

Abstract
Growth hormone (GH) induces pleiotropic effects on growth and metabolism via binding and subsequent activation of the growth hormone receptor (GHR) and its downstream signaling pathways. Growth hormone insensitivity (GHI) describes a group of disorders in which there is resistance to the action of GH and resultant insulin-like growth factor I (IGF-I) deficiency. GHI is commonly due to genetic disorders of the GH receptor causing GH receptor deficiency (e.g. Laron Syndrome (LS)), decreased activation of GHR, or defects in post-receptor signaling molecules. Genetically altered mouse lines have been invaluable to better understand the physiological impact of GHI due to the ability to do invasive and longitudinal measures of metabolism, growth, and health on a whole animal or in individual tissues/cells. In the current review, the phenotype of mouse lines with GHI will be reviewed. Mouse lines to be discussed include: 1) GHR−/− mice with a gene disruption in the GHR that results in no functional GHR throughout life, also referred to as the Laron mouse, 2) mice with temporal loss of GHR (aGHRKO) starting at 6 weeks of age, 3) mice transgenic for a GHR antagonist (GHA mice), 4) mice with GHI in select tissues or cells generated via Cre-lox or related technology, and 5) assorted mice with defects in post-receptor signaling molecules. Collectively, these mouse lines have revealed an intriguing role of GH action in health, disease, and aging.
1. Introduction
Understanding the function of hormones and how they may contribute to the metabolic or physiologic processes of complex systems requires in vivo models. That is, hormones affect multiple organs simultaneously; thus, in vivo mammalian models provide specific advantages over in vitro models, which are limited to investigation of isolated systems. To that end, genetically engineered mice have played a vital role in our understanding of many hormones due to their ease in genetic tractability, shorter lifespan, and genetic similarity to humans. ‘Transgenic animals’ that carry a foreign or over-expressed gene and ‘knockout mice’ in which the expression of a particular gene is disrupted globally, temporally or tissue-specifically can yield valuable information about the physiological or metabolic function of that protein.
Many mouse lines with alterations in GH action have been generated and have uncovered novel roles for GH. In the current review, we will discuss several mouse lines with varying levels of GH insensitivity (Figure 1). Specifically, we will discuss the phenotype of: 1) mice with complete GHI throughout life via GHR gene disruption (GHR−/− mice); 2) mice with partial GHI starting at 6 weeks of age due to temporal disruption of GHR (aGHRKO); 3) mice with decreased GH action via transgenic expression of a GHR antagonist (GHA mice); 4) mice with loss of GHR function in particular cells or tissues; and 5) mice with transgenic over-expression or deletion of targets that are activated or produced upon GH binding to GHR. Note that several of these mouse lines share features with clinical conditions (e.g. GHR−/− mice and (LS)), while others, such as the tissue specific lines, have no comparable clinical correlate. Regardless, these mice collectively provide valuable insight into the role of GH and its receptor in physiological and metabolic processes at the cell, tissue, and organismal level. While mice are vital for studying biological processes or tissue function that are evolutionarily conserved, caution is required when extrapolating the findings from mice to clinical populations due to vast differences in size, in response to experimental interventions, in adaptation to very distinct environments, in metabolic rate, and in differences in domestication and breeding/housing [1]. Despite this caveat, one mouse line that will be discussed later in this review (GHA mice), provided the foundational knowledge for the development of a drug that alters GH action, reaffirming the translatability of the findings from mice to humans.
Figure 1.
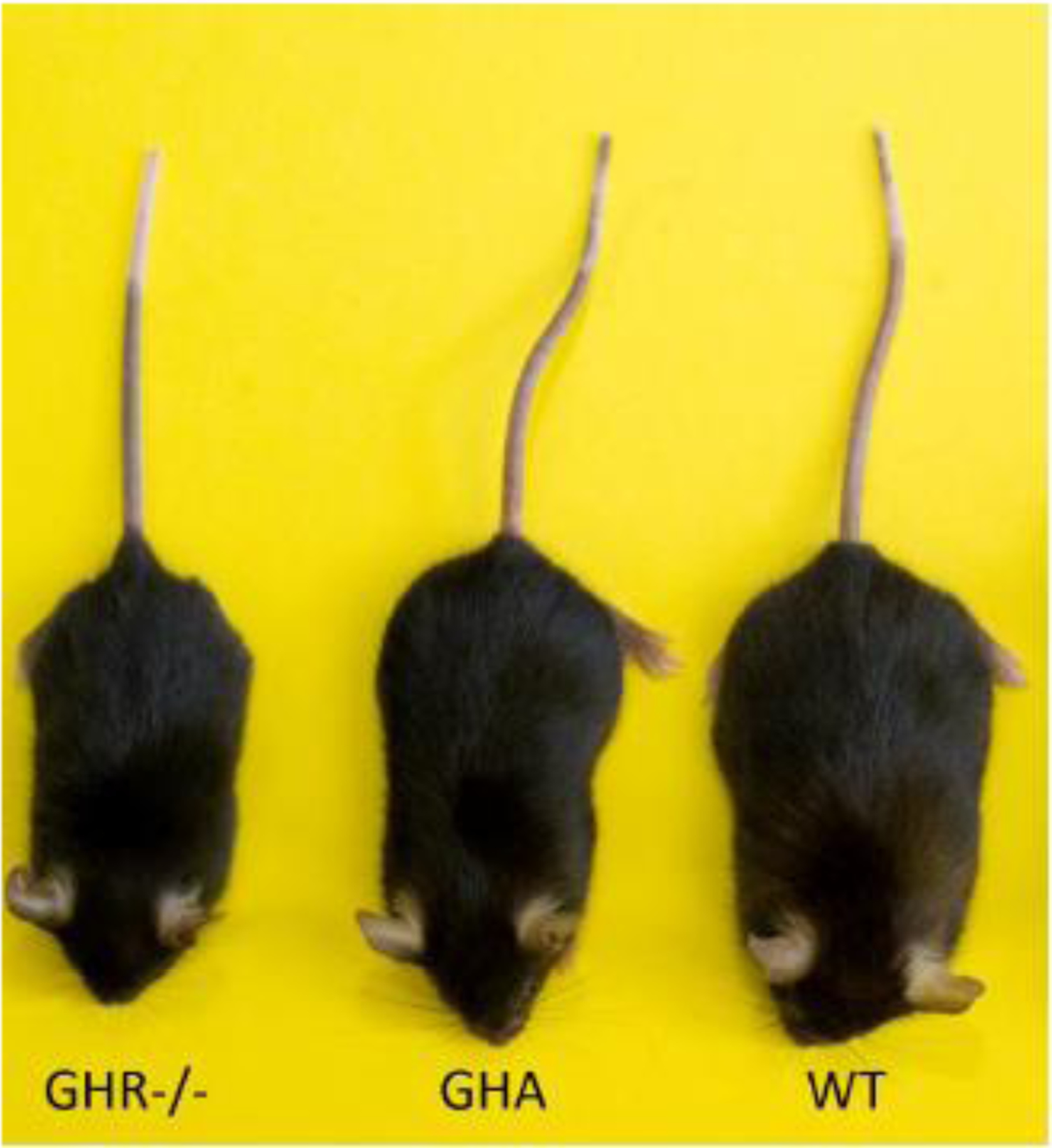
Relative size of mouse models of GHI. This representative sample of GHR−/− mice, GHA mice, and WT controls demonstrates the size change that occurs when a mouse is GH insensitive (GHR−/−) or expresses a GHR antagonist (GHA)
2. GHR−/− Mice
The GHR−/− mouse line was generated in the laboratory of Dr. John Kopchick in 1991 using homologous recombination to create a global knockout of the growth hormone receptor/binding protein gene in the mouse germline [2]. As such, the mouse line serves as an excellent model of GHI, known as Laron Syndrome (LS), due to postnatal growth retardation, short stature, elevated serum GH concentrations, and decreased serum IGF-1 concentrations [2]. The GHR−/− mouse line has since become an important in vivo research tool in distinguishing the importance of somatotropic (GH/IGF-1) action in growth, longevity, obesity, reproduction, insulin and glucose metabolism, to name a few [3]. To date, these mice have been characterized in over 200 papers, illustrating the widespread application of GHR−/− mice in studying GHI and the somatotropic axis.
