

Upregulation of N-type Ca2+ channel dependent subunits increases functional connections and synchronization for pain formation.
Abstract
Sustained neuropathic pain from injury or inflammation remains a major burden for society. Rodent pain models have informed some cellular mechanisms increasing neuronal excitability within the spinal cord and primary somatosensory cortex (S1), but how activity patterns within these circuits change during pain remains unclear. We have applied multiphoton in vivo imaging and holographic stimulation to examine single S1 neuron activity patterns and connectivity during sustained pain. Following pain induction, there is an increase in synchronized neuronal activity and connectivity within S1, indicating the formation of pain circuits. Artificially increasing neuronal activity and synchrony using DREADDs reduced pain thresholds. The expression of N-type voltage-dependent Ca2+ channel subunits in S1 was increased after pain induction, and locally blocking these channels reduced both the synchrony and allodynia associated with inflammatory pain. Targeting these S1 pain circuits, via inhibiting N-type Ca2+ channels or other approaches, may provide ways to reduce inflammatory pain.
INTRODUCTION
Pain is an essential perception that allows us to escape from and avoid potentially damaging stimuli. However, sustained, neuropathic pain is debilitating and remains a substantial burden on society. A 2011 commissioned government report estimated more than 100 million Americans suffering chronic pain costing about 600 billion U.S. dollars in direct health care costs and lost productivity (1). This and other commissions have recommended further research to improve the understanding of the causative neural mechanisms and identifying new therapeutic targets (1, 2). Patients with chronic pain perceive strong pain against non-noxious tactile stimuli (allodynia) as well as spontaneous pain and enhanced responses to noxious stimuli (hypersensitivity). A wealth of studies has revealed changes at the circuit and molecular level in pain circuits that are associated and even causative for allodynia and hypersensitivity. In the dorsal horn of the spinal cord, neural circuit plasticity results in enhanced afferent responses and/or efferent outputs (3) resulting from activation of transient receptor potential channels (4, 5), purinergic receptors (6, 7), and/or activation of microglia to release neurotrophic factors (8, 9) and a subsequent loss of chloride homeostasis (10). Structural changes in dorsal horn neurons (11, 12) and loss of descending inhibitory inputs have also been associated with chronic pain (13–15). More recently, structural and functional plasticity in cortical regions associated with nociception has also been reported to contribute to, or be associated with, chronic pain states. This includes increased excitability or synapse remodeling in the primary (S1) and secondary (S2) somatosensory cortex (16, 17), insula, and anterior cingulate cortex (18, 19) after chronic pain induction. The increased recognition of the role of neural circuit plasticity in cortical regions represents a significant step forward in understanding the sustained pain. The subjective response to pain in both acute and chronic states is strongly modulated by emotional, cognitive, homeostatic states (20), emphasizing the important contributions of cortical associations in pain processing.
Despite these mechanistic insights, no studies have yet tracked individual neurons in cortical circuits during sustained pain. Such information can answer basic questions about whether all neurons increase their activity or not during sustained pain, how pain changes interactions or connections between neurons, and, if these do change, whether specific activity patterns, or microcircuits, form during sustained pain. Such large-scale analysis has been invaluable for mapping out neural circuit plasticity and function during, for example, learning processes where specific circuit activity patterns or neural engrams emerge (21, 22). Using in vivo two-photon Ca2+ imaging in awake mice, we have examined individual neuron activity patterns within the S1 cortex during the development of sustained postoperative pain (23) and inflammatory pain (24). We show that S1 layer 2/3 (L2/3) neurons undergo a prolonged period of increased basal activity during sustained pain states that is associated with an increase in synchronization or coupling between neuron pairs. Furthermore, using chemogenic and pharmacological approaches, we demonstrate that this increased neuronal synchronicity is a critical variable for the induction of sustained pain behaviors. Furthermore, blocking N-type Ca2+ channels in the cortex prevents both the neural activity changes and the development of allodynia. Our data trace single neural activity during sustained pain and demonstrate the importance of local circuit networks in pain behaviors. These cortical networks provide the basis for investigating new therapeutic targets to treat pain.
RESULTS
Mapping out S1 neuronal responses to thermal stimulation in both WT and pain model mice
We used the hind paw incision acute postoperative pain model (23) and complete Freund’s adjuvant (CFA) hind paw injection as an inflammatory pain model (24), both in 6- to 8-week-old C57BL6 male mice. Both pain models induce a transient mechanical and thermal allodynia that can last for about 5 to 10 days and which we define as sustained pain. For 5 days after surgical incision, postoperative pain was evident as a significantly lower withdrawal threshold to hind paw stimulation with von Frey filaments (fig. S1A) and by a reduced latency for paw withdrawal in response to thermal stimulation (50° to 54.5°C; fig. S1B) (25). The inflammatory pain model showed more persistent behavioral changes; we observed significant decreases in both the hind paw mechanical withdrawal thresholds, and the thermal stimuli induced withdrawal latency for up to 10 days after the CFA injection (fig. S1, C and D).
To examine how neuronal activity changes in response to these pain stimuli, we conducted in vivo two-photon Ca2+ imaging of L2/3 neurons in the contralateral primary sensory cortex of the hindlimb paw area (S1HL) (Fig. 1, A and B) in mice with prior injection of an adeno-associated virus (AAV) carrying the GCaMP6f gene into the contralateral S1HL (Fig. 1A). We used the synapsin promotor, where GCaMP6f is mainly expressed in excitatory neurons (see also below). Many GCaMP6f-expressing neurons showed Ca2+ transients before, during, and after thermal stimulation (15 s at 50°C; Fig. 1, C and D; note that mice could escape from the thermal stimuli). We defined three time periods for analysis: 0 to 150 s of prestimulation as the baseline control, 0 to 30 s during the thermal stimulation, and for 120 s after returning temperature back to control levels. The correlation coefficient (C.C.) and power of Ca2+ transients (pCa2+; see Materials and Methods for definitions) were compared for each of these three time periods, in control mice and at 3 days after CFA injection into the right hind paw. The C.C. was increased by thermal stimulation of the right paw, for both control and CFA-injected mice indicating an increase in synchronization among S1HL neurons (Fig. 1, E to G and J). Consistent with the CFA-injected mice being in a hypersensitive state (see also Fig. 2 below), the mice withdrew their paw from the hot plate with a reduced latency (Fig. 1S versus Fig. 1V; see below) and displayed an increase in the absolute values of C.C. both during the “pre-” baseline period and during the stimulus, although the relative extent of increase was similar (fig. S1, F and G). The power (pCa2+) and the frequency of Ca2+ transients were also significantly increased during the thermal stimulation as compared to prestimulation values, for both mouse groups. The increase in pCa2+ and C.C. during the acute thermal stimulation also occurred with a reduced latency in the CFA mice as compared to the control mice (fig. S1H). After thermal stimulation, the C.C. and frequency of Ca2+ transients decreased, but not completely back to the baseline level (i.e., prestimulation); pCa2+ did not decrease after thermal stimulation (Fig. 1, G to I and J to L). Using a sliding window analysis to measure the time course of changes in C.C. and pCa2+ in control (Fig. 1, M and N) and in post-CFA mice (Fig. 1, O and P) also confirmed a significant increase during thermal stimulation. For both control and CFA-injected mice, the extent of thermal-induced increase in both pCa2+ and in the frequency of Ca2+ transients was inversely correlated with the baseline values, suggesting that neurons with lower baseline activity showed a higher relative response to the thermal stimulation (Fig. 1, Q, R, T, and U). Hence, thermal activity induces a clear increase in activity and synchronicity during and after a thermal stimulus across CFA and control mice and shows that neurons with low basal activity are enhanced relatively more. The strong inverse correlation between C.C. and the hindlimb paw escape latency during acute thermal stimulation also supports that neuronal synchronicity is associated with pain responses (Fig. 1, S and V). As the thermal stimulation induces a withdrawal response and sometimes more continuous paw shaking, we monitored mice to separate imaging periods into moving and resting epochs and compare the Ca2+ transients within these time periods. We did not detect any effect of movement on our measured C.C. and pCa2+ (fig. S1, I and J).
Fig. 1. Populational neuronal activity occurred both in WT and inflammatory pain model with the thermal stimulation.
