

Mirroring the obesity pandemic, nonalcoholic fatty liver disease (NAFLD) has become the most common liver disease affecting populations worldwide. Approximately 30% of the adult U.S. population is estimated to be afflicted with NAFLD. NAFLD is a spectrum of liver disease ranging from nonalcoholic fatty liver (NAFL), the nonprogressive subtype, to nonalcoholic steatohepatitis (NASH), the progressive subtype, which can lead to progressive liver disease, leading to cirrhosis and hepatocellular carcinoma (HCC).(1) NAFLD is considered the hepatic manifestation of the metabolic syndrome and is commonly associated with other diseases including type 2 diabetes, kidney disease, and cardiovascular disease (CVD). Emerging data suggest that NAFLD is not only linked to hepatic outcomes but also to extrahepatic outcomes such as CVD, cancer, and all-cause mortality. Understanding the risk factors associated with both hepatic as well as extrahepatic consequences of NAFLD is an area of major scientific interest.
In this issue of Hepatology, Unalp-Arida and Ruhl(2) examined the relationship between NAFLD and mortality in 13,298 participants from the Third National Health and Nutrition Examination Survey (NHANES III), a multiethnic sample of individuals from the general U.S. population collected between 1988 and 1994. The mean age of the participants was 53 years, and 53% were women. For each participant, the likelihood of having hepatic steatosis and/or fibrosis at baseline was estimated based on a handful of known risk factors (age, body mass index, and metabolic and biochemical parameters), combined into weighted risk scores.(3,4) A subset of the cohort (n = 5,662) was genotyped for PNPLA3 p.I148M, a common variant known to associate robustly with NAFL as well as NASH.(5) The participants were followed for mortality from enrollment in 1988–1994 until December 2015 by linkage with national death registries.
During a median follow-up of 23.2 years, 196 (1.1%) of the participants died of liver-related causes, 2,200 (14.6%) of CVD, and 5,570 (33.2%) of any cause. Compared to participants with a low liver fat score at baseline, those with intermediate or high scores had approximately 3-fold and 8-fold higher liver-related mortality, respectively, during follow-up. The corresponding estimates for the fibrosis score were comparable (approximately 2-fold and 7-fold higher liver-related mortality, respectively). These relatively large effect sizes for fibrosis are not unexpected, given the well-established causal role of hepatic fibrosis in the development of cirrhosis and HCC. However, the longitudinal association between hepatic steatosis and liver-related mortality is a novel and previously underappreciated finding. Furthermore, high liver fat and fibrosis scores were also associated with up to 2-fold increased cardiovascular and all-cause mortality.
Although these results are compelling and align with those from previous studies, two caveats should be noted. First, the associations could have been influenced by confounding, an inherent limitation to observational studies. Despite the extensive multivariate adjustments for potential confounders, residual confounding by unknown, unmeasured, or imprecisely measured confounders remains hard to rule out. Second, the liver fat and fibrosis scores did not include actual measurements of liver fat or fibrosis. Although the scores captured steatosis and fibrosis relatively well in the cohorts in which they were derived and validated (area under the curve: 0.86 and 0.82 in the validation cohorts),(3,4) the degree to which they reflect steatosis or fibrosis in NHANES III is not precisely clear.
The most striking data in the study are those pertaining to PNPLA3 p.I148M. Among the genotyped participants, 52% were homozygous for the common I variant (II homozygotes), 36% were heterozygous for the M variant (IM heterozygotes), and 12% were MM homozygotes. The M variant was associated with liver-related death, with hazard ratios of 2.9 (95% confidence interval [CI], 0.9 to 9.8) for IM heterozygotes, and 18.2 (95% CI, 3.5 to 93.8) for MM homozygotes, as compared with II homozygotes. The 18-fold increased risk of liver-related death seen in MM homozygotes is remarkable. It is unusual that common genetic variants confer effect sizes of this magnitude. In previous studies, MM homozygotes had an approximately 2-fold higher risk of steatosis,(5) a 3-fold to 4-fold higher risk of NASH(6) and cirrhosis,(7) and an up to 12-fold higher risk of HCC,(8) as compared with II homozygotes. The 18-fold higher liver-related mortality seen in MM homozygotes versus II homozygotes in the present study confirms and extends the pattern of increasing relative effect sizes with increasing severity of the NAFLD-related endpoint (Fig. 1). The mechanism underlying this phenomenon is unclear. It could be a simple consequence of the more severe liver endpoints being “cleaner,” more extreme phenotypes, allowing for stronger detection of associations. Another (more speculative) explanation could be that the hepatotoxic effect of PNPLA3 p.I148M gets amplified with increasing severity of NAFLD as it progresses from NAFL to NASH to cirrhosis and HCC.
