

Abstract
Rationale:
Hypertriglyceridemia (HyperTG) and low high-density lipoprotein cholesterol (HDL-C), both of which are regulated by lipoprotein lipase (LpL) activity, associate with increased cardiovascular disease (CVD). Genetic regulators of LpL actions track with CVD risk in humans. Whether this is due to changes in HDL-C or function, or circulating triglyceride (TG) levels is unresolved.
Objective:
We created HyperTG and HDL-C reduction in atherosclerotic mice to allow the assessment of how HyperTG and reduced HDL-C affect regression of atherosclerosis and the phenotype of plaque macrophages.
Methods and Results:
Atherosclerosis regression was studied in control LpL floxed (Lplfl/fl) mice and tamoxifen-inducible whole-body LpL KO (iLpl−/−) mice with HyperTG (~500mg/dL) and reduced HDL-C (~50% reduction). Atherosclerosis regression was studied using two models in which advanced plaques resulting from hypercholesterolemia are exposed to normal LDL-C levels using aortic transplantation or treatments with oligonucleotides. In a subset of mice, we expressed human cholesterol ester transfer protein (hCETP) to humanize the relationship between apoB-lipoproteins and HDL. HDL particle number (HDL-P), cholesterol efflux capacity (CEC) and HDL proteome were measured in HyperTG mice and humans. Surprisingly, HyperTG and reduced HDL-C levels due to loss of LpL did not affect atherosclerosis lesion size or macrophage content (CD68+ cells) in either model. Expression of hCETP and further reduction of HDL-C did not alter lesions. Sera from iLpl−/− mice had a decrease in total CEC, but not ABCA1-mediated CEC. HyperTG humans, including those with LpL deficiency, had greater ABCA1-mediated CEC and total CEC per HDL-P.
Conclusion:
Atherosclerosis regression in mice is driven by LDL-C reduction and is not affected by HyperTG and plasma HDL-C levels.
Keywords: Triglyceride, lipolysis, lipoprotein lipase lipids, lipids and lipoproteins, Atherosclerosis, Basic Science Research, Cardiovascular Disease, Clinical Studies, Lipids and Cholesterol
Graphical Abstract

INTRODUCTION
Although high-density lipoprotein cholesterol (HDL-C) levels are inversely correlated with cardiovascular disease (CVD), recent genetic studies have questioned whether this correlation reflects causation.1, 2 Fasting levels of HDL-C and triglyceride (TG) are inversely related, thus it is difficult to dissect the importance of each of these lipids. Postprandial TG excursions also inversely correlate with HDL-C3, 4 and some investigators consider HDL-C as a marker for average circulating TG levels.2 While several recent genome-wide association studies (GWAS) failed to directly link HDL-C modulating genes with CVD, genes affecting TG levels—lipoprotein lipase (LpL) and angiopoietin-like proteins 3 and 4—do associate with clinical CVD endpoints1, with reduced LpL activity appearing to increase CVD.
There are limited experimental models linking hypertriglyceridemia (HyperTG) and atherosclerosis, but a large series of experimental studies have documented benefits of raising HDL-C and apolipoprotein A1 (ApoA-I levels; reviewed in5, 6). For example, preclinical studies have shown that increased expression of ApoA-I, the major protein in HDL, reduces atherosclerosis6–8 and normalizes the defect in atherosclerosis regression that occurs with diabetes.9 Some clinical studies have infused HDL or HDL-like particles and shown reduced atherosclerosis development10 with acute beneficial changes in plaque composition (e.g. reduced lipid and macrophage content).11 Methods to markedly induce ApoA-I expression in humans are not available. Other methods to increase HDL-C, such as blocking the exchange of cholesterol and TG between TG-rich lipoproteins and HDL by cholesteryl ester transfer protein (CETP) inhibition, were not beneficial when corrected for the expected benefits of LDL-C reduction by this treatment.12–14
One postulated reason for the lack of benefits with CETP inhibition in humans is that the increase in HDL-C does not reflect an increase in cholesterol efflux capacity (CEC) or reverse cholesterol transport (RCT). In humans, CEC has been touted as a better CVD risk marker than HDL-C.15–17 Moreover, the regression of lesions after marked LDL-C reduction is widely believed to rely on HDL-CEC to remove accumulated cholesterol. However, kinetic studies performed nearly 4 decades ago showed that the variation in HDL-C levels found in most humans did not affect overall cholesterol balance, which implied that CEC does not differ among most people.18 Thus, the role of HDL particles and their functionality in atherosclerosis are unclear.