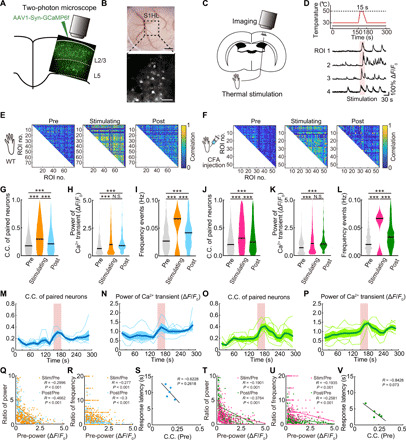
(A) Scheme for virus injection. (B) Light microscopic (top; scale bar, 500 μm) and two-photon images (bottom; scale bar, 100 μm) through a cranial window. (C and D) Experimental protocol (C) and representative calcium traces from four typical neurons (D) in response to 15 s, 50°C thermal stimulation applied to the footpad pink shaded area in (D). (E and F) Representative examples of correlation between Ca2+ activity in pairs of individual neurons in control and CFA mice and the effects of thermal stimulation. (G to I) C.C. of paired neurons (n = 14,257 pairs per five mice) and pCa2+ and frequency of Ca2+ transients (n = 372 neurons per five mice) in control mice. (J to L) C.C. (n = 12,847 pairs per five mice) and pCa2+ and frequency of Ca2+ transients (n = 357 neurons per five mice) in inflammatory pain model (CFA) mice. N.S., not significant; ***P < 0.001; paired t test. Violin plots show median (black lines) and data distributions. (M to P) Time course of changes in C.C. and pCa2+ in control [blue, mean; light blue, ±SEM; n = 5 mice, (M) P < 0.001, (N) P = 0.115; one-way analysis of variance (ANOVA)] and in CFA mice [green, mean; light green, ±SEM; n = 5 mice, (O) P = 0.009, (P) P = 0.089; one-way ANOVA]. (Q, R, T, and U) Scatterplots of relative changes in pCa2+ and frequency of Ca2+ transients induced by thermal stimulation, plotted against the resting pCa2+, in control mice (orange, Stim/Pre; blue, Post/Pre; n = 5 mice) and in CFA mice (pink, Stim/Pre; green, Post/Pre; n = 5 mice, Pearson’s correlation test). (S and V) Correlations between C.C. and the withdrawal response latency during thermal stimulation, in control mice and in CFA mice (n = 5 mice each, Pearson’s correlation test).
Fig. 2. Tracking neuronal activity in individual neurons during the progression of persistent pain.
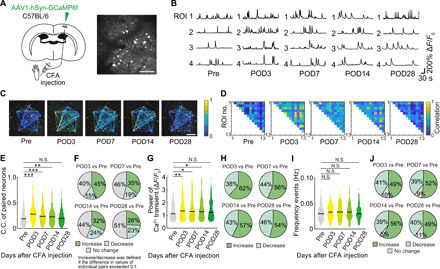
(A) Schematic diagram showing virus injection. WT mice were injected with AAV1-hSyn-GCaMP6f into the hindlimb region of primary somatosensory cortex (S1HL) to enable in vivo Ca2+ imaging of S1HL neurons. Responses from the same neurons were traced pre- and post-CFA injection (pre and days 3, 7, 14, and 28). Scale bar, 100 μm. (B) Representative calcium traces from four typical neurons, traced at different times before and after CFA injection. (C and D) Representative examples of C.C. (color coded) for specific neuronal pairs as indicated by connected lines in (C), from pre- to post-CFA injection. (E, G, and I) C.C. of paired neurons, pCa2+, and frequency of Ca2+ transients (n = 643 pairs, 97 neurons per seven mice) before and at specified times post-CFA. *P < 0.05, **P < 0.01, and ***P < 0.001; paired t test. (F, H, and J) Pie charts of the distribution of changes in C.C., pCa2+, and frequency of Ca2+ transients at times before and after CFA using the same dataset as in panels (E), (G), and (I). An increase or decrease in C.C. was defined if the difference was greater than 0.1.
Tracking neuronal activity in individual neurons during the progression of persistent pain
We next focused on the levels of spontaneous activity within the neuronal population in wild-type (WT) mice and in the two pain model mice and combined chronic imaging and behavior (fig. S2, A and B). We measured and quantified the frequency and pCa2+ of Ca2+ transients in single neurons and the correlation among the activities in single neurons within the neuronal population (the C.C.), using both the CFA injection model (Fig. 2, A to J) and the hind paw incision model (fig. S3, A to J). We first determined the nature of the neurons visualized with GCaMP6f expression driven by synapsin promotor (fig. S4, A to C). A small proportion of neurons with GCaMP6f fluorescence also subsequently stained positive for parvalbumin (PV; 5.6% of all GCaMP6f+ neurons). The averaged intensity of fluorescence in these PV-positive inhibitory neurons was only about half of that in non-PV+ excitatory neurons (PV− neurons, 1.000 ± 0.032; PV+ neurons, 0.585 ± 0.073). We suggest that the population response dynamics in our imaging data described below are largely related to excitatory neurons. As indicated above, injection of CFA into the right hind paw causes a persistent (10 to 14 days) decrease in mechanical withdrawal threshold and latency (fig. S1, A to D). During this period of hypersensitivity, there were marked increases in the proportion of neurons in the contralateral S1 whose activity became correlated (Fig. 2, A to E). The mean C.C. of neuronal pairs significantly increased from about 0.23 before CFA injection to between 0.25 and 0.30 at 3, 7, and 14 days postoperation (POD; Fig. 2E). This same pattern of changes in C.C. was seen if we decreased the Ca2+ transient detection baseline (fig. S10, B and C). About 45% of neurons showed an increase in their extent of correlation at POD3 (cf about 15% showing a decrease), and by 28 days after injection (POD28), equal number of neurons (26 and 23%) showed increases or decreases in C.C. relative to Pre, with about half the neurons (51%) showing no change (Fig. 2F). The pCa2+ of spontaneous transients in individual neurons also significantly and transiently increased following CFA injection (Fig. 2G), with the relative proportion of neurons increasing their pCa2+ modestly becoming more frequent (up to 62%) after CFA injection, before returning to more balanced proportions (46% decrease, 54% increase) at POD28 (Fig. 2H). There was no change in the frequency of spontaneous Ca2+ transients at POD3, POD7, POD14, and POD28 after CFA injection (Fig. 2I). Approximately equal proportions of neurons (49 to 56%) increased or decreased Ca2+ transient frequency at POD3, POD7, POD14, and POD28 after CFA injection (Fig. 2J). A similar pattern of changes in C.C., pCa2+, and frequency of Ca2+ transients was seen after paw incision, although the extent of changes was generally shorter, returning toward control levels by 7 days after paw incision, paralleling the shorter behavioral hypersensitivity (fig. S1). The C.C. of contralateral neurons increased at POD1, POD3, and POD7 with an increased proportion of neurons increasing their C.C. most marked at POD1 and POD3 after paw incision (fig. S3, A to F). Similarly, the mean spontaneous pCa2+ was increased at POD1 and POD3 after paw incision (fig. S3, G and H) while there were no changes in the frequency of spontaneous Ca2+ transients (fig. S3, I and J).
In summary, both pain-inducing models showed marked increases in the extent and number of neurons whose activity was correlated and with stronger Ca2+ transients, with the duration of these effects correlating to the time course of behavioral hypersensitivity. In control mice, either devoid of any injury or following saline injection, there were no changes in C.C., pCa2+, or Ca2+ transient frequency (fig. S5, A to H). The C.C. and pCa2+ in the ipsilateral S1 was actually reduced following CFA injection (fig. S5, I to L), consistent with previous work (26) and likely reflecting stronger cross-hemispheric inhibition secondary to enhanced ipsilateral S1 activity. To further verify that the changes we observed in synchronicity were in excitatory neurons, we also used the calmodulin kinase II α (CaMKIIα) promoter for the expression of GCaMP6f. The data were the same as seen when using the synapsin promoter, with C.C. and pCa2+ being significantly increased on POD3 after CFA injection (fig. S6, A to D).
Last, we examined whether inhibitory neurons in the contralateral S1 also showed changes after CFA injection, using a Cre-dependent GCaMP6f AAV to measure Ca2+ in PV+ inhibitory neurons (Fig. 3A). The C.C of these inhibitory neurons transiently decreased at POD3 and POD7 after CFA injection, while the pCa2+ and frequency did not change over the 14 days period of behavioral hypersensitivity (Fig. 3, B to D). Hence, while excitatory neurons increase the extent of their correlated firing and the amplitude of their Ca2+ transients, inhibitory neurons decrease their correlation over this same period.
Fig. 3. Tracking PV+ neuronal activity in individual neurons during the progression of persistent pain.
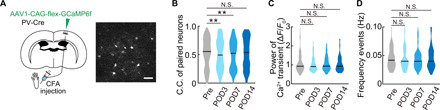
(A) Schematic diagram showing virus injection to image inhibitory neurons. PV-Cre mice were injected with AAV1-CAG-flex-GCaMP6f into S1HL to enable in vivo Ca2+ imaging of PV neurons. The same neurons were traced pre- and post-CFA injection (pre- and postoperative days 3, 7, and 14). Scale bar, 50 μm. (B to D) C.C. of paired neurons (n = 293 pairs per five mice), pCa2+, and frequency of Ca2+ transients (n = 53 neurons per five mice) from pre- to post-CFA injection. **P < 0.01, paired t test. Violin plots show median (black lines) and distribution of the data.
