

Summary
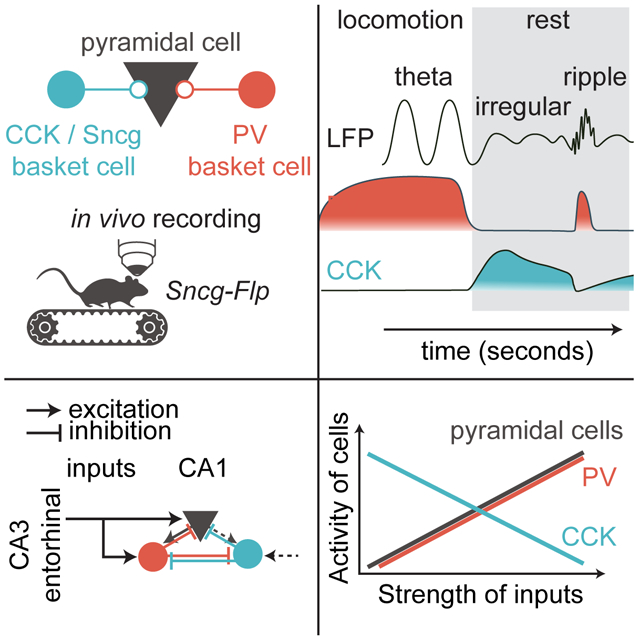
Hippocampal cholecystokinin (CCK-) and parvalbumin (PV-) expressing interneurons constitute two key GABAergic controllers of pyramidal cell output. While the temporally precise and millisecond-scale inhibitory regulation of neuronal ensembles delivered by PV interneurons is well established, the in vivo recruitment patterns of CCK- expressing basket cell (BC) populations has remained unknown. We show in the CA1 of the mouse hippocampus that the activity of CCK BCs inversely scales with both PV and pyramidal cell activity at the behaviorally relevant time scales of seconds. Intervention experiments indicated that the inverse coupling of CCK and PV GABAergic systems arises through a mechanism involving powerful inhibitory control of CCK BCs by PV cells. The tightly coupled complementarity of two key microcircuit regulatory modules demonstrates a novel form of brain state-specific segregation of inhibition during spontaneous behavior.
Graphical Abstract
eTOC blurb
Dudok et al show a complementary perisomatic inhibitory system consisting of CCK- and PV- expressing interneurons that are active at distinct times in an alternating fashion during spontaneous behaviors. Due to PV to CCK inhibition, CCK basket cells are the most active when PV and pyramidal cells are relatively silent.
Introduction
Basket cells (BCs) are GABAergic inhibitory interneurons (INs) that target the somata and proximal dendrites, enabling efficient control of the timing and rate of spiking of their postsynaptic targets. Complementary roles in circuit function were proposed for two types of BCs, expressing either parvalbumin (PV) or cholecystokinin (CCK) based on their striking developmental, molecular, anatomical and physiological dichotomy (Armstrong and Soltesz, 2012; Bartos and Elgueta, 2012; Freund, 2003; Freund and Katona, 2007; Klausberger et al., 2005; Pelkey et al., 2017). Fast-spiking PV INs originate from the medial ganglionic eminence and are ideally suited to synchronize the activity of pyramidal cells (PCs) during network oscillations, due to their fast integration of strong excitatory inputs, reliable action potential firing, and generation of precisely timed postsynaptic currents (Hu et al., 2014). In contrast, regular-spiking CCK INs derive from the caudal ganglionic eminence, receive fewer excitatory inputs that are integrated over longer time scales, and fire action potentials in an accommodating pattern (Armstrong and Soltesz, 2012; Bartos and Elgueta, 2012; Freund and Katona, 2007). Postsynaptic currents evoked by CCK BCs are variable in strength, imprecisely in timed, and often asynchronous (Glickfeld and Scanziani, 2006; Hefft and Jonas, 2005).
The availability of a PV-Cre transgenic driver line, while labeling heterogenous PV IN types (predominantly basket, but also bistratified and axo-axonic cells) (Pelkey et al., 2017), has proven to be invaluable for elucidating the role of PV BCs in cortical circuits, including their function in cortical oscillations, learning and memory, and sensory processing (Hu et al., 2014). Recent studies have indicated that hippocampal CCK IN synapses are regulated by sensory experience and that interventions that induce CCK IN dysfunction can lead to impaired learning (Hartzell et al., 2018; del Pino et al., 2017). However, in contrast to PV BCs, the circuit role of CCK BCs has remained elusive, primarily due to the lack of appropriate transgenic lines. In fact, while connectivity and in vitro electrophysiological data suggested that CCK INs may serve an important role in tuning network excitability as a function of the behavioral state of the animal (Freund, 2003), our knowledge about CCK BC activity in vivo has remained limited to a few individual cells (4 in the CA1, 7 total) recorded from anaesthetized rats (Klausberger et al., 2005; Lasztóczi et al., 2011). The latter studies showed that, during theta oscillations (4-10Hz), PV and CCK BC firing preferentially occurs at distinct phases of the oscillatory cycle, resulting in a temporal segregation of their activities on the scale of tens of milliseconds (Klausberger et al., 2005). However, the fundamental question of how inhibition by CCK INs is organized on the behaviorally relevant time scale of seconds remains critically unknown (Cardin, 2018).
Here, utilizing a transgenic mouse line with genetic access to CCK BCs, we demonstrate in the CA1 region of the hippocampus that CCK and PV INs scale their activities with respect to ensemble neuronal activity in a diametrically opposed manner. While PV IN activity scales with network activity on behavioral time scales (100 ms to seconds), the activity of CCK BCs in the CA1 is instead inversely scaled with network activity, both within brain states and during brain state transitions, by a mechanism involving CCK IN inhibition by PV INs.
Results
Sncg allows genetic targeting of CCK/CB1 BCs in the CA1
Recent classifications of cortical IN types using single-cell transcriptomics have shown that the Sncg gene (gamma-synuclein) is selectively expressed by CCK INs but not by other major IN types, including PV, somatostatin (SST) and non-CCK vasoactive intestinal polypeptide-expressing (VIP) neurons (Gouwens et al., 2020; Tasic et al., 2016; Yao et al., 2020). Although the role of SNCG protein in brain function remains poorly understood (Kuhn et al., 2007), we hypothesized that Sncg promoter activity could be used to specifically target CCK INs in a transgenic mouse line. Indeed, the recently developed Sncg-IRES2-FlpO mouse line (Sncg-Flp), when transfected with conditional viral vectors injected locally into the CA1, labeled predominantly proCCK positive INs in both sexes (75 ± 15%, Fig. 1A-B), while overlap with PV was negligible (2 ± 2%, Fig. 1B, S1A). Sncg labeled 58 ± 17% of all proCCK-positive cells (Fig. S1B). There was no clear indication of any sex-specific differences in proCCK expression in CA1 Sncg cells (72% in female, 76% in male mice). The distribution of labeled axons was restricted to the CA1 pyramidal layer (Fig. 1C), indicating that BCs, but not dendritically targeting CCK INs were labeled (Klausberger et al., 2005; Tricoire et al., 2011). The labeled axons were also immunopositive for CB1 (Fig. 1C-F), a cannabinoid receptor that is highly expressed by CCK INs, but not other IN types (Katona et al., 1999).
Figure 1. Sncg-Flp transgenic mice allow genetic targeting of CCK/CB1 expressing BCs.

A. eYFP was expressed under the control of Sncg-Flp, with AAV vector injected in the CA1. Confocal micrograph (maximum intensity projection of a 60 μm coronal slice). White arrows show colocalization between eYFP and proCCK immunostaining.
B. Somatic colocalization of proCCK (75 ± 15%, n = 244 cells, 6 mice, 2 females, 4 males) and PV (2 ± 2%, n = 177 cells, 4 male mice, Fig. S1). Markers show animals, box plots show median ± confidence interval (CI) and range.
C. Labeled axons were specific to the pyramidal and proximal radiatum layers, and were immunopositive for CB1 receptor (colocalization appears white).
D. High powered confocal micrograph (maximum intensity projection) showing CB1 immunopositive axonal varicosities in the str. pyramidale.
E. Scatterplot of voxel intensities showing elevated CB1 intensities in eYFP-positive voxels. White lines show thresholds and the regression line.
F. eYFP and CB1 colocalization exceeded chance (n = 3 male mice, t(2) = 4.42, p = .048, paired t-test).
G. Whole cell patch clamp recordings of Sncg cells in acute hippocampal slices revealed regular spiking, adapting firing patterns during ±75 pA current steps. See also Fig. S1.
H. Representative biocytin filled cell with axons in the str. pyramidale (axonal varicosities from a single 60 μm slice shown in light grey) and dendrites extending from the str. oriens to the lacunosum-moleculare (dark blue).
I. The soma from panel h was proCCK immunopositive.
J. The axon from panel h was CB1 immunopositive.
K. Schematic of the targeted Patch-seq approach.
L. Dendrogram of the sequenced cells mapped onto a consensus transcriptomic taxonomy. Dark shades identify three “subclasses”. Light shades identify “supertypes” and end branches show “clusters” (Yao et al., 2020, also see Methods).
M. Expression of common IN marker genes in individual Sncg cells (sorted by supertype).
N. Laminar distribution of cell bodies by transcriptomic supertype. Colored markers show the location of the cell body normalized to the thickness of the pyramidale layer (grey shading). Cell type had no effect on soma location. Kruskal-Wallis test, H(7) = 6.47, p = .486, n = 102 cells.
We carried out four sets of experiments to thoroughly characterize the electrophysiological, morphological and transcriptomic types of the cells labeled in the Sncg-Flp line within the CA1 area, as well as their postsynaptic effects. First, we performed targeted patch-clamp recordings in acute hippocampal slices to assess the electrophysiological properties of Sncg cells, combined with biocytin fills of the recorded cells to assess their morphology (Fig. 1G-J); these experiments focused on cells located in the pyramidale and oriens layers, where our subsequent in vivo recordings were targeted. All filled Sncg cells exhibited the adapting, regular spiking firing pattern typical of CCK/CB1 BCs (Fig. 1G,S1E). Analysis of Sncg cell intrinsic electrophysiological properties indicated that distributions of parameter values for the electrophysiological excitability attributes were typically unimodal. During repeated firing, the cells exhibited action potential broadening (Fig. S1D), as reported for CCK INs (Hefft and Jonas, 2005). The cells that were successfully visualized after electrophysiology exhibited basket cell-like axonal arborization largely restricted to the pyramidal layer (Fig. 1H; 19/21 cells), with their somata showing positivity to proCCK (Fig. 1I; 7/9 cells tested) and axons displaying immunoreactivity for CB1 (Fig. 1J; 6/8 tested). While these data confirmed that the Sncg line indeed labeled CCK BCs as suggested by the expression pattern (Fig. 1A), the cell type selectivity was not exclusive, as 2/21 (9.5%) of the visualized cells had axons in the dendritic (radiatum) layer of the CA1 (in agreement with a degree of transcriptomic and functional heterogeneity in a minority of Sncg cells described below).
Next, to assess the correlated electrophysiological- and transcriptomic type of the labeled cell population, we have carried out targeted patch-clamp recordings combined with single-cell whole transcriptome RNAseq (Patch-seq, Fig. 1K). Cells were mapped to a consensus taxonomy previously constructed from transcriptomic data of dissociated cortico-hippocampal INs using a bootstrapping approach (Gouwens et al., 2020; Yao et al., 2020). While cells mapped to clusters scattered across multiple distinct transcriptomic types, the majority of the cells (64%) belonged to a single type, Sncg Krt73 (Fig. 1L). In agreement with our prior immunolabeling results (Fig. 1I-J), Sncg cells consistently expressed Cck (95%) and Cnr1 (CB1, 97%), but not Pvalb (PV, 15%) or Sst (14%, Fig. 1M). Previous studies have reported the non-overlapping expression of either the neuropeptide VIP or the vesicular glutamate transporter vGluT3 by CCK INs (Somogyi et al., 2004). Among the labeled Sncg INs, more cells expressed Slc17a8 (vGluT3, 41%) than Vip (10%, Fig. 1M). The laminar location of somata within the CA1 was similar between all transcriptomic types of Sncg cells (Fig. 1N). Cells with successfully visualized axons after Patch-seq (11/114) were predominantly basket cells (10/11). In addition, Sncg cells recorded in both sets of experiments (i.e. Fig. 1G-H and Fig. 1K-N) had adapting firing patterns, as indicated by their similar adaptation scores (U = 1245, p = .17, n=122, Mann-Whitney U test). Interestingly, additional analysis of the Patch-seq datasets revealed differences between the intrinsic excitability properties of Htr3a and Slc17a8-expressing Sncg cells (Fig. S1F).
The next series of experiments aimed to determine whether the cells labeled in Sncg-Flp line effectively inhibit PC firing in the CA1, as expected from perisomatic INs. We virally expressed the channelrhodopsin hChR2(H134R) (referred to as ChR below) in Sncg cells to optogenetically drive spiking and neurotransmitter release. Whole-cell voltage clamp recordings from ChR-negative PCs in acute hippocampal slices revealed optically evoked inhibitory postsynaptic currents (oeIPSCs, Fig. 2A). These IPSCs were consistent with those arising from CCK/CB1 BCs, featuring characteristics of CCK BC synapses: 1) a distribution of IPSC onset times consistent with adapting presynaptic spiking, 2) fast IPSC rise times consistent with perisomatic inhibition (Lee et al., 2010), and 3) frequent failures and high rate of asynchronous IPSCs (Daw et al., 2009; Hefft and Jonas, 2005) persisting after the light pulse (Fig. 2B). Additionally, oeIPSCs were suppressed after brief depolarization of the PC (Fig. 2C-D), a phenomenon known as depolarization-induced suppression of inhibition (DSI), which is known to be mediated by retrograde endocannabinoid signaling acting on CB1 receptors expressed by CCK BCs (Freund et al., 2003; Katona et al., 1999; Wilson et al., 2001).
Figure 2. Sncg INs inhibit PCs.

A. Sncg INs channelrhodopsin (ChR) expression produced PC oeIPSCs in response to light pulses (50 ms, 0.5 Hz). Individual sweeps shown in grey, average in black or orange.
B. Asynchronous IPSCs persisted following light pulses. One-sided Wilcoxon signed rank test, n = 12 cells. Box plots show median, IQR and range. Asterisks show bins with IPSC rate greater than baseline (*: p < .05; **: p < .01).
C. Brief PC depolarization (to 0 mV for 1 s) resulted in depolarization-induced suppression of inhibition (DSI), evident in reduced oeIPSC amplitude.