In the present report, we tested whether reduced HDL-C due to HyperTG resulting from LpL deficiency would affect atherosclerosis regression, as well as CEC, in mice and humans. We hypothesized that LpL-deficiency would decrease CEC by reducing HDL particle numbers (HDL-P) and that this in turn would impair atherosclerosis regression. We chose to study regression, in part, because during this process, cholesterol must exit from atherosclerotic plaques, the first step of CEC. Moreover, current approaches to CVD in humans have a greater focus on the regression of atherosclerotic coronary lesions through marked cholesterol reduction.
Our surprising results show that though LpL deficiency induced a reduction in HDL-C, the now TG-rich HDL particles maintained CEC. This likely allowed normal atherosclerosis regression and illustrates that normal repair of lesions can occur in the setting of HyperTG and reduced HDL-C.
METHODS
Data Availability.
The authors declare that all supporting data are available within the article and its online supplementary files.
Animals.
Global inducible LpL knockout mice were generated by crossing floxed Lpl (Lplfl/fl) mice19 with β-actin driven tamoxifen-inducible Cre (MerCreMer) transgenic mice (Jackson Laboratory) to obtain the β-actin-MerCreMer/LpLfl/fl offspring designated as iLpl−/− mice, as previously described.20 Mice received intraperitoneal (i.p.) injections of tamoxifen (Toronto Research Chemicals, #T006000) or 4-hydroxytamoxifen (Tocris) in corn oil (Sigma) at a dose of 40 mg/kg BW/day for 5 consecutive days. On the day of sacrifice, mice were anesthetized via intraperitoneal (i.p.) injection of ketamine (100mg/kg) and xylazine (10mg/kg). Blood was collected via cardiac puncture in EDTA-containing tubes and mice were perfused with 10% sucrose in saline solution (0.9% NaCl). All procedures were approved by the Institutional Animal Care and Use Committee at New York University Langone Health approved protocol #160907 (“Lipoprotein Lipase and ApoB”). Mice were maintained in a temperature-controlled (25°C) facility with a 12 h-light/dark cycle and given free access to water and food, except when fasting blood was obtained. Mice were fed a laboratory rodent chow diet or western diet (WD; 0.3% cholesterol; Dyets Inc. D101977Gi) as indicated. A mix of female and male mice were studied, and no gender differences have been detected. Figures 1,2,4,5 and Supplemental Figures I, II: Female: Lplfl/fl (+LacZ) n=10, Lpl−/− (+LacZ) n=14, Lplfl/fl + hCETP n= 5, Lpl−/− + hCETP n=4; Male: Lplfl/fl (+LacZ) n=9, Lpl−/− (+ LacZ) n=4, Lplfl/fl +CETP n=6, iLpl−/− +CETP n=3. Figure 3, Male: Lplfl/fl (baseline) n=15; Lplfl/fl (regression) n=19; iLpl−/− (regression) n=11, Supplemental Figure II: Female: Lplfl/fl n= 2, Lpl−/− n=2; Male: Lplfl/fl n=4, Lpl−/− n=2, and Supplemental Figure IV: Female: Lplfl/fl Tie2LPL (baseline) n=4; iLpl−/− Tie2LPL (baseline) n=4; Lplfl/fl Tie2LpL (regression) n=4; iLpl−/− Tie2LpL (regression) n=3.
Figure 1. LpL deficiency increased VLDL-TG and reduced HDL-C, which was further reduced with hCETP expression.