Basal characteristics of the neurons involved in the increased spontaneous activity following pain
The single-neuron activity tracking during postinjury or inflammation pain hypersensitivity revealed a proportion of neurons whose connections and strength appeared to increase. We therefore examined whether this was a particular subpopulation of S1 neurons that differentially responded to CFA injection and paw incision. We stratified neurons based on their basal, pre-injury activity levels and plotted the relative increase in pCa2+ (using the ratio of pCa2+ post-pain to pCa2+ pre-pain, “ratio of power”) against the basal averaged level of pCa2+ (“pre-power”) for each neuron (Fig. 4A and fig. S7). There was an inverse correlation for both pain models seen after injury, with the slope of this relationship steeper for the more severe CFA model (Fig. 3A). This suggests that the neurons with weak activity levels are more likely to show a large relative increase following incision (fig. S7) or CFA-induced inflammation (Fig. 4A). The increase in C.C. of paired S1 neurons was seen for all neuronal groups when we stratified them into those with either low- or high-basal C.C. at day 3 after CFA injection (Fig. 4B). Together, this indicates that, although neurons with weaker basal responses show greater relative increases after inflammatory or surgical injury, the increase in synchrony between pairs of neurons is seen across all neurons regardless of initial activity strength. To further examine the contribution of C.C. to the pain behavior, we plotted the averaged C.C. at different times after CFA with mechanical threshold and withdrawal response latency and found a strong negative correlation between these values, suggesting that each incremental increase in C.C. is associated with an incremental increase in pain behavior (Fig. 4C).
Fig. 4. Relationship between resting activity and increases in power and C.C.
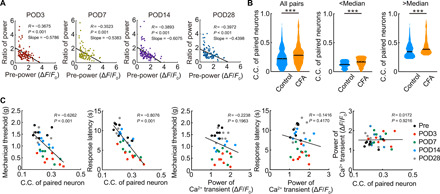
(A) Scatterplot of the relative increase in neuronal pCa2+ for each neuron during inflammatory pain, plotted against the basal pCa2+ before pain, and shown at four different days postoperation (n = 97 neurons per seven mice, Pearson’s correlation test). (B) Comparison of C.C. of paired neurons in control and inflammatory pain model mice (day 3 after CFA injection). In the center and right columns, the data were stratified into neurons with low-basal pCa2+ and higher pCa2+ (using median pCa2+ as the cutoff). A similar difference in C.C. was seen in both these cohorts. ***P < 0.001, Mann-Whitney U test. Violin plots show median (black lines) and distribution of the data. (C) Scatterplots of pain behavioral responses (mechanical threshold or escape response latency during thermal stimulation) against the neuronal C.C. or pCa2+. A significant negative correlation existed for C.C. against pain behaviors but not for pCa2+. There was no relationship between C.C. and pCa2+. Data from pre- and post-CFA neuronal responses, as shown in Fig. 2 [n = 7 mice for (C), regression lines show by Pearson’s correlation test].
Direct evidence for increase in functional connections within L2/3 neurons in the primary sensory cortex after pain induction
To more directly examine the functional connectivity within L2/3 neurons in S1 during the period of sustained pain, we generated holographic patterns of optogenetic stimulation using a spatial light modulator (SLM) and combined this holographic stimulation with two-photon imaging (Fig. 5A). We stimulated a single neuron and simultaneously quantified the responses of surrounding neurons in the contralateral S1HL (Fig. 5, B to E). To do this, we coexpressed both GCaMP6m with the red-shifted channel rhodopsin variant ChRmine using AAV injection (27). We defined a stimulation-evoked response as a Ca2+ transient with a peak time within 1 s after holographic stimulation (Fig. 5B, bottom left). We confirmed that evoked responses were detected above basal rates of spontaneous activity by showing that the C.C. following stimulation was significantly higher than the C.C. due to spontaneous activity measured before the stimulation period (Fig. 5B, bottom right). We then stimulated a target neuron and measured GCaMP6m responses in the stimulated and in surrounding neurons to define functional connectivity (Fig. 5B). The number of surrounding neurons responding to stimulation was small before the CFA injection that transiently increased on POD3 of CFA injection with the same activation protocol and that recovered to the basal level by POD7 (Fig. 5, C and D). This result was similar when all data were grouped (Fig. 5C) or when we quantified for individual mice at the cellular level over the 4 weeks of imaging (Fig. 5D). Similarly, when we plotted the proportion of stimulated cells that could evoke responses in adjacent cells at the individual mouse level, we also saw a transient but significant increase in connectivity (Fig. 5E). These results demonstrate directly that CFA injection resulted in increased functional local connectivity. This increase in connectivity, seen in this small sample of neurons with suprathreshold responses to a single stimulus applied to an adjacent neuron, was less sustained than observed across the larger and dispersed population responding spontaneously (as measured by increases in C.C.; Fig. 2E). Although these are quite different measures of connectivity to compare, it may be that enhanced connectivity at each neuron pair in the S1 neural circuits is additive.
Fig. 5. Increased functional connectivity of L2/3 S1HL neurons in CFA-injected mice.
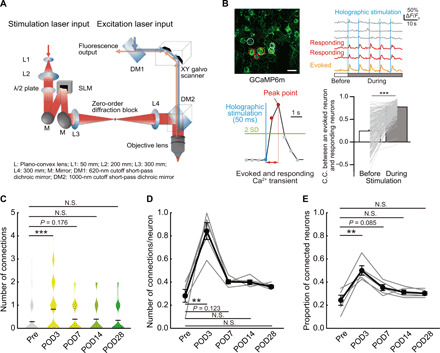
(A) Schematic diagram of holographic stimulation combined with two-photon microscope. (B) Representative images of S1 neurons expressing GCaMP6m. The stimulated neuron is circled in orange; responding and nonresponding neurons are circled in red and gray, respectively. Scale bar, 10 μm. Corresponding Ca2+ traces in these neurons, with the blue vertical lines (holographic stimulations) are shown (top). A schematic evoked Ca2+ response is shown. The C.C. between the stimulated and responding neurons is compared under spontaneous conditions and following stimulation (bottom; n = 230 pairs per five mice, paired t test). (C) Distributions of responding neurons from pre- to post-CFA injection [n = 5 mice, n = 72 (Pre), 75 (POD3), 77 (POD7), 77 (POD14), and 77 (POD28) neurons, Mann-Whitney U test]. Violin plot shows mean (black lines) and distribution of the data. (D) Proportion of neurons responding to each single neuron stimulation, measured pre- and post-CFA injection in the same animals (n = 5 mice, paired t test). (E) Proportion of stimulated neurons that could elicit responses in adjacent neurons. Changes in these connected neurons compared from pre- to post-CFA injections in the same animals (n = 5 mice, paired t test). **P < 0.01 and ***P < 0.001.
Chemogenic activation or inhibition of L2/3 neurons in S1 can modify pain behaviors
Paw inflammation and surgical injury presumably lead to the increased activity and connectivity in S1 secondary to concurrent enhanced spinal and peripheral activity. To probe whether the increase in S1 activity (pCa2+ power and synchronicity) per se may be causally linked to pain sensation, we used a chemogenic approach to selectively activate S1 neurons in the absence of injury. We used the hM3Dq designer receptor, a modified human M3 muscarinic (hM3) receptor activated by the inert clozapine metabolite clozapine N-oxide (CNO) that subsequently engages the Gq signaling pathway to increase neuronal activity. AAV-coded hM3Dq was combined with mCherry to identify transfected neurons following injection into the S1 hind paw area, and this was co-injected with the AAV encoding GCaMP6f to measure Ca2+ responses (Fig. 6, A and B).
Fig. 6. Chemogenetically increased neuronal activity in S1HL was associated with a decrease of the threshold of pain.
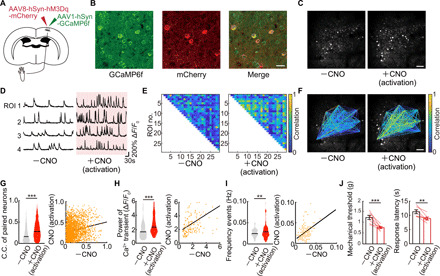
(A) Schematic diagram showing virus injection. WT mice were injected with AAV1-hSyn-GCaMP6f and AAV8-hSyn-hM3Dq-mCherry into the hindlimb region of primary somatosensory cortex (S1HL) to enable in vivo Ca2+ imaging and increased neuronal activity in S1HL neurons by administered CNO. (B) Immunostaining image of S1HL neurons. Left, GCaMP6f; middle, mCherry; right, merged image. Scale bar, 20 μm. (C) Representative example of images of the same neurons before and after a single dose of CNO. Scale bar, 100 μm. (D) Representative calcium traces of typical four neurons. Each neuronal activity was traced from before to after a single dose of CNO. (E and F) Representative example of paired neuronal C.C. from before to after a single dose of CNO. Scale bar, 100 μm. (G to I) Change in C.C. of paired neurons, pCa2+, and frequency of Ca2+ transients (n = 2292 pairs, 151 neurons per five mice) in before and after a single dose of CNO. **P < 0.01 and ***P < 0.001; paired t test. Violin plots show median (black lines) and distribution of the data. (J) Change in pain threshold before and after a single dose of CNO (n = 7 mice). **P < 0.01 and ***P < 0.001; paired t test. Error bars show means ± SEM.