D. oeIPSC amplitude across consecutive sweeps, normalized to baseline. Depolarization was delivered between sweeps 6 and 7. Maximal DSI was observed at sweep 8, Wilcoxon signed rank test, W = 1, p = .004, n = 11 cells from 3 animals, 1 male and 2 females, pooled after no significant difference between animals was revealed by Kruskal-Wallis test (p > .05). Asterisks show sweeps that were significantly different from baseline.
E. Schematic of the experiment design. Spike waveforms of an example unit (putative pyramidal cell) are shown. See also Fig. S2.
F. Raster plot of spikes from the same unit across all trials (20 ms light pulses at 2 Hz), aligned on stimulus onset. Note the suppression of spiking persisting beyond the light pulse.
G. Average z-scored firing rate (gaussian filtered) from all units ± CI. Vertical dashed lines indicate the time windows averaged for statistical analysis.
H. Light pulses suppressed firing rates in ChR mice compared to controls by −0.68 ± 0.08 σ between 0 to 40 ms, χ2(1) = 7.55, p = .006, likelihood ratio test, n = 289 units, 6 mice, 4 ChR and 2 YFP, 2 hemispheres per mice). Sex had no effect on response in ChR mice (χ2(1) = 0.5, p = .49, likelihood ratio test, n = 194 units, 2 females, 2 males. There was no significant rebound effect (40 to 80 ms, χ2(1) = 0, p = 1, likelihood ratio test).
To determine whether Sncg INs can control the firing rates of pyramidal cells in vivo, we expressed ChR (or control virus) in Sncg INs. We then performed extracellular silicon probe recordings in the CA1 pyramidal layer of awake mice that were head-restrained on a linear treadmill (Fig. 2E). Brief optogenetic stimulation reduced the mean firing rate of the total population, but not in no-opsin controls (Fig. 2F-H), significantly suppressing the firing rate of 30.4% of the units. The suppression lasted longer than the light pulse without producing a rebound effect (Fig. 2F-H). Such responses are consistent with the expected effect of CCK BC activation, due to the jitter in the onset of IPSCs and asynchronous release (Glickfeld and Scanziani, 2006; Hefft and Jonas, 2005). The responses were distinct from the reported effect of optogenetic PV IN activation, which can produce rebound activity and excess spiking of PCs following rhythmically delivered light pulses (Stark et al., 2013). Due to the high density of Sncg IN axons, rather than cell bodies in the str. pyramidale (Fig. 1C), the placement of the optical fiber immediately dorsal to str. pyramidale likely produced the observed response by directly exciting ChR-expressing perisomatic axons.
Together, these observations indicate that the Sncg-Flp mouse line predominantly labels a specific subpopulation of CA1 Sncg cells, namely, CCK/CB1 expressing BCs that have been well characterized in vitro (Cea-del Rio et al., 2010; Glickfeld and Scanziani, 2006; Hefft and Jonas, 2005). Since only a rather limited number of such cells have yet been recorded in vivo and solely in anaesthetized rats (Klausberger et al., 2005; Lasztóczi et al., 2011), we next examined the recruitment of CA1 Sncg INs in vivo.
Divergent recruitment of PV- and CCK-expressing interneurons during brain state transitions in vivo
Due to the relatively sparse glutamatergic inputs of CCK INs (Mátyás et al., 2004), it has been proposed that their activity is primarily determined by the brain state-dependent activity of subcortical projections, via the various neuromodulatory receptors that CCK BCs express (Armstrong and Soltesz, 2012; Freund, 2003; Freund and Katona, 2007). Locomotion is a strong and straightforward behavioral correlate of an actively engaged (“on-line”) brain state, characterized by theta waves (Buzsáki, 2002). When animals stop running, theta oscillation gives way to irregular local field potential (LFP) activity, interspersed with intermittent sharp wave-ripples (SWRs) that are transient synchronous network events linked to replaying memory traces off-line (Buzsáki, 2015; Karlsson and Frank, 2009). Notably, locomotion modulates the recruitment of interneurons in a cell type-specific manner (Arriaga and Han, 2017; Lapray et al., 2012). However, even the simple question of whether CCK BC activity increases or decreases during running has remained unknown. The Sncg-Flp mouse line provides, for the first time, an efficient tool for interrogating CCK BC function in awake, behaving animals using genetically encoded indicators.
In the first series of experiments designed to understand the activity patterns of CCK BCs, we performed in vivo 2-photon calcium imaging of Sncg INs with simultaneous LFP recordings in the CA1 region of the hippocampus while head fixed mice spontaneously ran or rested on a linear treadmill (Fig. 3A). Surprisingly, these calcium imaging experiments showed that Sncg IN activity was minimal during running, but then dramatically increased for several seconds after the mouse came to a stop (Fig. 3B-C; note that we observed similar changes in Sncg IN firing rates after run-stop in our silicon probe recordings; Fig. S2). These transient increases in Sncg IN activity were likewise not correlated with SWR events, which also occur during immobility. Indeed, in agreement with juxtacellular recordings of CCK BCs in anesthetized rats (Klausberger et al., 2005; Lasztóczi et al., 2011), the majority of Sncg INs showed decreased calcium activity during SWRs in the awake resting state (Fig. 3B-C). Only a minor portion (12 ± 13%) of Sncg cells were activated upon running and SWRs (Fig. 3C-D).
Figure 3. Recruitment of Sncg INs during spontaneous behavior.

A. Schematic of the experimental design for in vivo calcium imaging of Sncg IN activity with correlated detection of SWRs. Asterisk shows the same event on panels A and B. Voltage traces are shown in black, filtered trace in blue, envelope in orange, horizontal lines depict thresholds (3 and 5 SD, see Methods). T = 0 (dashed line) is where the envelope crosses the first threshold.
B. Example trace of in vivo calcium imaging of a single representative Sncg IN. Vertical lines show the onset of SWR events detected in the contralateral CA1.
C. Event-triggered average activity of individual Sncg INs aligned on Run – Stop and SWR events. Cells are sorted by response magnitude. All recordings were made in awake mice, SWRs were analyzed during rest. Note the different time scale and dynamic range for the two plots. T = 0 is the time of run-stop (treadmill speed drops to zero, top) or SWR onset (bottom).
D. The run- and SWR response scores of Sncg INs. Data was pooled from both experiments shown on this figure and Fig. 4. No effect of experimental group or sex was detected on the run response (likelihood ratio test, p(group) = 0.91, p(sex) = .054, n = 141 cells, 28 sessions, 8 animals, 5 females and 3 males) or the SWR response (p(group) = .11, p(sex) = .20).
The negative modulation of Sncg IN activity by locomotion is in stark contrast to the reported positive modulation of PV INs (Lapray et al., 2012). This observation prompted us to systematically compare Sncg and PV IN activity patterns during brain state transitions in awake mice, with intact sensory inputs and ascending neuromodulator systems. To this end, we used Sncg-Flp × PV-Cre double transgenic mice to genetically label both cell types within the same animals (Fig. 4A-B). We performed dual imaging of green (GCaMP6s) and red-shifted (jRGECO1a) calcium indicators, with simultaneous LFP recording to detect SWRs. Additionally, video recordings tracked pupil diameter, as well as non-locomotory events such as whisking and oro-alimentary movements (Fig. 4C). These behavioral attributes are correlates of variations in arousal levels and concomitant brain-wide fluctuations in neuronal activity (Reimer et al., 2016; Stringer et al., 2019). Overlaying the cell type-specific event-triggered average calcium traces revealed a striking negative correlation between the two IN types (Fig. 4D), most prominently observed in PV IN activation and simultaneous Sncg IN suppression when mice started running (run – start, Fig. 4E-F). In contrast, the opposite changes occurred when mice stopped running (run – stop, Fig. 4G-H).
Figure 4. Divergent recruitment of Sncg and PV INs during brain state transitions in vivo.

A. PV × Sncg double transgenic mice targeted distinct genetically encoded reporters to INs.
B. Fluorescent intensity (FI) in IN somata (log scale). Markers show individual cells, colored by FI ratio. N = 373 cells from 6 mice, 1 male, 5 females. Pearson’s R, p < .001.
C. Experimental strategy.
D. Example traces of a single Sncg and a PV IN. See also Fig. S3.
E. Event triggered average traces of Sncg and PV INs aligned to the start of running, along with the pupil diameter, running speed and face movement attributes. Traces show mean ± SEM values across all recorded cells.
F. PV IN responses to running (post-pre; pre = −10 to 0 s, post = 0 to 5 s) were greater than Sncg IN responses by 46 ± 2% DF/F, likelihood ratio test, χ2 = 294, p < .001, n = 469 cells from 3 male mice.
G. Response of Sncg and PV INs during run – stop events.
H. Sncg IN stop responses (post-pre; pre = −10 to 0 s, post = 0 to 5 s) were greater than in PV INs by 43 ± 2% DF/F, likelihood ratio test, χ2 = 372, p < .001, n = 469 cells from 3 male mice. See also Fig. S3.
I. Response of Sncg and PV INs during SWRs.
J. The SWR response of PV INs (post-pre; pre = −1 to 0 s, post = 0 to 128 ms) was greater than the response of Sncg INs by 6 ± 0.7% DF/F, likelihood ratio test, χ2 = 57, p < .001, n = 262 cells from 2 male mice.
K. Response of Sncg and PV INs to face movements. The events shown here were detected in the absence of running. Note that the value of the face movement attribute was also high during running (panels e, g, different y axis scale).
L. The response of PV INs to face movement events (post-pre; pre = −10 to 0 s, post = 0 to 1 s) was greater than the response of Sncg INs by 15 ± 1% DF/F, likelihood ratio test, χ2 = 245, p < .001, n = 469 cells from 3 male mice. Box plots show median ± IQR, whiskers show range, notches represent CI.
During SWR events, rapid and transient activation of PV INs coincided with a rapid decrease of Sncg IN activity after a gradual increase before the SWR (Fig. 4I-J). Conversely, at the onset of bursts of facial movements during immobility (with coincident transient pupil dilation), PV IN activation occurred together with Sncg IN suppression, followed by the opposite changes after the events. These facial movement-related changes were in general similar to the PV and Sncg activity patterns at the start and stop of running, except that they were faster and smaller (Fig. 4K-L). The consistent pattern across these activity changes was that PV IN responses positively correlated with behavioral readouts of attentive brain states and with SWRs (note that SWRs are involved in the reactivation of PC assemblies, Buzsáki, 2015), whereas Sncg IN responses negatively correlated both with these attentive brain states and PV IN activity.
To ascertain that the opposite recruitment of PV and CCK INs was not arising from the use of distinct calcium sensors in the preceding experiments, we additionally examined both INs populations simultaneously using GCaMP6f, performing post hoc identification of PV- and CCK-expressing cells through immunostaining. To achieve this, we used a double conditional strategy exploiting the interneuron-specific expression of the hDlx enhancer sequence in CCK-Cre mice (Fig. S3A, Taniguchi et al., 2011; Dimidschstein et al., 2016). We have detected sizable populations of both CCK and PV INs using this strategy (14% and 25%, respectively, Fig. S3B-D). Such a distribution is in agreement with reports that genetic targeting based on the Cck gene labels diverse interneuron populations, including PV-positive fast-spiking cells in the amygdala and the neocortex (Rovira-Esteban et al., 2019; Whissell et al., 2019), as well as with single cell RNAseq studies showing Cck expression across various interneuron types (Tasic et al., 2016; Zeisel et al., 2015). While most INs increased their activity in these experiments, a small subset of cells became strongly suppressed during running (Fig. S3E). Post-hoc identification of units based on proCCK and PV immunoreactivity confirmed that CCK- and PV-positive INs were activated in a remarkably anticorrelated fashion (Fig. S3), as with the experiments in Fig. 4. Note that negative correlation with running was also reported for a subset of CCK/VIP INs (Turi et al., 2019).
Consistently across experiments and transgenic lines, Sncg/CCK INs often displayed a characteristic response pattern. CCK IN activity was suppressed during running, then peaked at 3.52 ± 2.76 s after stop (mean ± SD of individual events), before gradually decreasing over the course of several seconds. Throughout the rest of the paper, we will refer to this response pattern as the “run – stop” response. While the response amplitude was greater after longer periods of running, robust run – stop responses were observed regardless of the parameters of the preceding running episode (Fig. S3L). The majority of Sncg cells (80 ± 22%) located in the str. pyramidale displayed a significant run – stop response, while fewer of the str. oriens Sncg cells did so (66 ± 16%, U = 57, p = .03, n = 14 mice, Mann-Whitney U test, Fig. S3J). The run – stop response of axons in the str. pyramidale was highly homogenous (Fig. S3K), indicating that Sncg BCs displayed a predominantly positive run – stop response, and that the minority of cells with a different run - stop response are likely not BCs. In the following experiments, we have focused our recordings on the str. pyramidale.
Control of Sncg cell recruitment involves inhibition by PV interneurons
To discern the relative explanatory power of behavioral state and PV IN activity on the calcium signals in the CA1 of Sncg mice, we have constructed linear regression models (see Methods) to predict average Sncg IN calcium signals within 1-second time bins from features of PV IN activity and behavior (speed, pupil diameter, face movements). These models yielded a highly accurate fit (R2 = 0.87, Fig. 5A, S4C) when all features were used as input, including copies of the features delayed by up to 10 seconds to account for potential delayed interactions (see Methods). Next, we tested the contribution of various features to prediction accuracy by removing them from the input. Importantly, PV cell activity alone allowed significantly better prediction accuracy than behavioral attributes alone (Fig. 5B). The response of Sncg cells during run/rest state transitions was predicted from behavioral attributes even if the activity of PV INs was ignored, but only if delayed interactions were modeled (Fig. 5B, S4D-E). However, fluctuations during immobility were not accurately predicted without features of PV IN activity (Fig. 5B, S4E). These results suggest that PV IN activity is a strong predictor of Sncg IN recruitment on the time scale of seconds, both during run-rest transitions and during resting state fluctuations. Thus, we next tested how effectively inhibition by PV INs regulates Sncg IN activity.
Figure 5. Sncg IN activity is suppressed by PV INs.

A. Example dual calcium imaging of Sncg and PV IN populations (top), with simultaneous recordings of treadmill movement, pupil diameter and face movements (bottom). The dotted line shows a linear model fit of Sncg IN activity using PV IN activity and the displayed behavioral attributes as input features (R2 = 0.87 ± 0.06, n = 16 sessions, 3 male mice).