2.1. Growth and Organ Size
The GHR−/− mice are distinct from wild-type (WT) mice in body weight and tissue weight. That is, both male and female GHR−/− mice are significantly smaller in mean body weight at all time points measured, from 53% of the size of WT controls at 6 weeks of age, to 56% for males and 44% for females of WT controls at 104 weeks of age [4]. Body length follows the same trend. However, there is no difference in body size or weight at birth [2]. In accordance with their diminutive size, the absolute weights of most tissues (e.g. liver, bone, stomach, testis, muscle, spleen, heart, most adipose depots and kidney) are decreased when compared to the WT control [4]. Most of these tissues are proportional to their smaller body size; however, there are exceptions. For example, several reports show kidney mass is reduced in both absolute and normalized weights in GHR−/− mice, while the brain [5] and interscapular brown fat [6] are decreased in absolute weight but greater when normalized to body weight. In the pancreas, islet beta-cell mass compared to body weight is 50% lower in GHR−/− mice compared to WT controls, though the pancreas itself exhibits no significant decrease [7]. Interestingly, subcutaneous fat is greater in absolute and relative weight in males despite their extreme short stature, and strikingly greater when normalized to body weight in both sexes [4].
Body composition has also been assessed over two years in male and female GHR−/− mice. Once again, because of the significant difference in body weights for GHR−/− mice compared to WT mice, it is important to consider fat mass changes relative to body weight [4]. When normalized to body weight, GHR−/− mice have a greater percent fat mass with no significant difference in absolute fat mass throughout life, though this difference is far more pronounced in male than female mice [3]. Lean mass, however, shows an opposite trend with percent lean mass relatively proportional to size between genotypes, but absolute lean mass is reduced in GHR−/− mice [4].
2.2. Endocrine and Metabolic Phenotype
The metabolic phenotype of GHR−/− mice has received significant attention. As expected with GHI, the GH/IGF-1 axis is perturbed. GHR−/− mice exhibit high serum GH levels and circulating IGF-1 levels less than 20% of the levels found in WT controls [5]. Additionally, circulating levels of various IGF binding proteins (proteins that attenuate the action of IGF) are significantly reduced at most adult time points. These include IGFBP-1, IGFBP-3, and IGFBP-4. In contrast, IGFBP-2 levels are significantly increased in the GHR−/− mice relative to WT controls [5].
Counterintuitive to this animal displaying obesity and in line with GH’s diabetogenic action, GHR−/− mice show improved glucose homeostasis. Fasting glucose levels in young GHR−/− mice are reduced to 65–86% the level of WT controls, though these levels are not sustained into adulthood [5]. Insulin levels, however, are significantly reduced to 10–26% the level of WT control mice [5] and remain lower than WT controls for their entire life, which may be partially explained by the decreased beta cell mass of the GHR−/− mice [7]. The altered glucose metabolism in GHR−/− mice is clarified by glucose and insulin tolerance tests. That is, when administered a glucose tolerance test (GTT), the GHR−/− mice exhibit decreased glucose tolerance compared to WT controls [3]; however, they exhibit elevated insulin sensitivity when administered an insulin tolerance test [7]. When fed a high fat diet, GHR−/− mice gain additional fat mass and display increased circulating insulin, but insulin levels still remain lower than WT mice and GHR−/− mice do not experience significant alterations in fasting glucose levels [8] nor alterations in insulin sensitivity [9].
As might be expected with the severe obese phenotype, adipokine levels are altered in these mice. Serum leptin levels are elevated, consistent with the phenotype of the GHR−/− mice [10]. Although adiponectin is normally low in obesity, adiponectin levels in the GHR−/− mice are high, which is consistent with adiponectin’s positive correlation to insulin sensitivity [10].
In addition to obesity, there are several other notable morbidities of the GHR−/− mouse line. The GHR−/− mice exhibit hepatic steatosis, even when fed a normal chow diet [11]. One study reports a marked decrease in GHR−/− osteocyte mitochondrial function, which is correlated to changes in bone matrix composition and decreases in skeletal health [12]. Additionally, there are sexual maturation consequences in the GHR−/− mouse as puberty is delayed in both sexes. For example, the male mouse fertility is reduced, and the female mouse fertility is preserved, though preovulatory follicles and corpora lutea are decreased [13].
2.3. Disease and Aging
The GHR−/− mice are incredibly long-lived, despite obesity and the aforementioned morbidities. In fact, one GHR−/− mouse holds the record for the longest-lived laboratory mouse at nearly 5 years old [14]. When compared to WT controls, the GHR−/− mice have significantly longer lifespans for both sexes, though to varying degrees (depending on the genetic background strain used), and this has been replicated in numerous laboratories [5]. In the GHR−/− mice, caloric restriction, which improves longevity in numerous other conditions, has no effect on longevity or insulin sensitivity [15].
The remarkable longevity of the GHR−/− mice is hypothesized to be a result of several interacting mechanisms. Some studies consider altered energy metabolism and improved glucose homeostasis, which correlates to a decreased respiratory quotient and increased oxygen consumption, as key factors in the longevity of the GHR−/− mice [16]. Others look to alterations in mTOR signaling, depletion of senescent cells (a pro-inflammatory cytokine source) or increased expression of anti-inflammatory cytokines, like adiponectin, to explain the fascinating relationship between decreased GH signaling and slowed aging [17]. Interestingly, the longevity experienced by global GHR deletion is not fully replicated in targeted deletion of GHR in tissues such as liver, muscle and adipose, despite metabolic parameters still being impacted [17], as will be discussed below.
In addition to longevity, GHR−/− mice experience protection from several diseases associated with aging. In a study done by Wolf et al., when compared with the WT control, GHR−/− mice demonstrate a significant delay in the development of age-related cataracts [18]. When type 1 diabetes is induced using streptozotocin, GHR−/− mice show no evidence of diabetic kidney disease, but their WT counterparts exhibit glomerulosclerosis among other kidney morbidities [19]. A handful of studies have highlighted that, in addition to a delay in physical aging, GHR−/− mice experience delays in mental aging [20]. Suppression of GH signaling is also shown to decrease the incidence of tumors, decrease the tumor burden (the number of different tumors found), and delay the progression of fatal neoplastic disease in GHR−/− mice when compared to WT counterparts [21]. Overall, GHR−/− mice provide striking evidence, at least in a controlled laboratory atmosphere, that complete GHI in mice is beneficial to healthspan and lifespan.
3.0. Adult GHR−/− Mice
Global knockout of the GHR in the germline creates some fascinating effects as detailed in the previous section. This generated interest in what the possible effects of GHR ablation would be if initiated at an older age. To answer this question, the Kopchick laboratory generated in 2014 an adult model of GHR−/− (aGHRKO) using an inducible Cre-lox system [22]. The GHR gene in these mice is flanked with LoxP sites, allowing Cre recombinase (activated by tamoxifen injection in this mouse line) to excise the sequence [23]. In this scenario, tamoxifen binds to the mutated estrogen receptor ligand domain, releasing the Cre recombinase to the nucleus where the floxed gene is recognized and disrupted. Unlike standard Cre-lox systems, the tamoxifen inducible system has the capability of being induced at any time point.
Currently, only one published paper characterizes the phenotype of aGHRKO mice [22]. In this paper, the GHR gene is disrupted at six weeks of age, at which mice are fertile but are not yet fully grown or considered fully mature adults [24]. Based on GHR gene expression data using quantitative PCR, the GHR gene is successfully, but not completely, disrupted. GHR mRNA expression is significantly reduced, albeit still detectable, in all tissues except for the heart, indicating at least partial GHI. Reductions in tissue-level IGF-1 expression are also indicative of GHI. For example, tissue IGF-1 levels are reduced up to 99% in the liver of aGHRKO mice compared to controls. A 29% and 57% reduction of IGF-1 is also observed in the retroperitoneal and perigonadal fat pads of both sexes, highlighting the differential level of GHI between tissues.
As would be expected with GHI, GH levels are much higher and IGF-1 levels much lower in the serum of male and female aGHRKO mice. Specifically, at 9 months of age male and female aGHRKO have an increase of circulating GH level of 2748% and 626%, respectively, compared to controls. Further evidence of reduced GH action is given by the severe reduction in serum IGF-1 levels, with levels approximately 9% of control mice in both sexes at 9 months of age in aGHRKO mice. This trend for GH and IGF-1 levels is maintained in the 19 month old cohort. Similar to GHR−/− mice, IGFBP-2 is significantly increased and IGFBP-3 decreased in both sexes of the aGHRKO mice when compared to littermate WT controls.
3.1. Body Composition and Metabolism
A significant reduction in the overall weight of aGHRKO mice is observed starting at 8 weeks of age in males and at 10 weeks of age in females (2 and 4 weeks post injection, respectively) compared to controls. The weight gap gradually widens throughout life. By dissection at 9 and 19 months of age, male mice have an average reduction in body weight of 14.8% and 16.2%, respectively, while female mice have a reduction in body weight of 18.6% and 12.6%, respectively. Body length is also reduced in both sexes when compared to WT controls.