In S1 L2/3 neurons expressing both GCaMP6f and hM3Dq, basal activity was recorded for an hour under resting conditions, and then a single CNO dose (5 mg/kg) was injected intraperitoneally, resulting in activation of S1 L2/3 neurons (Fig. 6, C and D). The synchrony of neuronal activation, the pCa2+, and the frequency of Ca2+ transients were all strongly enhanced following CNO-induced activation (Fig. 6, E to G). Although the extent of the increase in pCa2+ and C.C. was similar to that observed by pain stimuli, the patterns of activation differed in that there were positive correlations between the basal parameters and those seen post-CNO activation (Fig. 6, G to I). A single injection of CNO also resulted in a decreased mechanical threshold for paw withdrawal tested using von Frey hairs and a decreased escape latency in the hot plate test (Fig. 6J). Notably, these behavioral changes were observed at the same time period as when the neuronal responses were altered. CNO injection without hM3Dq expression did not affect pCa2+ and C.C. or behavior (fig. S8, A and B). We next examined the behavioral effects of chronic CNO injections over seven consecutive days, performing the von Frey and hot plate tests at the same time (≈1 hour) after each CNO injection and for another 7 days after the CNO injections were finished (Fig. 7A). Notably, we found a significant reduction in the hind paw withdrawal threshold (Fig. 7B) and in the response latency to the thermal stimulus (Fig. 7C), which was sustained for up to a week after CNO injections ceased. We also imaged Ca2+ events before, at the end of 7 days of CNO administration, and 7 days later after stopping CNO (Fig. 7, D to G). C.C., pCa2+, and frequency of Ca2+ transients were all significantly increased at 7 days and returned to baseline 1 week later. Therefore, the increased synchronicity and pCa2+ of the S1 neuronal responses are correlated, suggesting their contribution to the sustained changes in pain behavior, even in the absence of any peripheral damage. As a control, CNO administration to mice expressing only the tdTomato construct showed no changes in Ca2+ transients or pain behavior upon CNO administration (Fig. 7, B and C, and fig. S8, A and B). Furthermore, we still observed the CNO-induced increases in C.C. when Ca2+ transients were digitized, to eliminate any impacts of changes in Ca2+ event duration that may have otherwise contributed to C.C. (fig. S8C).
Fig. 7. Chronic increased neuronal activity by chemogenetics in S1HL was associated with a decrease of the threshold of pain.
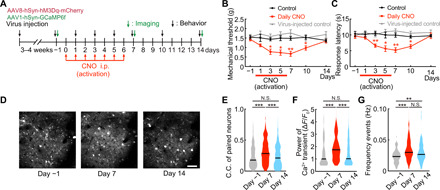
(A) Time course of chronic experiments. CNO was administered daily for 7 days. Behavioral evaluation was performed pre, day 1, day 3, day 5, day 7, day 10 (CNO off, day 3), and day 14 (CNO off, day 7). (B and C) Changes in pain mechanical threshold and thermal escape latency, plotted from pre to 14 days with daily administration of CNO occurring between days 0 and 6 (black, control; red, daily CNO; gray, virus-injected control; *P < 0.05 and **P < 0.01; two-way ANOVA followed by Bonferroni test, versus control). Error bars show means ± SEM. (D) Representative images of the same neurons shown from pre through to daily administration of CNO and 7 days after. Scale bar, 50 μm. (E to G) Distributions of C.C. of paired neurons, pCa2+, and Ca2+ transient frequency (n = 458 pairs, 70 neurons per five mice) from pre, following 7 days of CNO administration, and 7 days after CNO. **P < 0.01 and ***P < 0.001, paired t test. Violin plots show median (black lines) and distribution of the data.
Increase N-type Ca2+ channel expression with the pain induction
The results above indicate that selectively modulating S1 neural activity could modulate pain sensation, so we next sought to identify a molecular target that may contribute to this effect. Previous studies have suggested that L-, N-, and P/Q-type Ca2+ channel antagonists are effective drugs for chronic pain (28, 29), which led us to focus on whether a specific Ca2+ channel may be involved. We used fluorescence-activated cell sorting (FACS) to detect any changes in Ca2+ channel subunits in c-Fos+, NeuN+ cells (neurons) isolated from the contralateral S1 of control and CFA-injected mice (Fig. 8A). The population of c-Fos+ neurons was increased in the CFA inflammatory pain model, validating our selection approach. An array of voltage-gated Ca2+ channel subunits was detected in this c-Fos+ population. We found, however, that those responsible for N-type Ca2+ channel currents [Cav2.1-Allophycocyanin (APC)] and P/Q-type Ca2+ channel currents (Cav2.2-APC) were specifically increased in S1 neurons from the CFA-injected mice, while the L-type Ca2+ channel current subunits were not significantly different from control (Fig. 8B). The ratio of the mean fluorescence intensity (MFI) of Cav2.2 in NeuN, c-Fos double-positive neurons was also significantly increased compared with control, while the MFI for Cav2.1 was not different from control, suggesting increased expression of Cav2.2 after CFA (Fig. 8C). The increase in these Ca2+ channel subunits did not generalize to other channels, with no increases in some potassium and sodium channel subunits (Kv1.1, Kir6.2, Nav1.1, and Nav1.6; fig. S9, A to C).
Fig. 8. Increase N-type Ca2+ channel expression in S1 with the pain induction.
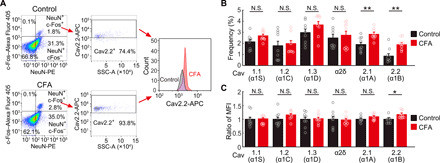
(A) A representative example of FACS pseudo-color plot. Removal of doublets and debris was performed. Each experiment was obtained from 105 cells. Left: Neurons are gated from non-neurons on the basis of the expression of NeuN-PE, and active neurons are gated from inactive neurons on the basis of the expression of c-Fos–Alexa Fluor 405. Middle: Cav2.2 (α1B)–APC (voltage-dependent N-type Ca2+ channel)–positive neurons are gated/counted in active neurons in control and inflammatory pain model of CFA injection. Right: Histogram of expression of Cav2.2 (α1B) in NeuN, c-Fos, and Cav2.2 (α1B) triple-positive neurons in both models (black, control; red, CFA). (B) Frequency of NeuN, c-Fos, and each ion channel triple-positive neurons in various calcium ion channels in cells gated with (A) (black, control; red, CFA; n = 9 mice for each; **P < 0.01, paired t test. Error bars show means ± SEM.). (C) The ratio of the MFI of each Ca2+ channel in NeuN, c-Fos double-positive neurons in inflammatory pain model compared with control (black, control; red; CFA; n = 9 mice for each; *P < 0.05, paired t test. Error bars show means ± SEM.).
To further examine whether Cav2.2-APC overexpression has functional consequences, we administered a selective N-type Ca2+ channel antagonist (PD173212) via intraventricular injection and examined its effects on neuronal activity (Fig. 9A) and pain sensation in mice following CFA injection. Spontaneous neuronal activity was decreased following PD173212 injections (Fig. 9, B and C, and movie S1), and subsequent quantification showed that the pCa2+ and C.C. of neurons were significantly reduced when compared with that in CFA mice (Fig. 9D). Consistently, the mechanical threshold for pain sensation and the response latency were increased with the intraventricular application of PD173212 (Fig. 9E and movie S2). Last, we implanted the slow release vehicle Elvax containing PD173212 on to the cortical surface in S1 to examine the effects of prolonged Cav2.2 channel block (Fig. 9F). Elvax containing PD173212 when applied on S1 significantly increased mechanical threshold and response latency compared with the CFA without PD173212 (Fig. 9G). In contrast, application of the same Elvax-PD173212 had no effect on pain thresholds on to the cortical surface in V1 as an area of unrelated pain. Although the absolute changes in C.C. and pain behaviors induced by PD173212 were modest, the changes in C.C. were similarly negative correlated with changes in pain behavior (fig. S10A), as we observed with CFA treatment (Fig. 4C).
Fig. 9. Topical administration of N-type Ca2+ channel antagonist improved pain threshold.
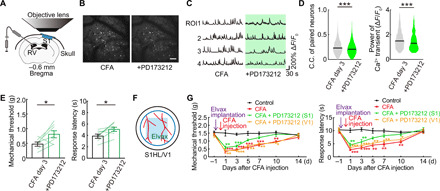
(A) Schematic diagram showing method of the intraventricular PD173212 (N-type Ca2+ channel antagonist) administration and in vivo imaging. (B) Typical in vivo two-photon images of before and after a single dose of PD173212 in inflammatory pain model mice (3 days after CFA injection). Same neurons traced. Scale bar, 100 μm. (C) Calcium traces of typical four neurons. Each neuronal activity was traced before and after administration of PD173212. (D) Change in paired neuronal C.C., pCa2+ of neurons (n = 4295pairs, 203 neurons per five mice) in before and after a single dose of PD173212 in inflammatory pain model. ***P < 0.001, paired t test. Violin plots show median (black lines) and distribution of the data. (E) Change in pain threshold before and after a single dose of PD173212 in inflammatory pain model (n = 7 mice). *P < 0.05, paired t test. Error bars show means ± SEM. (F) Schematic diagram showing method of administration of the drug-soaked Elvax (PD173212; 2.5 mM) to S1HL through an open-skull cranial window. (G) Change in pain threshold by chronic local administration of PD173212 to S1HL/V1 [black, control; red, CFA; green, CFA + PD173212 (S1HL); orange, CFA + PD173212 (V1); *P < 0.05, **P < 0.01, and ***P < 0.001; two-way ANOVA followed by Bonferroni test, versus control]. Error bars show means ± SEM.