FIG. 1.
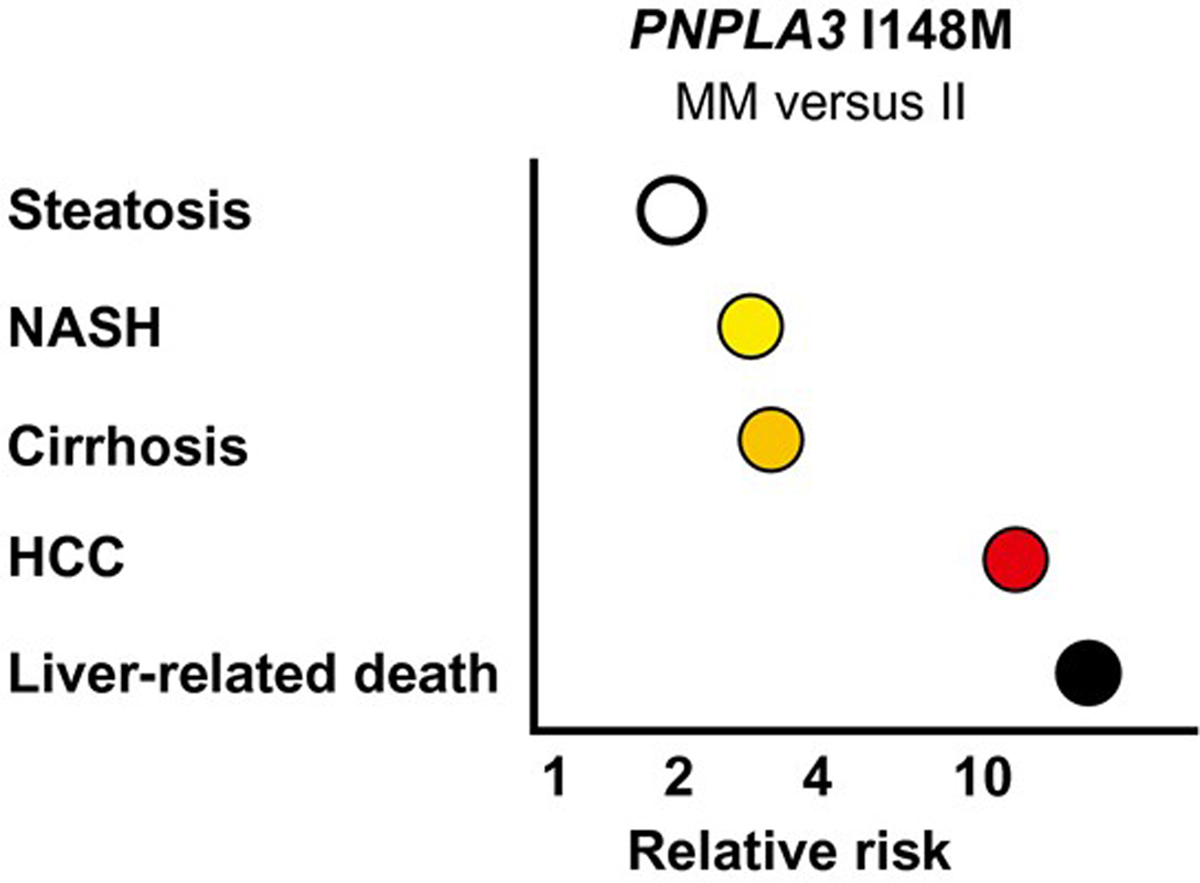
Homozygosity for PNPLA3 I148M and risk of NAFLD-related endpoints. The relative risk estimates for MM homozygotes versus II homoygotes for steatosis, NASH, cirrhosis, and HCC are derived from the literature.(5,6,8,10) The 18-fold increased risk of liver-related death is based on the point estimate reported by Unalp-Arida and Ruhl.(2)
Intriguingly, PNPLA3 p.I148M was also associated with higher all-cause mortality in the cohort, with hazard ratios of 1.16 (95% CI, 1.03 to 1.30) for IM heterozygotes, and 1.38 (95% CI, 1.05 to 1.82) for MM homozygotes, as compared with II homozygotes. This finding is important and somewhat unexpected and therefore warrants discussion.