B. Prediction scores of models assessing the effect of removing certain features from the inputs. During run-rest transitions (green bars), delay (effect size = 0.24 ± 0.02, χ2 = 112, p < .001, n = 16 sessions, 3 animals, likelihood ratio test) and PV (0.25 ± 0.02, χ2 = 68, p < .001) were significant factors, while behavior was not (χ2 = 1, p = .46). Similarly, during immobility, delay (0.2 ± 0.02, χ2 = 89 p < .001) and PV (0.28 ± 0.02, χ2 = 104 p < .001) are significant factors, while behavior is not (χ2 = 4, p = .06). ***: PV IN activity allowed more accurate prediction than behavioral parameters (p < .001, n = 16, Wilcoxon signed rank test on paired sample). Box plots show median ± IQR, whiskers show range, notches represent CI. See also Fig. S4.
C. ChR-associated mCherry expression in PV INs and GCaMP in Sncg INs. The Sncg IN somata were surrounded by the mCherry-expressing axons (white arrows) of PV INs.
D. Appositions of PV IN axons with the somata and proximal dendrites of Sncg IN. N = 37 cells from 6 mice, 1 male, 5 females. Colored markers show cells, black markers show animals, error bar shows median ± IQR.
E. Opsin (C1V1) expression in PV INs resulted in oeIPSCs in Sncg INs in response to light pulses (200 ms, 0.5 Hz). Individual sweeps shown in grey, average in blue. The maximal response during the stimulus window was averaged across sweeps. N = 6 Sncg INs from 3 mice.
F. During calcium imaging, trains of light pulses (3 s total, 15 ms each at 15.6 Hz) were delivered in a closed loop system, triggered at 50% probability 1 s after running ceased. No light trials did not alter the run - stop response in Sncg INs. The baseline (before run - stop) is subtracted from these traces.
G. The run - stop response of Sncg INs was suppressed by light in mice expressing ChR in PV INs.
H. Closed loop driving of PV INs suppressed Sncg INs during the stop response (effect of ChR with light compared to all controls: −33 ± 11% DF/F, χ2(1) = 10, p = .0017, likelihood ratio test, n = 128 cells * stimulus type, 16 sessions, 6 animals, 5 males, 1 female). **: Wilcoxon rank sum test for effect of light between Sncg cells from ChR and control mice, W = 247, p = .002, n =61, effect size r = 0.38. n.s.: no light trials, W = 443, p = .32.
Although CCK INs densely receive local inhibitory synapses (Mátyás et al., 2004), and there is anatomical evidence suggesting synapses between PV and CCK INs (Karson et al., 2009), previous reports demonstrated only CCK-to-PV, but not PV-to-CCK BC functional connectivity (Acsády et al., 2000; Karson et al., 2009; Savanthrapadian et al., 2014). Notably, PV axonal boutons were detected impinging on all tested Sncg cell bodies (37/37, Fig. 5C-D). To directly test functional connectivity in vitro, we have expressed the opsin C1V1 and eYFP in PV INs using the PV-specific E2 promoter (Vormstein-Schneider et al., 2020) in Sncg-Flp mice (Fig. 5E). In the presence of glutamate receptor blockers, optogenetically driving PV INs evoked robust compound IPSCs in all of the tested tdTomato-expressing Sncg INs (Fig. 5E; n = 6), demonstrating functional synaptic inputs from PV to Sncg INs. As expected, cells expressing C1V1-eYFP were fast spiking (n = 3, not shown), and PV-oeIPSCs on PCs were not suppressed by depolarization (Fig. S4F; compare with the DSI of Sncg-oeIPSCs, Fig. 2C-D), indicating the specificity of the viral targeting. To test whether PV INs are capable of suppressing Sncg IN activity in vivo, we have expressed either ChR or no-opsin control (mCherry) in PV INs, in combination with GCaMP in Sncg INs. During calcium imaging, pulses of blue light were delivered through the imaging objective to record Sncg IN calcium responses to optogenetically driving PV INs. During immobility, Sncg cell calcium signals were suppressed by blue light in ChR-expressing mice (by −24 ± 11% DF/F, χ2(1) = 5, p < .03, likelihood ratio test, n = 41 cells, 17 sessions, 6 animals). In order to determine whether PV IN inhibition remains effective during the run – stop response when Sncg IN activity is maximal, we modified the protocol to trigger the light 1 s after the end of running in a closed-loop manner, with 50% probability. Sncg cells displayed the characteristic run – stop response under both no-light and no-opsin control conditions, which was instead robustly suppressed by blue light in mice expressing ChR in PV INs (Fig. 5F-H). Optogenetic stimulation suppressed Sncg IN activity to levels below the baseline observed during running (Fig. 5G), corroborating our earlier electrophysiological observation that the firing rate of Sncg INs is greater than zero during running (Fig. S2). Given the specificity of axo-axonic cells for PCs (Buhl et al., 1994), the numerous appositions of PV IN boutons on Sncg IN somata (Fig. 5C,D) suggests that the PV IN cell type involved in controlling Sncg cell activity are likely BCs. Despite the strong average inhibitory effect of PV INs, some Sncg INs were unaffected by the stimulus (Fig. 5H), which may be due to limited optogenetic activation or variable innervation of Sncg cells by PV INs.
Having established the capacity of inhibitory PV IN inputs to robustly control Sncg IN responses in vivo, we next investigated whether specific excitatory inputs may be involved in driving Sncg IN activity during run – stop responses. The main inputs to CA1 arrive either from CA3 via the Schaffer collaterals (predominantly in the stratum radiatum) or from the lateral and medial entorhinal cortex (LEC and MEC, respectively) via the temporoammonic pathway (in the lacunosum-moleculare). CA2 also innervates CA1 (predominantly in the stratum oriens), and certain CA2 PCs are involved in synchronous events during immobility that are distinct from SWRs (Kay et al., 2016). Exploiting the reproducible run – stop responses of Sncg INs, we assessed the correlation of these various inputs with Sncg IN activity during immobility. The axons of PCs in distant areas were labeled with GCaMP and we determined the run – stop response of the axons in CA1 by calcium imaging (Fig. S5). While the recruitment of CA3 and MEC axons during run – stop responses did not exceed chance levels, a small but significant subpopulation of axons from both the LEC and CA2 were recruited specifically after stop (7% and 13%, respectively, Fig. S5H). Thus, while small subsets of excitatory inputs became activated during the run – stop response, potentially contributing to Sncg IN recruitment, the vast majority of axons of these major glutamatergic inputs remained silent, further underlining the particular importance of the PV IN control of CA1 Sncg cells.
Sncg interneuron activity scales inversely with network activity during spontaneous behavior on the time scale of seconds
Thus far, we have shown that Sncg IN activity is negatively modulated by locomotion (Fig. 3, 4), and that the activity of Sncg and PV INs are inversely correlated (Fig. 4, 5). These observations and the reported positive correlation between brain-wide average neuronal activity and spontaneous behaviors (Stringer et al., 2019) imply that Sncg IN activity is also inversely correlated to the average activity of the overall network. To directly test this prediction, we performed dual calcium imaging of the total CA1 neuronal population and Sncg INs (Fig. 6A-B). Indeed, we found that average neuronal activity was remarkably anticorrelated to Sncg IN activity (Fig. 6C). To quantify this correlation across a wide range of time scales, we have designed an approach to compute the average activity of Sncg cells as the function of the average activity of all neurons. Recordings were segmented into equally sized non-overlapping time bins and then sorted according to the DF/F values of all neurons (Fig. 6D-E; see also S6A). Such analysis revealed that when considering entire recording sessions that span across brain states, Sncg IN activity monotonically decreases with increasing activity of the overall neuronal ensemble (Fig. 6E). This analysis was performed across a range of temporal resolutions (0.1 to 10 s), as well as with various temporal offsets between the cell type-specific averages, and a quantitative metric of correlation was computed for each combination of resolution and offset (Fig. S6A, Methods). The data showed negative correlation between Sncg INs and all neurons that was maximal at a resolution of 1.0 s (Fig. 6F; note that while example traces such as in Fig. 6C are displayed using temporal smoothing throughout the paper, all analysis was performed on the raw data). Importantly, the negative correlation between the Sncg cells and all neurons was observed both during the entire recording as well as during specific subsets of the recording (e.g., run-rest transitions or immobility, Fig. S6D).
Figure 6. Sncg IN activity scales inversely with network activity during spontaneous behavior, while PV IN activity scales positively.

A. Labeling strategy for the simultaneous recording of the activity of Sncg INs and all neurons.
B. Example average time projection from a cropped region of a calcium movie. The arrow highlights an Sncg IN (cyan) among other neurons (red).
C. Example calcium traces (average of 5 Sncg cells and 1677 unidentified neurons). Note the coordinated inverse changes in fluorescence of Sncg INs and all neurons during running (shaded areas) and immobility (arrows highlight the run-stop response and fluctuations of activity during immobility).
D. DF/F traces were averaged in 1 s time bins, and the bins were sorted based on the intensity value of all neurons. The average running speed in each bin is also shown.
E. Normalized activity of Sncg INs and all neurons after averaging the traces into 25 equally sized bins according to rank by DF/F of all neurons (n = 13 sessions, 5 male mice). The curves and shaded areas show mean ± SEM.
F. Maximal absolute correlation was negative at 1.0 s resolution and zero offset, black dashed lines show the location of the maximum. See Fig. S6 and Methods for the description of the correlation metric.
G. Labeling strategy for the simultaneous recording of the activity of PV INs and all neurons.
H. Example image as in B. Arrows highlight PV cells coexpressing GCaMP (green) and mCherry (orange).
I. Example calcium traces (average of 21 PV cells and 951 unidentified neurons). Note the coordinated increase of activity during running (shaded areas). Arrows highlight coordinated fluctuations of activity during immobility.
J. Averaged and sorted 1 s time bins reveal strong correlation between PV IN and overall average neuronal activity.
K. Normalized activity of PV INs as a function of rank by DF/F of all neurons (n = 4 sessions, 4 male mice).
L. Maximal absolute correlation was positive at 0.78 s resolution and zero offset.
PV BCs are recruited by the excitatory inputs that drive PCs (feed-forward inhibition) and also by the PCs themselves (feed-back inhibition). This connectivity predicts coordinated changes in excitatory and inhibitory synaptic activity as input levels fluctuate (Isaacson and Scanziani, 2011; Markram et al., 2004). However, extensive evidence shows that PV BCs and PCs are active in distinct phases of hippocampal network oscillations segregated on a time scale of 5ms to 100 ms, corresponding to fast ripple and slower theta waves, respectively (Klausberger et al., 2003; Varga et al., 2012). To determine the correlation of PV IN activity to ensemble network activity on the time scale of seconds, we employed the analysis approach we just used for Sncg INs above, applying it to data obtained from simultaneous recordings of PV IN and overall neuronal population activity (Fig. 6G-H). Overlaying the calcium traces from all cells compared to PV INs revealed a striking correlation in the fluctuations of ensemble neuronal activity with PV IN activity both during running and immobility (Fig. 6I). Further analysis revealed that PV IN activity monotonically increases with rising overall neuronal activity (Fig. 6J-K), with positive correlation across the range of temporal resolutions (Fig. 6L). The data showed that the correlation was maximal at a resolution of 0.78 s and zero offset between the two cell types. Importantly, the positive correlation was not simply caused by the fact that the total neuronal population includes PV cells, as similar results were obtained when PV INs were excluded from the all neurons group, or when only place cells were included (Fig. S6B-C, see Fig. 7).
Figure 7. Sncg IN activity is inversely scaled to the activity of place cells and PV INs.

A. The activity of identified place cells in an example recording session are shown in frames when the animal was running (n = 884 units, sorted by preferred location).
B. The position of the mouse was accurately decoded from the activity of spatially selective cells (R = 0.83 ± 0.12 on held out laps, n = 8 sessions from 5 male mice).
C. Example trace showing average in-field place cell activity (see Methods).
D. Activity of in-field place cells sorted by Sncg IN activity.
E. Negative correlation between the activity of in-field place cells and Sncg INs. See also Fig. S6.
F. Maximal absolute correlation was negative at 0.8 s resolution and 0.2 s offset.
G. Relative activity of PV INs is plotted as a function of relative Sncg IN activity.
H. Maximal absolute correlation was negative at 1.0 s resolution and 0.5 s offset, n = 21 sessions, 3 animals, from the same set of experiments as on Fig. 4C-L. See also Fig. S7.
Due to the relatively sparse, spatially selective activity of CA1 PCs, average network activity is not necessarily representative of the activity of place cells coding for the location of an animal. Is it possible that Sncg IN activity is monotonically decreasing in the face of increasing average network activity, but at the same time is correlated to place cell activity? To test this, we have identified place cells in recordings performed while mice ran for water rewards (reward locations randomized in each lap) on a cue-rich treadmill (Fig. 7A). Identified place cells became activated in sequences repeating lap-by-lap, enabling a decoder to accurately predict the held-out position of the mouse from calcium traces (R = 0.83 ± 0.12), indicating that a spatial map of the treadmill context was formed (Fig. 7A,B). In contrast, Sncg IN traces did not encode location (R = 0.13 ± 0.11). In-field place cells (see Methods) had maximal calcium signals during running, while Sncg INs were suppressed (Fig. 7C, D). Similarly to average network activity, the activity of in-field place cells decreased monotonically with increasing Sncg IN activity (Fig. 7E,F), including during periods restricted to immobility or run-rest transitions (Fig. S6E).
Sncg IN activity is negatively correlated with the activity of the neuronal ensemble as a whole (Fig. 6E), whereas PV IN activity is positively correlated (Fig. 6K). This predicts that Sncg IN activity is also inversely correlated to PV IN activity throughout the trials and not only during specific brain state transitions (Fig. 4E-L). Indeed, we found a strong negative correlation between Sncg and PV IN responses on the time scale of seconds when we considered the entire recording sessions regardless of brain states (Fig. 7G,H). These results demonstrate a fundamentally distinct temporal organization of inhibition by CCK BCs and PV INs in awake, behaving animals, not only in relation to brain state changes, but also more generally during fluctuations of network activity on the time scale of seconds (see arrows in Fig 6C,I).
Discussion
Our study addresses the question of how the complementary inhibitory systems of CCK and PV INs are temporally organized on behaviorally relevant time scales (0.1 s to several seconds). We demonstrate that a novel Sncg-Flp mouse line allows specific genetic targeting of CCK BCs, and show that while PV IN activity scales with the activity of the CA1 neuronal ensemble, CCK BC activity is instead inversely scaled. The opposite scaling of IN cell types results in a pronounced brain state-specific temporal segregation of inhibitory systems during spontaneous behavior. During running, non-locomotory movements and SWR episodes, PV INs are recruited and CCK BCs are suppressed. Efficient inhibition of CCK BCs by PV INs inversely couples the activity of the two cell types, resulting in a “hand-off” of inhibition at transitions between brain states.