DISCUSSION
Although the detection of pain is triggered by neuronal activity conveying sensory signals through the peripheral and spinal nociceptive pathways (30, 31), perception of the features and severity of pain involves additional central processing. Human magnetic resonance imaging (MRI) and other studies over the past decade have recognized a “pain matrix,” where cortical and subcortical structures are activated during nociceptive stimuli and, furthermore, can participate in modulating the perception of pain (32–34). The extent to which all these discrete neural substrates contribute to the transition to chronic pain is not clear. Peripheral and spinal changes in chronic pain have been well documented, including altered nociceptors responses, increased excitability in spinal networks, and activation of glial cells (35, 36). MRI of patients with chronic pain has, more recently, identified, changes in the connectivity among central pathways in the pain matrix (37) and increased excitability of central circuits in preclinical rodent models (38, 39). For example, astrocyte-mediated growth of dendritic spines in S1 accompanies the initial phase of the transition to persistent allodynia following sciatic nerve injury (40). The current work broadens the evidence that changes in S1 accompany sustained pain. Single neurons in S1 showed enhanced synchronization and pCa2+ during spontaneous periods of activity following both surgical excision and inflammatory peripheral injury models, and single neurons showed greater functional synaptic connectivity to surrounding neurons in the days following these acute injuries. The behavioral characteristics of pain—mechanical allodynia and thermal hypersensitivity—could be reproduced by selective chemogenic activation of cortical activity patterns. Notably, this behavioral hypersensitivity was apparent with chemogenic stimulation of S1 in the absence of any peripheral injury, as was also previously observed when S1 astrocytes were selectively activated to induce spine plasticity (40) and has also been seen with sustained optogenetic activation of the midcingulate cortex (41). Therefore, evidence is building that changes in cortical circuits, in themselves, can be causative for chronic hypersensitivity, although whether this enhanced S1 circuit connectivity also leads to changes in spinal or other central circuits will be important to resolve. Of additional interest is whether the enhanced S1 circuit activity we observed here predisposes to greater stress or emotional regulation of pain via interactions with the prefrontal cortex and/or insular cortex [e.g., (42)].
We initially considered that greater basal neuronal activity may in itself predispose to increased synchronicity. However, the basal frequency of Ca2+ transients in S1 neurons was not increased following pain nor was there an increase in the amplitude of the sensory-evoked S1 responses (although they did occur faster). Furthermore, increases in C.C. occurred even when Ca2+ levels were thresholded as a single event, minimizing any effects on C.C. due to increases in Ca2+ event amplitude or duration. Last, the correlation only existed between pain and C.C. and not, for example, with Ca2+ event frequency or with power. Hence, we propose an increase in synchronously active excitatory neurons during sustained pain. Responses in inhibitory neurons during sustained inflammatory pain were different. We did not see major changes in PV interneuron activity, as observed in the nerve constriction model of chronic pain (16). We did, however, observe a transient decrease in C.C. in PV neurons consistent with the reduced connectivity observed using autofluorescent imaging in S1HL cortex in a different inflammatory pain (EAE) model (16, 43). We also did not see global increases in neuronal firing as proposed for CFA inflammatory pain (39), but different GABAergic neuronal phenotypes will respond differently during chronic pain (16). More subtle changes in excitation and inhibition, combined with altered functional synaptic wiring, are likely to determine the increase in synchronicity during pain. Neurons with low basal activity were more likely to increase their responses following pain induction, yet neurons that increased their synchronous firing were found among populations of both low and high basal activities. Hence, it appears that there is some specific pattern of increased activity, consistent with the formation of synchronized neural circuits in the S1 following peripheral injuries. This is akin to formation of specific circuits, or engrams, that accompany forms of learning and/or addictive behaviors; sustained pain shares a number of features similar to addiction and learned behaviors (21, 44).
Our results indicate functional up-regulation of Cav 2.2 in cortical neurons with inflammatory pain, and ventricular injection or topical cortical infusion of a selective N-type Ca2+ channel blocker effectively reduced mechanical hypersensitivity. N-type Ca2+ channel blockers have emerged as some of the more recent effective treatments for neuropathic pain (45). Pregabalin binds to voltage-gated Ca2+ channel accessory binding proteins, while Ziconotide, derived from structurally related cone shell toxins, directly binds the pore-forming a-subunit. The mechanism of action is based on targeting spinal circuits that become enhanced in pain states. Intrathecal administration of Ziconotide or related N-type Ca2+ channel antagonists is effective in restoring pain thresholds in both rat models of acute and chronic pain (46) and in patients with chronic severe pain (47, 48). However, the central side effect profile of Ziconotide limits its systemic use, although better tolerated derivatives may evolve (45, 47). The current research suggests an alternate cortical target for chronic pain therapy, and the particular importance of identifying the mechanisms mediating the cortical up-regulation of N-type Ca2+ channel subunits can be identified.
In conclusion, our studies have identified the development of synchronized circuits in the S1 cortex associated with the development of pain hypersensitivity following peripheral injury. Increased basal activity and synchronicity of cortical neurons in itself could trigger pain hypersensitivity, while targeting S1 circuit hyperactivity could reduce the enhanced pain sensation in the inflammatory pain model. Manipulation of cortical circuits involved in the generation of pain sensation could provide treatment pathways for intractable postoperative and neuropathic pain, and our results suggest an N-type voltage-dependent Ca2+ channel up-regulation in cortical circuits as one such molecular target.
MATERIALS AND METHODS
Animals
Experimental protocols were approved by the Animal Care and Use Committees of Kobe University Graduate School of Medicine and Nagoya University Graduate School of Medicine. We used male mice for all experiments to avoid potential variability arising due to the estrus cycles. All of the animals in this study were given free access to food and water and housed under 12 hours of light/dark cycle. We used C57BL/6 (WT) mice and PV-Cre mice (C57BL/6 background) (49). We used two mouse models as follows: One had an incision of the hind paw as a postoperative pain model (23) and the other received an injection of 40 μl of CFA (Sigma-Aldrich, F5881) into the hind paw as an inflammatory pain model (24).
Surgery and AAV injection
The first surgery was performed in mice at 6 to 8 weeks of age. Under anesthesia with ketamine [74 mg/kg, intraperitoneally (i.p.)] and xylazine (10 mg/kg, i.p.), the skin was disinfected with 70% (w/v) ethanol, the skull was exposed and cleaned, and a custom-made head plate was firmly attached to the skull by dental cement (G-CEM ONE; GC, Tokyo, Japan). The surface of the intact skull was subsequently coated with an acrylic-based dental adhesive resin cement (Super Bond; Sun Medical, Shiga, Japan) to avoid drying. This head plate allowed us to securely attach the mouse to a stainless frame to enable both the circular craniotomy and subsequent two-photon imaging in the awake state. The circular craniotomy (2.5 mm in diameter) was performed 1 or 2 days after plate attachment under isoflurane (1%) anesthesia, as previously described (50), and was over the contralateral hind paw area of the primary somatosensory cortex (S1, centered at 0.5 mm posterior and 1.5 mm lateral from bregma). After the craniotomy, to visualize neuronal activity in L2/3 excitatory neurons or PV neurons of the S1, a total of 1 μl of recombinant AAV encoding the synapsin promoter driven calcium indicator protein GCaMP6f was pressure injected at three sites in the L2/3 (at a depth of 150 to 200 μm below the cortical surface) using a glass pipette (tip diameter, 10 μm). The specific AAV vector solutions were AAV1-hSyn-GCaMP6f-WPRE-SV40 [Penn Vector Core; 9.0 × 1012 vector genomes/ml, diluted 1:1 in phosphate-buffered saline (PBS)], AAV1-CaMKII-GCaMP6f-WPRE-SV40 (3.0 × 1012 vector genomes/ml, diluted 1:1 in PBS), or AAV1-CAG-flex-GCaMP6f-WPRE-SV40 (Addgene; 1.0 × 1013 vector genomes/ml, diluted 1:1 in PBS). After injections, the brain surface was covered with 2% (w/v) agarose L (Nippon Gene, Tokyo, Japan) in saline and with a glass window composed of two coverslips (2 and 4.5 mm in diameter, respectively; Matsunami Glass, Osaka, Japan) joined with ultraviolet curable adhesive (NOR-61, Norland). The edges of the cranial window were sealed with a combination of dental cement and dental adhesive resin cement (Super Bond; Sun Medical, Shiga, Japan). Mice were subsequently housed individually, and imaging experiments started around 3 to 4 weeks after the surgical treatments (i.e., 9 to 12 weeks old).
Two-photon imaging
Two-photon images were acquired from the left S1 using a laser scanning system (LSM 7 MP system; Carl Zeiss, Oberkochen, Germany) with two types of water immersion objective lenses [10×, numerical aperture (N.A.) 0.5; 20×, N.A. 1.0; Carl Zeiss] and a Ti:sapphire laser (Mai Tai HP; Spectra-Physics, Santa Clara, CA) operating at a 950-nm wavelength. The imaged fields were 848.54 μm × 848.54 μm (original scan) or 565.69 μm × 565.69 μm (1.5× digital zoom), at a depth of 150 to 200 μm below the cortical surface. The pixel size was 1.657 or 1.104 μm (1.5× digital zoom). Frame duration was 390 or 300 ms (heat stimulation experiment). Continuous 1000-frame imaging was repeated for each imaging field. In fig. S1 (I and J), we monitored mice to visually separate imaging periods into moving and resting epochs with a charge-coupled device (CCD) camera recording.