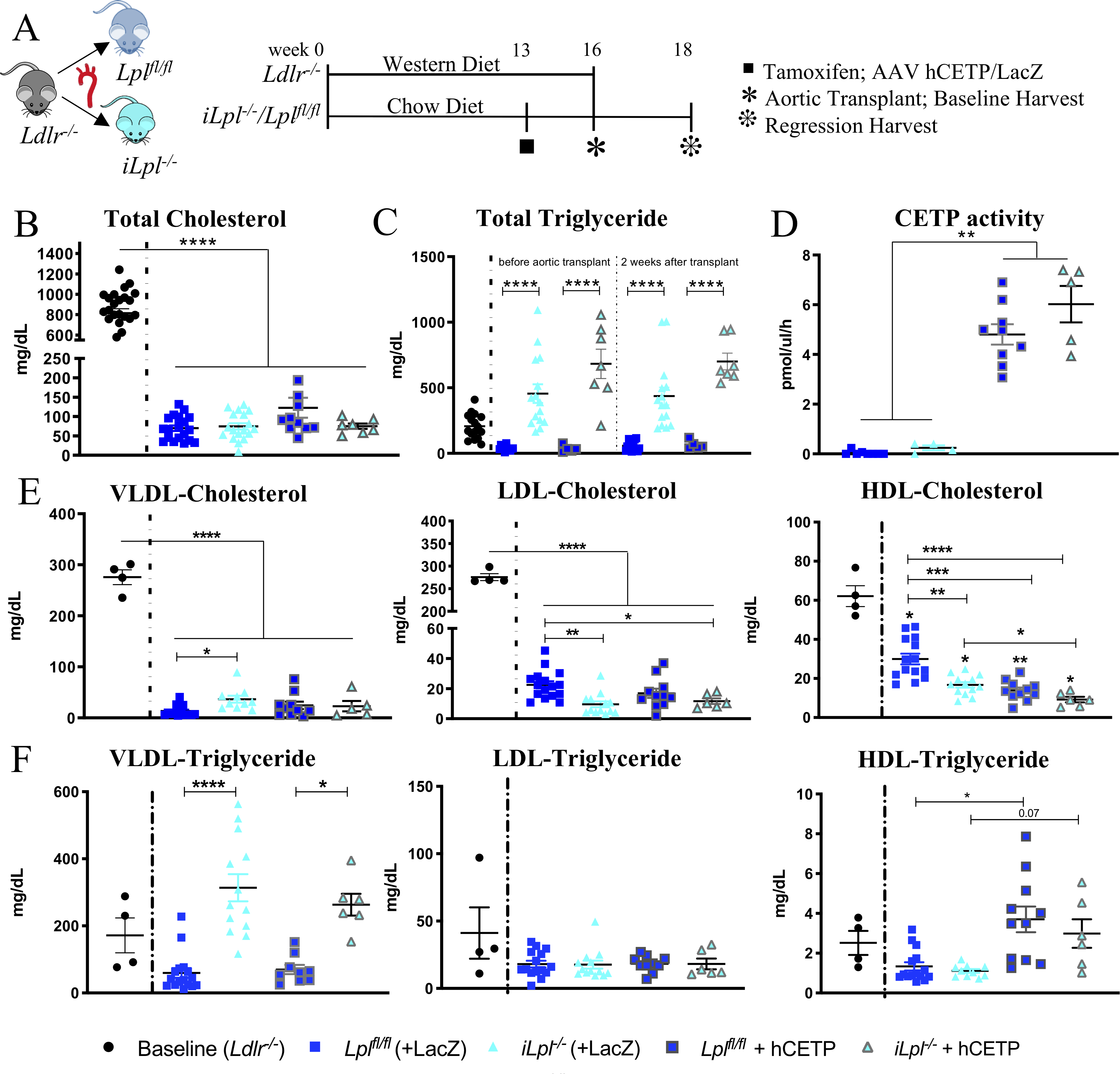
(A) Study Setup (B) Total plasma cholesterol levels at time of harvest (C) Total triglyceride (TG) levels in baseline and regression groups before and after the 2 weeks atherosclerosis regression period (D) CETP activity measured in circulation. Cholesterol (E) and triglyceride levels (F) of isolated lipoproteins. N= (B, C) Baseline 26, Lplfl/fl 19, Lpl−/− 18, Lplfl/fl +hCETP 11, Lpl−/− + hCETP 7; (D) Lplfl/fl 8, Lpl−/− 4, Lplfl/fl +hCETP 9, Lpl−/− + hCETP 5; (E, F) Baseline 4, Lplfl/fl 15, Lpl−/− 13, Lplfl/fl +hCETP 10, Lpl−/− + hCETP 6. Data represented as mean ± SEM, * P < 0.05, ** P<0.01, *** P < 0.001, **** P < 0.0001, 1-way ANOVA with Tukey’s multiple comparison test; HDL-C and HDL-TG Welch ANOVA with Games-Howell’s multiple comparison test; VLDL-TG & LDL-TG Kruskal-Wallis Test with Dunn’s multiple comparison test.