The majority of cells labeled by the Sncg-Flp line were regular spiking, adapting BCs (Fig. 1H), immunopositive for proCCK and CB1 (Fig. 1I-J). This makes the line an extremely valuable tool for genetically targeting CCK BCs, which was previously unresolved because all prior known markers of these cells (e.g. CCK, CB1, vGluT3) are also expressed by PCs or some non-CCK INs (Tasic et al., 2016; Zeisel et al., 2015). In fact, even intersectional labeling approaches yield only a minority of CCK cells (Fig. S3). While rare outliers were observed in our transcriptomic, morphological and functional data from the Sncg line, the predominance of CCK BCs among the Sncg INs indicates that the in vivo population characteristics of the recorded cells in this novel mouse line reflect CCK BC activity as opposed to other CCK IN types. However, additional subpopulations of CCK BCs, unlabeled by the Sncg-Flp approach, may exist. On the other hand, the inverse coupling of PV IN activity to Sncg INs is likely representative of PV BCs, as 1) BCs constitute the majority of cells labeled in PV-Cre, and 2) similar results were found when specifically analyzing PV INs activated in SWR events (Fig. S7A-F), a subsample expected to exclude axo-axonic cells that fire less during SWRs in vivo (Varga et al., 2014; Viney et al., 2013).
The network-wide average activity of neurons is not static but rather fluctuates depending on behavioral context, attentional level or general motor activity (Poulet and Crochet, 2019). Therefore, how does inhibition scale with these fluctuations? At the most fundamental level, the mode of scaling can be positive, negative, or constant (Fig. S7G). Both feedback and feed-forward inhibition are expected to scale positively with the strength of excitatory inputs and the activity of local principal neurons driven by these inputs (Isaacson and Scanziani, 2011; Markram et al., 2004). We show that while PV INs indeed scale positively with PC activity, CCK BCs instead scale negatively (Fig. 6, S7H). The inverse coupling of these two IN types results in the segregation of their activity into distinct temporal domains. Such temporal segregation on the time scale of seconds is unexpected in light of the reported firing patterns in anaesthetized animals, which indicate that both CCK and PV INs are spiking during each cycle of the theta oscillations, albeit separated by tens of milliseconds (Klausberger et al., 2005). In spite of the seemingly conflicting observations, our results showing the relative, but not complete suppression of CCK INs during locomotion (Fig. 5, S2) thus extend, rather than contradict those earlier reports about theta-related firing patterns of CCK and PV INs. Caveats of our results obtained using GCaMP imaging are the lack of single-spike resolution, the potential calcium buffering effect of the sensor, as well as the differential expression of calcium-binding proteins by CCK and PV INs. However, evidence of increased firing rate upon run-stop events was also observed in Sncg neurons using extracellular recording (Fig. S2).
While current models of hippocampal CCK BC function build on the specific phase preference of CCK BCs during theta oscillations (Freund et al., 2003; Klausberger et al., 2005; Valero and de la Prida, 2018), our results highlight the recruitment of CCK BCs in non-theta brain states. In contrast to the extensive research on hippocampal network function during rhythmic brain activity (such as theta, gamma and SWR oscillations) (Buzsáki, 2002; Colgin, 2016; Roumis and Frank, 2015), the ways in which the same network operates during irregular activity remains poorly understood. One recent insight into the latter question concerns an immobility-associated functional subnetwork in CA2 that recruits a subset of CA1 PCs during a non-theta, non-SWR brain states (Kay et al., 2016). Further research will be necessary to determine whether such subnetworks are preferentially innervated by CCK BCs that are active in the same temporal domain; the existence of a subset of CA2 fibers active during the run – stop period when the CCK BCs are also active seems to be consistent with the latter possibility. Potential subnetworks involving CCK BCs (Valero and de la Prida, 2018) may be in turn regulated by the immediate-early gene Npas4 that promotes the innervation of PCs by CCK BCs in an experience-dependent manner (Hartzell et al., 2018; Sun et al., 2020).
What mechanisms drive CCK BC activity outside of rhythmic (e.g., theta-dominated) brain states? In contrast to their relatively low number of excitatory synaptic inputs compared to PV INs (Mátyás et al., 2004), CCK INs are thought to receive strong modulatory inputs, based on the diversity of receptors they express (Armstrong and Soltesz, 2012; Freund, 2003; Freund and Katona, 2007). However, how these modulatory inputs influence CCK IN activity in awake, behaving animals remains unknown. The activity of distinct subcortical pathways (e.g. septum, raphe) is reported to increase during locomotion (Fuhrmann et al., 2015; Luchetti et al., 2020), but due to the expression of multiple receptor types with potentially opposing effects (such as both nicotinic and muscarinic acetylcholine receptors), it is not known whether the net effect of these inputs on CCK IN activity is depolarizing or hyperpolarizing in vivo. Here, we show that the activity of CCK INs is suppressed by locomotion, due to inhibition by local PV INs (Fig. 5). CCK INs may also receive long-range inhibitory inputs. Indeed, septo-hippocampal GABAergic inputs targeting CCK INs are active during running (Gulyás et al., 1990; Kaifosh et al., 2013) and inhibitory projections from the EC can also target certain CCK INs (Basu et al., 2016). In contrast, the nature of the mechanisms providing depolarizing drive to CCK INs are less clear. While our data suggest that small specialized subsets of CA2 and LEC axons may play a role in the temporal domain-specific excitation of CCK BCs (Fig. S5), future research will be needed to determine the possible contribution of other glutamatergic afferents (e.g., thalamic) and various potential neuromodulatory inputs (e.g., cholinergic, noradrenergic). Moreover, while our results highlight the complementary recruitment of hippocampal PV and CCK INs in head fixed mice during spontaneous behavior and a random foraging task on a linear treadmill, whether a similar compartmentalization of these inhibitory systems is present in other brain areas and during alternative locomotory and non-locomotory tasks remains unknown.
Interneurons inhibiting other interneurons display a unique mode of inhibition in cortical circuits. Such a disinhibitory role is well known for hippocampal VIP- or calretinin-expressing INs (Klausberger and Somogyi, 2008; Pelkey et al., 2017). While PV INs are known to inhibit other PV INs, it has been suggested that they may not significantly inhibit other IN types (Pfeffer et al., 2013). However, the prior lack of tools for specific genetic targeting prevented testing of whether PV INs inhibit CCK BC populations. We show that PV IN activity is a very strong negative predictor of CCK BC activity, demonstrate functional synaptic connection in vitro, and show that optogenetic driving of PV INs in vivo is sufficient to suppress CCK BC activity even during the run – stop response when CCK INs are otherwise maximally active (Fig. 5). These results suggest a previously underestimated role of PV INs in the disinhibitory control of the CA1 circuit. This disinhibitory motif is likely reciprocal, as suggested by the reported innervation of PV BCs by CCK BCs (Karson et al., 2009; del Pino et al., 2017). Therefore, it seems feasible that reciprocal disinhibitory mechanisms create a push-pull dynamic between these two major inhibitory systems, facilitating the temporal segregation of their activities on the time scale of seconds. On longer time scales, however, a negative feedback limiting CCK IN activity may also exist due to the effects of the CCK neuropeptide which has been shown to robustly depolarize and enhance the output of PV BCs, while simultaneously suppressing the output of CCK BCs (Földy et al., 2007; Lee et al., 2011).
Our results showing diametrically opposite scaling of CCK BC and PV IN activity with ensemble neuronal activity raise the possibility that two BC types with distinct synaptic properties are needed to provide unique forms of perisomatic inhibition in discrete brain states. The complementary intrinsic and synaptic properties of BC types enable PV cells to provide precise, millisecond-scale fast inhibition and the CCK cells to provide a sustained inhibitory tone (Freund, 2003; Armstrong and Soltesz, 2012; Hu et al., 2014). During brain states with rapidly alternating rhythmic activity (such as theta, gamma and ripple oscillations), perisomatic inhibition by PV BCs efficiently suppresses PC activity at specific oscillatory phases with high temporal precision, while in some cases also promoting subsequent rebound spiking (Klausberger et al., 2003; Cardin et al., 2009; Lapray et al., 2012; Varga et al., 2012; Stark et al., 2013; Amilhon et al., 2015; Gan et al., 2017). However, during episodes of irregular circuit activity, a perisomatic inhibitory tone provided by CCK BCs may exert prolonged control of PC firing, which is necessary even in the absence of strong excitatory inputs to suppress spurious noise correlations (Cardin, 2018; Renart et al., 2010). The strong feedforward activation of PV BCs, combined with disinhibitory connectivity between PV and CCK BCs, may establish a mechanism to seamlessly switch between sources of perisomatic inhibition during brain state transitions. The insights gained from the current study will be helpful in the design of future studies concerning CCK BC recruitment during brain states with irregular activity, both regarding normal cognitive functions as well as in various disease states.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Barna Dudok (bdudok@stanford.edu).
Materials Availability
The Sncg-IRES2-FlpO mouse line has been deposited with JAX. The AAV.hDlx.GCaMP6f plasmid has been deposited with Addgene.
Data and Code Availability
The datasets generated during the current study are available from the Lead Contact on reasonable request. Single cell gene expression (RNA-seq) data generated at the Allen Institute for Brain Science will be deposited in the Cell Types database (https://portal.brain-map.org/explore/classes/multimodal-characterization). Analysis scripts used in this study are available from the Lead Contact. Source data for the figures is available on Mendeley Data (http://dx.doi.org/10.17632/3yy6g6v7wp.1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures were carried out in accordance with NIH guidelines and with the approval of the Administrative Panel on Laboratory Animal Care of Stanford University or the Institutional Animal Care and Use Committees of the Allen Institute for Brain Science or Columbia University. A variety of transgenic heterozygous mice of both sexes, on a C57BL/6J background, were used between postnatal days 60 – 150 unless stated otherwise. Sncg-Flp: The Sncg-IRES2-FlpO knock-in driver line (JAX Stock No. 034424) was generated as previously described (Daigle et al., 2018; Madisen et al., 2010, 2015). Validation of this mouse line for the CA1 region is provided in Figs. 1 and 2 of the current study (description of cellular expression patterns in the rest of the brain will be communicated elsewhere, Daigle et al., in preparation). CCK-Cre: Ccktm1.1(cre)Zjh/J (JAX stock # 012706, MGI:5014096) was maintained by back-crossing heterozygous mice with BL/6J animals to avoid functional knockdown of CCK expression (see e.g. Su et al., 2019). PV × Sncg: Heterozygous Sncg-Flp mice were crossed with homozygous PV-Cre (129P2-Pvalbtm1(cre)Arbr/J, JAX stock # 008069, MGI:3590684) animals. Single transgenic PV-Cre mice were homozygous. Axon imaging experiments were performed using wild type and transgenic (pOxr1-Cre, JAX stock #030484; Amigo2-Cre, JAX stock #030215; Grik4-Cre, JAX stock #006474) mice. Mice were housed in groups (2-5 animals, unless single housing was necessitated by veterinary concerns regarding the well-being of the animal) on a normal diet and a normal 12/12 light dark cycle.
METHOD DETAILS
Virus injections
Stock AAV virus preparations were obtained from Stanford Neuroscience Gene Vector and Virus Core (AAV-DJ-Ef1a-fDIO-GCaMP6s, AAV-8-EF1a-fDIO-TdTomato-WPRE, AAV-DJ-EF1a-fDIO-hChR2(H134R)-eYFP-WPRE, AAV-DJ-EF1a-fDIO-eYFP-WPRE), Penn Vector Core (AAV1.Syn.Flex.GCaMP6f.WPRE.SV40, AAV1.Syn.NES-jRGECO1a.WPRE.SV40, AAV1.Syn.Flex.NES-jRGECO1a.WPRE.SV40) or UNC Vector Core (AAV5-EF1a-DIO-hChR2(H134R)-mCherry-WPRE-pA, AAV5-EF1a-DIO-mCherry-WPRE-pA). The AAV.hDlx.Flex.GCaMP6f.WPRE.SV40 virus was generated by Gibson Assembly using the AAV.hDlx. GCaMP6f.WPRE.SV40 (Addgene plasmid #83895) construct as a backbone and was then produced in house by the Dimidschstein laboratory at the Broad Institute using a standard production and purification protocols. AAV-S5E2-C1V1-eYFP was also produced in the Dimidschstein laboratory. Mice were injected with one or more of the viruses (300-400 nl each, full titer) into the right CA1 (2.3mm posterior, 1.5 mm lateral, 1.35mm ventral to bregma) using a Hamilton syringe as previously described (Bui et al., 2018). The different GCaMP6 variants used for Flp- or Cre- dependent experiments (s and f, respectively) were determined by virus availability. The run-stop activity of CCK IN was similar using both approaches.
Slice Electrophysiology
Mice were deeply anesthetized by Ketamine/Xylazine and then transcardially perfused with an ice-cold protective recovery solution containing (in mM): 92 NMDG, 26 NaHCO3, 25 glucose, 20 HEPES, 10 MgSO4, 5 Na-ascorbate, 3 Na-pyruvate, 2.5 KCl, 2 thiourea, 1.25 NaH2PO4, 0.5 CaCl2, titrated to a pH of 7.3-7.4 with HCl (Ting et al., 2014). Coronal slices (300 μm) containing the hippocampus were cut in ice-cold protective recovery solution using a vibratome (VT 1200 S, Leica Biosystems). Brain slices were then incubated in 35°C protective recovery solution for 12 minutes. Hippocampal slices were then maintained in room temperature aCSF consisting of (in mM): 126 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4. All solutions were equilibrated with 95% O2/5% CO2.
Intracellular recordings were performed in a submerged chamber perfused with oxygenated aCSF at 2.5 ml/min and maintained at 33°C by a chamber heater (BadController V, Luigs and Neumann). Hippocampal neurons were visualized using DIC illumination on an Olympus BX61WI microscope (Olympus Microscopy) with an sCMOS camera (Flash 4.0 LT+, Hamamatsu). Epifluorescence illumination from a mercury lamp was used to identify Sncg neurons based on tdTomato fluorescence. Recording pipettes were pulled from thin-walled borosilicate capillary glass (King Precision Glass) using a P97 puller (Sutter Instruments) and were filled with (in mM): 126 K-gluconate, 10 HEPES, 4 KCl, 4 ATP-Mg, 0.3 GTP-Na, 10 phosphocreatine (pH-adjusted to 7.3 with KOH, osmolarity 290 mOsm), as well as 0.2% biocytin. Pipettes had a 3-5 MΩ tip resistance.