In vivo two-photon imaging with holographic stimulation
To use holographic light patterns for activation of a specific, single neuron within a small three-dimensional (3D) volume of the S1 cortex, while simultaneously imaging Ca2+ transients across a wide field using two-photon imaging based on C2 plus (Nikon, Japan), two different excitation light beams (one is for imaging and the other is for holographic stimulation) used a Ti:sapphire laser (Coherence, Santa Clara, CA) tuned to 920 and 1040 nm, respectively (Fig. 5A). A digital hologram generates multispots to activate ChRmine and sequentially stimulate a single identified cell. We used a phase-mode SLM (Santec SLM-200) to display the digital hologram and illuminated this using a Ti:sapphire laser (1040-nm fixed wavelength; Coherent, Santa Clara, CA) in combination with a beam expander (L1 and L2) and relay lenses (L3 and L4) to match the size of generated SLM hologram to the exit pupil of the water immersion objective lens (16×, N.A. 0.8; Nikon). A 5× beam expansion magnitude was chosen to couple the 2-mm laser input beam to the 9.6 mm × 15.36 mm effective area of the SLM field. A λ/2 half wave plate was used to adjust the polarization direction to the optic axis of SLM, and a small carbon steel beam block was placed at an intermediate plane to suppress the residual light component that is not effectively modulated by the SLM. To generate controlled foci, we carefully calibrated the coordinates of holographic and imaging planes using the affine transformation (51), with a registration error typically within 2 μm on the sample. We also adopted the Gerchberg-Saxton algorithm to generate the desired shape and size (52). We used the MATLAB platform to calculate and project the phase distribution to the SLM. Two-photon images (512 × 512 pixels, 2 Hz, 2× digital zoom) were acquired from the left S1HL (L2/3) using a custom-made laser scanning system based on C2 plus (Nikon, Japan) with a 16× objective (N.A. 0.8; Nikon) and a mode-locked Ti:sapphire laser (Coherence, Santa Clara, CA) tuned to 920 nm. Images consisted of a 10-s baseline period followed by five times of 50-ms duration of holographic stimuli (1040 nm) presented at 0.125 Hz and accompanied by simultaneous imaging. Ca2+ transient responses, when present, were evoked by this holographic stimulation and with their peak appearing within 1 s (two frames) after stimulation (Fig. 5B). For the detection and analysis of Ca2+ transients, baseline fluorescence was defined as the mean of the baseline fluorescence intensity histogram obtained during baseline imaging period (F0). Ca2+ transients were defined as ΔF/F0 (ΔF = F − F0), where F is the instantaneous fluorescent signal and where ΔF exceeded 2 SDs of the baseline fluorescence (F0).
Withdrawal threshold test
Using von Frey filaments (Neuroscience Co., Japan), we examined the nociceptive threshold of the hind paw to mechanical stimulus. Animals were placed in a box with a wire grid floor and habituated to the environment for 10 to 15 min. Filaments were applied in either ascending or descending strengths (Dixon’s up-down method), and each filament was applied X times/only once for a maximum of 2 s. Paw withdrawal during the stimulation was considered a positive response, and the 50% response withdrawal threshold was calculated as the strength of filament that gave (53).
To evaluate thermal withdrawal thresholds, we used the hot plate test (plate temperature was set to 50° to 54.5°C). We defined the response latency as the time required for the appearance of the first escape response—licking, shaking, or jumping. A maximal cutoff of 30 s was used to prevent tissue damage (25). To quantify acute neural responses to thermal stimulation (Fig. 1), mice were secured in the imaging system and a small Peltier heat plate (UDH-300; UNIQUE MEDICAL, Tokyo, Japan) was applied to the right hind paw footpad while simultaneously imaging neural activity. The plate was rapidly and transiently heated to 50°C for 15 s, and responses were quantified during the heating ramp stage (Fig. 1D and fig. S1E). This transient heating evoked a reflex paw withdrawal escape response, whose timing was measured by simultaneously recording with a CCD camera. Only a single heating trial was recorded in each mouse.
Chemogenic manipulation
For chemogenic activation of S1 L2/3 excitatory neurons (54, 55), AAVs encoding GCaMP (AAV1-hSyn-GCaMP6f-WPRE-SV40; 9.0 × 1012 vector genomes/ml; Penn Vector Core) and encoding the hM3D DREADD (designer receptors exclusively activated by designer drugs) [AAV8-hSyn-hM3D(Gq)-mCherry; 2.0 × 1012 vector genomes/ml; Addgene] were diluted in PBS solution and injected at three sites in L2/3 of S1. CNO (Sigma-Aldrich) was used to activate these DREADDS and was dissolved in a saline stock solution of (0.5 mg/ml). In Fig. 6C, Ca2+ imaging of excitatory neurons was performed during a quiet resting state without CNO, and the same region was again imaged from 1 hour after CNO intraperitoneal injection (5 mg/kg). Withdrawal threshold tests were also assessed before and 1 hour after CNO injection. In Fig. 7 (A to G), the same dose of CNO was injected daily over a week (a total of seven injections). Daily CNO injections were spaced 24 hours apart, and withdrawal thresholds were evaluated 30 min before each CNO injection. For the virus-injected control experiment, we used AAV1-CaMKII 0.4-Cre (Penn Vector Core; 2.94 × 1013 vector genomes/ml) and AAV1-CAG-flex-tdTomato (Penn Vector Core; 7.6 × 1012 vector genomes/ml).
Image analysis
Images were analyzed using ImageJ (National Institutes of Health) and MATLAB (MathWorks, Natick, MA) software packages. Movies and 3D images were corrected for focal plane displacement using ImageJ plugin TurboReg and StackReg (56). To estimate S1 neuronal activity, regions of interest (ROIs) in L2/3 were determined using an automated algorithm (http://github.com/simonsfoundation/CaImAn) that defines an ROI as a discrete region showing a change in fluorescence at some stage during the analysis. Completely silent neurons (and/or those not expressing GCaMP6f) will not be detected using our approach. For the detection and analysis of calcium transients, baseline fluorescence was defined as the 35th percentile of the total fluorescence intensity histogram obtained during all imaging period (F0). Ca2+ transient was defined as ΔF/F0 (ΔF = F − F0), where F is the instantaneous fluorescent signal and where ΔF exceeded 2 SDs of the baseline fluorescence (F0) (57, 58). We used an F0 set at the 35th percentile of the total fluorescence distribution, except when we reanalyzed the results of Fig. 2 with F0 set to the 15th percentile (which gave us similar results to F0 set at the 35th percentile; fig. S10, B and C). The frequency of Ca2+ transients was calculated as the ratio of the total number of transients over the imaging period (s). The power of each Ca2+ transients (ΔF/F0) was subsequently computed using the mean squared amplitude of the Ca2+ during the transient as follows
where xi(1 ≤ i ≤ T) represents the Ca2+ level at each time during the transient of length, T.
C.C. between the activities of two neurons over time was measured using the cosine instead of the Pearson’s correlation, which is commonly used, because the Pearson’s correlation does not correctly measure correlation if time interval without two neurons firing is long. The Pearson’s C.C., rp, between two Ca2+ transients of length T, xi and yi (1 ≤ i ≤ T), in two ROIs in the imaging field, is given by
where μx and μy represent the averaged Ca2+ fluorescence in two ROIs. In the above equation, each Ca2+ transient is centered; that is, each average is subtracted from the corresponding Ca2+ transient. Since the Ca2+ transients are basically positive during neuronal firing and around zero without neuronal firing, the centered Ca2+ transients become negative in the case without neuronal firing. Thus, the Pearson’s correlation becomes large if the interval without two neurons firing is long, although neurons do not fire almost simultaneously. In this study, the correlation between neurons was measured using the following equation without centering
As rc can be regarded as the cosine of the angle between two T-dimensional vectors whose elements correspond to xi and yi, it is called the cosine correlation. rc is in the range from −1 to 1. The cosine correlation becomes positively large if two neurons fire simultaneously and close to zero if either neuron does not fire. In addition, rc can be negative due to the negative values of noise or oscillatory components in the Ca2+ transients.
Immunohistochemistry
Immunohistochemistry was performed to validate the expression of AAV1-hSyn-GCaMP6f, AAV8-hSyn-hM3D(Gq)-mCherry, and the proportion of PV neurons in the population of GCaMP6f-positive neurons driven by synapsin promotor. Animals were deeply anesthetized with ketamine and xylazine and transcardially perfused with 4% paraformaldehyde solution in PBS. Fixed brains were extracted from the skull and equilibrated in 30% sucrose solution in PBS. The brains were cut into 30-μm-thick sections using a microtome (Leica Microsystems, Wetzlar, Germany). After blocking and permeabilization for 1 hour in 5% bovine serum albumin and 0.5% Triton X-100 in PBS, the slices were incubated at 4°C overnight with primary antibody diluted in PBS. After PBS wash, slices were subsequently incubated with secondary antibody in PBS at room temperature for 3 hours. Slices were mounted on glass slides in Fluoromount-G (Southern Biotech, Birmingham, AL). Fixed tissue was imaged using a Zeiss LSM510 Meta confocal microscope (Carl Zeiss) with a 20× objective (N.A. 1.0; Carl Zeiss). The following antibodies were used for staining: anti-PV antibody (1:500; ab11427, Abcam) and goat anti-rabbit Alexa Fluor 647 (1:500; A-21245, Thermo Fisher Scientific).