Similar to the global GHR−/− mice, these mice have a significant increase in total adiposity when adjusted for body weight as well as an overall reduction in lean mass. Like GHR−/− mice, most of the fat mass increase is due to expansion of the subcutaneous fat pad in both sexes. Perigonadal fat is increased in male aGHRKO but not female, brown adipose tissue is significantly increased in the female aGHRKO mice but not males, and neither group shows a significant increase in the size of the mesenteric or retroperitoneal fat pads. Relative brain size is increased in the aGHRKO mice of both sexes. All the other measured tissues including liver, kidney, heart, spleen, lung, and quadriceps muscle have significant decrease in total size when compared to controls. The general decrease in organ sizes and increase in fat pads are similar to, but less extreme than, those seen in the germline GHR−/− mice.
Regarding glucose homeostasis and hormone levels, the phenotype of aGHRKO mice resembles the global GHR−/− in many respects. That is, aGHRKO mice are less glucose tolerant than control mice at both 5mo and 13mo of age even though they are more insulin sensitive via insulin tolerance tests. Total circulating insulin is lower in the plasma of male aGHRKO mice but not females. Overall, levels of adiponectin are elevated in both sexes, but leptin does not differ from controls. Resistin and IL-6 levels are higher in the female aGHRKO mice but not male. Despite increased adiposity, liver triglycerides are not increased in aGHRKO mice and are in fact lower in the female mice when compared to controls.
3.2. Aging
As increased longevity has been observed at several institutions, in both sexes, and in multiple background strains of global GHR−/− mice [3], the authors evaluated lifespan in aGHRKO mice. Median and maximal survival is not significantly different between male control and aGHRKO mice. For females, while there is no significant difference in median survival between controls and aGHRKO mice (125.5 vs 124 weeks, respectively), female aGHRKO mice do have a significant (P=0.025) increase in maximal lifespan relative to controls (177 weeks vs 150 weeks, respectively. While lifespan extension is not as extreme as in global GHR−/− mice and is specific to a particular sex, the aGHRKO mice provide evidence that GHR disruption does not have to initiate at conception to receive the benefits to lifespan. As these aGHRKO mice (with GHR gene disruption initiated at 6 weeks of age) are smaller in stature throughout life, there remains a debate as to whether the diminutive size of these mice may confer the longevity advantage. As such, studies are underway to disrupt the GHR gene at older ages when the mice are nearly fully grown.
4.0. GHA Mice
The growth hormone receptor antagonist (GHA) mouse was developed in the laboratory of Dr. John Kopchick in 1991. This group modified amino acids in the third alpha helix of bovine GH [25] to produce a complete amphipathic configuration and potentially a more efficacious GH [26]. Unexpectedly, the alterations to the 3rd alpha helix produced mice small in stature compared to WT mice [27]. Subsequent studies identified the glycine at position 119 to be the critical amino acid for the antagonistic effect. If glycine in this position is changed to anything except alanine, improper/dysfunctional binding of GH to the second receptor within the GHR homodimer occurs, thus creating a classic GHR antagonist. This discovery was the first to suggest that the GH molecule had two regions or sites (now referred to as Site 1 and Site 2) necessary for activating the GHR. Thus, this GH analog competitively inhibits the binding of endogenous GH to the GH receptor (GHR), blocking its function [28]. Mice transgenic for the mutated bovine GH gene in which the codon for glycine at position 119 is replaced with lysine are referred to as the growth hormone receptor antagonist or GHA mice.
4.1. Body Composition and Metabolism
GHA mice are quite distinct from their WT counterparts as well as from GHR−/− mice. Like GHR−/− mice, GHA mice have significant decreases in lean mass and significant increases in adipose tissue mass, which tends to accumulate mostly in their subcutaneous fat pad at early ages but increases in all depots at advanced ages [29]. As for body size, they are significantly smaller than WT mice (mean growth ratio of 0.7 compared to WT littermates) [27], but notably larger than GHR−/− mice (Figure 1). However, this weight phenotype disappears in later life, as the GHA mice catch up with WT in body weight but not length [30]. Organ weights are also affected, with significant decreases in the weights of kidney, liver, heart, and gastrocnemius observed when normalized to body weight. GHA appears to have a negative effect on the production of brown adipose tissue (BAT), as the size of the interscapular BAT depot is decreased in GHA mice when compared to WT mice [6].
Many studies have evaluated hormone levels or glucose homeostasis in these mice, with levels changing with advancing age. Despite increased serum GH, there is a 75–80% decrease in serum IGF-1, while levels of IGFBP-3 are also 30% lower in these mice at 11 months of age, while other IGFBPs (−1,−2, and −4) are unchanged [5]. Fasting glucose, fasting insulin and glucose tolerance are similar to controls for both males and females for the first year of life, although insulin levels are increased by 72 weeks of age in GHA males compared to controls [30]. Interestingly, GHA mice are protected from hyperinsulinemia and glucose intolerance when placed on a high fat diet despite increases in body fat [31]. Adiponectin levels are increased in the GHA mice and increase to a greater extent with advancing age and obesity, strongly correlating with leptin levels [32]. Similarly to the GHR−/− mice, GH antagonism seems to protect the kidneys of GHA mice, as glomerular hypertrophy and upregulated albumin excretion do not develop in streptozotocin-induced diabetic GHA mice [33].
4.2. Aging
Although GHR−/− mice and GHA mice both have a reduction in GH action, they are still phenotypically distinct in several ways. For example, GHA mice have no significant changes in lifespan of either male or female mice compared to littermate WT controls [5]. This lack of lifespan extension in GHA mice has garnered significant attention as most mouse lines that have a decrease in IGF-1 action and an increase in adiponectin see significant increases in lifespan [34]. However, it is important to note that GHR−/− mice have a lifelong progressive accumulation of adipose tissue in all adipose depots, which is typically considered detrimental to lifespan. Accordingly, GHA have increased insulin at advanced ages [30], contrasting the insulin profile of aged GHR−/− mice [35]. Thus, the fact that they have a normal lifespan despite obesity and elevated insulin is noteworthy and suggests some benefits to GHR antagonism. Further evidence for beneficial effects of GHR antagonism is shown in the failure of GHA mice to develop cellular senescence in white adipose tissue (WAT) of 18-month old female GHA mice [35]. As senescence has emerged as a potent contributor to aging and age-related diseases, normal levels of senescence in WAT of GHA mice in the context of increased adiposity/insulin could be interpreted as a positive outcome given that obesity is known to accelerate WAT senescence [36].
4.3. Legacy
The most important legacy of the GHA mice is the creation of the GHR antagonist drug Somavert™ (Pegvisomant for injection; hereafter referred to as pegvisomant). Pegvisomant was approved by the FDA for treatment of patients with acromegaly in 2003 and shortly thereafter in Europe and Japan. Currently sold by Pfizer, pegvisomant is marketed worldwide and is used as a treatment of acromegaly and could have other potential therapeutic uses such as a treatment against cancer, diabetic nephropathy, and diabetes [37, 38]. The drug has also been suggested as a possible intervention to slow aging in humans [39]. Importantly, the discovery of pegvisomant through the generation of genetically modified mice highlights the translatability and value of mouse models to examine the function of GH and its receptor.
5.0. Tissue-specific GHR−/− Mice
Discussion up until this point has focused on models in which GH signaling is altered systemically, but GH’s effects differ depending on the tissue in which it is acting. For example, GH acts on the liver to induce IGF-1 production while its primary effect in bone is longitudinal bone growth and has catabolic effects on adipose tissue, increasing lipolysis and inhibiting adipogenesis. Thus, multiple groups have used Cre-lox technology to create mouse lines in which GH insensitivity is isolated to a specific tissue or cell type. Consequently, the GHR gene has been disrupted in classical GH target tissues such as liver, muscle, adipose, and bone as well as brain, heart, macrophages, pancreatic beta cells, the intestine, and hematopoietic stem cells. A brief summary of the phenotypes of these mouse lines is presented in Table 1, and these mice have been more fully characterized in several recent reviews [40, 41]. As might be expected, none of the tissue-specific lines recapitulate the extreme short stature, longevity or other characteristics of the global GHR−/− mice.
Table 1.
