received two vehicle [phosphate-buffered saline (PBS)] injections of the same volume. Notably, 6 weeks post injection (p.i.), some Venus fluorescence was observed in the PBS-injected hemisphere due to neuronal projections of ZFP-TF.89v–expressing ipsilateral neurons.
Fig. 2. Tau-targeted ZFP-TFs reduce tau mRNA and protein expression in vivo.
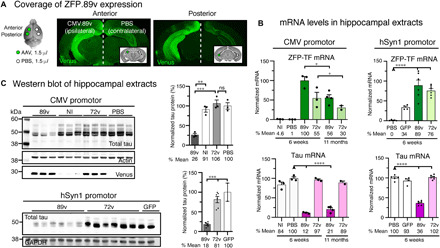
(A) Location of hippocampal injection sites and subsequent ZFP-TF.89v expression in the mouse brain. The left hemisphere received two 1.5-μl injections of AAV9 CMV.89v (CMV.72v) into the anterior and posterior hippocampus, and the right hemisphere received two injections of PBS of the same volume into the same coordinates. Some hippocampi did not receive an injection and served as noninjected (NI) controls. Representative images of coronal brain sections show the widespread expression of the Venus reporter protein in the left hippocampus. Venus labeling in the right hemisphere results from contralateral projections of CMV.89v-transduced neurons in the left hippocampus. (B) mRNA levels of ZFP-TF (ZFP; green) and tau (pink) in CMV.89v- and hSyn1.89v-injected hippocampi. After 6 weeks, CMV.89v reduces tau mRNA down to 12% (by 88%) and hSyn1.89v down to 36% (by 64%), whereas control CMV.72v- or hSyn1.72v-injected hippocampi have the same amount of tau mRNA as noninjected and PBS-injected mice. After 11 months, tau was still reduced by 80% in CMV.89v hippocampi. Data are presented as means ± SEM. One-way ANOVA with Sidak’s test for multiple comparisons, reference for significance indicated by arrowhead, n = 3 mice per group. (C) Tau protein reduction in hippocampal extracts of CMV.89v- and hSyn1.89v-injected animals demonstrated by Western blot of lysates 6 weeks p.i. CMV.89v reduced total tau protein expression by ~74% and hSyn1.89v by ~82%. CMV.72v, noninjected, and PBS-injected hippocampi had similar tau protein levels. One-way ANOVA with Sidak’s posttest, n = 3 mice per group.
To quantify the reduction in tau expression, we extracted the hippocampi of mice injected with CMV.89v, CMV.72v, hSyn1.89v, or hSyn1.72v (CMV.ZFP-TFs: 1.1 × 1010 vg in 3 μl; hSyn1.ZFP-TFs: 2.37 × 1010 vg in 3 μl) and measured tau mRNA levels by RT-qPCR (reverse transcription quantitative polymerase chain reaction) and tau protein levels by Western blot. Six weeks after injection, we observed a remarkable reduction of tau mRNA by 88% (Fig. 2B) and tau protein by 74% in the CMV.89v- and hSyn1.89v-injected hippocampi (Fig. 2C) when compared to PBS-injected hippocampi. Control ZFP-TF.72v–injected hippocampi showed no changes in tau mRNA or protein expression. Even 11 months after a single AAV CMV.89v administration, we found that tau mRNA was reduced by 80% (Fig. 2B). Notably, the detected ZFP transcripts as well as tau reduction in hSyn1.GFP-injected hippocampi likely result from retrograde trafficking of AAV ZFP particles that were injected in the contralateral hippocampus in the same mice (30).
Notably, we also constructed AAVs encoding ZFP-TF.89v under two other neuronal promoters: the human calcium/calmodulin-dependent protein kinase 2a (CaMK2a) promoter, which induces strong expression primarily in excitatory neurons (31), and the methyl CpG-binding protein 2 promoter (MeCP2) (32), which drives lower ZFP-TF levels. All three neuron-specific expression vectors (hSyn1, CaMK2a, and MeCP2) led to similar or even greater tau mRNA reduction in primary cortical neurons and in vivo when compared to CMV.89v (Fig. 1D and fig. S2). MeCP2.89v, which produced ~6-fold less ZFP-TF mRNA compared to hSyn1.89v and CaMK2a.89v but produced comparable ZFP protein levels, led to the greatest tau mRNA and protein reduction in vivo.
We then investigated whether the presence of the Venus reporter in ZFP-TF.89v would alter ZFP-TF expression or tau reduction. However, CMV.89 and CMV.89v produced similar expression levels of ZFP mRNA and comparable tau mRNA reduction in vivo (fig. S3, A and B).
Despite the high level of tau reduction, stereological counting of Venus-expressing cells (Venus+/DAPI+) in the hippocampal formation suggested a low transduction rate in CMV.89v- and CMV.72v-injected mice (fig. S3C), suggesting that even very low amounts of Venus (i.e., indistinguishable from background autofluorescence) and ZFP-TF expression are sufficient to repress tau expression. To test this hypothesis, we performed fluorescence-activated cell sorting (FACS) of freshly dissociated hippocampi from CMV.89v- and CMV.72v-injected mice (8 weeks p.i.) and measured tau mRNA reduction in the sorted cells (fig. S3, D to G). In Venus+ sorted cells, tau mRNA was reduced by 80 to 90%, even at barely detectable cellular Venus expression levels.
Furthermore, by in situ hybridization of mouse tau mRNA in combination with immunofluorescence of Venus (immuno-FISH using RNAscope technique) in fixed brain slices, we found substantial tau mRNA reduction in hSyn1.89v-injected hippocampal neurons already 2 weeks p.i. but not in the noninjected hippocampus (Fig. 3A). On the single-cell level, the number of tau mRNA foci per neuronal cell body in 89v-expressing neurons was reduced by >50% (median per mouse; n = 3 mice; Fig. 3, B and C); when determining the most probable value from the distribution of tau mRNA foci per cell body by Gaussian fits, an even greater ~7-fold reduction of tau mRNA foci was detected (x = most probable; 19.6 ± 0.5 foci per cell for 72v and 3.3 ± 1.3 foci per cell for 89v; Fig. 3D). Notably, 72v- and 89v-expressing neurons had similar Venus fluorescence intensity, which did not correlate with the number of tau mRNA foci, again suggesting that minimal ZFP-TF.89v was sufficient to repress tau mRNA at the single-cell level. In summary, these data show that hSyn1.89v efficiently reduced tau mRNA in individual neurons in vivo after only 2 weeks, whereby even low amounts of ZFP-TFs per cell (associated with very low Venus fluorescence) appeared sufficient to drive tau repression.
Fig. 3. Effective tau mRNA repression in individual ZFP-TF–expressing neurons.
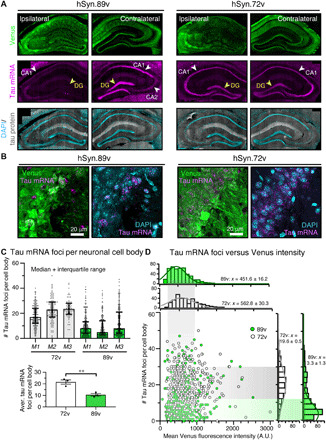
(A) Coronal brain sections from hSyn1.89v-injected mice immunolabeled for Venus (green) and tau protein (gray) and hybridized with fluorescently labeled mouse tau-specific RNA probe (pink). Already after 2 weeks, hSyn1.89v-injected hippocampi show a pronounced reduction in neuronal tau mRNA signal in the dentate gyrus (DG) and CA1 compared to hSyn1.72v-expressing hippocampi. (B) Individual neuronal cell bodies (=soma including nucleus) in CA2 were analyzed for tau mRNA foci. (C) Number of tau mRNA foci per cell body in CA2 and CA3: hSyn1.72v-injected mice: 18 to 24 foci per cell body, with 21.6 ± 1.8 (means ± SEM) foci across mice; hSyn1.89v-injected mice: 9 to 13 foci per cell body with 10.6 ± 1.1 foci per cell body across mice. Data are presented as means ± SEM for each mouse (M1 to M3) and each group, n = 60 to 215 neurons per mouse, three animals per group. Two-tailed Student’s t test. (D) Two-dimensional histogram of the distribution of tau mRNA foci per Venus+ neuron (y axis) and the corresponding neuronal Venus intensity (x axis). The most probable number (from Gaussian fit to distribution; means ± SD) of foci is ~20 in hSyn1.72v- and ~3 in hSyn1.89v-expressing neurons. No correlation between tau mRNA foci and Venus fluorescence intensity is apparent in hSyn.89v neurons. Same data as in (C). A.U., arbitrary units.
Tolerability of tau reduction by AAV ZFP-TF.89v in vivo
AAV ZFP-TF.89v–induced expression of ZFP-TF and Venus and down-regulation of tau could lead to unintended neuroinflammation, neurotoxicity, and axonal or synaptic alterations. We first tested for potential adverse effects of ZFP-TF.89v in the brain by measuring hippocampal mRNA transcript levels of glia cells and neuronal markers.
Six weeks after AAV injection, for both CMV and hSyn1 promoter–driven ZFP-TF expression, some increase in GFAP (glial fibrillary acidic protein) and Iba1 transcripts was detected in ZFP-TF.89v– and in ZFP-TF.72v–injected mice but not in PBS-injected mice (not significant for CMV.89v; Fig. 4A). This increase in glial markers may be due to AAV-mediated protein overexpression per se. Immunohistochemistry and stereology in brain slices confirmed mildly elevated numbers of hippocampal immunolabeled astrocytes and microglia upon ZFP-TF.89v, ZFP-TF.72v, and GFP (green fluorescent protein) expression (fig. S4, A and C). Colocalization analysis of Venus and GFAP fluorescence in CMV.89v-injected hippocampi indicated no expression of ZFP-TFs in astrocytes (fig. S4B) but showed a few GFAP-negative tufted cells that were Venus positive. However, most of the Venus signal was detected in the neuron-dense layers of the hippocampal formation, in which Venus-filled cell bodies also morphologically resembled neurons. In AAV9 hSyn1.89v–injected mice ZFP-TF, Venus expression was restricted to neurons because of the neuronal promoter specificity.
Fig. 4. Transient glia cell activation without neurotoxicity upon ZFP-TF.89v tau reduction in vivo.
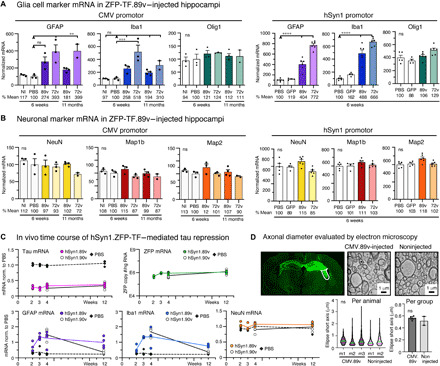
(A) mRNA levels of GFAP, Iba1, and Olig1 in hippocampal extracts after ZFP-TF injection. After 6 weeks, GFAP and Iba1 were elevated in all AAV ZFP-TF–injected mice, indicating mild to moderate gliosis. 72v expression generally caused stronger glia activation. After 11 months, compared to 6 weeks, CMV.89v–injected mice showed reduced Iba1 levels. Means ± SEM, one-way ANOVA with Sidak’s test, reference group for significance indicated by arrowhead, n = 3 mice per group. (B) mRNA levels of NeuN, Map1B, and Map2 in hippocampal extracts after AAV ZFP-TF injection. No changes occurred after 6 weeks and 11 months across all groups. mRNA levels are normalized to PBS. Means ± SEM, one-way ANOVA with Sidak’s test, n = 3 mice per group. (C) Time course of ZFP, tau, and cell type–specific mRNA levels after hSyn1.89v compared to PBS injection. A second tau-targeted ZFP-TF, hSyn1.90v, was tested at 4 and 12 weeks p.i. Tau mRNA was stably repressed by ~70% in hSyn1.ZFP-TF–injected mice after 2 weeks. GFAP and Iba1 mRNA increased until 4 weeks and then declined. hSyn1.90v showed substantially less glia transcripts after 12 weeks. NeuN mRNA was unaffected. Lines connect means of hSyn1.89v-, hSyn1.90v-, or PBS-injected hippocampi, and data points represent individual animals, n = 4 mice per time point. (D) Transmission electron microscopy micrographs of axon cross sections in the fornix (white outlined area) of CMV.89v-injected (12 months) and noninjected mouse. No difference in axon diameter was detected, neither across animals [violin plot; median (green) and interquartile range (pink)] nor across groups (bar graph; means ± SEM, two-tailed Student’s t test, n = 3 CMV.89v and n = 2 noninjected mice). Myelination (dark layer around axons) appeared variable across individual axons in each animal.