Flow cytometry
Animals were anesthetized with ketamine and xylazine and transcardially perfused with PBS. Fresh brain tissues (size of 1 mm × 1 mm × 1 mm) in the contralateral hind paw region of primary somatosensory cortex were dissected from control mice and inflammatory pain model mice (3 days after injection of CFA into the right hind paw) while on ice. After fine mincing of dissected tissue, the samples were transferred to centrifuge tubes and centrifuged by 2000g for 5 min at 4°C. After removing the supernatant, a papain dissociation enzyme solution containing also collagenase and deoxyribonuclease (Worthington Biochemical Corporation, #LK003170) was added, and the tubes were rotated again for 30 min at 37°C. The supernatant was again removed, and the remaining pellet was washed with FACS buffer (containing 5% fetal bovine serum in PBS). This procedure was repeated three times. The final pellet was fixed by 2% paraformaldehyde for 10 min, washed with FACS buffer two to three times, and processed by blocking solution (5% goat serum) and permeabilizing agent (0.5% Triton X-100 in PBS) for 60 min. The processed material was filtered through a 40-μm cell strainer and divided into several microtubes. Each sample was incubated with primary antibodies at room temperature for 1 hour in FACS buffer. After washing with the FACS buffer, the samples were subsequently incubated in secondary antibodies at room temperature for 1 hour in FACS buffer. The following antibodies were used for staining: anti-chicken NeuN (1:200; ab134014, Abcam), anti-rabbit c-Fos (1:200; #3168266, Millipore), anti-mouse c-Fos (1:200; ab208942, Abcam), anti-mouse Cav1.1 (1:200; GTX22862, GeneTex), anti-rabbit Cav1.2 (1:200; GTX54754, GeneTex), anti-mouse Cav1.3 (1:200; ab85491, Abcam), anti-mouse Cavα2δ (1:200; GTX22864, GeneTex), anti-rabbit Cav2.1 (1:200; GTX54753, GeneTex), anti-rabbit Cav2.2 (1:200; PA5-77296, Thermo Fisher Scientific), anti-rabbit Kv1.1 (1:200; ab177481, Abcam), anti-rabbit Kir6.2 (1:200; ab79171, Abcam), anti-rabbit Nav1.1 (1:200; ab24820, Abcam), anti-rabbit Nav1.6 (1:200; ab65166, Abcam), goat anti-chicken Phycoerythrin (PE) (1:200; ab72482, Abcam), goat anti-rabbit Alexa Fluor 405 (1:400; A-31556, Thermo Fisher Scientific), goat anti-mouse Alexa Fluor 405 (1:400; A-31553, Thermo Fisher Scientific), goat anti-rabbit APC (1:400; A-10931, Thermo Fisher Scientific), and goat anti-mouse APC (1:400; A-865, Thermo Fisher Scientific). Samples were run through a BD FACSVerse automated cell counter (BD Biosciences), and data were analyzed with FlowJo software (FlowJo LLC).
Elvax application of N-type Ca2+ channel blocker
Elvax is a noninflammatory polymer used for the slow release of drug into tissues, including brain. As described previously (59), Elvax beads (DuPont; 20 mg) were dissolved in dichloromethane (200 μl) and mixed with 5 μl of dimethyl sulfoxide (DMSO) containing 100 mM PD173212 (N-type Ca2+ channel blocker; ab141832, Abcam). The mixture was stirred with a vortex mixer for 1 hour after which the solution was placed on a glass dish and kept at −70°C overnight and then at −20°C for another night to allow the dichloromethane to evaporate. The final concentration of PD173212 adsorbed into the Elvax was approximately 2.5 mM. A small piece of drug-soaked Elvax (2 mm × 2 mm) was placed on the dura matter through an open-skull cranial window. The drug-soaked Elvax was left on the dura matter for 2 weeks while behavioral measurements were obtained.
Intraventricular drug injection
For the intraventricular injection of PD173212 (N-type Ca2+ channel blocker), we implanted a guide (CXG-8; Eicom, Kyoto, Japan) and dummy cannula (CXD-8; Eicom) into the right lateral ventricle following cranial window surgery. Thirty minutes before imaging and/or behavior, we gave a single injection of PD173212 (1 μl; 1 μg/μl in DMSO) into the ventricle via an injection cannula (CXMI-8; Eicom) attached to an electrically driven pump (UMP3; WPI, Sarasota, FL).
Data analysis and statistics
Data were analyzed using GraphPad Prism 8 statistical software (GraphPad Software Inc., La Jolla, CA). All data are presented as the means ± SEM. Unpaired and paired t tests, Pearson’s correlation test, and analysis of variance (ANOVA) followed by Bonferroni post hoc tests were used to test for statistical significance.
Supplementary Material
Acknowledgments
We thank D. Kim of the GINIE Project at Janelia Farm Research Campus (HHMI) for providing AAV1-hSyn-GCaMP6f-WPRE-SV40 and B. Roth at the University of North Carolina for providing AAV8-hSyn-hM3D(Gq)-mCherry and AAV8-hSyn-hM4D(Gi)-mCherry and K. Deisseroth at the Stanford University for providing pAAV-CaMKIIa-GCaMP6m-p2A-ChRmine-Kv2.1-WPRE. Funding: This work was supported by Grants-in-Aid for Young Scientists (18K16482 to T.O.), by Grants-in-Aid for Scientific Research on Innovative Areas (15H01300, 16H01346, 17H05747, 19H04753, 19H05219, and 25110732 to H.W.), by Grants-in-Aid for Transformative Research Areas (A) (20H05899 to H.W. and 20H05885 to O.M.), by Grants-in-Aid for Young Scientists (A) (26710004 to H.W.), by Grant-in-Aid for Scientific Research (B) (18H02598 to H.W.), by Japan Agency for Medical Research and Development (grant number: JP20ak0101150), and by JST CREST (grant number: JPMJCR1755). Author contributions: T.O., S.M., and H.W. designed the research. T.O., D.K., Y.N., N.O., X.Q., A.M., H.Y., Z.G., Y.A., Y.T., O.M., and T.T. performed the research and analyzed the data. T.O., A.J.M., S.M., and H.W. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabd8261/DC1
REFERENCES AND NOTES
- 1. Relieving pain in America: A blueprint for transforming prevention, care, education, and research. Mil. Med. 181, 397–399 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Price T. J., Basbaum A. I., Bresnahan J., Chambers J. F., De Koninck Y., Edwards R. R., Ji R.-R., Katz J., Kavelaars A., Levine J. D., Porter L., Schechter N., Sluka K. A., Terman G. W., Wager T. D., Yaksh T. L., Dworkin R. H., Transition to chronic pain: Opportunities for novel therapeutics. Nat. Rev. Neurosci. 19, 383–384 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji R.-R., Chamessian A., Zhang Y.-Q., Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tominaga M., Caterina M. J., Malmberg A. B., Rosen T. A., Gilbert H., Skinner K., Raumann B. E., Basbaum A. I., Julius D., The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Caterina M. J., Leffler A., Malmberg A. B., Martin W. J., Trafton J., Petersen-Zeitz K. R., Koltzenburg M., Basbaum A. I., Julius D., Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Tsuda M., Shigemoto-Mogami Y., Koizumi S., Mizokoshi A., Kohsaka S., Salter M. W., Inoue K., P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424, 778–783 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Beggs S., Trang T., Salter M. W., P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 15, 1068–1073 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coull J. A. M., Beggs S., Boudreau D., Boivin D., Tsuda M., Inoue K., Gravel C., Salter M. W., De Koninck Y., BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Peng J., Gu N., Zhou L., Eyo U. B., Murugan M., Gan W.-B., Wu L.-J., Microglia and monocytes synergistically promote the transition from acute to chronic pain after nerve injury. Nat. Commun. 7, 12029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L., Chen S.-R., Chen H., Wen L., Hittelman W. N., Xie J.-D., Pan H.-L., Chloride homeostasis critically regulates synaptic NMDA receptor activity in neuropathic pain. Cell Rep. 15, 1376–1383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woolf C. J., Shortland P., Coggeshall R. E., Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature 355, 75–78 (1992). [DOI] [PubMed] [Google Scholar]
- 12.Todd A. J., Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 11, 823–836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leong M. L., Gu M., Speltz-Paiz R., Stahura E. I., Mottey N., Steer C. J., Wessendorf M., Neuronal loss in the rostral ventromedial medulla in a rat model of neuropathic pain. J. Neurosci. 31, 17028–17039 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall T. M., Herman D. S., Largent-Milnes T. M., Badghisi H., Zuber K., Holt S. C., Lai J., Porreca F., Vanderah T. W., Activation of descending pain-facilitatory pathways from the rostral ventromedial medulla by cholecystokinin elicits release of prostaglandin-E2 in the spinal cord. Pain 153, 86–94 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.François A., Low S. A., Sypek E. I., Christensen A. J., Sotoudeh C., Beier K. T., Ramakrishnan C., Ritola K. D., Sharif-Naeini R., Deisseroth K., Delp S. L., Malenka R. C., Luo L., Hantman A. W., Scherrer G., A brainstem-spinal cord inhibitory circuit for mechanical pain modulation by GABA and Enkephalins. Neuron 93, 822–839.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cichon J., Blanck T. J. J., Gan W.-B., Yang G., Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat. Neurosci. 20, 1122–1132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhuo M., Cortical excitation and chronic pain. Trends Neurosci. 31, 199–207 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Talbot J. D., Marrett S., Evans A. C., Meyer E., Bushnell M. C., Duncan G. H., Multiple representations of pain in human cerebral cortex. Science 251, 1355–1358 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Bliss T. V., Collingridge G. L., Kaang B.-K., Zhuo M., Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci. 17, 485–496 (2016). [DOI] [PubMed] [Google Scholar]