Tissue-specific GHR knockout mouse lines. Summary of the reported tissue specific knockout lines, including information about the promoter used to drive gene disruption and selected phenotypes, including weight, length, serum GH and IGF-1 levels, and body composition. Blank cells indicate parameters that were not reported. (ND = normal diet; CR = calorie restriction)
| Tissue/Cell | Name | Promoter | Weight | Length | GH | IGF-1 | Body composition | References |
|---|---|---|---|---|---|---|---|---|
| Liver | GHRLD | Albumin | ↔ | ↔ | ↑ 233% | ↓ 94% | ↔ | [42] |
| LiGHRKO | Albumin | ↓ | ↓ | ↑♂:~275% ♀:~333% |
↓♂:~94% ♀:~88% |
↑ Lean, ↓ Fat | [43, 74] | |
| L-Ghr−/− | Albumin | ↔ | ↑ ~200% | ↔ | [45] | |||
| Li-GHRKO | Albumin | ↔ | [44] | |||||
| Liver (Adult) | aLivGHRkd | AAV-TBG | ↔ | ↓♂:~89% ♀:~38% |
[46] | |||
| Muscle | ΔGHR | mef-2c-73k | ↑ | ↔ | ↔ | ↑ Fat | [47] | |
| mGHRKO | MCK | ↓ | ↔ | ↔ | ↔ | ↓ Lean & Fat | [48] | |
| MuGHRKO | MCK | ↓ ♂, ↑ ♀ | ↔ | ↔ | ↔ | ↓ Lean & Fat ♂, ↑ Lean & Fat ♀ | [49] | |
| Fat | FaGHRKO | Fabp4 | ↑ | ↔ | ↑♂:21.9%; ↔ ♀ |
↑ Fat & Fluid | [50] | |
| AdGHRKO | AdipoQ | ↔ | ↔ | ↔ | ↔ | ↑ Lean ♂; ↑ Fat ♂ ♀ | [51] | |
| Fat-Ghr−/− | AdipoQ | ↔ | ↔ | [45] | ||||
| Bone | DMP-GHRKO | DMP-1 | ↔ | ↑ 8 wk: ~300%; ↔ 13 wk |
[53] | |||
| Brain | AgRP GHR | AgRP | ↔ ND; ↓ CR | ↔ | ↔ ND; ↓ Lean & Fat CR | [54] | ||
| LepR GHR KO | LepR | ↑ ND; ↓ CR | ↑ | ↔ Lean ↓ Fat | ||||
| Brain GHR KO | Nestin | ↑ ND; ↓ CR | ↑ | ↑ Lean ↔ Fat | ||||
| LeprFYFPΔGHR | LepR | ↔ | ↔ | ↔ | ↔ | ↔ | [55] | |
| Heart (Adult) | iC-GHRKO | Myh6-Mer | ↔ | ↔ | ↔ young; ↓ old:~25% |
↑ Lean ↓ Fat | [56] | |
| Macrophage | MacGHRKO | LysM | ↔ | ↔ | [58] | |||
| β Cell | βGHRKO | Rat Insulin | ↔ | [59] | ||||
| Intestine | IntGHRKO | Villin | ↔ | ↔ | ↔ | [60] | ||
| Hematopoietic Stem Cell | Vav1 | [61] |
5.1. Liver-specific GHR knockout
As perhaps the major effect of GH is its stimulation of IGF-1 in the liver, the first reported tissue-specific GHR disruption was in the liver by Fan et al. in 2009 [42]. Liver-specific GHR disruption has subsequently been reported by List et al. in 2014 [43], Liu et al. in 2016 [44], and Fang et al. in 2019 [45]. All the reported models use Cre-lox technology to disrupt the GHR gene in the liver using the albumin promoter to drive Cre recombinase expression. A unique feature of these mice is that they have high serum GH but low serum IGF-1, while still being sensitive to GH in most tissues. The phenotypes of the mice are reported in a variety of ages (List and Liu were the only to include data from mice over 6 months of age), and only the paper by List reports both sexes, while the other articles are limited to male mice only.
The reported phenotypes of liver specific GHR knockout mice are broadly similar, with minor variations that may be due to the differences in ages and sexes reported. Surprisingly, despite decreased serum IGF-1, body weight is unchanged, except in the List cohort. Body composition also differs in the List cohort, with decreased fat mass and increased percent lean mass observed in both males and females. This may be due to the longitudinal observation of body composition in the List cohort, capturing changes seen at multiple time points. The other cohorts have no change in overall body composition, although the epididymal fat mass is increased in the Fang cohort. Glucose metabolism is impaired except for the Fang cohort, with either high fasting glucose, glucose intolerance and/or insulin resistance. More evidence of metabolic dysfunction consistently reported in male liver GHR knockout mice is liver steatosis and increased liver triglycerides, presumably due to the loss of the lipolytic and anti-lipogenic effects of GH in the liver, and consistent with an increase in liver weight.
Other phenotypes are reported in select cohorts. For example, the mice from the Fan cohort have decreased bone density, while the List cohort have increased grip strength and increased circulating adipokine (leptin, resistin, and adiponectin). Overall, the liver-specific GHR knockout animals show that despite dramatic decreases in serum IGF-1, they do not have a decrease in body weight, with only one cohort showing any decrease at all [43]. These results demonstrate that local GH action and IGF-1 production are important for growth. Liver-specific GHR knockout mice also lack the increased lifespan seen in global GHR−/−, as they have a normal lifespan.
The work using mice with a lifelong liver GHR disruption was expanded through the generation of an adult-onset liver GHR knockout (aLivGHRkd) model by Cordoba-Chacon et al [46]. The Cre-lox system was also used for these mice, but Cre expression was induced using an adeno-associated virus with the thyroxine binding globulin promoter. These mice also have decreased circulating IGF-1 (decreased by ~89% in males, ~38% in females), but no change to circulating GH. aLivGHRkd mice have increased hepatic de novo lipogenesis, leading to steatosis in males. Both sexes show increased liver weight, while females have increased epididymal fat weight. In this study, the mice are dissected only seven days after liver GHR disruption, which demonstrates the potent effect GH signaling has on hepatic lipid accumulation but does not give an indication of longer-term effects in this model.
5.2. Muscle-specific GHR Knockout
GH and IGF-1 are central to muscle development and function, but individual contributions to muscle physiology were uncertain, leading to the development of muscle-specific GHR knockout mice. Three laboratories independently developed muscle GHR knockout mice, and the results were reported by Mavalli et al. in 2010 [47], Vijayakumar et al. in 2012 [48], and List et al. in 2015 [49].
The Cre-lox system was used in each case, but two different promoters were used to drive Cre recombinase expression. The Mavalli cohort used the mef-2c-73k promoter, while the other two groups use the muscle creatine kinase (MCK) promoter. The difference in promoter can be observed in the phenotypes reported, with the two models using the MCK promoter having phenotypes that are more similar to each other than they are to the mef-2c-73k-generated model. The Mavalli cohort have increased body weight and body fat with glucose intolerance and insulin resistance, while the other cohorts have the opposite, with decreased body weight, lean and fat mass, and improved glucose tolerance and insulin sensitivity. None of the three mouse lines have changes in serum GH or IGF-1. The Vijayakumar cohort are protected from high-fat diet induced hepatic steatosis and have normal muscle structure, while the Mavalli cohort have smaller and weaker muscles. In terms of longevity, the List cohort is the only one to assess longevity with only male mice having an increased lifespan.
5.3. Adipose-Specific GHR knockout
The GH axis is well known to have lipolytic and anti-lipogenic properties, so mice with GHR knocked out specifically in the adipose tissue were developed by two labs and with two different promoters driving Cre recombinase expression. List et al. first used the Fabp4 promoter in 2013 (called FaGHRKO mice) [50] and subsequently used the adiponectin (AdipoQ) promoter in 2019 (called AdGHRKO mice) [51], while Fang et al also used the AdipoQ promoter in 2019 [45]. The newer mouse lines developed using the AdipoQ promoter are considered to be more specific to adipocytes within the tissue and more uniformly impact various depots [52].
As with the muscle knockouts, there are differences among mice with different promoters driving Cre, but there are also similarities in the case of the adipose GHR knockout lines. The FaGHRKO mice, despite increased body weight, fat mass, and a male-specific 21.9% increase in serum IGF-1, have an otherwise normal metabolic phenotype, with normal glucose metabolism and no hepatic steatosis. The AdGHRKO mice also have increased fat mass and males have increased lean mass, but no significant change in body weight. AdGHRKO mice have improved insulin sensitivity and decreased hepatic steatosis, and normal circulating GH and IGF-1 levels. Both FaGHRKO and AdGHRKO have increased circulating adiponectin levels, while other adipokines (leptin, resistin, adipsin) differ between the two mouse lines. The Fang cohort are reported to have normal body composition, glucose metabolism, and serum GH levels.