Whole cell recordings were performed on tdTomato-positive (Sncg, red) or YFP-positive (PV, green) neurons in the CA1 dorsal hippocampus. Firing properties were assessed during current injection steps (−200 to 750 pA, 1s). Recordings were excluded for neurons with a resting membrane potential above −55 mV or where the series resistance increased by >20% of baseline. Pipette capacitance was neutralized for all recordings. Input resistance was calculated from the change in steady-state membrane potential resulting from hyperpolarizing current injections, while sag was measured as the difference between the steady-state and peak negative potential during a −100 pA hyperpolarizing current injection. Action potential threshold was the voltage where the dV/dt prior to a detected event first exceeded 3 times the standard deviation. Width was the time an action potential, resampled at 100 kHz, exceeded the half-height between threshold and peak voltages. Action potential properties were only measured in the first spike evoked by a depolarizing current for each neuron. Action potential adaptation was measured as the average proportional step-wise change between the first 5 interspike intervals for each neuron in response to depolarizing current injections. After recording, brain slices were transferred into a fixative solution containing 4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer.
For experiments with optogenetic stimulation, glutamate receptor blockers were included in the bath to isolate inhibitory currents (in micromoles): 20 D-APV (Cayman Chemical, #14539) and 5 NBQX (Tocris, #1044). For DSI experiments recording pipettes were filled with (in mM): 130 KCl, 10 HEPES, 2 MgCl2, CaCl2, 1 EGTA, 2 ATP-Mg (pH-adjusted to 7.3 with KOH, osmolarity 290 mOsm). Whole cell recordings were performed on unlabeled pyramidal cells in the CA1 dorsal hippocampus. Optically-evoked IPSCs (oeIPSCs) were measured as inward currents in response to 50 or 200 ms of 465 ± 15 nm (for ChR) or 530 ± 20 nm (for C1V1) illumination provided by a DG-4 arc lamp (Sutter Instruments) in neurons held at −65 mV. The use of different opsins was dictated by virus availability, and both were confirmed to be effective in vitro to drive opsin-expressing IN spiking. Spontaneous IPSCs were recorded at the end of the sweeps. DSI was induced by depolarizing neurons to 0 mV for 1 sec. oeIPSC amplitude was measured as the maximum inward current during the period of optical stimulation. Successful oeIPSCs were defined as having an amplitude > 5 SD beyond the mean current in the 100 ms prior to light stimulation. Neurons with a success rate < 60% during 30 initial light stimulations were not included in DSI measurements. Optically-evoked IPSC amplitudes during DSI experiments were normalized to the average response of each cell to light stimulation prior to depolarization.
Data were acquired in pClamp software (Molecular Devices) using a Multiclamp 700B amplifier (Molecular Devices), low-pass filtered at 2 kHz, and digitized at 10 kHz (Digidata 1440A, Molecular Devices). Data analysis was performed using Clampfit (Molecular Devices) and custom written Python scripts.
Patch-Seq
Tissue processing.
Mice (male and female, Sncg-IRES2-FlpO-neo;Ai65F and Slc32a1-IRES-Cre;Sncg-IRES2-FlpO;Ai65) between the ages of P45-70 were anesthetized with 5% isoflurane and intracardially perfused with 25 or 50 mL of ice-cold slicing artificial cerebral spinal fluid (slicing aCSF), consisting of (in mM): 92 NMDG-HCl, 25 NaHCO3, 25 D-glucose, 20 HEPES, 12 N-acetyl-L-cysteine, 10 MgSO4, 5 Na-ascorbate, 3 Na-pyruvate, 3 myo-inositol, 2.5 KCl, 2 thiourea, 1.25 NaH2PO4, 0.5 CaCl2, 0.01 taurine, pH 7.3, equilibrated with 95% O2/5% CO2. Coronal slices (350 μm) were generated (VT 1200 S Vibratome, Leica Biosystems). Slices were transferred to an oxygenated and warmed (34 °C) slicing aCSF for 10 min, then maintained until recording in room temperature holding aCSF, consisting of (in mM): 84 NaCl, 25 NaHCO3, 25 D-glucose, 20 HEPES, 12.3 N-acetyl-L-cysteine, 5 Na-ascorbate, 3 Na-pyruvate, 3 myo-inositol, 2.5 KCl, 2 thiourea, 2 MgSO4, 2 CaCl2, 1.25 NaH2PO4, 0.01 taurine, pH 7.3, equilibrated with 95% O2/5% CO2).
Patch-clamp recording.
Slices were bathed in warm (34 °C) recording aCSF, consisting of (in mM): 126 NaCl, 26 NaHCO3, 12.5 D-glucose, 2.5 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgSO4, pH 7.3, sterile filtered, and equilibrated with 95% O2/5% CO2. The bath aCSF contained blockers of fast glutamatergic (1 mM kynurenic acid) and GABAergic synaptic transmission (0.1 mM picrotoxin). Thick-walled borosilicate glass (Warner Instruments, G150F-3) electrodes were manufactured (Narishige PC-10) with a resistance of 4–5 MΩ. Before recording, the electrodes were filled with ~1.0-1.5 μL of internal solution, consisting of (in mM): 110 K-gluconate, 10 HEPES, 10 phosphocreatine-Na2, 4 KCl, 1 ATP-Mg, 0.3 GTP-Na, 0.2 mM EGTA, with 20 μg/mL glycogen, 0.5 U/μL RNAse inhibitor (Takara, 2313A) and 0.5% biocytin (Sigma B4261), pH 7.3. Internal solution was sterile filtered and batch-tested for content that could be amplified prior to use.
The pipette was mounted on a Multiclamp 700B amplifier headstage (Molecular Devices) fixed to a micromanipulator (PatchStar, Scientifica). Electrophysiology signals were recorded using an ITC-18 Data Acquisition Interface (HEKA). Commands were generated, signals processed, and amplifier metadata were acquired using MIES (https://github.com/AllenInstitute/MIES/), written in Igor Pro (Wavemetrics). Data were Bessel filtered at 10 kHz and digitized at 50 kHz. Data were reported uncorrected for the measured −14 mV liquid junction potential between the electrode and bath solutions. After formation of a stable seal and break-in, the resting membrane potential of the neuron was recorded (typically within the first minute). A bias current was injected, either manually or automatically using algorithms within the MIES data acquisition package, for the remainder of the experiment to maintain that initial resting membrane potential. Bias currents remained stable for a minimum of 1 s before each stimulus current injection. Upon completion of electrophysiology recordings, the nucleus was slowly extracted from each neuron. The pipette containing internal solution, cytosol and nucleus was removed from the pipette holder and the contents were expelled into a PCR tube containing lysis buffer (Takara, 634894). The pipette was broken on the side wall of the tube while being observed to minimize bubbles. Periodic ‘mock recording’ controls (electrode is inserted into slice, retracted with slight negative pressure) showed negligible contamination compared to the signal obtained from an extracted nucleus.
cDNA amplification and library construction.
We used the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara, 634894) to reverse transcribe poly(A) RNA and amplify full-length cDNA according to manufacturer instructions. We performed reverse transcription and cDNA amplification for 21 PCR cycles in 8-well strips, in sets of 12–24 strips at a time. At least 1 control strip was used per amplification set, which contained 4 wells without cells and 4 wells with 10 pg of control RNA. Control RNA was either Mouse Whole Brain Total RNA (Zyagen, MR-201) or control RNA provided in the SMART-Seq v4 kit. All samples proceeded through Nextera XT DNA Library Preparation (Illumina FC-131-1096) using the Nextera XT Index Kit V2 Set A (FC-131-2001). Nextera XT DNA Library prep was performed according to manufacturer instructions, except that the volumes of all reagents (including cDNA input) were decreased by 50-60%. Subsampling was conducted to a read depth of 0.5 million reads per cell.
Biocytin histology and slice imaging.
Tissue was stained, then mounted sections were imaged as described previously (Gouwens et al., 2019). Briefly, a horseradish peroxidase (HRP) enzyme reaction using diaminobenzidine (DAB) as the chromogen was used to visualize the filled cells after electrophysiological recording. After mounting, operators captured images on an upright AxioImager Z2 microscope (Zeiss, Germany) equipped with an Axiocam 506 monochrome camera and 0.63x optivar. Two-dimensional tiled overview images were captured with a 20X objective lens (Zeiss Plan-NEOFLUAR 20X/0.5) in brightfield transmission and fluorescence channels. Tiled image stacks of individual cells were acquired at higher resolution in the transmission channel only for the purpose of automated and manual reconstruction. Light was transmitted using an oil-immersion condenser (1.4 NA). High-resolution stacks were captured with a 63X objective lens (Zeiss Plan-Apochromat 63x/1.4 Oil or Zeiss LD LCI Plan-Apochromat 63x/1.2 Imm Corr) at an interval of 0.28 μm (1.4 NA objective) or 0.44 μm (1.2 NA objective) along the Z-axis. Tiled images were stitched in ZEN software and exported as single-plane TIFF files.
In vivo extracellular recording in the CA1
Sncg-Flp mice were injected with a virus expressing either ChR-eYFP or eYFP only in both hemispheres of dorsal CA1, as described above. Metal head bars were implanted on the skulls of mice using Super Glue and dental cement afterwards. In vivo extracellular recording experiments were carried out at least 3 weeks post virus injection. On the recording day, a craniotomy was performed at the injection site on isoflurane anesthetized mice. Next, the mice were head-restrained on a treadmill and allowed to recover from anesthesia while a silicon probe with an attached optical fiber (Buzsaki32 or Poly10mm, NeuroNexus) was gradually lowered into dorsal CA1. An Open Ephys recording system (https://open-ephys.org) was used together with Intan 32 channel headstages to acquire data continuously at 30 kHz. 20 ms blue laser pulses (430-490 nM, 4.5 mW power) were delivered at 2 Hz within the first minute of recording sessions to observe the response of single units to light. Offline spike sorting was performed using MountainSort (Chung et al., 2017), followed by visual inspection using the open source software Phy (https://github.com/cortex-lab/phy). Active units with mean firing rate above 1 Hz were included in subsequent analysis (n=194 for ChR group and n=95 for YFP group). To compute the light-triggered average firing rate, timestamps of each unit’s spikes were first aligned and were then sorted into 1 ms time bins. Total spike counts for each unit were summed across trials with a smoothing gaussian kernel (σ = 3 ms) followed by division of total time to obtain average firing rates. To combine data across units with different baseline firing rates, average firing rates for each unit were then normalized with z-scoring to obtain grand normalized average firing rates for each group of mice (i.e. ChR group and YFP group). To estimate the percentage of units suppressed, we determined the z score threshold at which <5% of cells in the YFP group was suppressed (−0.91), and applied it to the ChR group.
Surgery and training for in vivo microscopy
After recovery from virus injection, the cortex above the injection site was aspirated and a stainless steel cannula with attached coverglass was implanted over the hippocampus as described earlier (Kaifosh et al., 2013; Lovett-Barron et al., 2014), followed by a stainless steel headbar. Following recovery, mice were placed on a fluid restriction schedule, and were then trained to run on a linear treadmill with a cue-less belt (2 m) for randomly available water rewards (Danielson et al., 2016). After the mice learned the task (typically running 1m / min), they were returned to an ad libitum water schedule, and recordings were completed without reward presentation. For experiments with place cell recordings (Fig. 7), mice were housed with an in cage running wheel to facilitate running behavior. Mice in place cell experiments were fluid restricted prior to testing on cued belts (constructed of 4 segments featuring different materials/textures and with objects of various sizes/textures affixed to the belt every 20-30 cm). Randomly available water rewards were provided during place cell recording sessions to motivate sufficient running for reliable place field analysis. One day before recording, a chronic electrode (either single-shank silicone probe, NeuroNexus, used in CCK-Dlx mice, or bipolar wire electrode (tungsten, 0.002”, 0.5 mm tip separation, A-M systems), used in Sncg mice, was implanted into the left hippocampus, mirroring the virus injection site. Virus expression and electrode placement was verified post hoc in each mouse included in the study.
In vivo imaging of cell bodies
Head-fixed mice were imaged using a resonant scanner 2-photon microscope (Neurolabware), equipped with a pulsed IR laser (Mai Tai, Spectra-Physics), gated GaAsP PMT detectors (H11706P-40, Hamamatsu), and a 16x objective (0.8 NA WI, Nikon). 2-photon image acquisition was controlled by a Scanbox (Neurolabware) system, which also synchronized video acquisition (Mako, Allied Vision), treadmill speed monitoring and field potential recording via a DAC (National Instruments) to the imaging frames. Treadmill position was recorded and rewards were controlled by a system consisting of Arduino microcontrollers, custom electronics and software (BehaviorMate) (Ahmed et al., 2020; Kaufman et al., 2020; Turi et al., 2019; Zaremba et al., 2017), that were synchronized to the image acquisition system. Speed was determined from change in position, negative speed means the mouse is backing up. Silicone probes were connected (single electrode sites at a time) to an amplifier (ELC-03XS, NPI), and referenced against an immersion bath. Bipolar electrodes were recorded using a differential amplifier (Model 1700, A-M Systems). For optogenetic stimulation, 470 nm light from a 400 mW LED (ThorLabs) was delivered through the imaging objective at maximal intensity (≈25 mW/mm2 before entering the brain). Optogenetic stimuli consisted of 15 ms pulses at the beginning of each imaging frame acquisition, and the PMT detectors were gated for the duration of the stimulus. Data were then recorded following the pulse in the remainder of each frame, at normal frame rate. In closed loop experiments, stimulus trains were triggered by a state machine algorithm with 50% probability when sustained running was followed by >1 s of immobility. Recording sessions lasted 7-15 minutes, during which the mice typically ran 3-10 laps (6-20 meters).
In vivo imaging of axons in the CA1
LEC targeting injections were performed in two male C57BL/6J mice with GCaMP6s expressed in the EC using a recombinant adeno-associated virus (rAAV) driven by a Synapsin promoter (Syn-GCaMP6s-WPRE-SV40). Viral delivery to the LEC was performed by stereotaxically injecting 64 nL of rAAV (2x 32-nL pulses) at three dorsoventral locations using a Nanoject syringe (−3.0 mm AP; −4.7 mm ML; −2.6, −2.5 and −2.4mm DV relative to bregma).
For MEC targeting, two C57BL/6J and one pOxr1-Cre male mice were used. In the C57BL/6J mice, the same GCaMP6s construct was used as the LEC (Syn-GCaMP6s-WPRE-SV40). Meanwhile, the pOxr1-Cre animal was injected with a Cre-dependent AAV1-Syn-FLEX-GCaMP6s-WPRE-SV40 virus. Viral delivery was performed by stereotaxically injecting 64 nL of rAAV at four locations while holding the animal’s head at a 7 degree angle (+ 0.2 mm rostral of the transverse sinus; −3.1 mm ML relative to bregma; −1.2, −1.1, −0.9, and −0.8 DV relative to bregma).