Figure 2. HyperTG due to global LpL deficiency does not affect atherosclerosis regression in the aortic transplant mouse model.
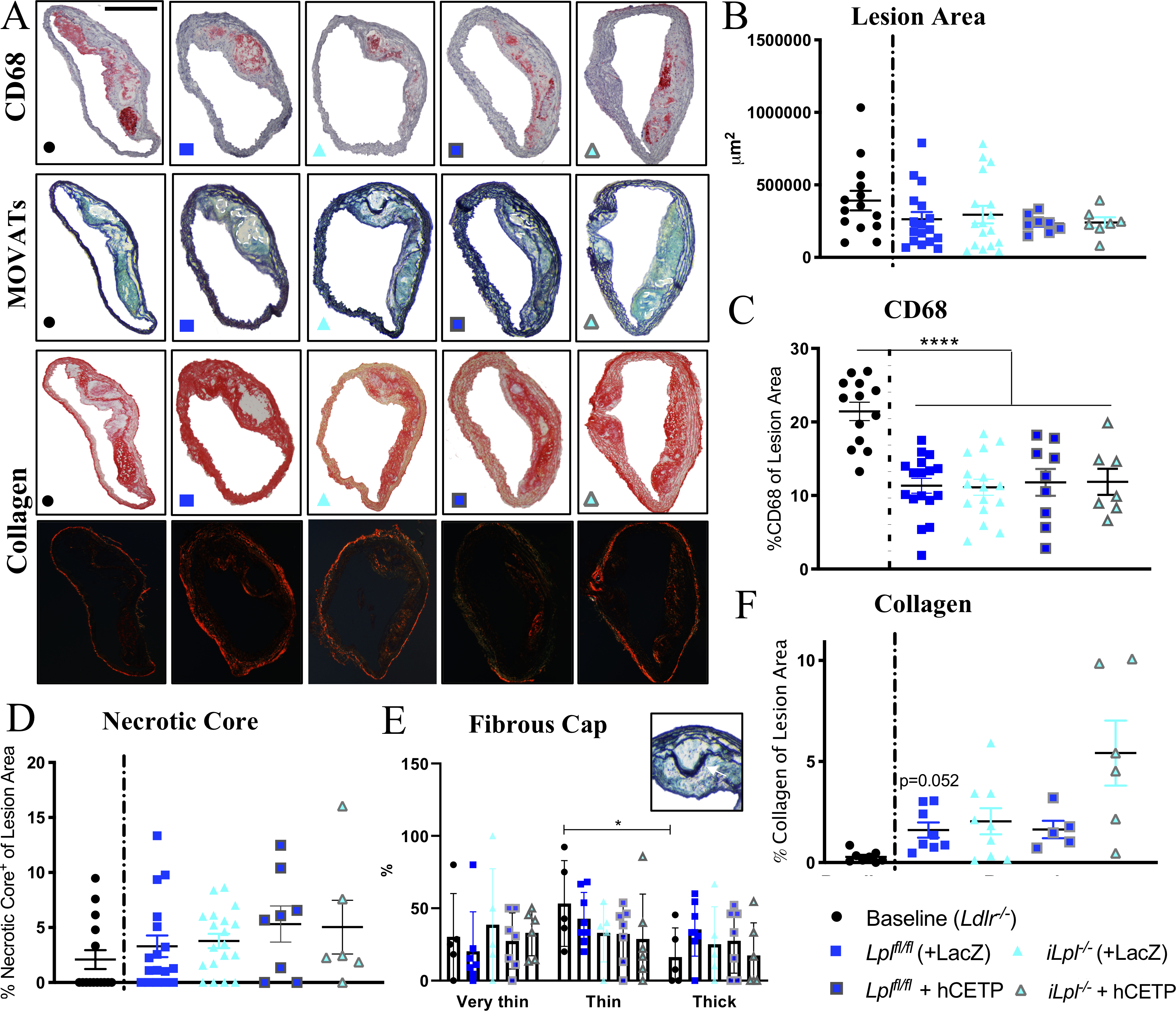
(A) Representative stainings; CD68, MOVATs, Picrosirius Red Staining (brightfield & polarized light); scale bar = 500 μm. Quantification of (B) Lesion Area (μm2) and (C) CD68 Area (% of Lesion Area ) (D) Necrotic Core (% of Lesion Area) (E) Fibrous Cap determined by number of layers above the Necrotic Core, Thick >10layers, Thin 5–10layers, Very Thin <5 layers; scale bar = 100 μm (F) Collagen (% of Lesion Area); N= (B, C, D) Baseline 15, Lplfl/fl 18, Lpl−/− 16, Lplfl/fl +hCETP 8, Lpl−/− + hCETP 6; (E) Baseline 5, Lplfl/fl 7, Lpl−/− 5, Lplfl/fl +hCETP 7, Lpl−/− + hCETP 6 (F) Baseline 8, Lplfl/fl 8, Lpl−/− 9, Lplfl/fl +hCETP 5, Lpl−/− + hCETP 6. Data represented as mean ± SEM, **** P < 0.0001, 1-way ANOVA with Tukey’s multiple comparison test; (E) 2-way ANOVA with Tukey’s multiple comparison test (F) Welch ANOVA with Games-Howell’s multiple comparison test.