5.4. Bone-specific GHR knockout
To separate GH and IGF-1 effects on bone development and physiology, Liu et al created a bone-specific GHR knockout mouse in 2016 [53] using the Cre-lox system with the dentin matrix protein-1 promoter driving Cre expression. Despite lacking GHR in the bone and having increased serum GH at 8 weeks of age, these mice have no change in body weight or composition. However, knockout animals had impaired bone development, including radial growth and mineral acquisition. Other metabolic phenotypes were not reported.
5.5. Brain-specific GHR knockout
Although GH is produced in the pituitary gland, the brain was not considered to be a major target tissue of GH except in the hypothalamus for feedback inhibition of GH production. More recently, GHR has been shown to be expressed on a variety of brain cells including areas that regulate metabolism. To better understand the role that GH signaling in the brain plays in metabolism, GHR was knocked out by two different laboratories using three different promoters with the Cre-lox system. GHR has been knocked out in brain cells expressing agouti-related protein (AgRP) [54], leptin receptor (LepR) [54, 55], and nestin [54].
The AgRP-expressing cell-specific GHR knockout (AgRP GHR KO) animals have a normal metabolic phenotype, with normal body weight, body length, body composition, glucose tolerance and insulin tolerance. When stressed with caloric restriction, AgRP GHR KO mice exhibit decreased body weight, fat mass, and lean mass and decreased glucose. LepR GHR knockout animals have been generated in two laboratories and reported in 2017 by Cady et al. [55] and in 2019 by Furigo et al. [54]. The Cady cohort have normal body weight, length, and composition as well as normal serum GH, IGF-1 and adiponectin. These mice have impaired glucose metabolism, with glucose intolerance and insulin resistance. In contrast, the Furigo cohort mice have increased body length and weight, with increased fat mass and no change to lean mass. Despite the increased fat mass, this cohort has decreased circulating leptin. Under caloric restriction, the Furigo cohort have decreased body weight and fasting glucose compared to controls on caloric restriction. The third type of brain-specific knockout is the nestin promoter-driven GHR knockout, called “Brain GHR KO” by the authors. Brain GHR KO mice have increased body weight and body length; the increase in body weight seems to come from lean mass, which is increased while fat mass is unchanged. Under caloric restriction, body weight is decreased, but no change to glucose metabolism is reported.
5.6. Heart-specific GHR knockout
Because systemic GH alteration was previously shown to affect cardiac function, an inducible cardiac-specific GHR knockout (iC-GHRKO) mouse line was developed by Jara et al. [56]. By inducing GHR knockout in adult mice, the possible developmental defects of the heart are avoided. Interestingly, iC-GHRKO mice have no changes to cardiac function but have an unexpected metabolic phenotype. That is, iC-GHRKO mice have normal body weight and body length but have altered body composition with increased lean mass and increased fat mass. Glucose metabolism changes with age, as 6 month old iC-GHRKO mice have normal glucose tolerance and increased insulin sensitivity, and mice over a year of age have glucose intolerance and insulin resistance. The changes in glucose metabolism coincide with changes in serum IGF-1 levels: IGF-1 is normal in the younger mice and decreased in the aged mice.
5.7. Macrophage-specific GHR knockout
The expansion of adipose tissue in obesity is accompanied by infiltration of pro-inflammatory macrophages into adipose tissue. GH is also known to regulate macrophage cytokine expression, and GH alterations are associated with obesity. To clarify the role of GH signaling in macrophages during obesity, Lu et al generated a macrophage-specific GHR knockout mouse line [57, 58] and placed them on a high fat diet. Despite the high fat diet, the macrophage GHR knockout mice have no change in body weight or body composition. They do, however, have impaired glucose metabolism with glucose intolerance and insulin resistance. Despite impaired adipocyte differentiation and unchanged total fat mass, epididymal depot weight is increased, and the adipose tissue shows increased expression of inflammatory cytokines such as IL-1B, IL-6, and TNF-A.
5.8. Pancreatic beta cell-specific GHR knockout
As demonstrated by almost every model of GH alteration, GH and glucose metabolism are closely linked. To further investigate this relationship and determine the GH-specific effects on part of the endocrine pancreas, Wu et al generated a beta cell-specific GHR knockout mouse line [59]. No change in body weight is observed in these animals, but they have decreased beta cell mass. They also have impaired glucose metabolism (glucose intolerance and decreased glucose-stimulated insulin secretion) when placed on a high fat diet, cementing the relationship between glucose metabolism and the GH axis.
5.9. Intestine-specific GHR knockout
GHR is expressed throughout the intestine, and recombinant GH is FDA-approved to treat short bowel syndrome. Evidence from mouse studies of intestinal inflammation and off-label use of recombinant GH on clinical populations also show that GH improves outcomes in cases of intestinal inflammation. To explore the relationship between GH and intestinal function, Young et al generated an intestinal epithelial cell-specific GHR knockout (IntGHRKO) mouse [60] using the villin promoter for Cre expression. IntGHRKO mice have no change in body weight, length or composition, but females have glucose intolerance and insulin resistance. Additional sex specific differences are seen in gut barrier function and fat absorption.
5.10. Hematopoietic stem cell specific GHR knockout
GH alterations can affect hematopoiesis, but whether this effect is direct on hematopoietic stem cells or indirect through another target cell is undetermined. To address this, Stewart et al developed a hematopoietic stem cell-specific GHR knockout mouse [61]. In this model, hematopoietic stem cell function was normal, and no growth or metabolic phenotypes were reported. Thus, the function of GH in these cells remains unresolved.
6. Disruption of downstream signaling that interrupts GH action
As discussed above, GHI is a group of rare genetic disorders in which the body is not responsive to GH, leading to a reduction or absence of the biological effects of GH though there are normal or even elevated GH levels [62–65]. All mouse lines discussed thus far have alterations in GHR levels or GH binding; however, GHI can also be caused by mutations in downstream signaling proteins, such as Janus kinase 2 (JAK2), or signal transducer and activator of transcription 5b (STAT5b). GHI can also be studied by disrupting the products of GH action, including IGF-1, IGFBP-3 and acid-labile subunit (ALS) [66, 65].
There are multiple mouse lines established in the past decade with disruption of GH downstream signaling, exhibiting the phenotypes of GHI, which are summarized in Table 2 [65]. Mice with disruptions in GH signaling pathways include the knockout of JAK2, STAT5b or suppressor of cytokine signaling-2 (SOCS2). Although JAK2 and STAT5b make up the canonical signaling pathway of GH, they are not specific to GH action and may also be activated by other cytokines or growth factors [67–70]. SOCS2, a negative regulator of STAT5b signaling, may also be induced by other factors, so it is not specific to GH signaling either [67], but disruption of SOCS2 has some interesting growth phenotypes. Curiously, different doses of SOCS2 have opposite effects in GHR downstream signaling and activities in vitro [71]. In addition, the acceleration of growth of SOCS2 knockout mouse has been shown to be STAT5b-dependent [71].
Table 2.
Mouse lines with downstream disruption of GH action. Summary of the reported mouse lines with a disruption downstream of GHR that inhibits normal GH signaling. Selected phenotypes are included, such as serum GH and IGF-1 levels, body size and weight, bone growth, insulin sensitivity, reproductive capacity, and cancer incidence. Blank cells indicate parameters that were not reported.
| Mouse lines | GH level | IGF-1 level | Body size / Weight | Body composition | Bone growth | Insulin sensitivity | Reproductive capacity | Cancer incidence | Other notes | References |
|---|---|---|---|---|---|---|---|---|---|---|
| JAK2 KO | Lethal | Erythropoiesis impaired | [75] | |||||||
| STAT5b KO | ↑ (male only) | ↓ (both sexes) | ↓ (males only) | ↓ adipose tissue | ↓ (males only) | Poor lactation; deficient pregnancy | Varies by genetic background | Delay of hair growth; GH pulse-resistant | [69, 70, 76, 77] | |
| SOCS2 TG | ↑ | ↓ adipose tissue | Fertile | [71] | ||||||
| SOCS2 KO | ↑ local IGF-1; no difference in serum | ↑ | ↔ | ↑ | ↔ | Thickening of dermis; excessive collagen deposition | [78, 79] | |||
| ALS KO | ↔ | ↓ serum IGF-1 and IGFBP3 | ↓ | ↓ bone mineral density | ↔ | ↔ | [80, 81] | |||
| IGF-1 KO | Below detection limits | ↓ | ↓ | Infertile | High neonatal lethality (less than that of IGFR −/−), only 68% reach adulthood | [82, 83] | ||||
| IGFBP3-KO | ↔ | ↔; ↓ | No change at birth; ↑ overall weight | ↑ muscle mass; ↓ WAT mass | ↑ fasting blood glucose, ↑ insulin, impaired GTT | ↔ | ↑lung cancer progression | ↓ plasma TG adiponectin levels;↓ metabolic rate | [84–86] |
GH action induces the production of three major important products - IGF-1, IGFBP-3, and ALS - in the liver [72], and these factors have also been disrupted in mouse lines. IGF-1, as a potent growth factor involved in growth and development, is predominantly synthesized in the liver in response to GH [67]. IGFBP-3 and ALS are produced to bind to IGF-1 and form complexes, thereby extending the half-life of IGF-1 in serum and extending its biological activities [73, 72]. In Table 2, we also briefly summarize the phenotypes of these GHI mouse lines. Overall, short stature, elevated serum GH and decreased serum IGF-1 levels, and decreased bone growth are observed in some of these GHI mouse lines, sharing similar characteristics of GHI in humans [65].