CA2 injections were performed in three Amigo2-Cre male mice that were injected with 64 nL of AAV1-Syn-FLEX-GCaMP6f-WPRE-SV40 virus at three dorsoventral locations (−1.6 mm AP; −2.1 mm ML; −1.2, − 1.1 and −1.0mm DV relative to bregma).
For CA3 axon labeling, two Grik4-Cre male mice were stereotaxically injected with 64 nL of AAV1-Syn-FLEX-GCaMP6f-WPRE-SV40 virus at three dorsoventral locations (−2.3 mm AP; −2.1 mm ML; −2.4, −2.5 and −2.6mm DV relative to bregma).
Axon imaging was conducted using a two-photon microscope equipped with an 8 kHz resonant scanner (Bruker) acquiring 512 x 512 pixels at 30Hz using either a 16x (0.8 NA, 3.0 mm WD Nikon) or 40x (0.8 NA, 3.5 mm WD Nikon) objective. For excitation, we used 50-100 mW laser power (Chameleon Ultra II, Coherent) tuned to 920 nm. Fluorescence signals were collected with photomultiplier tubes (GaAsP PMT, Hamamatsu Model 7422P-40). A dual stage preamp (1.43105 dB, Bruker) was used to amplify signals prior to digitization. Animals were head-fixed under the microscope and trained to run for water rewards on a 2 m cued treadmill belt. Cues where either small objects attached to the treadmill belt or virtual provided by 6.5” LCD panels surrounding the animal. Treadmill velocity as well as reward delivery were controlled by custom electronics and software (BehaviorMate).
Post hoc tissue processing and imaging
Following in vivo recordings, mice were anaesthetized by isoflurane, prior to an intraperitoneal injection of a mixture of ketamine (100 mg/kg) and Xylazine (10 mg/kg) in saline. Following this, the animals were transcardially perfused with saline (0.9% NaCl for 1 minute) and then with fixative solution (4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer). Perfused brains were then post-fixed in the same fixative solution for 24 h at 4°C, prior to slicing on a vibratome (VT 1200 S, Leica Biosystems) to allow for the verification of virus expression pattern and electrode placement, as well as for immunostaining. Note that anti-GFP antibody is used to label GCaMP, and anti-DsRed to label mCherry, as these reporters are derived from the respective fluorescent proteins. Immunostaining was carried out on free-floating coronal sections (60 μm). Sections were washed in 0.1 M phosphate buffer, prior to blocking in 0.05 M tris-buffered saline (TBS) containing 0.03% Triton X-100 and 1% bovine serum albumin. Sections were incubated overnight in one or more primary antibodies diluted in TBS: rabbit anti-proCCK (1:1000, Frontiers Neuroscience #AF350, RRID: AB_2571674); rabbit anti-CB1 (1:3000, Immunogenes, RRID: AB_2813823); mouse anti-PV (1:1000, Sigma #P3088, RRID: AB_477329); sheep anti-PV (1:1000, Invitrogen #PA5-47693, RRID: AB_2609239); chicken anti-GFP (1:3000, Aves #GFP-1020, RRID: AB_10000240); Rabbit anti-DsRed (1:1000, Takara #632496, RRID: AB_10013483). After washing with TBS, sections were incubated for 3 h in secondary antibodies diluted in TBS (1 μg/ml, conjugated with CF405S, CF488A, CF568 or DyLight647, highly cross-absorbed, by Biotium or Thermo Fisher). Sections were then washed and mounted in Vectashield (Vector Laboratories). Confocal images were acquired on a Zeiss LSM 710 imaging system using a 20x 0.8 NA or 63x 1.4 NA objective and analyzed using ImageJ (NIH). Partial reconstruction of a representative neuron was done by loading and aligning maximum intensity projections of confocal z-stacks in Adobe Illustrator, and drawing lines along dendrites, and circular markers on axonal varicosities. Colocalization coefficients and randomized images were computed using the Colocalization Test function of FIJI analysis software. The quantification of cell body colocalization with multiple markers was done by first labeling all reporter-expressing cells within a targeted field (between the alveus and the lacunosum-moleculare of the CA1), blinded to the marker immunostaining channels, and then displaying the included cells one by one and labeling immunopositive cells based on the presence of somatic immunostaining. This manual method was necessitated by the various staining patterns and signal-to-noise ratios of markers. For the analysis of colocalized reporter expression in PV × Sncg mice, every fluorescent cell (in either channel, between the alveus and the lacunosum-moleculare of the CA1) was included. A freehand ROI was drawn on the soma, and the mean fluorescence intensity was measured in both channels. The image acquisition was configured for optimal histograms in both channels, therefore the raw intensity values reflect the relative expression levels. For the quantification of PV inputs impinging on Sncg somata, first the included Sncg somata were labeled on maximal intensity projections of the z-stacks, blinded to the PV channel. Next, each included soma was located in the stack, and all optical sections containing the soma were inspected. PV varicosities that contacted the soma (no dark pixels between the soma and the varicosity) were marked using the ROI Manager in ImageJ, and the coordinates of each varicosity were saved to allow subsequent summary analysis. Figures showing microscopy images were prepared using ImageJ and Adobe Photoshop. Image stacks were processed using the Despeckle and Subtract background commands in ImageJ, maximum intensity projections were then created and exported as 8-bit RGB images. Pseudocoloring and adjustments of gamma were performed for each channel using the Hue and Curves commands in Photoshop. A Gaussian blur filter (radius < 1 pixel) was applied to high-power images obtained with the 63x objective as these were acquired with spatial oversampling. All adjustments were made adhering to common image integrity standards, and were applied uniformly to the entire image.
QUANTIFICATION AND STATISTICAL ANALYSIS
Processing of calcium imaging data
Calcium imaging data were processed and analyzed using Python scripts. Movies were initially motion corrected by rigid translation, followed by non-rigid correction (HiddenMarkov2D function of sima) (Kaifosh et al., 2014). Binary regions of interest (ROIs) were selected in a semi-automated manner to include single cell bodies. For the initial automated detection, movies were divided into segments of 100 frames each, the average intensity projection of each segment was computed and the resulting resampled movie was used for detection. Interneurons were detected using the STICA method of sima, filtered based on size, and inspected against the maximum intensity projection and running-triggered average of each movie. Cells that were not detected with the automated approach were then manually added by drawing additional ROIs. For the detection of pyramidal cells, the PlaneCA1PC method of sima was run on the inverted resampled movie, which resulted in detection of the hollow nuclei of cells. These ROIs were filtered based on size, and binary dilation was performed to include the cytoplasm around the nuclei. In a subsequent step, ROIs were detected in the non-inverted resampled movie, filtered based on size and those that did not overlap with existing ROIs were added to the set. This approach detected a large number of ROIs that corresponded well to individual cells, although some cells may be represented by more than one ROI and some ROIs may contain overlapping parts of multiple cells. ROIs outside the str. pyramidale were excluded. Next, the fluorescence intensity traces were extracted for each ROI by averaging the included pixel intensities within each frame. These raw traces were processed following standard steps for obtaining DF/F traces (Jia et al., 2011), but with a modified approach to determine the time-dependent baseline. Instead of using the minimum of a sliding window that may result in “jumps” of baseline (especially with positively running-modulated cells if the length of running periods exceeds the length of the sliding window), a 3rd degree polynomial was fit on the trace after applying temporal smoothing, removing peaks (detected using continuous wavelet transform by scipy.signal), eliminating periods of running, and ignoring the beginning and end of the recording. The calculated polynomial was then used as a baseline. This approach resulted in reasonably estimated baselines throughout the entire recording. Note that with this approach, negative DF/F values are possible. For inspecting and displaying traces, an exponentially weighted moving average was calculated (decay parameter set to 1 s). However, quantitative analyses were carried out from the non-smoothed traces. Z-scored traces were obtained after determining the standard deviation (SD) of each cell’s baseline by excluding events exceeding 2 SD in two iterations. Deconvolved spike probabilities (for estimating place fields) were computed using the oasis package (Friedrich et al., 2017). Before further analyses, the processed traces of cells were filtered based on unified quantitative quality control criteria to exclude ROIs that were exceedingly noisy or contained no peaks (lack of deconvolved spikes). The traces from pyramidal cells (typically several hundred per field of view) were not curated manually. The traces of interneurons (1 - few dozen per field of view) were further inspected individually by visualizing the traces, the morphological image and the running-triggered average image. ROIs that were affected by movement artifacts, or contained large abrupt shifts of baseline, were excluded from analysis.
Run – stop events were defined as the beginnings of uninterrupted periods of immobility (>50 frames, 7.8 s) that followed uninterrupted periods (>50 frames) of running. To estimate the ratio of Sncg cells displaying run – stop response, the response of a cell to each individual run – stop event was determined in every recording session that contained the given cell. This was compared to a control dataset consisting of 30 randomly picked frames per imaging session. Cells with a positive average response that exceeded chance level determined from the 95 percentile of random events were considered run – stop cells.
Event-triggered averages were calculated after automatically detecting the frames with event onsets by fixed criteria across all sessions. Masks around the event frames were determined such that there was no overlap allowed between consecutive events (that is, a given frame was only included once in the average even if it was close to multiple events). For immobility-associated events, traces were only included until the mouse stayed immobile. A matrix including all events from all cells in all sessions was constructed, and the average was computed in two steps according to the experiment design (i.e. events were first averaged by cell, then averaged by cell type or opsin expression). The number used to calculate SEM was the number of cells in which at least one observation was recorded in the given time point. The process of event detection and averaging was fully automated to remove investigator bias. For the purpose of averaging, behavioral attributes (such as speed, pupil diameter and non-locomotory movements) were weighted according to the number of included cells per session.
LFP traces were automatically processed to detect SWR events as described earlier (Varga et al., 2014). Detected events were then manually curated in a blinded manner by displaying events (1 s-long snippets of LFP) in the order of decreasing ripple-band power, without any context of behavioral or calcium imaging data.
Cropped video recordings containing the mouse face were analyzed by a fully automated pipeline to determine pupil diameter and movement maps in each frame. To determine pupil diameter, the region of the movie containing the eye was cropped and the center of the pupil was labeled (note that the eyeball of head fixed mice is not moving). Horizontal and vertical line profiles were computed across the center of the pupil, and the resulting four distances to maximum negative difference (where the signal intensity drops most sharply) were averaged. To determine facial movements, a region of the snout including the whiskers and excluding the eyes was cropped and processed to compute motion maps in each frame (Powell et al., 2015). The average value of the motion map was computed for each frame and processed into z-scored traces.
For the analysis of axon imaging experiments, the Suite2p (Pachitariu et al., 2017) software package was used to identify ROIs within the resulting 2-photon datasets, as well as to extract fluorescence signals and correct for neuropil contamination. The ROIs identified by suite2p were manually curated post-hoc to ensure both the spatial footprints resembled axon segments or boutons. Additionally, ROIs with no identified calcium events were excluded from analysis as likely false-positive detections. Detected fluorescence was smoothed and DF/F was calculated. Then calcium events were identified by finding imaging frames where DF/F increased to over 2 SD above the median and were considered to end when DF/F returned below 0.5 SD beyond baseline. Calcium events were restricted to those that lasted at least 250 ms for GCaMP6f or 1000 ms for GCaMP6s recordings. Run – stop responsive ROIs were identified by comparing the probability of a calcium event occurring in a 6s window before and after termination of a running bout, excluding reward-related run-stops and comparing to shuffled data for each ROI.
Analysis of Patch-Seq data
Sequencing data processing.
Fifty-base-pair paired-end reads were aligned to GRCm38 (mm10) using a RefSeq annotation gff file retrieved from NCBI on 18 January 2016 (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/all/). Sequence alignment was performed using STAR v2.5.3 (Dobin et al., 2013) in two pass mode. PCR duplicates were masked and removed using the STAR option "bamRemoveDuplicates." Only uniquely aligned reads were used for gene quantification. Gene counts were computed using the R Genomic Alignments package (Lawrence et al., 2013) summarizeOverlaps function, using "IntersectionNotEmpty" mode for exonic and intronic regions separately.
Identifying transcriptomic types.
Since expression levels of individual genes in single cell RNAseq data are inherently noisy, cell types were determined by the total transcriptome as opposed to the expression of selected marker genes. Transcriptomic type mapping of Patch-seq cells was performed as described previously (Gouwens et al., 2020), except using the transcriptomes of dissociated cells from Yao et al. (Yao et al., 2020) as reference dataset. A feedforward neural network classifier was built that receives gene expression data as an input and then outputs the predicted cell type for each individual cell. The reference cells and their corresponding types were used to train the classifier. First, the reference cells were divided into two groups, consisting of training (80%) and test (20%) sets. The input was the log2(CPM+1) gene expression data and the output was a probability vector that predicted the type of each cell. After training, the model was used to predict the cell type for the cells with Patch-seq recordings. Training on dissociated cells and the subsequent label prediction for each cell (with a Patch-seq recording) was performed 100 times (with the training always done on a different, randomly selected 80% of dissociated cells). At the end, the percentage of times a cell was mapped to a given leaf or branch point in the reference taxonomy was defined as the corresponding mapping probability. For each cell, the mapping probabilities were sorted and the highest probability cell type was assigned as the cell type of that cell. Cells successfully mapped to an end branch were plotted on a dendrogram of caudal ganglionic eminence-derived GABAergic cell types defined by Yao et al., 2020. Cells not mapped to end branches with high confidence were omitted. Note that certain supertypes include clusters that are not adjacent on this dendrogram (such as Sncg Krt73 and Sncg Serpinf1) (Yao et al., 2020).
Statistical analysis and modeling
Statistical analysis and modeling were performed using Python and R. Plots were generated using Python and edited for style using Adobe Illustrator. Appropriate statistical tests were selected based on study design and data was checked for meeting the assumptions of the applied tests. The presence of extreme outliers (values above Q3 + 3*IQR or below Q1 - 3*IQR) was identified. Normal distribution of data (or differences in case of paired tests) was tested using a Shapiro-Wilk test and inspecting the Q-Q plot. Reported measures of central tendency and error are mean ± standard error, unless otherwise specified. Confidence intervals on plots represent bootstrapped 95% CI around the indicated mean or median. Tests are 2-sided unless otherwise specified. Sample sizes don’t reflect repeated measures unless otherwise specified. Statistical parameters are reported in the Results or in the figure legends.