To examine whether the observed transient increases in astrocyte and microglia markers were related to viral dose, we performed bilateral hippocampal injections of hSyn1.89 at three doses (3 × 109, 1 × 1010, and 3 × 1010 vg per hemisphere) and found that only the highest dose of hSyn1.89v resulted in elevated glial transcript levels (fig. S4D). Tau mRNA reduction was significant and similar across the 10-fold viral dosage range, consistent with the idea that even low ZFP-TF expression levels are sufficient to drive potent tau reduction.
Notably, no changes in mRNA expression of the oligodendrocyte marker Olig1 or in the number of hippocampal oligodendrocytes were detected (Fig. 3A and fig. S4E). mRNA and protein levels of neuronal signature proteins—NeuN, Map1A, Mab1b, and Map2—were also not affected by ZFP-TF.89v (Fig. 4B and fig. S5A), indicating no neuronal loss and no compensatory up-regulation of other microtubule-associated proteins (MAPs) upon repression of tau. As noted below, electron microscopy did not reveal any morphological alterations in axons. Additional data on neuronal and glia cell mRNA transcript levels and immunohistology data for AAV ZFP-TF.89v with the neuronal promoters CaMK2a and MeCP2 are summarized in fig. S6; they confirm the data for CMV- and hSyn1-driven ZFP-TF.89v expression and tau reduction.
The elevation of glial mRNAs in CMV.89v and CMV.72v mice decreased after prolonged expression of ZFP-TFs for 11 months (Fig. 4A). To determine the kinetics of glia activation in more detail, we injected mice with hSyn1.89v (3 × 1010 vg per hemisphere) and determined GFAP and Iba1 mRNA levels at 2, 3, 4, and 12 weeks. Both GFAP and Iba1 levels increased during the first 4 weeks and then started to slowly decrease between 4 and 12 weeks p.i., consistent with gliosis related to the AAV injection rather than tau repression (Fig. 4C). In the same mice, tau mRNA levels remained at a constant low level, whereas ZFP mRNA levels continued to slowly increase between 2 and 12 weeks after injection. To decrease the glial activation observed for hSyn1.89v, we developed an improved tau-targeted ZFP-TF, hSyn1.90v, which showed superior in vitro dose response (fig. S1C) and no glial activation after long-term ZFP-TF expression (Fig. 4C). These studies confirmed the rapid and sustained tau reduction 2 weeks after AAV ZFP-TF injection, as well as the reversion of the initial glial cell response after 12 weeks.
A decreased hippocampal volume and neuronal cell layer thickness of hippocampal subregions are often used as indicators of neuronal degeneration. We did not observe changes in hippocampal dimensions (hippocampal area; thickness of dentate gyrus, CA1, and CA2) upon ZFP-TF.89v expression, regardless of the promoter used (fig. 5B).
To further investigate the effect of ZFP-TF-89v–driven tau reduction on axonal integrity, we performed transmission electron microscopy on hippocampal tissue to evaluate axons from long-term (12 months) CMV.89v-expressing neurons in the fornix (Fig. 4D and fig. S5C). Comparing CMV.89v-expressing mice with noninjected WT mice, we did not detect differences in axon diameter or the morphological appearance of axon bundles.
Since neurotoxicity could also manifest as synaptic changes, we investigated whether synaptic proteins were altered in hippocampal extracts from mice injected with ZFP-TFs. CMV.89v and hSyn1.89v mice (but not CaMK2a and MeCP2 mice) showed a mild increase in hippocampal presynaptic synapsin-1 as determined by Western blot (means ± SEM: 1.37 ± 0.07 for CMV.89v-injected; 1.04 ± 0.03 for PBS-injected, P = 0.029; 1.05 ± 0.08 for CMV.72v-injected, P = 0.097; fig. S7). Protein levels of the postsynaptic marker PSD-95 were not altered.
In summary, these data demonstrate that a single hippocampal AAV ZFP-TF.89v injection can achieve hippocampal tau reduction of 70 to 80%, whereby no overt neuroinflammation or neurotoxicity occurs, even after long-term expression in the brain. Furthermore, tau reduction can be achieved using different ubiquitous and neuronal promoters and over a range of AAV doses, which makes AAV ZFP-TFs versatile tools not only for translational tau reduction in the brain but also as research tools.
Rapid brain-wide tau reduction after single intravenous AAV-PHP.B ZFP-TF delivery
In transgenic mouse models, a brain-wide reduction of tau by ~50% has been shown to markedly reduce neurotoxicity in the context of Aβ and seizures (7, 33, 34). To translate these conditions into a therapeutic approach, we made use of recent advances in AAV engineering that yielded vector variants capable of widespread CNS transduction following intravenous injection of the AAV particles. For example, AAV9-PHP.B was developed to cross the blood-brain barrier with high efficiency and to enable widespread transduction of cells in the brain (20, 35). We chose to combine the AAV9 PHP.B system with our tau-targeted ZFP-TFs to achieve brain-wide long-term tau repression in the adult mouse brain, in which tau knockdown could be induced with a single systemic injection of AAVs, without a direct brain intervention. While realizing that the use of PHP.B itself is not feasible in humans, this approach nonetheless provides a proof of principle that brain-wide knockdown may be possible for tau-related human neurodegenerative brain diseases.
We generated AAV9-PHP.B versions of ZFP-TF.89v and ZFP-TF.90v with both CMV and hSyn1 promoters and compared their tau mRNA repression efficiency to the already characterized AAV ZFP-TFs in vitro in primary neurons. Compared to their conventional AAV9 versions, both AAV-PHP.B ZFP-TFs, PHP.B 89v and PHP.B 90v, showed superior neuronal transduction, ZFP expression, and hence tau repression in vitro, especially with ZFP-TF expression regulated under the hSyn1 promoter (fig. S8A).
For the following in vivo studies, we first examined the transduction efficiency of the AAV-PHP.B serotype in vivo. We injected PHP.B hSyn.GFP (retro-orbital intravenous injection of 1 × 1012 vg per mouse) into WT mice and found brain-wide coverage of GFP-expressing neurons after 4 weeks (Fig. 5A). Then, to validate that AAV-PHP.B ZFP-TFs can facilitate brain-wide neuronal tau reduction, we delivered PHP.B viruses (2.5 × 1012 vg per mouse) encoding hSyn1.89v, hSyn1.90v, hSyn1.72v, or hSyn1.GFP through retro-orbital intravenous injections into adult WT mice (Fig. 5B) and determined tau mRNA (by qPCR) and tau protein [by enzyme-linked immunosorbent assay (ELISA)] in different brain regions at different time points p.i. (three to five animals per time point), as well as tau protein in the cerebrospinal fluid (CSF) after 24 weeks. A rapid ~50% reduction in cortical, hippocampal, and cerebellar tau mRNA levels was observed as early as 1 week after hSyn1.89v or hSyn1.90v administration (Fig. 5C). Tau protein levels dropped more slowly (significant after 2 weeks)—as expected given the relatively long half-life of tau protein—and continued to decrease until 10 weeks p.i. down to ~20 to 30% of the tau levels measured in hSyn1.72v- and hSyn1.GFP-injected control animals (Fig. 5D). Notably, tau mRNA and protein reduction were sustained in all three brain regions out to 24 weeks.
Fig. 5. Rapid brain tau reduction after a single systemic injection of AAV-PHP.B ZFP-TFs.
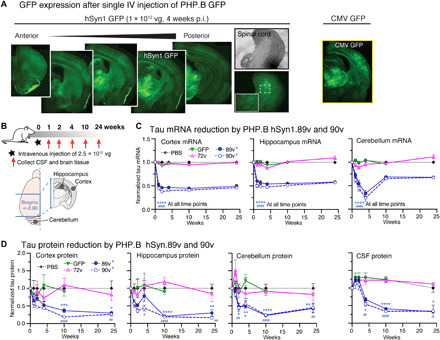
(A) Brain sections from mice with a single intravenous (IV) injection of PHP.B hSyn1.GFP show transduction of neurons in various brain regions and spinal cord. Injection of the same dose of AAV-PHP.B CMV.89v also achieves brain-wide expression. (B) Experimental overview of time course of tau reduction using AAV-PHP.B hSyn1.89v and hSyn1.90v. (C) Tau mRNA levels in cortex, hippocampus, and cerebellum of PHP.B hSyn1.89v– and 90v-injected animals from 0 to 24 weeks. PHP.B hSyn1.GFP and PHP.B hSyn1.72v did not reduce tau mRNA. Means ± SEM, two-way ANOVA versus PBS, n = 3 to 8 mice per group and time point, three tissue samples per region and measurement. P value indication: * for 89v; # for 90v. Note that the unexpected low tau mRNA levels in the cerebellum after 4 weeks do not reflect in tau protein levels. (D) Tau protein levels (by ELISA) in cortex, hippocampus, cerebellum, and CSF of PHP.B hSyn1.89v– and 90v-injected animals from 0 to 24 weeks. PHP.B hSyn1.GFP and PHP.B hSyn1.72v did not reduce tau protein. Means ± SEM, two-way ANOVA versus PBS, n = 3 to 8 mice per group and time point, three tissue samples per region and measurement. P value indication: * for 89v; # for 90v.
When investigating the effect of long-term AAV-PHP.B hSyn1.ZFP-TF expression, we found that, even after 6 months, both hSyn1.89v and hSyn1.90v repressed tau by ~50% across multiple brain regions and the entire spinal cord (Fig. 6A). Animals injected with PBS or with AAV-PHP.B hSyn1.72v did not show tau reduction. For all AAV-PHP.B ZFP-TFs, the highest AAV-PHP.B transduction and ZFP-TF expression occurred in the midbrain (thalamus), whereas the lowest expression levels were detected in the cortex and hippocampus; this is in accordance with the original report on AAV9-PHP.B brain transduction (20).
Fig. 6. Sustained tau repression across brain regions without off targets after systemic AAV-PHP.B ZFP-TF injection.
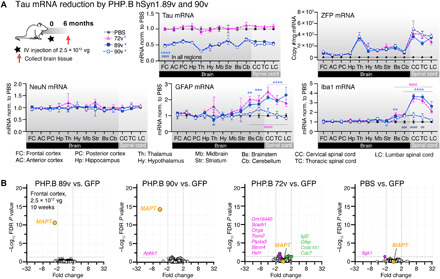
(A) Tau, ZFP, and cell type–specific (GFAP, Iba1, and NeuN) mRNA levels across brain regions after long-term (6 months) expression of hSyn1.89v, hSyn1.90v, or hSyn1.72v. Mice received one retro-orbital injection of 2.5 × 1012 vg per mouse. Data for tau mRNA are normalized to mice treated with PBS. Data are presented as means ± SEM, two-way ANOVA, n = 6 to 8 mice per group. P value indication: * for 89v; # for 90v. (B) RNA array data from PHP.B hSyn1.89v–, hSyn1.90v-, hSyn1.72v-, and PBS-injected mice compared to PHP.B GFP–injected animals show no significant off-target gene deregulation after 10 weeks (2.5 × 1012 vg per mouse). Threshold for significant down-regulation (pink circles) or up-regulation (green circles) at false discovery rate (FDR)–adjusted P values, P < 0.05. n = 8 to 10 mice per group and n = 19,464 transcript clusters assessed. Raw data are available in table S4.