Figure 4. LpL deficiency (+/− hCETP) does not affect macrophage phenotype.
Macrophage phenotype was assessed by isolating mRNA from atherosclerotic CD68+ macrophages using Laser Capture Microdissection and mRNA expression measured for (A) Metabolism-related genes (B) Inflammatory genes (C) Anti-inflammatory genes. Additionally, immunofluorescence staining of atherosclerotic arches was performed for (D-E) Arginase-1 (%cells per total cells) and (F-G) iNOS (%cells per total cells), scale bars = 500 μm. N= (A-C) Lplfl/fl +hCETP 5, Lpl−/− + hCETP 5; (E, G) Lplfl/fl 6, Lpl−/− 5, Lplfl/fl +hCETP 6, Lpl−/− + hCETP 5. Data represented as mean ± SEM, * P < 0.05, (A-C) unpaired t-test, (E, G) 1-way ANOVA with Tukey’s multiple comparison test.
Figure 5. LpL deficiency-mediated HyperTG reduces HDL-C and HDL-P, but does not impair CEC.

(A) Total HDL-P and its subfractions. (B) Total and ABCA1-mediated HDL Cholesterol Efflux Capacity (CEC). (C) Total and ABCA1-mediated CEC per HDL-P. (D) Volcano-plot of HDL Proteomics in iLpl−/− versus Lplf//fl. (E) Main regulated proteins are part of plasma lipoprotein remodeling. (F) Volcano plot iLpl−/− + hCETP vs. Lplfl/f + hCETP. (G) Regulated proteins in the plasma lipoprotein remodeling pathway. Proteomics normalized to spiked ApoA-I; complete list of proteins can be found in Supplemental Table II–V. N= (A) Lplfl/fl 19, Lpl−/− 17, Lplfl/fl +hCETP 9, Lpl−/− + hCETP 5; (B,C) Lplfl/fl 19, Lpl−/− 13, Lplfl/fl +hCETP 8, Lpl−/− + hCETP 5; (D-G) Lplfl/fl n=8, iLpl−/− n=8, Lplfl/f + hCETP n=17, iLpl−/− + hCETP n=10. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001, 1-way ANOVA with Tukey’s multiple comparison test; Medium HDL-P and (C) Kruskal-Wallis Test with Dunn’s multiple comparison test.