When observations were not independent (i.e. multiple cells were recorded from the same session and from the same animal), linear mixed effects models were used. For likelihood ratio tests, a mixed model was constructed (using the Imer function of lme4 in R), with the independent variables (such as cell type, virus, light stimulus or sex) as fixed effects and grouping variables (such as animal and session) as random effects (depending on the experiment design). In the case of nested groups, such as 2 hemispheres per mouse, random effects with random slopes were modeled. The test model was then compared to a null model (identical, but one fixed effect omitted) using the anova function in R to estimate a likelihood ratio by χ2 testing. The reported effect sizes are the fixed effect ± standard error as returned by the test model fitted with the REML approach. In our experiments, the above described method was more stringent compared to using common tests for comparing two groups after pooling animals and sessions (i.e. t-test or Mann-Whitney-Wilcoxon test). No power analysis was used to determine sample sizes. For in vivo experiments, to reduce the number of animals used, a minimum of 3 animals per group was chosen. Ample statistical power was achieved using mixed effects models due to the relatively large number of cells recorded in each animal. The number of cells per animal was not predetermined (all available cells in each animal were included).
To model Sncg cell activity (Fig. 5), input parameters were averaged in 1 second non-overlapping bins and standardized. Delayed interactions were accounted for by adding copies of the input shifted by increments of 1 second (for up to 10 seconds). In control models, parameters of either PV cell activity or behavior were omitted from the input. Bayesian ridge models (BayesianRidge function of scikit-learn in Python) were fitted on the entire session and then scored (coefficient of determination R2 of the prediction, computed by the score method of the model) against the observed data using either the entire session or concatenated segments of the session. Run/rest transitions were selected by including frames in which any running was observed in the previous or the following 10 seconds. Immobility was selected as the inverse of this. Scoring of held-out data was done by splitting the session in 3 equal chunks, fitting the model on 2 of the chunks and scoring the held out one. All 3 combinations were tested and the mean score was computed. To determine the contribution of factors to prediction accuracy, a linear mixed effects model was constructed using delay, behavior and PV as fixed effects, with animal and session as random effects. The dependent variable (prediction R2 score) was transformed sqrt(1 - R2) so that the residuals of the model were evenly distributed and indicated linearity. Then, null models were constructed by omitting either one of the fixed effects to perform likelihood ratio tests, corrected for multiple comparisons. Finally, pairwise comparisons of control groups were performed using Wilcoxon signed rank test on paired sample and correcting for multiple comparisons. The Wilcoxon effect sizes are reported.
To assess the response of cells to optogenetic stimulation (Fig. 5), as the effect of opsin and the effect of stimulus was not independent (i.e. we expect the stimulus to be effective only in the presence of ChR), a dummy variable was included to compare observations with ChR and light against all control conditions (ChR and no light, no opsin and light, no opsin and no light). To determine the effect of opsin and the effect of light separately, we have used unpaired Wilcoxon rank sum tests to compare ChR and mCherry responses within each stimulus group. Wilcoxon signed rank tests were performed on paired samples to compare light and no light responses in each virus group.
Correlation between the activity of two neuronal populations (a source and a target) was calculated from z-scored DF/F traces by averaging the traces from all cells, within each cell type, into equally sized time bins of various sizes (the lower bound was determined by imaging frame rate of 66 ms / sample; the upper bound was arbitrarily set to 10 s, since at very slow time-scales fluctuations will be averaged out, yielding trivial results in correlations between neuronal populations). Then the values for each cell type were linearly rescaled so that the minimum value was 0 and the maximum value was 1. A temporal shift was applied to the source trace (from −10 to 10 s, in 0.1 s increments) before the binning to allow the analysis of delayed correlations. The bins were then sorted based on the source values and the same sorting was applied to the values of the target cell type. Then, the sorted values were divided into 25 equally sized bins and averaged. Thus, between different sessions, the value in the same bin represents the relative activity at times when source cell type activity was at the same within-session percentiles. To compute the correlation score for each time binning resolution and offset settings, the null hypothesis was that both the source and target curves were flat lines (i.e. neuronal activity does not fluctuate over time). The strongest positive correlation is expected if both curves go linearly from 0 in the first bin to 1 in the last (i.e. neuronal activity fluctuates between its minimum and maximum in discrete steps that last longer than the bin size, in perfect synchrony between the source and target cell types). Therefore, let R(obs) be the mean square error between the curve and the unity line, with R(null) being the mean square error between the unity line and a flat line at the mean of the curve. The R values of the averaged curves were computed as 1 - R(obs) / R(null). The R values were floored at 0, to avoid confusing a correlation worse than the null case for a negative correlation. The source curve was always positively sloped (due to the sorting). In the case of a negatively sloped target curve, as the strongest negative correlation is expected if the source curve goes from 0 to 1 while the target curve goes from 1 to 0, the negative of the correlation with the inverse unity line was used. Next, the product of the source and target R values was computed for each window size and offset. Then, the location of maximum in the resulting matrix was determined by smoothing the matrix with a 0.3 s gaussian filter and determining the location of the maximal absolute value. To assess the contribution of run/rest transitions, frames were grouped based on the presence of any locomotion within ±10 s and the temporal binning as well as the rest of the analysis was performed on the resulting state-selective traces.
To identify place cells (Fig. 7), first the mutual information (MI) was computed for each cell’s deconvolved firing rate during running and mouse location on the treadmill (binned in 25 bins, 8 cm each). Cells were included if the MI exceeded the 95 percentile of MI values obtained by shuffling the position data. Each cell’s preferred location was determined by computing the distance-weighted average firing rate for each spatial bin, using exponentially decaying weights (weight decayed to half within 0.5m distance), and finding the bin with maximal firing rate. To decode position (binned in 25 bins) from place cell activity (standardized, smoothed DF/F), a multinomial Naive Bayes classifier (MultinomialNB function of scikit-learn) was trained (by holding out data from one lap at a time). The position in every lap was predicted by a model fit on all the other laps, and correlated with actual position. To compute the average in-field place cell activity in each frame, a weighted average was computed, with weights for each cell exponentially decaying with distance between the preferred location and the actual location in each frame (the weights dropped to zero within 20 cm).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-proCCK | Frontiers Neuroscience | #AF350, RRID: AB_2571674 |
| rabbit anti-CB1 | Immunogenes | Anti-CB1 (Rabbit) Polyclonal Antibody, RRID: AB_2813823 |
| mouse anti-PV | Sigma | #P3088, RRID: AB_477329 |
| sheep anti-PV | Invitrogen | #PA5-47693, RRID: AB_2609239 |
| chicken anti-GFP | Aves | #GFP-1020, RRID: AB_10000240 |
| Rabbit anti-DsRed | Takara | #632496, RRID: AB_10013483 |
| Bacterial and Virus Strains | ||
| AAV-DJ-Ef1a-fDIO-GCaMP6s | Stanford Neuroscience Gene Vector and Virus Core | #167 |
| AAV-8-EF1a-fDIO-TdTomato-WPRE | Stanford Neuroscience Gene Vector and Virus Core | #156 |
| AAV-DJ-EF1a-fDIO-hChR2(H134R)-eYFP-WPRE | Stanford Neuroscience Gene Vector and Virus Core | #087 |
| AAV-DJ-EF1a-fDIO-eYFP-WPRE | Stanford Neuroscience Gene Vector and Virus Core | #086 |
| AAV1.Syn.GCaMP6f.WPRE.SV40 | Penn Vector Core | N/A |
| AAV1.Syn.Flex.GCaMP6f.WPRE.SV40 | Penn Vector Core | N/A |
| AAV1.Syn.NES-jRGECO1a.WPRE.SV40 | Penn Vector Core | N/A |
| AAV1.Syn.Flex.NES-jRGECO1a.WPRE.SV40 | Penn Vector Core | N/A |
| AAV5-EF1a-DIO-hChR2(H134R)-mCherry-WPRE-pA | UNC Vector Core | N/A |
| AAV5-EF1a-DIO-mCherry-WPRE-pA | UNC Vector Core | N/A |
| AAV.hDlx.Flex.GCaMP6f.WPRE.SV40 | Dimidschstein laboratory | N/A |
| AAV-S5E2-C1V1-eYFP | Dimidschstein laboratory | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| NBQX | Tocris | #1044 |
| D-APV | Cayman Chemical | #14539 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Patch Seq data | This paper, Mendeley Data | http://dx.doi.org/10.17632/3yy6g6v7wp.1 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Sncg-IRES2-FlpO | Allen Institute | JAX # 034424 |
| Ccktm1.1(cre)Zjh/J | JAX | # 012706, MGI:5014096 |
| Pvalbtm1(cre)Arbr/J | JAX | # 008069, MGI:3590684 |
| Tg(Oxr1-cre)C14Stl | JAX | # 030484, MGI:5907471 |
| Tg(Amigo2-cre)1Sieg/J | JAX | # 030215, MGI:5897571 |
| Tg(Grik4-cre)G32-4Stl/J | JAX | # 006474, MGI:3699171 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| pClamp | Molecular Devices | v10.7.0.3 |
| Clampfit | Molecular Devices | v10.7.0.3 |
| MIES (Igor Pro) | Allen Institute | https://github.com/AllenInstitute/MIES/ |
| STAR | Dobin et al., 2013 | PMID: 23104886 |
| Genomic Alignments package (R) | Lawrence et al., 2013 | PMID: 23950696 |
| Open Ephys | open-ephys.org | https://open-ephys.org |
| MountainSort | Chung et al., 2017 | PMID: 28910621 |
| Matlab | Mathworks | R2019b |
| ZEN | Zeiss | 2012 SP5 FP1 v14 |
| Scanbox | Neurolabware | v4.6 |
| ImageJ/FIJI | NIH | v1.52p |
| Python | Python.org | v3.6 |
| sima (Python) | Kaifosh et al., 2014 | PMID: 25295002 |
| OASIS (Python) | Friedrich, Zhou and Paninski, 2017 | PMID: 28291787 |
| Suite2p (Python) | Pachitariu et al., 2017 | https://doi.org/10.1101/061507 |
| Other | ||
Highlights.
CA1 CCK basket cells are selectively labeled in the Sncg-Flp mouse line
CCK basket and PV cells display complementary activity during spontaneous behaviors
PV cells inhibit CCK basket cells in vivo
CCK basket cell activity inversely scales with the pyramidal cell ensemble activity
Acknowledgements
Research reported in this publication was supported by the NIH NINDS under Award Number R01NS99457 (to I.S), U19NS104590 (to I.S and A.L), K99NS117795 (to B.D), 5F31NS110316-03 (to J.C.B) and R01NS081297 and 5P01NS074972 (to G.F.), as well as NIMH grants R01MH124047 and R01MH124867 (to A.L.), R01MH111529 (to J.D.), UG3MH120096 (to J.D. and G.F.), and R01MH071679 (to G.F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was also supported by the American Epilepsy Society Postdoctoral Research Fellowship to B.D., the AES Junior Investigator Award to F.T.S., the JSPS Overseas Research Fellowship to S.T., the Canadian Institutes of Health Research Postdoctoral Research Fellowship to J.F., the Simons Foundation Award 566615 (to J.D. and G.F.) and a gift from the Friends-Of-FACES foundation (to J.D.).
We thank the Stanford Neuroscience Gene Vector and Virus Core for providing AAV vector services, and Andrew Olson and the Stanford Neuroscience Microscopy Service, supported by NIH NS069375, for providing microscopy support. Patch-seq experiments were funded by the Allen Institute for Brain Science. We thank the LAS, Tissue Processing, Histology, Imaging, and Molecular Biology teams at the Allen Institute for Brain Science for experiment support and Kristen Hadley, Rusty Mann, Lindsay Ng, Ram Rajanbabu, Jessica Trinh, and Katherine Baker at the Allen Institute for Brain Science for contributing towards Patch-seq recordings. We thank Kimberly Smith for help with transcriptomic processing.
We thank Josh Trachtenberg and Dario Ringach for advice on 2-photon microscopy, Alexandra Linker for contributing to confocal image analysis, Mikko Oijala for help with programming, and Sylwia Felong, Theresa Nguyen, Rika Kumar, Anna Ortiz, Ben Sitzer, Sandra Linder and Kyle Dinkins for technical and administrative support.