In regions of higher ZFP-TF expression (e.g., thalamus and spinal cord), the astrocyte marker GFAP and the microglia marker Iba1 were elevated accordingly for hSyn1.89v but not for hSyn1.90v. These data indicate that, at similar and sustained ZFP-TF expression and tau reduction, AAV-PHP hSyn1.90v did not lead to any glia cell activation in the brain. The mild gliosis observed for ZFP-TF.89v (with AAV9 hSyn1.89v after hippocampal injections and with AAV-PHP.B hSyn1.89v after intravenous administration) is thus not caused by ZFP-TF–mediated tau reduction itself.
To assess off-target gene alterations that might be caused by ZFP-TF–mediated tau repression in the brain, we performed Affymetrix array analysis of RNA isolated from hippocampal lysates (10 weeks p.i.) and found single-gene specificity for AAV-PHP.B hSyn1.89v and hSyn1.90v for MAPT down-regulation in vivo (Fig. 6B). We did not detect a significant increase in astrocytic GFAP or microglial Iba1 transcript levels (indicative of proinflammatory glial cell activation) or a decrease in neuronal NeuN transcripts (indicative of neuronal loss) upon AAV PHP.B hSyn1.89v– and hSyn1.90v-mediated tau reduction in the cortex and hippocampus after 10 and 24 weeks (fig. S8B). Moreover, no difference in the number of cortical glia cells and neurons was detected by stereological counting at 24 weeks after injection (fig. S8C). Notably, we detected a modest increase in GFAP and Iba1 transcripts in the cerebellum and spinal cord of hSyn1.89v- and hSyn1.72v-expressing mice after 24 weeks, which did not occur upon hSyn1.90v expression and therefore is unlikely to result from ZFP-TF–mediated tau reduction.
Because AAV-PHP.B is delivered through the bloodstream, viral particles can induce ZFP-TF gene expression not only in the brain but also in the spinal cord and peripheral tissues, depending on the specificity of the promoter. To determine the ZFP-TF expression pattern in the brain and other tissues, we dissected multiple brain regions, the spinal cord, and a variety of organs (fig. S8D) from AAV PHP.B CMV.89v– and 90v- and hSyn1.89v- and 90v-injected animals and determined the levels of ZFP-TF and tau mRNA at 4 weeks p.i. Even at a low viral dose (1 × 1012 vg per mouse), neuron-specific hSyn1 but not CMV-driven expression of 89v and 90v reduced tau across different brain regions and in the spinal cord up to ~20 to 40% (fig. S8, E and F). As expected, neuron-specific hSyn1.89v and 90v expression showed little accumulation of ZFP-TF transcripts in the peripheral tissues, whereas CMV.89v produced 5- to 10-fold higher ZFP-TF mRNA levels in peripheral tissue, likely due to the CMV promoter activity in different cell types.
We conclude that AAV9-PHP.B in combination with a neuron-specific promoter is a proficient tool to deliver ZFP-TFs to neurons in the CNS, whereby a single intravenous administration could achieve a sustained ~50% repression of neuronal tau mRNA and protein across multiple brain regions, the spinal cord, and in the CSF. Notably, our data suggest that the reduction of tau in bulk measurements could be the result of incomplete AAV transduction efficiency, since almost complete tau repression is achieved in neurons expressing even small amounts of tau-targeted ZFP-TFs (Fig. 3 and fig. S3, D to G).
Tau reduction by ZFP-TF.89v reduces dystrophic neurites in Aβ transgenic mice
It has previously been shown that the genetic ablation of endogenous tau is protective against Aβ–induced neurotoxicity in mice (7) and can reduce axonal pathology around plaques (36). To test whether the repression of tau using ZFP-TFs would have a similar neuroprotective effect against Aβ–induced neurotoxicity, we administered AAV-PHP.B hSyn1.89v or hSyn1. 90v into mice that produce human Aβ peptide and develop Aβ plaques in the cortex and hippocampus [APP/PS1 line; (37)]. In these mice, plaques are surrounded by neuritic dystrophies: small blebbing protrusion of dendritic and axonal processes filled with neuroplasmic molecules including tau (38). These dystrophies are one of the early Aβ–induced neuropathological changes in the brain (39, 40). In APP/PS1 mice, Aβ plaque pathology starts around 2 months of age with substantial neuritic dystrophies and plaques apparent at the age of 6 months (37).
AAV-PHP.B hSyn1.89v and hSyn1.90v were administered retro-orbitally into 4-month-old (at medium plaque pathology) APP/PS1 mice (1 × 1012 vg per mouse, n = 4 to 7 male animals; Fig. 7A) for 8 weeks, which reduced cortical and hippocampal tau mRNA and protein levels by ~30% (Fig. 7, B and C). To assess the baseline levels of tau and neuritic dystrophies in APP/PS1 mice, we injected mice with the same dose of AAV-PHP.B hSyn1.GFP viral particles as a control.
Fig. 7. Global tau reduction by AAV-PHP.B ZFP-TFs protects against Aβ plaque–induced dystrophies.
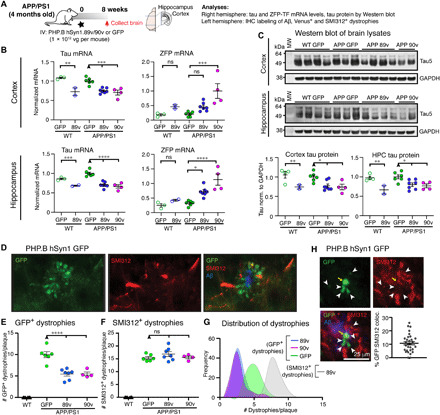
(A) Experimental overview: 4-month-old male APP/PS1 mice and age-matched control WT mice were retro-orbitally injected with a low dose (1 × 1012 vg per mouse) of PHP.B hSyn1.89v, PHP.B hSyn1.90v, or PHP.B hSyn1.GFP, and the brain tissue was analyzed for RNA and protein content by immunohistochemistry (IHC) after 8 weeks. (B) Tau and ZFP-TF transcripts in the anterior cortex and hippocampus (HPC) of injected WT and APP/PS1 mice. Means ± SEM, one-way ANOVA, n = 2 to 7 mice per group. (C) Tau protein in cortical and hippocampal lysates of injected WT and APP/PS1 mice analyzed by Western blot. Tau protein is reduced by 20 to 30% in all mice injected with PHP.B hSyn1.89v or 90v. Means ± SEM, two-way ANOVA with Sidak’s test, n = 2 to 7 mice per group. (D) GFP (green) and axonal phospho-neurofilament (SMI312, red)–containing dystrophies around cortical Aβ plaques (blue) in a PHP.B hSyn1.GFP–injected animal. (E) The number of GFP+ dystrophies (immunolabeled with anti-GFP antibodies) per cortical plaque is reduced by ~50% in 89v- and 90v- compared to GFP-expressing APP/PS1 mice. Means ± SEM, two-way ANOVA with Sidak’s test, n = 4 to 7 mice per group, four to five cortical sections per mouse. (F) The number of SMI312+ dystrophies per cortical plaque is similar in 89v- and 90v- compared to GFP-expressing APP/PS1 mice. Means ± SEM, two-way ANOVA with Sidak’s test, n = 4 to 7 mice per group and four to five cortical sections per mouse. (G) Distribution of dystrophy numbers per cortical plaques in all mice and sections [same data as in (E) and (F)]. (H) Colocalization analysis of GFP-filled (green) and SMI312-filled (red) dystrophies around Aβ plaques (blue) in PHP.B hSyn1.GFP–injected APP/PS1 mice reveals that only ~10% of SMI312+ dystrophies originated from neurons transduced with the AAV and expressing GFP. Means ± SEM, n = 6 mice. MW, molecular weight.
Venus-filled dystrophies around plaques were visualized in fixed brain sections of injected APP/PS1 mice after immunostaining for Venus and Aβ peptide (Fig. 7D), and the number of GFP+ dystrophies around 30 cortical plaques was counted throughout the cortex of each mouse. Excitingly, we detected a ~50% decrease in the number of Venus+ dystrophies in ZFP-TF.89v– and in ZFP-TF.90v–expressing mice (Fig. 7, D and E). These results are especially encouraging since cortical tau mRNA and proteins levels were reduced by only ~30% compared to GFP-expressing animals.
The number of dystrophies labeled with SMI312—a marker for phosphorylated axonal neurofilament proteins commonly used to label dystrophies around Aβ (41)—seems to be unaffected in 89v- and 90v- compared to GFP-expressing animals (Fig. 7, F and G). However, by immunofluorescence, only ~10% of SMI312+ dystrophies also contained GFP (Fig. 7H), which could explain why no reduction of SMI312+ dystrophies at plaques was observed in AAV-PHP.B hSyn1.89v– and 90v-injected animals. These data suggest that only neurons expressing ZFP-TF.89v or 90v were protected.
We were able to confirm the protective effect of tau reduction by ZFP-TF.89v in another cohort of APP/PS1 mice, in which we reduced cortical tau through a unilateral stereotactical injection of AAV9 hSyn1.89v into the somatosensory cortex (2.34 × 1010 vg in 3 μl; fig. S9, A to F). In these mice, the contralateral right hemisphere received the same amount of AAV hSyn1.tRFP, which encodes only the red fluorescent protein turbo-RFP (tRFP), to control for Aβ plaque–induced dystrophy formation in the presence of tau. At 11 weeks p.i. (at ~7 months of age), we compared the number of Venus-filled (green) dystrophies around cortical plaques in the hSyn1.89v-injected cortex with the number of tRFP-filled dystrophies in the other hemisphere, which thereby functioned as a within-animal control. In this experimental setup, we observed 35% fewer Venus-filled dystrophies per amyloid plaque when compared to tRFP-filled dystrophies in the same animal. Mice injected with hSyn1.72v did not show a reduction in cortical neuritic dystrophies. Notably, the average size of the remaining Venus+ dystrophies was similar to that of the tRFP-filled ones, and some tRFP+ dystrophies were positive for phospho-tau (pS396/pS404, PHF1 antibody); this was, as expected, not the case for hSyn.89v-expressing (Venus+) dystrophies (fig. S9F). The size and number of amyloid plaques were similar between the hSyn1.89v- and hSyn1.tRFP-treated hemispheres in each mouse, suggesting a specific effect of the ZFP-TF–mediated tau knockdown on tau-induced dystrophies.
To test whether the neuroprotective effect in AAV-PHP.B ZFP-TF–injected mice resulted directly from tau reduction or indirectly from changes in Aβ metabolism, we determined the transcript levels of APP and of β-secretase 1 (BACE1) and PS1, the two enzymes important for the cleavage of APP into Aβ peptide (36). Genetic ablation of tau in tau knockout mice has been shown to reduce BACE1 protein in Aβ transgenic mice and may thus be involved in the protective effect of tau reduction (36). In AAV-PHP.B 89v– and 90v-injected APP/PS1 and WT mice, compared to GFP control–injected animals, we did not detect significant changes in mRNA levels of APP processing enzymes. However, APP mRNA and protein appeared to be decreased in AAV-PhP.B 89v–injected mice (Fig. 8, A and B, and fig. S10A), but the ratio of Aβ:APP, a proxy for the degree of APP processing, did not change. At this point, the reason for the reduction of APP mRNA and protein associated with AAV-PhP.B ZFP-TF treatment remains unclear, and we can only speculate about some secondary effect or feedback mechanism that reduces APP transcription or increases the activity (but not levels) of APP processing enzymes.