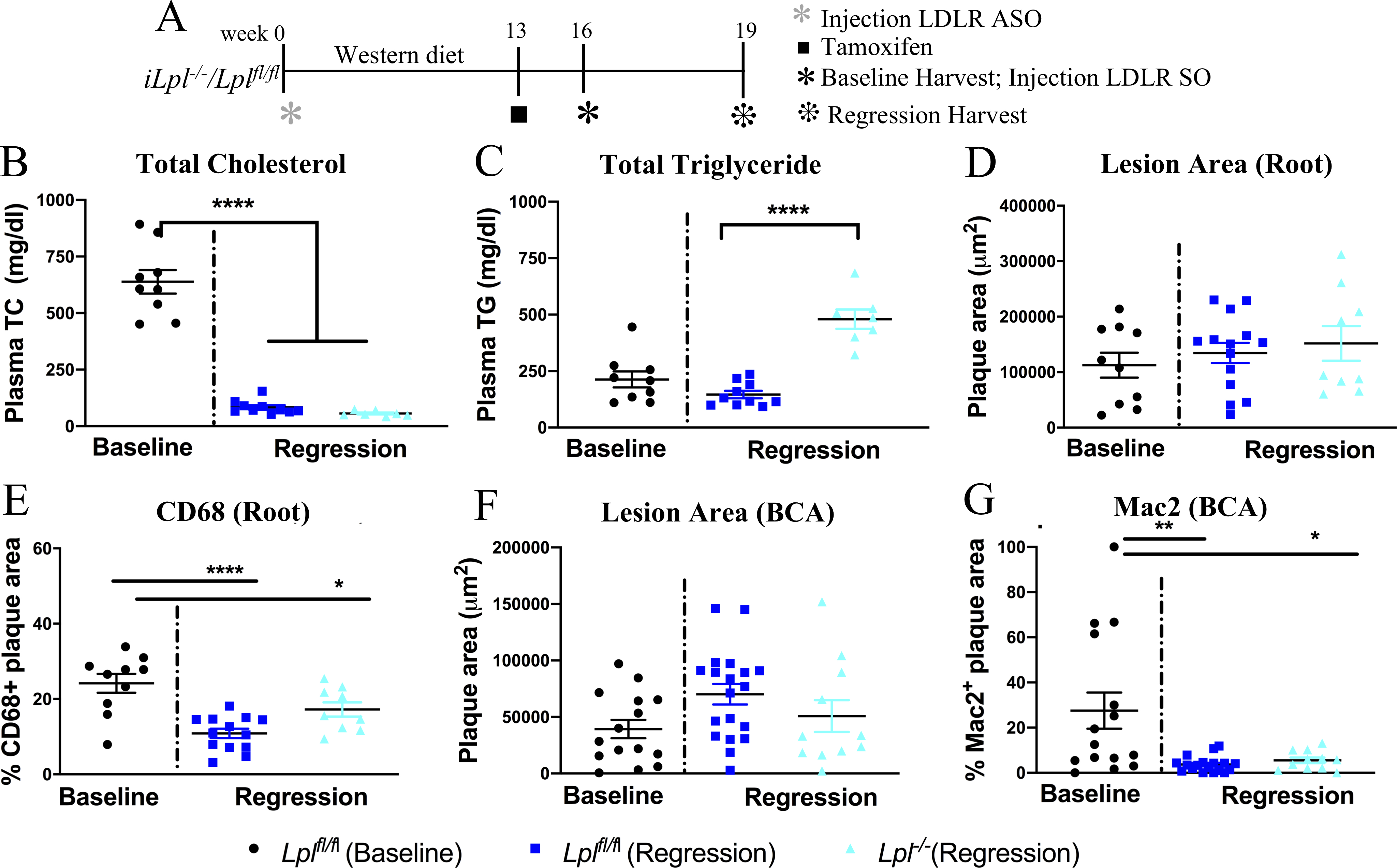
Figure 3. LpL deletion does not alter atherosclerosis regression in a non-invasive model of regression.

(A) Study design: atherosclerosis was created in Lplfl/fl and iLpl−/− mice with LDLR antisense oligonucleotides (ASO) and western diet feeding for 16 weeks. One set of mice were analyzed at 16 weeks as the baseline group and the rest of the mice were treated with SO to induce regression and were analyzed after 3 weeks. The mice in regression group were also treated with tamoxifen at week 13 to induce hypertriglyceridemia in iLpl−/− mice. (B) Plasma total cholesterol (TC) and (C) triglyceride (TG) levels in baseline and regression groups. (D) Total plaque area, (E) % of macrophages (CD68+) within aortic root lesions, (F) total plaque area, (G) % of macrophage (Mac2+) in plaques within the BCA in the baseline and regression groups. N=(B,C) Baseline Lplfl/fl 9, Regression Lplfl/fl 10 and iLpl−/− 7, (D,E) Baseline Lplfl/fl 10, Regression Lplfl/fl14 and iLpl−/− 9 and (F,G) Baseline Lplfl/fl 15, Regression Lplfl/fl 19 and iLpl−/− 11. Results are expressed as mean ± SEM. * P < 0.05, ** P<0.01 and **** P < 0.0001 using 1-way ANOVA with Tukey’s multiple comparison test.