7. Summary and Future Directions
The impact of GHI in laboratory mice is fairly consistent and well-documented, and these mice have allowed for more invasive measures than would be feasible in limited clinical populations. Overall, GHI commonly results in mice that are smaller in size with increased GH, decreased IGF-1 levels, increased adiposity, reduced lean mass, and improved glucose homeostasis. As discussed in a separate paper in this thematic issue, many of the findings obtained in GHI mice apply to other mammalian species, such as humans. In fact, and as noted previously, studies in these genetically engineered mice have resulted in the discovery of a highly efficacious drug, pegvisomant, which demonstrates the clinical relevance of findings from mice. Still, inconsistencies, controversies and further questions exist.
Among the various phenotypes of these mice, the increase in healthspan and longevity has likely garnered the most attention. Most studied are the GHR−/− mice, which have measurable delays or decreases in many established markers of aging and morbidity along with a robust increase in longevity, at least in the laboratory setting. Impressively, the health benefits observed in GHR−/− mice match or even exceed the effects of all reported genetic, environmental (e.g. diet, habitat), or pharmacological interventions to extend lifespan in mice, making them highly studied in the aging field. However, the mechanisms responsible for these observations and their relevance to humans are still debated. Afterall, multiple mechanisms have been implicated to contribute to their longevity, and patients with LS, despite many commonalities in phenotype, have not been reported to experience lifespan extension. Could the healthspan and longevity phenotype with GHI be species-specific? While definitely possible due to the known differences in gene expression, tissue function and structure of complex systems, it is also likely that the well-controlled laboratory setting for mice allows for more pronounced and measurable differences. The many confounding variables for humans (e.g. socioeconomic status, genetic variation, disease state, environment, habitat, diet, exercise, and drug/pharmaceutical use) undoubtedly mask underlying and detectable differences. Although one would expect studies across species to vary, species-specific differences in gene networks, metabolism, and pathophysiology appear irrevocably distinct between the species. These circumstances should be considered in all interpretations with these mice and clinical GHI.
Despite the limitations of working with mice to study GHI, many questions remain that can only be studied in mice due to the invasive nature of the research or their ease of genetic manipulation. We anxiously await the additional tissue- or cell-specific mouse lines, mice with GHR gene disruption at more advanced ages, mice that combine genetic manipulation of the GH/IGF-axis with other dietary or pharmacological agents, and mice that target other GH downstream molecules as we continue to unravel to the role of growth hormone on biological processes.
Funding
This work was funded by NIH grant R01 AG059779
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflicts of interest/Competing interests (include appropriate disclosures)
N/A
References
- 1.Perlman RL. Mouse models of human disease: An evolutionary perspective. Evol Med Public Health. 2016;2016(1):170–6. doi: 10.1093/emph/eow014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc Natl Acad Sci U S A. 1997;94(24):13215–20. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.List EO, Sackmann-Sala L, Berryman DE, Funk K, Kelder B, Gosney ES et al. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR−/−) mouse. Endocr Rev. 2011;32(3):356–86. doi: 10.1210/er.2010-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berryman DE, List EO, Palmer AJ, Chung MY, Wright-Piekarski J, Lubbers E et al. Two-year body composition analyses of long-lived GHR null mice. J Gerontol A Biol Sci Med Sci. 2010;65(1):31–40. doi: 10.1093/gerona/glp175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology. 2003;144(9):3799–810. doi: 10.1210/en.2003-0374. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Knapp JR, Kopchick JJ. Enlargement of interscapular brown adipose tissue in growth hormone antagonist transgenic and in growth hormone receptor gene-disrupted dwarf mice. Exp Biol Med (Maywood). 2003;228(2):207–15. doi: 10.1177/153537020322800212. [DOI] [PubMed] [Google Scholar]
- 7.Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ et al. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab. 2004;287(3):E405–13. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 8.Berryman DE, List EO, Kohn DT, Coschigano KT, Seeley RJ, Kopchick JJ. Effect of growth hormone on susceptibility to diet-induced obesity. Endocrinology. 2006;147(6):2801–8. doi: 10.1210/en.2006-0086. [DOI] [PubMed] [Google Scholar]
- 9.Robertson K, Kopchick JJ, Liu JL. Growth hormone receptor gene deficiency causes delayed insulin responsiveness in skeletal muscles without affecting compensatory islet cell overgrowth in obese mice. Am J Physiol Endocrinol Metab. 2006;291(3):E491–8. doi: 10.1152/ajpendo.00378.2005. [DOI] [PubMed] [Google Scholar]
- 10.Egecioglu E, Bjursell M, Ljungberg A, Dickson SL, Kopchick JJ, Bergstrom G et al. Growth hormone receptor deficiency results in blunted ghrelin feeding response, obesity, and hypolipidemia in mice. Am J Physiol Endocrinol Metab. 2006;290(2):E317–25. doi: 10.1152/ajpendo.00181.2005. [DOI] [PubMed] [Google Scholar]
- 11.Barclay JL, Nelson CN, Ishikawa M, Murray LA, Kerr LM, McPhee TR et al. GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology. 2011;152(1):181–92. doi: 10.1210/en.2010-0537. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, Solesio ME, Schaffler MB, Frikha-Benayed D, Rosen CJ, Werner H et al. Mitochondrial Function Is Compromised in Cortical Bone Osteocytes of Long-Lived Growth Hormone Receptor Null Mice. J Bone Miner Res. 2019;34(1):106–22. doi: 10.1002/jbmr.3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandrashekar V, Zaczek D, Bartke A. The consequences of altered somatotropic system on reproduction. Biol Reprod. 2004;71(1):17–27. doi: 10.1095/biolreprod.103.027060. [DOI] [PubMed] [Google Scholar]
- 14.Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol. 2013;9(6):366–76. doi: 10.1038/nrendo.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arum O, Bonkowski MS, Rocha JS, Bartke A. The growth hormone receptor gene-disrupted mouse fails to respond to an intermittent fasting diet. Aging Cell. 2009;8(6):756–60. doi: 10.1111/j.1474-9726.2009.00520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang Y, McFadden S, Darcy J, Hascup ER, Hascup KN, Bartke A. Lifespan of long-lived growth hormone receptor knockout mice was not normalized by housing at 30 degrees C since weaning. Aging Cell. 2020;19(5):e13123. doi: 10.1111/acel.13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartke A, List EO, Kopchick JJ. The somatotropic axis and aging: Benefits of endocrine defects. Growth Horm IGF Res. 2016;27:41–5. doi: 10.1016/j.ghir.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf N, Penn P, Pendergrass W, Van Remmen H, Bartke A, Rabinovitch P et al. Age-related cataract progression in five mouse models for anti-oxidant protection or hormonal influence. Exp Eye Res. 2005;81(3):276–85. doi: 10.1016/j.exer.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 19.Bellush LL, Doublier S, Holland AN, Striker LJ, Striker GE, Kopchick JJ. Protection against diabetes-induced nephropathy in growth hormone receptor/binding protein gene-disrupted mice. Endocrinology. 2000;141(1):163–8. doi: 10.1210/endo.141.1.7284. [DOI] [PubMed] [Google Scholar]