Fig. 8. Global tau reduction by PHP.B hSyn1.89v or 90v protects against Aβ plaque–induced dystrophies.

(A) Transcript levels of APP and its processing enzymes BACE1 and PS1 in hippocampal and cortical lysates of WT and APP/PS1 mice injected with AAV-PHP.B GFP, 89v, or 90v. Reduced APP mRNA was detected in the cortex of 89v-expressing APP/PS1 compared to GFP- and 90v-expressing mice. BACE1 and PS1 mRNA was similar across groups. (B) Cortical lysates from APP/PS1 mice with tau reduction contain similar levels of BACE1 protein but reduced APP protein compared to GFP-expressing controls. Aβ:APP ratios are similar across groups. (C) Transcript levels of Aβ peptide–degrading proteases in hippocampal and cortical lysates from AAV-PHP.B GFP–, 89v-, or 90v-expressing mice: mRNA of the endopeptidases neprilysin and Ece1 and the matrix metalloprotease MMP9 are similar across groups. Data in (A) to (C) are presented as means ± SEM, n = 4 to 6 mice, one-way ANOVA with Sidak’s test for multiple comparison.
mRNA levels of the metalloendopeptidases neprilysin and endothelin-converting enzyme-1 (Ece1) that degrade intracellular neuronal Aβ (42) did not change upon tau reduction in APP/PS1 mice (Fig. 8C). The transcript levels of matrix metalloproteases MMP2 (matrix metalloproteinase 2) but not MMP9, which both degrade extracellular Aβ, were up-regulated in the hippocampus of AAV-PHP.B.90v–injected animals (Fig. 8C and fig. S10B).
Since neuronal health—at baseline and in the context of Aβ—largely depends on the activation state of glia cells in the brain, we tested for alterations in the activation state of astrocytes and microglia upon tau reduction in APP/PS1 mice; this could contribute to the observed reduction of neuritic dystrophies in AAV-PHP.B ZFP-TF–injected mice. In cortical and hippocampal lysates, GFAP mRNA was increased in AAV-PHP.B 90v– but not 89v-injected animals; this may also explain the increase in MMP2 transcripts in these mice (fig. S10B). Furthermore, we found discordant up- and down-regulation of astrocyte genes reported for the “neuroprotective” A2 state (Gbp2 and Ggta1) and the “neurotoxic” A1 state (Emp1 and CD14) of astrocytes (43, 44). For microglial transcripts, the housekeeping protein Iba1 was not altered upon tau reduction in WT or APP/PS1 mice (fig. S10C). However, compared to GFP- and ZFP-TF.90v–expressing animals, APP/PS1 mice that received AAV-PHP.B 89v showed a significant decrease in signature genes of disease-associated microglia (DAM), such as TREM2, Cst7, and Clec7a (45, 46). Since dystrophic neurites were reduced also in ZFP-TF.90v–expressing animals, which show no microglia deactivation, we conclude that tau reduction was the primary cause for protecting neurons against these early signs of toxicity. Further evaluation is needed to determine whether the reduction of DAMs in mice that received ZFP-TF.89v contributes to neuronal health or counteracts Aβ clearance.
DISCUSSION
The down-regulation of endogenous tau has been shown to be beneficial in different neurodegenerative disease contexts in mouse models (7, 13, 22). However, therapeutic approaches to reduce tau in the human brain require careful design and rigorous safety evaluation; they should be tested for potential off-target gene dysregulation and should not result in neuroinflammation or overt pathological changes such as neuronal damage.
In this study, we describe tau-targeted AAV-delivered ZFP-TFs that fulfill these criteria and are promising new tools for the reduction of tau in the adult brain: A single administration of AAV ZFP-TFs achieved gene-specific, rapid, and sustained reduction of CNS tau, without overt changes in other MAPs or indications of neural damage. Furthermore, ZFP-TFs bind and do not edit a specific genomic DNA sequence and therefore can circumvent safety issues attributed to potential off-target genome alterations (47) and preexisting immunity (48) associated with gene editing strategies such as CRISPR-Cas9– or TALEN-based approaches. We showed that, for ZFP-TFs, off-target gene deregulation and glial activation—as we observed for the ZFP-TF identified in initial screening—can be diminished by advanced design and stringent screening of ZFP-TFs for dose response and off targets. Furthermore, preexisting immunity against viral capsid proteins, which may potentially inhibit efficient delivery of ZFP-TFs through AAVs for certain administration routes, could be mitigated by short-term immunosuppression. Using the advantages of ZFP-TFs, we recently were able to create a therapeutic approach for the treatment of Huntington’s disease through allele-selective repression of the mutant huntingtin gene with a single intrastriatal injection of AAV ZFP-TFs targeting the expanded CAG trinucleotide repeat (49).
We identified ZFP-TFs that are able to potently repress endogenous mouse tau mRNA in primary neurons and in the adult brain. ZFP-TF.89 and ZFP-TF.90 both showed very low off-target gene dysregulation in vitro and in vivo, while providing sustained ~70 to 85% tau reduction in the hippocampus upon direct AAV ZFP-TF injection and ~30 to 50% reduction upon intravenous administration of AAV PHP.B ZFP-TF variant, even after a nearly 1 year posttreatment. Mice that received tau-reducing ZFP-TF.89 or ZFP-TF.90 did not show signs of neurotoxicity and only transient (direct AAV9 ZFP-TF injection) or no (AAV9-PHP.B ZFP-TF) glial activation. These results demonstrate that a brain-wide adult-onset reduction of tau protein by ~50% has minimal impact on the expression levels of other proteins in the brain. The combination of highly specific tau-targeted ZFP-TFs with AAVs may thus provide the unique possibility for therapeutic CNS tau reduction following a single treatment. Other anti-tau therapies currently in clinical phase 1 (13, 50, 51) also suggest that tau reduction of up to 50% does not lead to overt structural or functional changes in the brain (13). However, in contrast to AAV-delivered ZFP-TFs, these approaches require multiple intrathecal injections (13, 52, 53) or are specific for unique epitopes or isoforms. For research purposes in mice, single intravenous injections of AAV-PHP.B particles have great benefits because they do not require the implantation of pumps, involve less monitoring (54), and have the potential for better widespread expression and long-lasting knockdown than multiple injections (55).
Safety is a critical consideration for any therapeutic approach seeking to lower tau in the brain. Evidence that the removal of tau is well tolerated comes from genetic tau knockout models that show only small nonpathological deficits in muscle function, sleep/awake behavior, and fear conditioning and reduced long-term potentiation in aged mice (56–60). However, these deficits can partially be attributed to the mouse genetic background (61) and to the absence of tau during development in tau knockout mice. Furthermore, other anti-tau therapies currently in phase 1 [for examples, see (13, 50, 51)] have demonstrated that tau reduction of up to 50% (13) does not lead to overt structural or functional changes in the brain. As this study was underway, a report suggested that acute knockdown of tau in adult C57BL/6 mice using AAV-driven inducible short hairpin RNA (shRNA) led to reversible behavioral changes in spatial memory and learning and to synaptic deficits (62). Unfortunately, the tolerability of the tau-targeted shRNA used in this study, including neuroinflammatory and off-target effects, remains unclear (63). When using highly specific ZFP-TF.90 to knock down tau in the same mouse background, we did not observe off-target gene deregulation and found only acute glia cell activation at the highest intrahippocampal AAV dose, which was likely caused mainly by the injection injury. After this initial phase of gliosis, astrocyte and microglia activation returned to baseline, notably in the setting of a sustained ZFP expression and tau repression. In systemic ZFP-TF–injected animals, no glia activation occurred in any brain region except the cerebellum, where elevated GFAP and Iba1 mRNA levels did not correlate with ZFP-TF expression or tau repression.
The overall behavior of all animals in this study was assessed daily over the course of the studies, and we did not observe any obvious changes in body weight, behavior (e.g., feeding, grooming, social behavior, spontaneous movement, motility, and lethargy), or physical health status in the animals. As noted above, minimal gliosis upon direct AAV ZFP-TF injections and no overt changes in axonal morphology or synaptic protein content were noted. However, a detailed behavioral characterization and the assessment of neurophysiological function upon ZFP-TF–induced tau reduction will be required. In summary, ZFP-TFs can induce tau protein knockdown by up to ~80% in the mouse brain. This reduction in tau was shown to persist for at least 11 months and was not associated with overt neurotoxic effects.
In this study, several lines of experimental evidence suggest that a low copy number of ZFP-TF molecules per cell is sufficient to drive efficient tau repression: (i) Low ZFP mRNA expression, for example, by using the weaker promoter MeCP2 instead of hSyn1 or CaMK2a for direct AAV ZFP-TF injections, yielded a comparable degree of tau mRNA reduction. This could be explained if relatively few ZFP-TF molecules per cell are needed to bind and repress the chromosomal MAPT locus. In this case, the level of tau mRNA repression would remain similar even at lower levels of ZFP-TF expression in a cell. (ii) Dual RNAscope/immunofluorescence analysis showed no correlation between ZFP expression (approximated from GFP intensity) and the amount of tau mRNA lowered at the single-cell level, suggesting that a saturation of cellular tau mRNA repression by ZFP-TFs was reached already after 2 weeks and that the per-cell dose could be further reduced while maintaining efficient tau reduction. (iii) FACS of hippocampal neurons revealed efficient ~80% tau knockdown even in cells with low ZFP-TF expression, and (iv) AAV MeCP2.89v injections yielded >80% tau repression at low ZFP-TF mRNA levels. Together, these findings suggest that AAV ZFP-TFs with even lower expression levels per cell could be efficacious, potentially including tunable or self-regulating elements.
Partial or full genetic knockout of endogenous tau has been shown to be beneficial in different disease contexts, including Aβ–induced synapse loss, hyperactivity, and cognitive decline in humanized Aβ transgenic mice (7, 63–65). In human FTD-mutant tau transgenic mice, the reduction of human tau in the CNS, for example, using ASOs (13) and doxycycline-induced transgene suppression (12, 66), reduces tau aggregation and associated neuronal loss. However, in AD, where no tau mutations are present, neuronal dystrophies are reflective of an early neurotoxic change; these aberrant “blob-like” axonal and dendritic swellings are filled with phosphorylated tau, neurofilament proteins, and ubiquitinated proteins (67, 68) and occur in the immediate vicinity of Aβ plaques and are thought to affect neuronal function. We think that repressing endogenous tau in amyloid-expressing mice is largely analogous to the human situation in AD. Therefore, we tested whether endogenous tau repression in the adult brain using ZFP-TFs could reduce these early pathological changes, which seem to be, at least partially, caused by endogenous tau in the context of Aβ production and deposition in mice (7, 36).
We observed that even a ~30% decrease in CNS tau in the adult brain, over a fairly short time frame (~2 months), was sufficient to alleviate Aβ–induced neuronal abnormalities in the brain: Repressing tau with ZFP-TF.89v and 90v significantly reduced the number of dystrophies in cortical neurons of APP/PS1 mice, suggesting that these neurons were, at least in part, rescued from Aβ–induced toxicity. This effect could be reproduced independently using different experimental paradigms (stereotactic hippocampal and systemic intravenous injection of ZFP-TF AAVs) and with ZFP-TFs (89v and 90v) having different DNA binding sites and affinities. Although we detected reduced levels of APP mRNA and protein in the hippocampus and cortex of APP/PS1 mice with brain-wide tau reduction, no overt changes occurred in key players of APP processing, BACE1 and PS1, that could have contributed to this effect. However, we did not determine the activation state of BACE1 and PS1 in the mice. At this point, a connection between ZFP-TF–mediated tau reduction and reduction of APP expression or activation of APP processing enzymes cannot be ruled out. The ratio between APP and Aβ remained unchanged, which excludes the possibility that enhanced APP processing might have produced even more Aβ in APP/PS1 mice with tau reduction. We did not find conclusive indications that astrocytes and microglia, for example, through changes in their activation state, may have contributed to the reduction of neuritic dystrophies upon tau repression.