Footnotes
Declaration of Interests
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acsády L, Katona I, Martínez-Guijarro FJ, Buzsáki G, and Freund TF (2000). Unusual target selectivity of perisomatic inhibitory cells in the hilar region of the rat hippocampus. J. Neurosci 20, 6907–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MS, Priestley JB, Castro A, Stefanini F, Solis Canales AS, Balough EM, Lavoie E, Mazzucato L, Fusi S, and Losonczy A (2020). Hippocampal Network Reorganization Underlies the Formation of a Temporal Association Memory. Neuron 107, 283–291.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amilhon B, Huh CYL, Manseau F, Ducharme G, Nichol H, Adamantidis A, and Williams S (2015). Parvalbumin Interneurons of Hippocampus Tune Population Activity at Theta Frequency. Neuron 86, 1277–1289. [DOI] [PubMed] [Google Scholar]
- Armstrong C, and Soltesz I (2012). Basket cell dichotomy in microcircuit function. J. Physiol 590, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriaga M, and Han EB (2017). Dedicated hippocampal inhibitory networks for locomotion and immobility. J. Neurosci 37, 1076–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, and Elgueta C (2012). Functional characteristics of parvalbumin- and cholecystokinin-expressing basket cells. J. Physiol 590, 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu J, Zaremba JD, Cheung SK, Hitti FL, Zemelman BV, Losonczy A, and Siegelbaum SA (2016). Gating of hippocampal activity, plasticity, and memory by entorhinal cortex long-range inhibition. Science 351, aaa5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl EH, Han ZS, Lorinczi Z, Stezhka VV, Karnup SV, and Somogyi P (1994). Physiological properties of anatomically identified axo-axonic cells in the rat hippocampus. J. Neurophysiol 71, 1289–1307. [DOI] [PubMed] [Google Scholar]
- Bui AD, Nguyen TM, Limouse C, Kim HK, Szabo GG, Felong S, Maroso M, and Soltesz I (2018). Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science (80-. ). 359, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G (2002). Theta Oscillations in the Hippocampus. Neuron 33, 325–340. [DOI] [PubMed] [Google Scholar]
- Buzsáki G (2015). Hippocampal sharp wave-ripple: A cognitive biomarker for episodic memory and planning. Hippocampus 25, 1073–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA (2018). Inhibitory Interneurons Regulate Temporal Precision and Correlations in Cortical Circuits. Trends Neurosci. 41, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai LH, and Moore CI (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cea-del Rio CA, Lawrence JJ, Tricoire L, Erdelyi F, Szabo G, and McBain CJ (2010). M3 muscarinic acetylcholine receptor expression confers differential cholinergic modulation to neurochemically distinct hippocampal basket cell subtypes. J. Neurosci 30, 6011–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JE, Magland JF, Barnett AH, Tolosa VM, Tooker AC, Lee KY, Shah KG, Felix SH, Frank LM, and Greengard LF (2017). A Fully Automated Approach to Spike Sorting. Neuron 95, 1381–1394.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL (2016). Rhythms of the hippocampal network. Nat. Rev. Neurosci 17, 239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, Takeno MM, Huang L, Gu H, Larsen R, et al. (2018). A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 174, 465–480.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson NB, Zaremba JD, Kaifosh P, Bowler J, Ladow M, and Losonczy A (2016). Sublayer-Specific Coding Dynamics during Spatial Navigation and Learning in Hippocampal Area CA1. Neuron 91, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Tricoire L, Erdelyi F, Szabo G, and McBain CJ (2009). Asynchronous transmitter release from cholecystokinin-containing inhibitory interneurons is widespread and target-cell independent. J. Neurosci 29, 11112–11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi G-A, Guo L, Xu Q, Liu R, Lu C, Chu J, et al. (2016). A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci 19, 1743–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Lee SY, Szabadics J, Neu A, and Soltesz I (2007). Cell type-specific gating of perisomatic inhibition by cholecystokinin. Nat. Neurosci 10, 1128–1130. [DOI] [PubMed] [Google Scholar]
- Freund TF (2003). Interneuron Diversity series: Rhythm and mood in perisomatic inhibition. Trends Neurosci. 26, 489–495. [DOI] [PubMed] [Google Scholar]
- Freund TF, and Katona I (2007). Perisomatic inhibition. Neuron 56, 33–42. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, and Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev 83, 1017–1066. [DOI] [PubMed] [Google Scholar]
- Friedrich J, Zhou P, and Paninski L (2017). Fast online deconvolution of calcium imaging data. PLoS Comput. Biol 13, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann F, Justus D, Sosulina L, Kaneko H, Beutel T, Friedrichs D, Schoch S, Schwarz MK, Fuhrmann M, and Remy S (2015). Locomotion, Theta Oscillations, and the Speed-Correlated Firing of Hippocampal Neurons Are Controlled by a Medial Septal Glutamatergic Circuit. Neuron 86, 1253–1264. [DOI] [PubMed] [Google Scholar]
- Gan J, Weng S, Pernía-Andrade AJ, Csicsvari J, and Jonas P (2017). Phase-Locked Inhibition, but Not Excitation, Underlies Hippocampal Ripple Oscillations in Awake Mice In Vivo. Neuron 93, 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, and Scanziani M (2006). Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat. Neurosci 9, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Sorensen SA, Berg J, Lee C, Jarsky T, Ting J, Sunkin SM, Feng D, Anastassiou CA, Barkan E, et al. (2019). Classification of electrophysiological and morphological neuron types in the mouse visual cortex. Nat. Neurosci 22, 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwens NW, Sorensen SA, Baftizadeh F, Budzillo A, Lee BR, Jarsky T, Alfiler L, Baker K, Barkan E, Berry K, et al. (2020). Integrated Morphoelectric and Transcriptomic Classification of Cortical GABAergic Cells. Cell 183, 935–953.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Görcs TJ, and Freund TF (1990). Innervation of different peptide-containing neurons in the hippocampus by gabaergic septal afferents. Neuroscience 37, 31–44. [DOI] [PubMed] [Google Scholar]
- Hartzell AL, Martyniuk KM, Brigidi GS, Heinz DA, Djaja NA, Payne A, and Bloodgood BL (2018). NPAS4 recruits CCK basket cell synapses and enhances cannabinoid-sensitive inhibition in the mouse hippocampus. Elife 7, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, and Jonas P (2005). Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat. Neurosci 8, 1319–1328. [DOI] [PubMed] [Google Scholar]
- Hu H, Gan J, and Jonas P (2014). Fast-spiking, parvalbumin+ GABAergic interneurons: From cellular design to microcircuit function. Science (80-. ). 345, 1255263–1255263. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, and Scanziani M (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Rochefort NL, Chen X, and Konnerth A (2011). In vivo two-photon imaging of sensory-evoked dendritic calcium signals in cortical neurons. Nat. Protoc 6, 28–35. [DOI] [PubMed] [Google Scholar]
- Kaifosh P, Lovett-Barron M, Turi GF, Reardon TR, and Losonczy A (2013). Septo-hippocampal GABAergic signaling across multiple modalities in awake mice. Nat. Neurosci 16, 1182–1184. [DOI] [PubMed] [Google Scholar]
- Kaifosh P, Zaremba JD, Danielson NB, and Losonczy A (2014). SIMA: Python software for analysis of dynamic fluorescence imaging data. Front. Neuroinform 8, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson MP, and Frank LM (2009). Awake replay of remote experiences in the hippocampus. Nat. Neurosci 12, 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karson MA, Tang A-H, Milner TA, and Alger BE (2009). Synaptic Cross Talk between Perisomatic-Targeting Interneuron Classes Expressing Cholecystokinin and Parvalbumin in Hippocampus. J. Neurosci 29, 4140–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, and Freund TF (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci 19, 4544–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman AM, Geiller T, and Losonczy A (2020). A Role for the Locus Coeruleus in Hippocampal CA1 Place Cell Reorganization during Spatial Reward Learning. Neuron 105, 1018–1026.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay K, Sosa M, Chung JE, Karlsson MP, Larkin MC, and Frank LM (2016). A hippocampal network for spatial coding during immobility and sleep. Nature 531, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, and Somogyi P (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JDB, Cobden PM, Buzsáki G, and Somogyi P (2003). Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature 421, 844–848. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, O’Neill J, Huck JHJ, Dalezios Y, Fuentealba P, Suen WY, Papp E, Kaneko T, Watanabe M, et al. (2005). Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J. Neurosci 25, 9782–9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Haebig K, Bonin M, Ninkina N, Buchman VL, Poths S, and Riess O (2007). Whole genome expression analyses of single- and double-knock-out mice implicate partially overlapping functions of alpha- and gamma-synuclein. Neurogenetics 8, 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapray D, Lasztoczi B, Lagler M, Viney TJ, Katona L, Valenti O, Hartwich K, Borhegyi Z, Somogyi P, and Klausberger T (2012). Behavior-dependent specialization of identified hippocampal interneurons. Nat. Neurosci 15, 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasztóczi B, Tukker JJ, Somogyi P, and Klausberger T (2011). Terminal field and firing selectivity of cholecystokinin-expressing interneurons in the hippocampal CA3 area. J. Neurosci 31, 18073–18093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Földy C, and Soltesz I (2010). Distinct endocannabinoid control of GABA release at perisomatic and dendritic synapses in the hippocampus. J. Neurosci 30, 7993–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Földy C, Szabadics J, and Soltesz I (2011). Cell-type-specific CCK2 receptor signaling underlies the cholecystokinin-mediated selective excitation of hippocampal parvalbumin-positive fast-spiking basket cells. J. Neurosci 31, 10993–11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, Turi GF, Hen R, Zemelman BV, and Losonczy A (2014). Dendritic inhibition in the hippocampus supports fear learning. Science 343, 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti A, Bota A, Weitemier A, Mizuta K, Sato M, Islam T, McHugh TJ, Tashiro A, and Hayashi Y (2020). Two functionally distinct serotonergic projections into hippocampus. J. Neurosci 40, JN-RM-2724-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, and Wu C (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci 5, 793–807. [DOI] [PubMed] [Google Scholar]
- Mátyás F, Freund TF, and Gulyás AI (2004). Convergence of excitatory and inhibitory inputs onto CCK-containing basket cells in the CA1 area of the rat hippocampus. Eur. J. Neurosci 19, 1243–1256. [DOI] [PubMed] [Google Scholar]
- Pachitariu M, Stringer C, Dipoppa M, Schröder S, Rossi LF, Dalgleish H, Carandini M, and Harris K (2017). Suite2p: beyond 10,000 neurons with standard two-photon microscopy. BioRxiv 061507. [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, and McBain CJ (2017). Hippocampal GABAergic Inhibitory Interneurons. Physiol. Rev 97, 1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, and Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci 16, 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pino I, Brotons-Mas JR, Marques-Smith A, Marighetto A, Frick A, Marín O, and Rico B (2017). Abnormal wiring of CCK+ basket cells disrupts spatial information coding. Nat. Neurosci 20, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet JFA, and Crochet S (2019). The Cortical States of Wakefulness. Front. Syst. Neurosci 12, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell K, Mathy A, Duguid I, and Häusser M (2015). Synaptic representation of locomotion in single cerebellar granule cells. Elife 4, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer J, McGinley MJ, Liu Y, Rodenkirch C, Wang Q, McCormick DA, and Tolias AS (2016). Pupil fluctuations track rapid changes in adrenergic and cholinergic activity in cortex. Nat. Commun 7, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renart A, de la Rocha J, Bartho P, Hollender L, Parga N, Reyes A, and Harris KD (2010). The Asynchronous State in Cortical Circuits. Science (80-. ). 327, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumis DK, and Frank LM (2015). Hippocampal sharp-wave ripples in waking and sleeping states. Curr. Opin. Neurobiol 35, 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira-Esteban L, Gunduz-Cinar O, Bukalo O, Limoges A, Brockway E, Müller K, Fenno L, Kim YS, Ramakrishnan C, Andrási T, et al. (2019). Excitation of Diverse Classes of Cholecystokinin Interneurons in the Basal Amygdala Facilitates Fear Extinction. ENeuro 6, ENEURO.0220-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savanthrapadian S, Meyer T, Elgueta C, Booker SA, Vida I, and Bartos M (2014). Synaptic Properties of SOM- and CCK-Expressing Cells in Dentate Gyrus Interneuron Networks. J. Neurosci 34, 8197–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi J, Baude A, Omori Y, Shimizu H, Mestikawy S. El, Fukaya M, Shigemoto R, Watanabe M, and Somogyi P (2004). GABAergic basket cells expressing cholecystokinin contain vesicular glutamate transporter type 3 (VGLUT3) in their synaptic terminals in hippocampus and isocortex of the rat. Eur. J. Neurosci 19, 552–569. [DOI] [PubMed] [Google Scholar]
- Stark E, Eichler R, Roux L, Fujisawa S, Rotstein HG, and Buzsáki G (2013). Inhibition-induced theta resonance in cortical circuits. Neuron 80, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer C, Pachitariu M, Steinmetz N, Reddy CB, Carandini M, and Harris KD (2019). Spontaneous behaviors drive multidimensional, brainwide activity. Science (80-. ). 364, eaav7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Ye W, Liu H, Zhang B, Jendrichovsky P, Hoang AD, Chen X, Lau CYG, Yu D, Xiong W, et al. (2019). Entorhinal Cholecystokinin Enables Theta-Burst Stimulation-Induced Hippocampal LTP and Transfer of Spatial Memory. SSRN Electron. J [Google Scholar]
- Sun X, Bernstein MJ, Meng M, Rao S, Sørensen AT, Yao L, Zhang X, Anikeeva PO, and Lin Y (2020). Functionally Distinct Neuronal Ensembles within the Memory Engram. Cell 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsani D, Fu Y, Lu J, Lin Y, et al. (2011). A Resource of Cre Driver Lines for Genetic Targeting of GABAergic Neurons in Cerebral Cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricoire L, Pelkey KA, Erkkila BE, Jeffries BW, Yuan X, and McBain CJ (2011). A Blueprint for the Spatiotemporal Origins of Mouse Hippocampal Interneuron Diversity. J. Neurosci 31, 10948–10970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turi GF, Li W-K, Chavlis S, Pandi I, O’Hare J, Priestley JB, Grosmark AD, Liao Z, Ladow M, Zhang JF, et al. (2019). Vasoactive Intestinal Polypeptide-Expressing Interneurons in the Hippocampus Support Goal-Oriented Spatial Learning. Neuron 101, 1150–1165.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero M, and de la Prida LM (2018). The hippocampus in depth: a sublayer-specific perspective of entorhinal–hippocampal function. Curr. Opin. Neurobiol 52, 107–114. [DOI] [PubMed] [Google Scholar]
- Varga C, Golshani P, and Soltesz I (2012). Frequency-invariant temporal ordering of interneuronal discharges during hippocampal oscillations in awake mice. Proc. Natl. Acad. Sci. U. S. A 109, E2726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga C, Oijala M, Lish J, Szabo GG, Bezaire M, Marchionni I, Golshani P, and Soltesz I (2014). Functional fission of parvalbumin interneuron classes during fast network events. Elife 3, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viney TJ, Lasztoczi B, Katona L, Crump MG, Tukker JJ, Klausberger T, and Somogyi P (2013). Network state-dependent inhibition of identified hippocampal CA3 axo-axonic cells in vivo. Nat. Neurosci 16, 1802–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vormstein-Schneider D, Lin JD, Pelkey KA, Chittajallu R, Guo B, Arias-Garcia MA, Allaway K, Sakopoulos S, Schneider G, Stevenson O, et al. (2020). Viral manipulation of functionally distinct interneurons in mice, non-human primates and humans. Nat. Neurosci 23, 1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whissell PD, Bang JY, Khan I, Xie YF, Parfitt GM, Grenon M, Plummer NW, Jensen P, Bonin RP, and Kim JC (2019). Selective activation of cholecystokinin-expressing GABA (CCK-GABA) neurons enhances memory and cognition. ENeuro 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, and Nicoll R. a (2001). Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31, 453–462. [DOI] [PubMed] [Google Scholar]
- Yao Z, Liu H, Xie F, Fischer S, Booeshaghi AS, Adkins RS, Aldridge AI, Ament SA, Pinto-Duarte A, Bartlett A, et al. (2020). An integrated transcriptomic and epigenomic atlas of mouse primary motor cortex cell types. BioRxiv 2020.02.29.970558. [Google Scholar]
- Zaremba JD, Diamantopoulou A, Danielson NB, Grosmark AD, Kaifosh PW, Bowler JC, Liao Z, Sparks FT, Gogos JA, and Losonczy A (2017). Impaired hippocampal place cell dynamics in a mouse model of the 22q11.2 deletion. Nat. Neurosci 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Manchado ABM, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science (80-. ). 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the Lead Contact on reasonable request. Single cell gene expression (RNA-seq) data generated at the Allen Institute for Brain Science will be deposited in the Cell Types database (https://portal.brain-map.org/explore/classes/multimodal-characterization). Analysis scripts used in this study are available from the Lead Contact. Source data for the figures is available on Mendeley Data (http://dx.doi.org/10.17632/3yy6g6v7wp.1).