- 20.Kinney-Forshee BA, Kinney NE, Steger RW, Bartke A. Could a deficiency in growth hormone signaling be beneficial to the aging brain? Physiol Behav. 2004;80(5):589–94. doi: 10.1016/j.physbeh.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 21.Ikeno Y, Hubbard GB, Lee S, Cortez LA, Lew CM, Webb CR et al. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J Gerontol A Biol Sci Med Sci. 2009;64(5):522–9. doi: 10.1093/gerona/glp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Junnila RK, Duran-Ortiz S, Suer O, Sustarsic EG, Berryman DE, List EO et al. Disruption of the GH Receptor Gene in Adult Mice Increases Maximal Lifespan in Females. Endocrinology. 2016;157(12):4502–13. doi: 10.1210/en.2016-1649. [DOI] [PubMed] [Google Scholar]
- 23.Silvente-Poirot S, de Medina P, Record M, Poirot M. From tamoxifen to dendrogenin A: The discovery of a mammalian tumor suppressor and cholesterol metabolite. Biochimie. 2016;130:109–14. doi: 10.1016/j.biochi.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Flurkey K, Currer J, Harrison D. The Mouse in Aging Research. The Mouse in Biomedical Research. 2007;3. doi: 10.1016/b978-012369454-6/50074-1. [DOI] [Google Scholar]
- 25.Chen WY, Wight DC, Wagner TE, Kopchick JJ. Expression of a mutated bovine growth hormone gene suppresses growth of transgenic mice. Proc Natl Acad Sci U S A. 1990;87(13):5061–5. doi: 10.1073/pnas.87.13.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharadadevi A, Sivakamasundari C, Nagaraj R. Amphipathic alpha-helices in proteins: results from analysis of protein structures. Proteins. 2005;59(4):791–801. doi: 10.1002/prot.20459. [DOI] [PubMed] [Google Scholar]
- 27.Chen WY, White ME, Wagner TE, Kopchick JJ. Functional antagonism between endogenous mouse growth hormone (GH) and a GH analog results in dwarf transgenic mice. Endocrinology. 1991;129(3):1402–8. doi: 10.1210/endo-129-3-1402. [DOI] [PubMed] [Google Scholar]
- 28.Okada S, Chen WY, Wiehl P, Kelder B, Goodman HM, Guller S et al. A growth hormone (GH) analog can antagonize the ability of native GH to promote differentiation of 3T3-F442A preadipocytes and stimulate insulin-like and lipolytic activities in primary rat adipocytes. Endocrinology. 1992;130(4):2284–90. doi: 10.1210/endo.130.4.1547740. [DOI] [PubMed] [Google Scholar]
- 29.Berryman DE, List EO, Coschigano KT, Behar K, Kim JK, Kopchick JJ. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm IGF Res. 2004;14(4):309–18. doi: 10.1016/j.ghir.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Berryman DE, Lubbers ER, Magon V, List EO, Kopchick JJ. A dwarf mouse model with decreased GH/IGF-1 activity that does not experience life-span extension: potential impact of increased adiposity, leptin, and insulin with advancing age. J Gerontol A Biol Sci Med Sci. 2014;69(2):131–41. doi: 10.1093/gerona/glt069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang T, Householder LA, Lubbers ER, List EO, Troike K, Vesel C et al. Growth hormone receptor antagonist transgenic mice are protected from hyperinsulinemia and glucose intolerance despite obesity when placed on a HF diet. Endocrinology. 2015;156(2):555–64. doi: 10.1210/en.2014-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lubbers ER, List EO, Jara A, Sackman-Sala L, Cordoba-Chacon J, Gahete MD et al. Adiponectin in mice with altered GH action: links to insulin sensitivity and longevity? J Endocrinol. 2013;216(3):363–74. doi: 10.1530/JOE-12-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flyvbjerg A, Bennett WF, Rasch R, Kopchick JJ, Scarlett JA. Inhibitory effect of a growth hormone receptor antagonist (G120K-PEG) on renal enlargement, glomerular hypertrophy, and urinary albumin excretion in experimental diabetes in mice. Diabetes. 1999;48(2):377–82. doi: 10.2337/diabetes.48.2.377. [DOI] [PubMed] [Google Scholar]
- 34.Atzmon G, Pollin TI, Crandall J, Tanner K, Schechter CB, Scherer PE et al. Adiponectin levels and genotype: a potential regulator of life span in humans. J Gerontol A Biol Sci Med Sci. 2008;63(5):447–53. doi: 10.1093/gerona/63.5.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Comisford R, Lubbers ER, Householder LA, Suer O, Tchkonia T, Kirkland JL et al. Growth Hormone Receptor Antagonist Transgenic Mice Have Increased Subcutaneous Adipose Tissue Mass, Altered Glucose Homeostasis and No Change in White Adipose Tissue Cellular Senescence. Gerontology. 2016;62(2):163–72. doi: 10.1159/000439050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H et al. Fat tissue, aging, and cellular senescence. Aging Cell. 2010;9(5):667–84. doi: 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopchick JJ. Discovery and mechanism of action of pegvisomant. Eur J Endocrinol. 2003;148 Suppl 2:S21–5. doi: 10.1530/eje.0.148s021. [DOI] [PubMed] [Google Scholar]
- 38.Muller AF, Kopchick JJ, Flyvbjerg A, van der Lely AJ. Clinical review 166: Growth hormone receptor antagonists. J Clin Endocrinol Metab. 2004;89(4):1503–11. doi: 10.1210/jc.2002-022049. [DOI] [PubMed] [Google Scholar]
- 39.Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C et al. Interventions to Slow Aging in Humans: Are We Ready? Aging Cell. 2015;14(4):497–510. doi: 10.1111/acel.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young JA, List EO, Kopchick JJ. Deconstructing the Growth Hormone Receptor(GHR): Physical and Metabolic Phenotypes of Tissue-Specific GHR Gene-Disrupted Mice. Prog Mol Biol Transl Sci. 2016;138:27–39. doi: 10.1016/bs.pmbts.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 41.List EO, Berryman DE, Jensen EA, Kulkarni P, McKenna S, Kopchick JJ. New insights of growth hormone (GH) actions from tissue-specific GH receptor knockouts in mice. Arch Endocrinol Metab. 2019;63(6):557–67. doi: 10.20945/2359-3997000000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ et al. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem. 2009;284(30):19937–44. doi: 10.1074/jbc.M109.014308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.List EO, Berryman DE, Funk K, Jara A, Kelder B, Wang F et al. Liver-specific GH receptor gene-disrupted (LiGHRKO) mice have decreased endocrine IGF-I, increased local IGF-I, and altered body size, body composition, and adipokine profiles. Endocrinology. 2014;155(5):1793–805. doi: 10.1210/en.2013-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Cordoba-Chacon J, Kineman RD, Cronstein BN, Muzumdar R, Gong Z et al. Growth Hormone Control of Hepatic Lipid Metabolism. Diabetes. 2016;65(12):3598–609. doi: 10.2337/db16-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fang F, Shi X, Brown MS, Goldstein JL, Liang G. Growth hormone acts on liver to stimulate autophagy, support glucose production, and preserve blood glucose in chronically starved mice. Proc Natl Acad Sci U S A. 2019;116(15):7449–54. doi: 10.1073/pnas.1901867116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cordoba-Chacon J, Majumdar N, List EO, Diaz-Ruiz A, Frank SJ, Manzano A et al. Growth Hormone Inhibits Hepatic De Novo Lipogenesis in Adult Mice. Diabetes. 2015;64(9):3093–103. doi: 10.2337/db15-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mavalli MD, DiGirolamo DJ, Fan Y, Riddle RC, Campbell KS, van Groen T et al. Distinct growth hormone receptor signaling modes regulate skeletal muscle development and insulin sensitivity in mice. J Clin Invest. 2010;120(11):4007–20. doi: 10.1172/JCI42447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vijayakumar A, Wu Y, Sun H, Li X, Jeddy Z, Liu C et al. Targeted loss of GHR signaling in mouse skeletal muscle protects against high-fat diet-induced metabolic deterioration. Diabetes. 2012;61(1):94–103. doi: 10.2337/db11-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.List EO, Berryman DE, Ikeno Y, Hubbard GB, Funk K, Comisford R et al. Removal of growth hormone receptor (GHR) in muscle of male mice replicates some of the health benefits seen in global GHR−/− mice. Aging (Albany NY). 2015;7(7):500–12. doi: 10.18632/aging.100766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.List EO, Berryman DE, Funk K, Gosney ES, Jara A, Kelder B et al. The role of GH in adipose tissue: lessons from adipose-specific GH receptor gene-disrupted mice. Mol Endocrinol. 2013;27(3):524–35. doi: 10.1210/me.2012-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.List EO, Berryman DE, Buchman M, Parker C, Funk K, Bell S et al. Adipocyte-Specific GH Receptor-Null (AdGHRKO) Mice Have Enhanced Insulin Sensitivity With Reduced Liver Triglycerides. Endocrinology. 