In conclusion, we demonstrate that ZFP-TFs are efficient DNA-targeted tools to selectively and durably down-regulate tau mRNA levels and protein expression in the adult brain following a single administration of AAV ZFP-TF into the hippocampus or AAV-PHP.B intravenously. The precision, efficiency, specificity, and tolerability of ZFP-TFs in mice support their continued development to achieve persistent tau down-regulation for the treatment of human tauopathies.
MATERIALS AND METHODS
Animals
For all studies characterizing the activity of ZFP-TFs, 3- to 4-month-old male WT mice (C57BL/6, the Jackson Laboratory) were used. For the study on tau repression in Aβ–expressing mice, 4-month-old APP/PS1 [in-house colony; (37)] mice of both genders were used. The number of animals used was kept at a minimum while still enabling statistical power (n = 3 to 5 animals per group per experiment). Primary cortical mouse neurons were purchased from Gibco. All procedures in this study are performed in compliance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals, the Office of Laboratory Animal Welfare, and the guidelines of Massachusetts General Hospital. The animals’ living conditions, including housing, feeding, and nonmedical care, were overseen by the internal animal facility (Center for Comparative Medicine) of Massachusetts General Hospital.
Zinc finger transcription factors
Design and assembly of ZFP-TFs
ZFP backbones are based on the human ZFP Zif268/EGR1, and ZFPs targeting the murine MAPT gene were assembled using an archive of prevalidated one- and two-finger modules as previously described (69). The resulting ZFPs were cloned into a mammalian expression vector (pVAX) to produce a fusion to the KRAB repression domain of KOX1. ZF sequence information is provided in fig. S1D. Amino acid sequences of the ZFP-TFs used are given in table S5.
mRNA production for transient transfection
Templates for in vitro transcription were generated from pVAX-ZFP or pVAX-GFP plasmids using PCR [forward primer: GCAGAGCTCTCTGGCTAACTAGAG; reverse primer: T(180)CTGGCAACTAGAAGGCACAG]. mRNA was synthesized using the mMESSAGE mMACHINE T7 ULTRA Transcription Kit (Thermo Fisher Scientific) as per the manufacturer’s instructions and purified using RNeasy96 columns (Qiagen).
N2a culture and ZFP mRNA transfection
Mouse neuroblastoma N2a cells show low tau protein expression compared to neurons, but tau mRNA is among the top 13% of all transcripts in these cells, which makes them suitable as a cell model for a screen of tau mRNA reduction by ZFP-TFs. N2a cells were maintained in Eagle’s minimum essential medium with 10% fetal bovine serum and penicillin/streptomycin at 37°C and 5% CO2. mRNA transfection was performed using a 96-well Shuttle Nucleofector (Lonza); 3 to 1000 ng of ZFP mRNA per 100,000 cells was transfected using Lonza program DS-150 in 20 μl of Amaxa SG transfection solution. Eighty microliters of complete media was added after transfection, and cells were plated onto 96-well plates at 30,000 cells per well. The transfection rate was >90% as determined from cotransfection of GFP encoding RNA. Twenty-four hours after transfection, cells were harvested for gene expression analysis by RT-qPCR. Alternatively, 300 ng of ZFP mRNA per 1.5 × 105 N2a cells was similarly transfected, and cells were harvested for Affymetrix analysis.
AAV production
rAAV vectors were generated by the triple-transfection method. Briefly, human embryonic kidney 293 cells were plated in 10-layer CellSTACK chambers (Corning, Acton, MA) and grown for 3 days to a density of 80%. Three plasmids, an AAV Helper plasmid containing the Rep and Cap genes, Adenovirus Helper plasmids containing the adenovirus helper genes, and a transgene plasmid containing the sequence to be packaged flanked by AAV2 inverted terminal repeats, were transfected into the cells using calcium phosphate (70). After 3 days, the cells were harvested. The cells were lysed by three rounds of freeze/thaw, and cell debris was removed by centrifugation. The rAAV was precipitated using polyethylene glycol. After resuspension, the virus was purified by ultracentrifugation overnight on a cesium chloride gradient. The virus was formulated by dialysis and then filter-sterilized. After adjusting the titer (virus genomes per milliliter) of all AAV batches by dilution with PBS and 0.001% Pluronic F-68, the AAVs were aliquoted to single-use doses and stored at −80°C until use. After thawing, no refreezing was done.
Primary neuron culture and AAV ZFP-TF infection
Primary mouse cortical neurons were purchased from Gibco. Cells were plated at 50,000 or 200,000 cells per well and maintained according to company specifications using poly-d-lysine substrate and Gibco Neurobasal Medium. Forty-eight hours after plating [at DIV2 (2 days in vitro)], 50,000 cells per well were infected with AAV ZFP-TFs at the indicated multiplicities of infection (MOIs) and harvested 10 days later (at DIV12; 50% media exchanges were performed every 3 to 4 days) followed by RNA isolation and gene expression analysis by RT-qPCR or protein isolation and analysis by Western blot. Alternatively, 200,000 neurons per well were treated with 1 × 105 vg per cell to ensure a 100% transduction rate and processed in a similar fashion for Affymetrix analysis at DIV12.
Nucleic acid isolation
Total RNA and genomic DNA were extracted from mouse tissue using Qiagen AllPrep kits according to the manufacturer’s protocol. Tissues were homogenized in 600 μl of Qiagen lysis (RLT) buffer supplemented with 0.5% (v/v) antifoaming reagent using a TissueLyser in 1.5-ml Eppendorf tubes for 1.3-min intervals at 25.1/s frequency. Homogenates were applied to AllPrep DNA mini spin columns and centrifuged at 20,000g for 30 s. Flow-through was collected for RNA extraction by AllPrep RNA columns. Column-based genomic DNA and mRNA were performed using the manufacturer’s protocol. Columns were eluted with 80 μl of elution buffer for DNA or with RNase-free dH2O for RNA. RNA isolation for Affymetrix analysis was performed using a High Pure RNA isolation kit (Roche) following the manufacturer’s protocol and eluting RNA with 55 μl of elution buffer. Cell lysates and RT for dose-titration N2a and primary neuron studies were generated using Ambion Power SYBR Green Cells-to-CT kits.
Microarray studies
Total RNA isolated from N2a cells, primary cortical neurons, or fresh-frozen brain and peripheral organ tissue was analyzed for gene expression changes by microarray. Each replicate (50 ng of total RNA) was processed according to the manufacturer’s protocol for sample preparation, hybridization, fluidics, and scanning (Affymetrix MTA1.0 GeneChip arrays, Affymetrix). Robust multi-array average (RMA) was used to normalize the raw signal from each probe set. Fold change analysis was performed using Transcriptome Analysis Console 4.0 (Affymetrix) with “analysis type – expression (gene)” and “summarization – RMA” options selected. ZFP-TF– or PBS-treated samples were compared to samples treated with a non–tau-targeted ZFP-TF that lacks a predicted gene target in the mouse genome (ZFP-TF.72). Change calls are reported for transcripts (probe sets) with a >2-fold difference in mean signal relative to control and a P value of <0.01 [one-way analysis of variance (ANOVA) analysis and unpaired t test for each probe set].
In vivo AAV injections
For hippocampal expression, AAVs encoding ZFP-TFs or vehicle (PBS and 0.001% Pluronic F-68) were stereotactically injected into the left and right anterior and posterior hippocampus (HPC; four injection sites) of 3- to 4-month-old male C57BL/6 mice. Injections were performed using standard aseptic surgical procedures under isoflurane anesthesia (3 to 4% induction, 1 to 2% maintenance) using a 10-μl gas-tight Hamilton syringe with a 30-gauge beveled needle that was coupled to an injector pump, which was directly attached to the stereotaxic frame into which the animal was placed. Following a midline incision and retraction of the skin, bregma was identified, and burr holes were drilled through the cranium at the desired coordinates. After lowering the needle into the brain to reach the injection location, the AAV solution was injected at a flow rate of 0.2 μl/min. After finishing the injection and before retracting the needle, the needle was left in place for 5 min to allow the diffusion of the injected volume. The skin was sutured over the injection site, and the animal was allowed to recover from anesthesia on a 37°C warming pad before it was returned to a clean home cage.
For hippocampal injections into WT mice, a total volume of 6 μl (3 μl per hemisphere, 1.5 μl per injection site) was injected using the following coordinates (from bregma): anterior HPC: anterior-posterior (AP), −2.0 mm; medial-lateral (ML), ±1.5 mm; DV, −1.6 mm; posterior HPC: AP, −3.3 mm; ML, ±3.0 mm; DV, −3.2 mm. For cortical injections into WT and APP/PS1 mice, a total volume of 6 μl (3 μl per hemisphere) was injected into the somatosensory cortex using the following coordinates (from bregma): left cortex: AP, −2.0 mm; ML, 2.0 mm; DV, −0.6 mm; right cortex: AP, −0.5 mm; ML, 2.0 mm; DV, −0.6 mm. Intravenous administration of AAV PHP.B vectors was performed by injecting the virus into the retro-orbital sinus. For analgesia, all mice were treated with Tylenol (in drinking water) for 3 days after dose administration and received a subcutaneous injection of buprenorphine (0.05 mg/kg) immediately after injection.
Brain and peripheral tissue harvest
To harvest the hippocampal tissue for subsequent RNA and protein analysis, mice were perfused with PBS, and the brain was extracted and dissected on ice. Hippocampi and other brain regions for RNA analysis were flash-frozen and placed into RNAlater (Thermo Fisher Scientific) and kept at 4°C for 2 days and then frozen at −80°C until RNA extraction. Hippocampi for protein analysis were homogenized in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) containing protease inhibitor (cOmplete Mini, Roche), centrifuged at 10,000g for 10 min, and the supernatant was analyzed by Western blot and ELISA for its tau content. For the immunohistological labeling of neurons, glia cells, or Aβ plaques and dystrophies, the respective brain hemispheres were drop-fixed in PBS/4% paraformaldehyde at 4°C for 3 to 5 days. Afterward, they were incubated for 2 to 3 days in PBS/30% (w/v) sucrose for cryoprotection, and 40-μm-thick brain coronal sections were cut on a freezing microtome and stored in PBS/50% glycerol at −20°C until use.
Immediately following the brain microdissection, the spinal cord was removed and dissected into cervical, thoracic, and lumbar regions and frozen on dry ice. Then, the peripheral tissues were also collected: lung, intestine, kidney, spleen, liver, testes (for males), and quadriceps muscle. All peripheral tissues were frozen on dry ice. All frozen tissue was stored at −80°C until use.
CSF collection
Immediately before euthanasia, mice were anesthetized with 5% isoflurane and maintained at 3% for CSF collection. For CSF collection, mice were placed on a heating pad to maintain body temperature, and CSF was drawn through the cisterna magna and immediately frozen on dry ice (71). CSF samples identified a priori as having significant blood contamination (CSF appeared red) were removed from analyses.
Gene expression analysis using RT-qPCR
RT-qPCR was performed using the SsoAdvanced Universal probes supermix (Bio-Rad) or Fast Multiplex PCR MM without ROX (Qiagen) depending on the probe sets analyzed. Each biological replicate was assessed in technical quadruplicate on a 384-well CFX real-time qPCR instrument (Bio-Rad). RT-qPCR data analysis was conducted using the Bio-Rad CFX Manager 3.1 software. The used qPCR probes are listed in table S6.