Why is the association between the M variant and increased all-cause mortality important? In contrast to the liver fat and fibrosis scores, PNPLA3 p.I148M is less likely to be affected by confounding, because genetic variants are randomly inherited at conception and remain constant throughout life. If the M variant associates with NAFLD and with all-cause mortality, it can reasonably be inferred that the effect on the latter is mediated through NAFLD (assuming that no pleiotropic effects of PNPLA3 p.I148M also influence mortality). In other words, the association of PNPLA3 p.I148M with all-cause mortality in this study provides genetic evidence that NAFLD per se increases all-cause mortality in the U.S. population. This hypothesis should be tested in future studies using a formal Mendelian randomization framework, an epidemiological approach that uses genetic proxies (here: PNPLA3) for exposures of interest (NAFLD) to infer causal effects on outcomes (mortality).
Why is it surprising that PNPLA3 p.I148M associates with increased all-cause mortality? First, the variant has not been implicated in any of the large-scale genome-wide association studies on mortality or parental life span that have been performed in recent years.(9) Second, if the M variant confers an increased mortality rate, one might expect it to be relatively depleted in older individuals, in turn causing frequencies of II, IM, and MM genotypes to diverge from those expected (i.e., violation of Hardy-Weinberg equilibrium). PNPLA3 p.I148M genotypes have been in Hardy-Weinberg equilibrium in several large cohorts,(10) again arguing against an effect on mortality. However, previous studies may have failed to detect the association due to insufficient lengths of follow-up time, or because of differences in participant characteristics. In the current study,(2) the increase in liver-related mortality seen in carriers of the M variant was more pronounced in the second decade of follow-up than in the first, supporting the importance of a long follow-up time for detecting the effects of PNPLA3 p.I148M on mortality. Moreover, the deleterious effect of the M variant is markedly amplified by obesity, an example of gene-environment interaction.(10) It is possible that the growing rate of obesity in the U.S. population during recent decades has augmented the harmful effects of PNPLA3 p.I148M in the study by Unalp-Arida and Ruhl,(2) and that it is only in the obese population that the variant confers a higher all-cause mortality. Finally, despite the careful and comprehensive sensitivity analyses performed in the present study,(2) we must also consider the possibility of a chance finding (i.e., a false-positive association), an ever-looming specter in genetic epidemiology. The association of PNPLA3 p.I148M with all-cause mortality is new and unexpected and therefore requires validation in independent cohorts in future studies.
Taken together, the epidemiological and genetic findings of the present study confirm NAFLD as a major risk factor for liver-related death and lend support to the notion that NAFLD increases all-cause mortality. These data emphasize the broad clinical impact of NAFLD as well as the unmet need for effective therapies against the disorder.
Acknowledgments
S.S. received funding support from Independent Research Fund Denmark (Sapere Aude Research Leader 9060-00012). R.L. received funding support from NIEHS (5P42ES010337), NCATS (5UL1TR001442), NIDDK (R01DK106419, P30DK120515), and DOD PRCRP (CA170674P2).
Footnotes
Potential conflict of interest: Dr. Loomba consults, advises, and received grants from Boehringer Ingelheim, Bristol-Myers Squibb, Cirius, Eli Lilly, Galmed, Gilead, Intercept, Janssen, Merck, NGM, Pfizer, Prometheus, and Siemens. He consults and advises Arrowhead, AstraZeneca, Bird Rock, Celgene, CohBar, Conatus, Gemphire, Glympse Bio, GNI, GRI Bio, Ionis, Metacrine, Novartis, Novo Nordisk, Sanofi, and Viking. He received grants from Allergan, Grail, Madrigal, NuSirt, and pH Pharma. He is co-founder of Liponexus, Inc.
REFERENCES
- 1).Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta-analysis. Hepatology 2017;65:1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Unalp-Arida A, Ruhl CE. PNPLA3 I148M and liver fat and fibrosis scores predict liver disease mortality in the United States population. Hepatology 2020;71:820–834. [DOI] [PubMed] [Google Scholar]
- 3).Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007;45:846–854. [DOI] [PubMed] [Google Scholar]
- 4).Kotronen A, Peltonen M, Hakkarainen A, Sevastianova K, Bergholm R, Johansson LM, et al. Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology 2009;137:865–872. [DOI] [PubMed] [Google Scholar]
- 5).Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Dai G, Liu P, Li X, Zhou X, He S. Association between PNPLA3 rs738409 polymorphism and nonalcoholic fatty liver disease (NAFLD) susceptibility and severity: a meta-analysis. Medicine (Baltimore) 2019;98:e14324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Shen JH, Li YL, Li D, Wang NN, Jing L, Huang YH. The rs738409 (I148M) variant of the PNPLA3 gene and cirrhosis: a meta-analysis. J Lipid Res 2015;56:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, Dufour JF, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol 2014;61:75–81. [DOI] [PubMed] [Google Scholar]
- 9).Deelen J, evans DS, Arking DE, Tesi N, Nygaard M, Liu X, et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat Commun 2019;10:3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]