2019;160(1):68–80. doi: 10.1210/en.2018-00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang ZV, Deng Y, Wang QA, Sun K, Scherer PE. Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology. 2010;151(6):2933–9. doi: 10.1210/en.2010-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z, Kennedy OD, Cardoso L, Basta-Pljakic J, Partridge NC, Schaffler MB et al. DMP-1-mediated Ghr gene recombination compromises skeletal development and impairs skeletal response to intermittent PTH. Faseb J. 2016;30(2):635–52. doi: 10.1096/fj.15-275859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furigo IC, Teixeira PDS, de Souza GO, Couto GCL, Romero GG, Perello M et al. Growth hormone regulates neuroendocrine responses to weight loss via AgRP neurons. Nat Commun. 2019;10(1):662. doi: 10.1038/s41467-019-08607-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cady G, Landeryou T, Garratt M, Kopchick JJ, Qi N, Garcia-Galiano D et al. Hypothalamic growth hormone receptor (GHR) controls hepatic glucose production in nutrient-sensing leptin receptor (LepRb) expressing neurons. Mol Metab. 2017;6(5):393–405. doi: 10.1016/j.molmet.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jara A, Liu X, Sim D, Benner CM, Duran-Ortiz S, Qian Y et al. Cardiac-Specific Disruption of GH Receptor Alters Glucose Homeostasis While Maintaining Normal Cardiac Performance in Adult Male Mice. Endocrinology. 2016;157(5):1929–41. doi: 10.1210/en.2015-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu C, Kumar PA, Fan Y, Sperling MA, Menon RK. A novel effect of growth hormone on macrophage modulates macrophage-dependent adipocyte differentiation. Endocrinology. 2010;151(5):2189–99. doi: 10.1210/en.2009-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu C, Kumar PA, Sun J, Aggarwal A, Fan Y, Sperling MA et al. Targeted deletion of growth hormone (GH) receptor in macrophage reveals novel osteopontin-mediated effects of GH on glucose homeostasis and insulin sensitivity in diet-induced obesity. J Biol Chem. 2013;288(22):15725–35. doi: 10.1074/jbc.M113.460212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu Y, Liu C, Sun H, Vijayakumar A, Giglou PR, Qiao R et al. Growth hormone receptor regulates beta cell hyperplasia and glucose-stimulated insulin secretion in obese mice. J Clin Invest. 2011;121(6):2422–6. doi: 10.1172/JCI45027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Young JA, Jensen EA, Stevens A, Duran-Ortiz S, List EO, Berryman DE et al. Characterization of an intestine-specific GH receptor knockout (IntGHRKO) mouse. Growth Horm IGF Res. 2019;46–47:5–15. doi: 10.1016/j.ghir.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stewart MH, Gutierrez-Martinez P, Beerman I, Garrison B, Gallagher EJ, LeRoith D et al. Growth hormone receptor signaling is dispensable for HSC function and aging. Blood. 2014;124(20):3076–80. doi: 10.1182/blood-2014-05-575308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marino G, Ugalde AP, Fernandez AF, Osorio FG, Fueyo A, Freije JM et al. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc Natl Acad Sci U S A. 2010;107(37):16268–73. doi: 10.1073/pnas.1002696107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laron Z Consequences of not treating children with laron syndrome (primary growth hormone insensitivity). J Pediatr Endocrinol Metab. 2001;14 Suppl 5:1243–8; discussion 61–2. [PubMed] [Google Scholar]
- 64.Wit JM, de Luca F. Atypical defects resulting in growth hormone insensitivity. Growth Horm IGF Res. 2016;28:57–61. doi: 10.1016/j.ghir.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 65.David A, Hwa V, Metherell LA, Netchine I, Camacho-Hubner C, Clark AJ et al. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. 2011;32(4):472–97. doi: 10.1210/er.2010-0023. [DOI] [PubMed] [Google Scholar]
- 66.Bernasconi A, Marino R, Ribas A, Rossi J, Ciaccio M, Oleastro M et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. 2006;118(5):e1584–92. doi: 10.1542/peds.2005-2882. [DOI] [PubMed] [Google Scholar]
- 67.Brooks AJ, Waters MJ. The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol. 2010;6(9):515–25. doi: 10.1038/nrendo.2010.123. [DOI] [PubMed] [Google Scholar]
- 68.Waters MJ. The growth hormone receptor. Growth Horm IGF Res. 2016;28:6–10. doi: 10.1016/j.ghir.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 69.Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A. 1997;94(14):7239–44. doi: 10.1073/pnas.94.14.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology. 2001;142(9):3836–41. doi: 10.1210/endo.142.9.8400. [DOI] [PubMed] [Google Scholar]
- 71.Greenhalgh CJ, Metcalf D, Thaus AL, Corbin JE, Uren R, Morgan PO et al. Biological evidence that SOCS-2 can act either as an enhancer or suppressor of growth hormone signaling. J Biol Chem. 2002;277(43):40181–4. doi: 10.1074/jbc.C200450200. [DOI] [PubMed] [Google Scholar]
- 72.Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E et al. The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr Connect. 2018;7(6):R212–R22. doi: 10.1530/EC-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Domene HM, Hwa V, Argente J, Wit JM, Camacho-Hubner C, Jasper HG et al. Human acid-labile subunit deficiency: clinical, endocrine and metabolic consequences. Horm Res. 2009;72(3):129–41. doi: 10.1159/000232486. [DOI] [PubMed] [Google Scholar]
- 74.Dominick G, Berryman DE, List EO, Kopchick JJ, Li X, Miller RA et al. Regulation of mTOR activity in Snell dwarf and GH receptor gene-disrupted mice. Endocrinology. 2015;156(2):565–75. doi: 10.1210/en.2014-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93(3):397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 76.Oberley CC, Bilger A, Drinkwater NR. Genetic background determines if Stat5b suppresses or enhances murine hepatocarcinogenesis. Mol Carcinog. 2015;54(10):959–70. doi: 10.1002/mc.22165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davey HW, Park SH, Grattan DR, McLachlan MJ, Waxman DJ. STAT5b-deficient mice are growth hormone pulse-resistant. Role of STAT5b in sex-specific liver p450 expression. J Biol Chem. 1999;274(50):35331–6. doi: 10.1074/jbc.274.50.35331. [DOI] [PubMed] [Google Scholar]
- 78.Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature. 2000;405(6790):1069–73. doi: 10.1038/35016611. [DOI] [PubMed] [Google Scholar]
- 79.Greenhalgh CJ, Bertolino P, Asa SL, Metcalf D, Corbin JE, Adams TE et al. Growth enhancement in suppressor of cytokine signaling 2 (SOCS-2)-deficient mice is dependent on signal transducer and activator of transcription 5b (STAT5b). Mol Endocrinol. 2002;16(6):1394–406. doi: 10.1210/mend.16.6.0845. [DOI] [PubMed] [Google Scholar]
- 80.Ueki I, Ooi GT, Tremblay ML, Hurst KR, Bach LA, Boisclair YR. Inactivation of the acid labile subunit gene in mice results in mild retardation of postnatal growth despite profound disruptions in the circulating insulin-like growth factor system. Proc Natl Acad Sci U S A. 2000;97(12):6868–73. doi: 10.1073/pnas.120172697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Domene HM, Bengolea SV, Jasper HG, Boisclair YR. Acid-labile subunit deficiency: phenotypic similarities and differences between human and mouse. J Endocrinol Invest. 2005;28(5 Suppl):43–6. [PubMed] [Google Scholar]
- 82.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993;75(1):59–72. [PubMed] [Google Scholar]
- 83.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75(1):73–82. [PubMed] [Google Scholar]
- 84.Wang YA, Sun Y, Palmer J, Solomides C, Huang LC, Shyr Y et al. IGFBP3 Modulates Lung Tumorigenesis and Cell Growth through IGF1 Signaling. Mol Cancer Res. 2017;15(7):896–904. doi: 10.1158/1541-7786.MCR-16-0390. [DOI] [PubMed] [Google Scholar]
- 85.Yamada PM, Mehta HH, Hwang D, Roos KP, Hevener AL, Lee KW. Evidence of a role for insulin-like growth factor binding protein (IGFBP)-3 in metabolic regulation. Endocrinology. 2010;151(12):5741–50. doi: 10.1210/en.2010-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lofqvist C, Chen J, Connor KM, Smith AC, Aderman CM, Liu N et al. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc Natl Acad Sci U S A. 2007;104(25):10589–94. doi: 10.1073/pnas.0702031104. [DOI] [PMC free article] [PubMed] [Google Scholar]