Protein analysis
For Western blot analysis of brain samples, frozen brain samples were homogenized in ice-cold RIPA buffer (Sigma-Aldrich) containing protease (cOmplete Mini, Roche) and phosphatase (PhosSTOP, Roche) inhibitors. After the homogenates were centrifuged at 3000g for 10 min at 4°C, the total protein content in the supernatant (=brain lysate) was determined by bicinchoninic acid assay (Pierce) and adjusted to 1 mg/ml. To analyze the tau content by Western blot, 20 μg of total protein per lane was loaded on a 4 to 12% bis-tris SDS–polyacrylamide gel electrophoresis gel (Invitrogen), separated using Mops running buffer (Invitrogen), and blotted on a nitrocellulose membrane (Amersham). The membrane was blocked in PBS-based blocking buffer (LI-COR) for 1 hour at room temperature, followed by incubation with primary antibodies in blocking buffer overnight at 4°C [primary antibodies: rabbit anti–total tau (1:2000; Dako, catalog no. A0024), chicken anti-GFP (1:1000; Aves, catalog no. GFP-1020), rabbit anti-ZFP (1:1000; LSBio, LS-C374589), rabbit anti-Map1a (1:1000; Abcam, ab184350), rabbit anti-Map1b (1:1000; Sigma-Aldrich, SAB2701260), rabbit anti-Map2 (1:1000; Abcam, ab32454), rabbit anti–synapsin-1 (1:1000; Cell Signaling Technology, #5297), rabbit anti–PSD-95 (1:1000; Cell Signaling Technology, #3450), rabbit anti-BACE1 (C terminus, amino acids 485 to 501; 1:1000; Merck, #195111-M), and mouse anti–Aβ peptide (clone 6e10; 1:1000; BioLegend, SIG-39320)]. Notably, for detection of Aβ, the membrane was boiled in PBS for 5 min immediately after the transfer, before incubation with blocking buffer. Following three washes in tris-buffered saline (TBS) and 0.1% Tween 20, blots were incubated with secondary antibodies in blocking buffer (LI-COR donkey anti-rabbit 680 and anti-chicken 800, each 1:5000) for 2 hours at room temperature. Detection of protein bands was achieved using a LI-COR infrared scanner at wavelengths of 680 and 800 nm. After the detection of tau, GFP, ZFP, Maps, and synaptic proteins, to probe for actin or GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as a loading control, the membrane was reprobed with rabbit anti-actin (1:1000; Sigma-Aldrich, catalog no. A2066) or chicken anti-GAPDH (1:1000; Millipore, AB2302) for 3 hours at room temperature, followed by another incubation with secondary antibodies as before.
Enzyme-linked immunosorbent assays
ELISAs for mouse tau were performed on the PBS-soluble fraction of brain homogenates or on CSF. For the total mouse tau ELISA [Meso Scale Discovery (MSD)], ELISAs were run following the manufacturer’s instructions. Briefly, brain homogenate samples were diluted to 1:250 or 1:500 in standard diluent (10% MSD buffer A in 1× MSD wash buffer), and 25 μl was added per well. Samples were run in duplicate. For CSF, 5 to 10 μl of CSF was loaded on the total mouse tau ELISA in a total volume of 25 μl. Plates were developed using an MSD plate reader, and samples were fit to an eight-point standard curve to determine total mouse tau concentrations in each sample.
Fluorescence-activated cell sorting
For the isolation and sorting of hippocampal neurons from CMV.89v- and CMV.72v-injected mice, the animals were sacrificed, the brains were extracted, and the left and right hippocampi were dissected on ice and immediately dissociated by papain digestion (Papain Dissociation System, Worthington Biochem Corp). The dissociated cells were resuspended and diluted in ice-cold Hanks’ balanced salt solution and kept at 4°C until sorting shortly after. To isolate Venus-expressing (GFP+) cells, the cell suspension was loaded into the FACS machine (Bio-Rad S3), and cells were sorted depending on their GFP fluorescence intensity in RNase-free tubes and stored at 4°C until >10,000 cells were sorted for each group. Following FACS sorting, cells were rerun to confirm purity of the sorted sample. Immediately after FACS was completed, TRIzol was added to the cells to preserve mRNA integrity, and cells were frozen and stored at −80°C for RNA analysis. For the calibration of FACS, noninjected WT mice were used to control for neuronal background fluorescence in the GFP channel, and mice expressing GFP instead of neuronal tau protein [MAPT0/0 eGFP; (72)] were used to define the gate for GFP fluorescence.
Immunohistochemistry of brain sections
For immunofluorescent labeling of transduced neurons, glia cells, and Aβ plaques, floating brain sections were rinsed in PBS, permeabilized with 0.2% Triton X-100/TBS for 20 min at room temperature, blocked in 3 to 5% normal goat serum (NGS) in PBS for 1 hour at room temperature, and then incubated with primary antibodies diluted in 3% NGS/PBS overnight at 4°C: chicken anti-GFP (1:1000; Aves, #GFP-1020), mouse anti-GFAP-cy3 (astrocytes; 1:1000; Sigma-Aldrich, #C9205), rabbit anti-Iba1 (microglia; 1:500; Wako, #019-19741), rabbit anti-Olig2 (oligodendrocytes; 1:500; Abcam, #ab136253), mouse anti-SMI312 (1:500; BioLegend, #837904), and mouse anti–Aβ (amyloid plaques; 1:200; clone 6F/3D, Dako). After washing three times with PBS, secondary antibodies diluted 1:1000 in 3% NGS/PBS were applied for 1.5 hours at room temperature: Alexa Fluor 488 anti-chicken, Cy3 anti-mouse, Alexa Fluor 647 anti-rabbit, and Alexa Fluor 450 anti-mouse (Thermo Fisher Scientific). After three washes in PBS, sections were mounted on microscope glass slides with mounting media containing DAPI (4′,6-diamidino-2-phenylindole; SouthernBiotech). Imaging of immunolabeled sections was done on a Zeiss Axiovert equipped with a QuickSnap camera or on an Olympus BX5.
Combined immunofluorescence and in situ hybridization by RNA scope
Animals (n = 3 per group) received a unilateral hippocampal dual injection of 2× 1.5 μl of AAV hSyn-1.89v or hSyn-1.72v (2.34 × 1010 vg per hippocampus), and after 14 days, the animals were transcardially perfused with PBS, and their brains were drop-fixed in PBS containing 4% paraformaldehyde for 24 hours. After equilibration in 30% sucrose overnight, the brains were cut into 40-μm-thick sections on a freezing microtome, transferred into 10% glycerol as cryoprotectant, and immediately mounted on Superfrost Plus slides for immuno-FISH. The detection of mouse tau mRNA in the brain sections was done using the RNAscope FISH for fixed-frozen tissue following the manufacturer’s manual. The slides were dehydrated and then exposed to RNAscope hydrogen peroxide solution at room temperature for 10 min. After incubation in RNAscope Target retrieval solution at 98° to 102°C for 15 min, the slides were incubated with RNAscope Protease III at 40°C for 30 min using the HybEZ II hybridization system. The mouse MAPT mRNA probe (RNAscope probe-MmMAPT-no-X-Hs) was added onto the brain sections, and the slides were incubated at 40°C for 2 hours. Consecutive amplification was done by incubating the samples at the same temperature with RNAscope Multiplex FL v2 AMP1 for 30 min, with RNAscope Multiplex FL v2 AMP2 30 min, and with RNAscope Multiplex FL v2 AMP3 for 15 min. The HRC-C1 signal was then developed by incubating the samples with RNAscope Multiplex FL v2 HRP-C1 at 40°C, followed by 30 min of incubation with TSA Plus Cy5 (Akoya Biosciences). The developing horseradish peroxidase (HRP) signal was stopped by adding RNAscope Multiplex FL v2 HRP blocker for 15 min. The RNAscope materials used are listed in table S7.
The samples were then processed for immunolabeling for Venus and tau. After blocking in 10% NGS in PBS for 1 hour, the tissue slides were incubated in primary antibodies [chicken anti-GFP (1:1000; Aves, #GFP-1020) and rabbit anti-total tau (1:2000; Dako, #A0024)] overnight at room temperature. After washing in PBS, the tissue slides were incubated with secondary antibodies [biotin-conjugated donkey anti-rabbit (1:200; Abcam, #ab7082) and donkey anti-chicken Alexa Fluor 488 (1:200; Abcam, #ab150173)] for 2 hours at room temperature. After washing in PBS, the samples were incubated with Cy7-streptavidin (1:200; AAT Bioquest, #16914), then washed again in PBS, and lastly coverslipped using mounting media with DAPI (Thermo Fisher Scientific).
For tiled overview images, the stained brain sections were scanned using an Olympus virtual slide microscope VS120 with a 40× objective. Confocal microscopy of CA2 and CA3 hippocampal neurons was performed on an Olympus confocal microscope Fv3000 with a 60× oil immersion objective. This allowed us to count tau mRNA foci per cell body (including soma and nucleus). For this, standard plug-ins in ImageJ [National Institutes of Health (NIH)] were used to define regions of interest (ROIs) manually for individual cell bodies of Venus-expressing neurons in the green channel (Venus) and then apply these ROIs to the far-red channel (tau mRNA-Alexa Fluor 747) to determine the number of mRNA foci in each ROI. Data were collected from three animals per group, two brain sections per animal, 30 to 150 neurons per brain section, and 60 to 215 cells per animal. Imaging and analysis were done blinded for treatment group.
Stereology of cell counts and hippocampal dimensions
The number of transduced cells, astrocytes, microglia, and nuclei in the hippocampi of AAV-injected mice were determined in 40-μm-thick coronal brain sections (starting at −2.0 mm from bregma and equally spaced by 400 μm, five sections per animal) with a stereology approach using the software CAST (Olympus). Using this method, the area of interest (hippocampus) is defined as ROI, and the immunofluorescently labeled cells distributed in 5 to 15% (10% for GFAP and Iba-1) were randomly counted in 10% of the ROI (by superimposing a grid with specified mesh size). The assessed cell numbers were extrapolated to estimate the number of cells in the entire hippocampus (=100% ROI) in three to four brain sections per mouse. The density of astrocytes (number of astrocytes per square micrometer of hippocampus) was calculated as follows: [average number of astrocytes per square micrometer across sections (in 10% HPC) × 10 = average number of astrocytes per square micrometer in 100% HPC]. Counting was done on an Olympus BX51 light microscope equipped with a 20× objective. Oligodendrocytes were counted in the entire hippocampal formation in Olig2-stained brain sections [manual ROI definition, automated thresholding, semiautomated particle detection, and counting procedures in ImageJ (NIH)]. The density of oligodendrocytes (number of oligodendrocytes per square micrometer of hippocampus) was compared in each brain section between the injected and noninjected hemisphere to account for animal-intrinsic variations and differences in the analyzed coronal brain section coordinates. Transduction efficiency of the AAV vectors was determined as the coverage of transduced cells in the hippocampal areas CA1, CA2, and CA3 and the dentate gyrus; the number of GFP+ cells was normalized to the total number of DAPI+ nuclei in the hippocampus. To evaluate the hippocampal dimensions, the area of the hippocampal formation was measured in overlays of 5× bright-field and GFP images of coronal brain sections (four to five sections per animal). To account for differences in the analyzed coronal brain section coordinates, we compared the injected to the noninjected hemisphere in the same section. Thicknesses of the dentate gyrus, CA1, and CA3 were determined by measuring the distances across neuronal cell layer in the respective region at five to six points per region, brain section, and hemisphere. Imaging, counting, and analysis were done on randomized images and blinded for treatment group and genotype.
Colocalization analysis of Venus and GFAP-Cy3 (fig. S4B) in hippocampal areas of AAV CMV.89v–injected mice was performed using the ImageJ plug-in Coloc 2. A Pearson’s R value of ~0 indicates no colocalization.