- 20.McWilliams L. A., Cox B. J., Enns M. W., Mood and anxiety disorders associated with chronic pain: An examination in a nationally representative sample. Pain 106, 127–133 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Peters A. J., Chen S. X., Komiyama T., Emergence of reproducible spatiotemporal activity during motor learning. Nature 510, 263–267 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Josselyn S. A., Tonegawa S., Memory engrams: Recalling the past and imagining the future. Science 367, eaaw4325 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brennan T. J., Vandermeulen E. P., Gebhart G. F., Characterization of a rat model of incisional pain. Pain 64, 493–502 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Newbould B. B., Chemotherapy of arthritis induced in rats by mycobacterial adjuvant. Br. J. Pharmacol. Chemother. 21, 127–136 (1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Callaghan J. P., Holtzman S. G., Quantification of the analgesic activity of narcotic antagonists by a modified hot-plate procedure. J. Pharmacol. Exp. Ther. 192, 497–505 (1975). [PubMed] [Google Scholar]
- 26.Ishikawa T., Eto K., Kim S. K., Wake H., Takeda I., Horiuchi H., Moorhouse A. J., Ishibashi H., Nabekura J., Cortical astrocytes prime the induction of spine plasticity and mirror image pain. Pain 159, 1592–1606 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Marshel J. H., Kim Y. S., Machado T. A., Quirin S., Benson B., Kadmon J., Raja C., Chibukhchyan A., Ramakrishnan C., Inoue M., Shane J. C., McKnight D. J., Yoshizawa S., Kato H. E., Ganguli S., Deisseroth K., Cortical layer-specific critical dynamics triggering perception. Science 365, eaaw5202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanegas H., Schaible H., Effects of antagonists to high-threshold calcium channels upon spinal mechanisms of pain, hyperalgesia and allodynia. Pain 85, 9–18 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Sluka K. A., Blockade of N- and P/Q-type calcium channels reduces the secondary heat hyperalgesia induced by acute inflammation. J. Pharmacol. Exp. Ther. 287, 232–237 (1998). [PubMed] [Google Scholar]
- 30.Tsuji T., Inui K., Kojima S., Kakigi R., Multiple pathways for noxious information in the human spinal cord. Pain 123, 322–331 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Craig A. D., Bushnell M. C., Zhang E.-T., Blomqvist A., A thalamic nucleus specific for pain and temperature sensation. Nature 372, 770–773 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Brooks J., Tracey I., From nociception to pain perception: Imaging the spinal and supraspinal pathways. J. Anat. 207, 19–33 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schweinhardt P., Bushnell M. C., Pain imaging in health and disease—How far have we come? J. Clin. Invest. 120, 3788–3797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis K. D., Flor H., Greely H. T., Iannetti G. D., Mackey S., Ploner M., Pustilnik A., Tracey I., Treede R.-D., Wager T. D., Brain imaging tests for chronic pain: Medical, legal and ethical issues and recommendations. Nat. Rev. Neurol. 13, 624–638 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Woolf C. J., Dissecting out mechanisms responsible for peripheral neuropathic pain: Implications for diagnosis and therapy. Life Sci. 74, 2605–2610 (2004). [DOI] [PubMed] [Google Scholar]
- 36.McMahon S. B., Malcangio M., Current challenges in glia-pain biology. Neuron 64, 46–54 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Denk F., McMahon S. B., Tracey I., Pain vulnerability: A neurobiological perspective. Nat. Neurosci. 17, 192–200 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Kim S. K., Nabekura J., Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J. Neurosci. 31, 5477–5482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eto K., Ishibashi H., Yoshimura T., Watanabe M., Miyamoto A., Ikenaka K., Moorhouse A. J., Nabekura J., Enhanced GABAergic activity in the mouse primary somatosensory cortex is insufficient to alleviate chronic pain behavior with reduced expression of neuronal potassium-chloride cotransporter. J. Neurosci. 32, 16552–16559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S. K., Hayashi H., Ishikawa T., Shibata K., Shigetomi E., Shinozaki Y., Inada H., Roh S. E., Kim S. J., Lee G., Bae H., Moorhouse A. J., Mikoshiba K., Fukazawa Y., Koizumi S., Nabekura J., Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J. Clin. Invest. 126, 1983–1997 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan L. L., Pelzer P., Heinl C., Tang W., Gangadharan V., Flor H., Sprengel R., Kuner T., Kuner R., A pathway from midcingulate cortex to posterior insula gates nociceptive hypersensitivity. Nat. Neurosci. 20, 1591–1601 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Alexandre C., Latremoliere A., Ferreira A., Miracca G., Yamamoto M., Scammell T. E., Woolf C. J., Decreased alertness due to sleep loss increases pain sensitivity in mice. Nat. Med. 23, 768–774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Potter L. E., Paylor J. W., Suh J. S., Tenorio G., Caliaperumal J., Colbourne F., Baker G., Winship I., Kerr B. J., Altered excitatory-inhibitory balance within somatosensory cortex is associated with enhanced plasticity and pain sensitivity in a mouse model of multiple sclerosis. J. Neuroinflammation 13, 142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elman I., Borsook D., Common brain mechanisms of chronic pain and addiction. Neuron 89, 11–36 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Burgess G., Williams D., The discovery and development of analgesics: New mechanisms, new modalities. J. Clin. Invest. 120, 3753–3759 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bowersox S. S., Gadbois T., Singh T., Pettus M., Wang Y. X., Luther R. R., Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic pain. J. Pharmacol. Exp. Ther. 279, 1243–1249 (1996). [PubMed] [Google Scholar]
- 47.Deer T. R., Pope J. E., Hanes M. C., McDowell G. C. II, Intrathecal therapy for chronic pain: A review of morphine and ziconotide as firstline options. Pain Med. 20, 784–798 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallace M. S., Charapata S. G., Fisher R., Byas-Smith M., Staats P. S., Mayo M., McGuire D., Ellis D.; David Ellis, Ziconotide Nonmalignant Pain Study 96-002 Group , Intrathecal ziconotide in the treatment of chronic nonmalignant pain: A randomized, double-blind, placebo-controlled clinical trial. Neuromodulation 9, 75–86 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Tanahira C., Higo S., Watanabe K., Tomioka R., Ebihara S., Kaneko T., Tamamaki N., Parvalbumin neurons in the forebrain as revealed by parvalbumin-Cre transgenic mice. Neurosci. Res. 63, 213–223 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Masamizu Y., Tanaka Y. R., Tanaka Y. H., Hira R., Ohkubo F., Kitamura K., Isomura Y., Okada T., Matsuzaki M., Two distinct layer-specific dynamics of cortical ensembles during learning of a motor task. Nat. Neurosci. 17, 987–994 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Sakata H., Sakamoto Y., Fast computation method for a Fresnel hologram using three-dimensional affine transformations in real space. Appl. Optics 48, H212–H221 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Gerchberg R. W., A practical algorithm for the determination of the phase from image and diffraction plane pictures. Optik 35, 237–246 (1972). [Google Scholar]
- 53.Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., Yaksh T. L., Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 (1994). [DOI] [PubMed] [Google Scholar]
- 54.Alexander G. M., Rogan S. C., Abbas A. I., Armbruster B. N., Pei Y., Allen J. A., Nonneman R. J., Hartmann J., Moy S. S., Nicolelis M. A., McNamara J. O., Roth B. L., Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armbruster B. N., Li X., Pausch M. H., Herlitze S., Roth B. L., Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U.S.A. 104, 5163–5168 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thévenaz P., Ruttimann U. E., Unser M., A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Tian L., Hires S. A., Mao T., Huber D., Chiappe M. E., Chalasani S. H., Petreanu L., Akerboom J., McKinney S. A., Schreiter E. R., Bargmann C. I., Jayaraman V., Svoboda K., Looger L. L., Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato D., Wake H., Lee P. R., Tachibana Y., Ono R., Sugio S., Tsuji Y., Tanaka Y. H., Tanaka Y. R., Masamizu Y., Hira R., Moorhouse A. J., Tamamaki N., Ikenaka K., Matsukawa N., Fields R. D., Nabekura J., Matsuzaki M., Motor learning requires myelination to reduce asynchrony and spontaneity in neural activity. Glia 68, 193–210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kakizawa S., Miyazaki T., Yanagihara D., Iino M., Watanabe M., Kano M., Maintenance of presynaptic function by AMPA receptor-mediated excitatory postsynaptic activity in adult brain. Proc. Natl. Acad. Sci. U.S.A. 102, 19180–19185 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabd8261/DC1