Electron microscopy
A naïve tau knockout mouse [the Jackson Laboratory, B6.Cg-MAPTtm1(EGFP)Klt Tg(MAPT)8cPdav/J], two naïve C57BL/6 mice (the Jackson Laboratory), and three C57BL/6 mice that received AAV CMV.89v were reserved for electron microscopy. Brains were perfused and fixed with 1% glutaraldehyde (Electron Microscopy Sciences) and 1% paraformaldehyde (Electron Microscopy Sciences) in 0.1 M sodium cacodylate buffer (pH 7.4; Electron Microscopy Sciences) and then stored at 4°C in fixative until further processing. To embed tissue for electron microscopy, a brain matrix was used to cut 1-mm coronal slices. Two slices containing the anterior hippocampus and fimbria were reserved for electron microscopy from each animal. Slices were rinsed in sodium cacodylate and then fixed in 1% osmium tetroxide (Electron Microscopy Sciences) overnight. The following day, slices were rinsed in sodium cacodylate and then distilled H2O and dehydrated in an ascending ethanol series (30, 50, 70, and 95% and twice in 100%, each for 10 min) followed by two rinses in propylene oxide (Electron Microscopy Sciences) for 10 min. Sections were equilibrated in increasing concentrations of Poly/Bed 812 (Polysciences Inc.) before being placed in open molds for 24 hours in a desiccator followed by baking at 60°C for 48 hours. Embedded tissues were first trimmed to a small 1- to 2-mm ROI containing the axons of the external capsule and fimbria lateral to the hippocampus. Tissue within this ROI was sectioned at 70 nm thick on a Leica Ultracut ultramicrotome using a diamond knife and placed on Formvar-coated 150-mesh copper grids (Electron Microscopy Sciences). Grids were poststained with 4% uranyl acetate and 4% lead citrate and imaged using a JEOL JEM 1011 transmission electron microscope.
For the analysis of axon diameters, electron microscopy micrographs were processed using standard plug-ins in ImageJ (NIH). The areas of all individual axon cross sections were selected manually in the electron microscopy image to create a binary mask of all axon cross sections. Ellipses were then fitted to the axonal cross section, and the short axis of the resulting fit was taken as axonal diameter. Two images each from three CMV.89v-injected and two noninjected animals were analyzed. Transmission electron microscopy micrograph recording and analysis were performed blinded for treatment group and genotype.
Dystrophy and plaque analyses in APP/PS1 mice
To assess the number and sizes of dystrophies and Aβ plaques in APP/PS1 mice injected with AAV 89v, AAV 72v, and tRFP, images of brain sections immunolabeled for Aβ were analyzed using ImageJ (NIH freeware). Dystrophies were visible through endogenous fluorescence of Venus and tRFP expressed by AAV-transduced neurons. For plaque and dystrophy sizes, plaques in the cortical area of AAV expression were outlined with ellipses, and dystrophies around these plaques were outlined with circles. The individual sizes of the plaques and dystrophies were measured as respective ellipse or circle areas. The average number of dystrophies per plaque was assessed for each animal as the average ratio of all visible dystrophies per individual plaque counted in four to five sections per animal. Imaging, ROI definition, and analysis were done on randomized sections and blinded for treatment group and genotype. Statistical comparisons were performed as two-tailed Student’s t test between treatment groups.
Statistical analysis for all studies
Statistical analysis in this work was performed using GraphPad Prism version 7 (GraphPad Software, San Diego, CA, USA; www.graphpad.com). The statistical test used for each experiment is provided in the associated methods and/or figure legend. In general, two-tailed Student’s t test was used when comparing two groups, and one-way ANOVA with Sidak’s posttest for multiple comparison was used when comparing three or more groups. Significance is indicated by * or #. The following standard abbreviations are used to reference P values: ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Supplementary Material
Acknowledgments
We thank S. Abrahamson, G. Atkinson, A. Hatami, F. Lorget, K. Meyer, T. Meyer, B. Souberbielle, and M. S. Jung for reviewing the manuscript. Funding: Funding for the study was supported by a sponsored research agreement to Massachusetts General Hospital by Sangamo Therapeutics (to B.T.H.). S.W. was partially funded by Massachusetts General Hospital and the German Center for Neurodegenerative Diseases (DZNE) of the Helmholtz Foundation. B.T.H. received additional funding from the JPB Foundation. S.L.D. was partially funded by NIA training grants to the Division of Medical Sciences at Harvard University (NIH/NIA T32 AG000222) and BrightFocus Foundation. Acknowledgment is also made to the donors of Alzheimer’s Disease Research, a program of BrightFocus Foundation, for support of this research. Author contributions: S.W., S.L.D., and B.Z. designed experiments and analyzed data. S.W., S.L.D., and K.M. performed most experiments; R.E.B. designed and performed EM experiments; Q.Y., H.-O.B.N., A.G., H.T., and R.S. helped with AAV manufacturing and neuron experiments. S.H., L.Z., S.L., N.S., R.A., and J.C.M. designed and tested ZFPs. B.Z., I.A., and Q.Y. conducted microarray experiments. M.P.-R., L.D., D.M., C.C., R.N.B., K.M., A.L., A.C., B.T.C., and A.B.R. helped with mouse and biochemistry experiments. S.W., S.L.D., B.Z., H.S.Z., B.R., and B.T.H. were responsible for study design. B.Z., E.J.R., F.D.U., H.S.Z., M.C.H., B.R., and B.T.H. supervised experiments. S.W. and S.L.D. wrote the manuscript draft and prepared the figures. S.W., B.Z., S.L.D., A.L., B.R., A.M.P., and B.T.H. reviewed and revised the manuscript to its final state. BTC contributed to data collection. Competing interests: FDU is a scientific co-founder of Tune Therapeutics and serves as a paid consultant to the company. BTH has a family member who works at and owns stock in Novartis. FDU is a scientific co-founder of Tune Therapeutics and serves as a paid consultant to the company. BTH serves on the SAB of Dewpoint and owns stock. He serves on a scientific advisory board or is a consultant for Biogen, Novartis, Cell Signaling, the US Dept of Justice, Takeda, Vigil, W20 group, and Seer; His laboratory is supported by Sponsored research agreements with Abbvie, F Prime, and research grants from the National Institutes of Health, Cure Alzheimer’s Fund, Tau Consortium, Brightfocus, and the JPB Foundation. Susanne Wegmann, Sarah L. DeVos, Bryan Zeitler, Annemarie Ledeboer, H. Steve Zhang, and Bradley T. Hyman are inventors on a pending United States patent application related to this work filed 01 Dec 2017 by The General Hospital Corporation and Sangamo Therapeutics, Inc. (US Application Number 15/828,931). Corresponding patent applications are pending in Australia (AU2017367722; 01 Dec 2017), Brazil (BR112019010014; 01 Dec 2017), Canada (CA3043635; 01 Dec 2017), China (CN201780084345.1; 01 Dec 2017), European Patent Office (EP2017876620; 01 Dec 2017), Hong Kong (HK62020003611; 02 Mar 2020), Israel (IL266862; 01 Dec 2017), Japan (JP2019529263; 01 Dec 2017), Korea, Republic of (KR1020197016870; 01 Dec 2017), Mexico (MX2019006426; 01 Dec 2017), and Russian Federation (RU2019120040; 01 Dec 2017). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The authors will review all requests for any materials, except for any requests for any materials that can be purchased, can made by the requestor from publicly available design specifications, such as sequences, or are publicly available through other means. Such permitted requests for materials may be granted, provided that the authors and/or Sangamo Therapeutics have the legal rights to provide such materials without breaching any obligations owed to a third party and the requestor and Sangamo Therapeutics are able to execute a material transfer agreement with mutually agreed terms. The datasets generated during and/or analyzed during the current study are available from the corresponding authors upon reasonable request. Correspondence and material requests should be addressed to the corresponding authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe1611/DC1
REFERENCES AND NOTES
- 1.Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T., Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639 (1992). [DOI] [PubMed] [Google Scholar]
- 2.Mitchell T. W., Mufson E. J., Schneider J. A., Cochran E. J., Nissanov J., Han L. Y., Bienias J. L., Lee V. M. Y., Trojanowski J. Q., Bennett D. A., Arnold S. E., Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer’s disease. Ann. Neurol. 51, 182–189 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Ghoshal N., García-Sierra F., Wuu J., Leurgans S., Bennett D. A., Berry R. W., Binder L. I., Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp. Neurol. 177, 475–493 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Arboleda-Velasquez J. F., Lopera F., O’Hare M., Delgado-Tirado S., Marino C., Chmielewska N., Saez-Torres K. L., Amarnani D., Schultz A. P., Sperling R. A., Leyton-Cifuentes D., Chen K., Baena A., Aguillon D., Rios-Romenets S., Giraldo M., Guzmán-Vélez E., Norton D. J., Pardilla-Delgado E., Artola A., Sanchez J. S., Acosta-Uribe J., Lalli M., Kosik K. S., Huentelman M. J., Zetterberg H., Blennow K., Reiman R. A., Luo J., Chen Y., Thiyyagura P., Su Y., Jun G. R., Naymik M., Gai X., Bootwalla M., Ji J., Shen L., Miller J. B., Kim L. A., Tariot P. N., Johnson K. A., Reiman E. M., Quiroz Y. T., Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 25, 1680–1683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goedert M., Jakes R., Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta - Mol. Basis Dis. 1739, 240–250 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Guerrero-Muñoz M. J., Gerson J., Castillo-Carranza D. L., Tau oligomers: The toxic player at synapses in Alzheimer’s disease. Front. Cell. Neurosci. 9, 464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberson E. D., Scearce-Levie K., Palop J. J., Yan F., Cheng I. H., Wu T., Gerstein H., Yu G.-Q., Mucke L., Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Lopes S., Vaz-Silva J., Pinto V., Dalla C., Kokras N., Bedenk B., Mack N., Czisch M., Almeida O. F. X., Sousa N., Sotiropoulos I., Tau protein is essential for stress-induced brain pathology. Proc. Natl. Acad. Sci. U.S.A. 113, E3755–E3763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gheyara A. L., Ponnusamy R., Djukic B., Craft R. J., Ho K., Guo W., Finucane M. M., Sanchez P. E., Mucke L., Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 76, 443–456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeVos S. L., Goncharoff D. K., Chen G., Kebodeaux C. S., Yamada K., Stewart F. R., Schuler D. R., Maloney S. E., Wozniak D. F., Rigo F., Bennett C. F., Cirrito J. R., Holtzman D. M., Miller T. M., Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 33, 12887–12897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng J. S., Craft R., Yu G.-Q., Ho K., Wang X., Mohan G., Mangnitsky S., Ponnusamy R., Mucke L., Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLOS ONE 9, e115765 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K. H., Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeVos S. L., Miller R. L., Schoch K. M., Holmes B. B., Kebodeaux C. S., Wegener A. J., Chen G., Shen T., Tran H., Nichols B., Zanardi T. A., Kordasiewicz H. B., Swayze E. E., Bennett C. F., Diamond M. I., Miller T. M., Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 9, eaag0481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeVos S. L., Miller T. M., Antisense oligonucleotides: Treating neurodegeneration at the level of RNA. Neurotherapeutics 10, 486–497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asuni A. A., Boutajangout A., Quartermain D., Sigurdsson E. M., Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 27, 9115–9129 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ittner A., Bertz J., Suh L. S., Stevens C. H., Götz J., Ittner L. M., Tau-targeting passive immunization modulates aspects of pathology in tau transgenic mice. J. Neurochem. 132, 135–145 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Herrmann A., Spires-Jones T., Clearing the way for tau immunotherapy in Alzheimer’s disease. J. Neurochem. 132, 1–4 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Yanamandra K., Kfoury N., Jiang H., Mahan T. E., Ma S., Maloney S. E., Wozniak D. F., Diamond M. I., Holtzman D. M., Anti-Tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80, 402–414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burger C., Gorbatyuk O. S., Velardo M. J., Peden C. S., Williams P., Zolotukhin S., Reier P. J., Mandel R. J., Muzyczka N., Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. 10, 302–317 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Deverman B. E., Pravdo P. L., Simpson B. P., Kumar S. R., Chan K. Y., Banerjee A., Wu W. L., Yang B., Huber N., Pasca S. P., Gradinaru V., Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassil F., Brown H. J., Pattabhiraman S., Iwasyk J. E., Maghames C. M., Meymand E. S., Cox T. O., Riddle D. M., Zhang B., Trojanowski J. Q., Lee V. M. Y., Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of lewy body disorders with Aβ pathology. Neuron 105, 260–275.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wegmann S., Maury E. A., Kirk M. J., Saqran L., Roe A., DeVos S. L., Nicholls S., Fan Z., Takeda S., Cagsal-Getkin O., William C. M., Spires-Jones T. L., Pitstick R., Carlson G. A., Pooler A. M., Hyman B. T., Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 34, 3028–3041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller R., Heinrich M., Heck S., Blohm D., Richter-Landsberg C., Expression of microtubule-associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res. 288, 239–249 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Lopresti P., Szuchet S., Papasozomenos S. C., Zinkowski R. P., Binder L. I., Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. U.S.A. 92, 10369–10373 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haery L., Deverman B. E., Matho K. S., Cetin A., Woodard K., Cepko C., Guerin K. I., Rego M. A., Ersing I., Bachle S. M., Kamens J., Fan M., Adeno-associated virus technologies and methods for targeted neuronal manipulation. Front. Neuroanat. 13, 93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kügler S., Kilic E., Bähr M., Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 10, 337–347 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Szymczak A. L., Workman C. J., Wang Y., Vignali K. M., Dilioglou S., Vanin E. F., Vignali D. A. A., Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide-based retroviral vector. Nat. Biotechnol. 22, 589–594 (2004). [DOI] [PubMed] [Google Scholar]
- 28.S. Wegmann, R. E. Bennett, A. S. Amaral, B. T. Hyman, in Methods in Cell Biology (2017), vol. 141, pp. 307–322. [DOI] [PubMed] [Google Scholar]
- 29.Lykken E. A., Shyng C., Edwards R. J., Rozenberg A., Gray S. J., Recent progress and considerations for AAV gene therapies targeting the central nervous system. J. Neurodev. Disord. 10, 16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green F., Samaranch L., Zhang H. S., Manning-Bog A., Meyer K., Forsayeth J., Bankiewicz K. S., Axonal transport of AAV9 in nonhuman primate brain. Gene Ther. 23, 520–526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dittgen T., Nimmerjahn A., Komai S., Licznerski P., Waters J., Margrie T. W., Helmchen F., Denk W., Brecht M., Osten P., Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc. Natl. Acad. Sci. 101, 18206–18211 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adachi M., Keefer E. W., Jones F. S., A segment of the Mecp2 promoter is sufficient to drive expression in neurons. Hum. Mol. Genet. 14, 3709–3722 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Ittner L. M., Ke Y. D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B. C., Christie M. J., Napier I. A., Eckert A., Staufenbiel M., Hardeman E., Götz J., Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Holth J. K., Bomben V. C., Graham Reed J., Inoue T., Younkin L., Younkin S. G., Pautler R. G., Botas J., Noebels J. L., Tau loss attenuates neuronal network hyperexcitability in mouse and drosophila genetic models of epilepsy. J. Neurosci. 33, 1651–1659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rincon M. Y., De Vin F., Duqué S. I., Fripont S., Castaldo S. A., Bouhuijzen-Wenger J., Holt M. G., Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene Ther. 25, 83–92 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Peters F., Salihoglu H., Pratsch K., Herzog E., Pigoni M., Sgobio C., Lichtenthaler S. F., Neumann U., Herms J., Tau deletion reduces plaque-associated BACE 1 accumulation and decelerates plaque formation in a mouse model of Alzheimer’s disease. EMBO J. 38, e102345 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radde R., Bolmont T., Kaeser S. A., Coomaraswamy J., Lindau D., Stoltze L., Calhoun M. E., Jäggi F., Wolburg H., Gengler S., Haass C., Ghetti B., Czech C., Hölscher C., Mathews P. M., Jucker M., Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kowall N. W., Kosik K. S., Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann. Neurol. 22, 639–643 (1987). [DOI] [PubMed] [Google Scholar]
- 39.Knowles R. B., Wyart C., Buldyrev S. V., Cruz L., Urbanc B., Hasselmo M. E., Stanley H. E., Hyman B. T., Plaque-induced neurite abnormalities: Implications for disruption of neural networks in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 96, 5274–5279 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudry E., Wu H. Y., Arbel-Ornath M., Hashimoto T., Matsouaka R., Fan Z., Spires-Jones T. L., Betensky R. A., Bacskai B. J., Hyman B. T., Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J. Neurosci. 32, 3176–3192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudinskiy N., Hawkes J. M., Betensky R. A., Eguchi M., Yamaguchi S., Spires-Jones T. L., Hyman B. T., Orchestrated experience-driven Arc responses are disrupted in a mouse model of Alzheimer’s disease. Nat. Neurosci. 15, 1422–1429 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ries M., Sastre M., Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 8, 160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das S., Li Z., Noori A., Hyman B. T., Serrano-Pozo A., Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflammation 17, 227 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liddelow S. A., Guttenplan K. A., Clarke L. E., Bennett F. C., Bohlen C. J., Schirmer L., Bennett M. L., Münch A. E., Chung W. S., Peterson T. C., Wilton D. K., Frouin A., Napier B. A., Panicker N., Kumar M., Buckwalter M. S., Rowitch D. H., Dawson V. L., Dawson T. M., Stevens B., Barres B. A., Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sala Frigerio C., Wolfs L., Fattorelli N., Thrupp N., Voytyuk I., Schmidt I., Mancuso R., Chen W. T., Woodbury M. E., Srivastava G., Möller T., Hudry E., Das S., Saido T., Karran E., Hyman B., Perry V. H., Fiers M., De Strooper B., The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 27, 1293–1306.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T. K., David E., Baruch K., Lara-Astaiso D., Toth B., Itzkovitz S., Colonna M., Schwartz M., Amit I., A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Maeder M. L., Gersbach C. A., Genome-editing technologies for gene and cell therapy. Mol. Ther. 24, 430–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner D. L., Amini L., Wendering D. J., Burkhardt L. M., Akyüz L., Reinke P., Volk H. D., Schmueck-Henneresse M., High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 25, 242–248 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Zeitler B., Froelich S., Marlen K., Shivak D. A., Yu Q., Li D., Pearl J. R., Miller J. C., Zhang L., Paschon D. E., Hinkley S. J., Ankoudinova I., Lam S., Guschin D., Kopan L., Cherone J. M., Nguyen H. O. B., Qiao G., Ataei Y., Mendel M. C., Amora R., Surosky R., Laganiere J., Vu B. J., Narayanan A., Sedaghat Y., Tillack K., Thiede C., Gärtner A., Kwak S., Bard J., Mrzljak L., Park L., Heikkinen T., Lehtimäki K. K., Svedberg M. M., Häggkvist J., Tari L., Tóth M., Varrone A., Halldin C., Kudwa A. E., Ramboz S., Day M., Kondapalli J., Surmeier D. J., Urnov F. D., Gregory P. D., Rebar E. J., Muñoz-Sanjuán I., Zhang H. S., Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 25, 1131–1142 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Yanamandra K., Jiang H., Mahan T. E., Maloney S. E., Wozniak D. F., Diamond M. I., Holtzman D. M., Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann. Clin. Transl. Neurol. 2, 278–288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee S. H., Le Pichon C. E., Adolfsson O., Gafner V., Pihlgren M., Lin H., Solanoy H., Brendza R., Ngu H., Foreman O., Chan R., Ernst J. A., DiCara D., Hotzel I., Srinivasan K., Hansen D. V., Atwal J., Lu Y., Bumbaca D., Pfeifer A., Watts R. J., Muhs A., Scearce-Levie K., Ayalon G., Antibody-mediated targeting of tau in vivo does not require effector function and microglial engagement. Cell Rep. 16, 1690–1700 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Wurster C. D., Ludolph A. C., Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord. 11, 175628641877693 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Combs B., Kneynsberg A., Kanaan N. M., Gene therapy models of Alzheimer’s disease and other dementias. Methods Mol. Biol. 1382, 339–366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bottros M. M., Christo P. J., Current perspectives on intrathecal drug delivery. J. Pain Res. 7, 615–626 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ingersoll K. S., Cohen J., The impact of medication regimen factors on adherence to chronic treatment: A review of literature. J. Behav. Med. 31, 213–224 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahmed T., Van der Jeugd A., Blum D., Galas M. C., D’Hooge R., Buee L., Balschun D., Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 35, 2474–2478 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Morris M., Hamto P., Adame A., Devidze N., Masliah E., Mucke L., Age-appropriate cognition and subtle dopamine-independent motor deficits in aged Tau knockout mice. Neurobiol. Aging 34, 1523–1529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ikegami S., Harada A., Hirokawa N., Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 279, 129–132 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Cantero J. L., Hita-Yañez E., Moreno-Lopez B., Portillo F., Rubio A., Avila J., Tau protein role in sleep-wake cycle. J. Alzheimers Dis. 21, 411–421 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Cantero J. L., Moreno-Lopez B., Portillo F., Rubio A., Hita-Yañez E., Avila J., Role of tau protein on neocortical and hippocampal oscillatory patterns. Hippocampus 21, 827–834 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Lei P., Ayton S., Moon S., Zhang Q., Volitakis I., Finkelstein D. I., Bush A. I., Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 9, 29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Velazquez R., Ferreira E., Tran A., Turner E. C., Belfiore R., Branca C., Oddo S., Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell 17, e12775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vossel K. A., Xu J. C., Fomenko V., Miyamoto T., Suberbielle E., Knox J. A., Ho K., Kim D. H., Yu G. Q., Mucke L., Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J. Cell Biol. 209, 419–433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dawson H. N., Cantillana V., Jansen M., Wang H., Vitek M. P., Wilcock D. M., Lynch J. R., Laskowitz D. T., Loss of tau elicits axonal degeneration in a mouse model of Alzheimer’s disease. Neuroscience 169, 516–531 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rapoport M., Dawson H. N., Binder L. I., Vitek M. P., Ferreira A., Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 99, 6364–6369 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Polydoro M., de Calignon A., Suárez-Calvet M., Sanchez L., Kay K. R., Nicholls S. B., Roe A. D., Pitstick R., Carlson G. A., Gómez-Isla T., Spires-Jones T. L., Hyman B. T., Reversal of neurofibrillary tangles and tau-associated phenotype in the rTgTauEC model of early Alzheimer’s disease. J. Neurosci. 33, 13300–13311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.K. S. Kosik, in Growth Factors and Alzheimer’s Disease, F. Hefti, P. Brachet, B. Will, Y. Christen, Eds. (Springer Berlin Heidelberg, 1991), pp. 234–240. [Google Scholar]
- 68.Vickers J. C., Mitew S., Woodhouse A., Fernandez-Martos C. M., Kirkcaldie M. T., Canty A. J., McCormack G. H., King A. E., Defining the earliest pathological changes of Alzheimer’s disease. Curr. Alzheimer Res. 13, 281–287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Urnov F. D., Rebar E. J., Holmes M. C., Zhang H. S., Gregory P. D., Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Jagannathan B., Elms P. J., Bustamante C., Marqusee S., Direct observation of a force-induced switch in the anisotropic mechanical unfolding pathway of a protein. Proc. Natl. Acad. Sci. U.S.A. 109, 17820–17825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barten D. M., Cadelina G. W., Hoque N., DeCarr L. B., Guss V. L., Yang L., Sankaranarayanan S., Wes P. D., Flynn M. E., Meredith J. E., Ahlijanian M. K., Albright C. F., Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J. Alzheimers Dis. 24, 127–141 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Tucker K. L., Meyer M., Barde Y. A., Neurotrophins are required for nerve growth during development. Nat. Neurosci. 4, 29–37 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe1611/DC1