Plasma lipid measurements.
Total cholesterol (TC) was measured using an enzymatic assay (Total Cholesterol E Kit, Wako Life Science, cat # NC9138103 or Infinity Total Cholesterol Reagent, Thermo Scientific, cat #TR13521). Plasma TG was measured using either enzymatic assay (L-Type Triglyceride M, Wako Life Science or Infinity Triglycerides, Thermo Scientific, cat #TR2242) or a fluorometric kit (BioVision, #K622–100). Plasma ApoA-I was assessed by mass spectrometry using spiked ApoA-I for calibration.
Lipoprotein fractionation.
Lipoprotein fractions were obtained by sequential density ultracentrifugation as described before20, and TC and TG measured as described above. Briefly, equal amounts of mouse plasma (70–100 μL) were used for sequential density ultracentrifugation to separate very-low-density lipoprotein (VLDL d<1.006 g/mL), LDL (d=1.006–1.063 g/mL), and HDL (d=1.063–1.21 g/mL) in a TLA 100 rotor (Beckmann Instruments, Palo Alto, CA).
hCETP.
hCETP expression was induced by injecting mice i.p. with either adeno- associated virus (AAV) hCETP (AAV8.TBG.PI.hCETP.SV40) or a LacZ control virus (AAV8.TBG.PI.LacZ.bGH) at a concentration of 5×1011 GC (genome copies)/mouse (Penn Vector Core) as done before.21 Plasma CETP activity was measured using a CETP Activity Assay Kit (Sigma Aldrich, MAK106). CETP expression in the liver was assessed by qPCR from homogenized liver samples (Primer sequence in Supplemental Table I).
Aortic transplant regression model.
The aortic transplant model has been described before.22–25 Briefly, a plaque burdened aortic arch from a hypercholesterolemic Ldlr−/− mouse (fed a WD for 16wks) was transplanted into a normocholesterolemic recipient mouse (iLpl−/− or Lplfl/fl), interpositioned with the abdominal aorta and blood flow was directed through the graft. All recipient mice were ~22 weeks old and maintained on standard chow diet until sacrificed 14 days after aortic transplantation.
LDL receptor (LDLR) antisense oligonucleotide (ASO) regression model.
This protocol was based on our published model.26 Briefly, GalNAc-conjugated Gen 2.5 ASO targeting mouse LDLR was developed and provided by Ionis Pharmaceuticals. LDLR ASO was injected intraperitoneally at a dose of 5 mg/kg body weight once a week for 16 weeks. GalNAc-conjugated sense oligonucleotide (SO) designed to bind and inactivate the LDLR ASO was injected once i.p. at a dose of 20 mg/kg body weight to accelerate the return of LDLR expression.
Bone marrow transplant (BMT).
Bone marrow cells were isolated from C57BL/6 mice by flushing tibiae and femur bones with DMEM (Dulbecco’s Modified Eagle Media, Fisher Scientific), followed by red blood cell lysis and BM cells suspended in DMEM. 5*106 cells in 200μl DMEM/mouse were injected retro-orbitally into 8 weeks old radiated (10.000 Gy) iLpl−/− and Lplfl/fl, followed by 4 weeks of antibiotic treatment administered in drinking water.
Atherosclerotic plaque assessment.
Aortic arches were removed after perfusion with saline + 10% sucrose, embedded in OCT (TissueTEK) and frozen at −80°C. Serial sections (6μm thick) were cut using a cryostat and stained for CD68 (Bio-Rad, cat. MCA1957; 2nd antibody: biotinylated rabbit anti-rat IgG, Vector Laboratories, cat. BA4000)25, inducible nitric oxide synthase (iNOS, 647-conjugated; Abcam, cat. ab209027)27 and Arginase1 (488-conjugated; Thermo Fisher Scientific, cat. 53–3697-82) to determine content and phenotype of macrophages. To distinguish target staining from background staining, we performed the same protocol, and replaced the primary antibody with an isotope control (Rat IgG2a, Abcam, cat. ab18450). For further plaque characteristics, Movats28 and Picrosirius Red (Abcam, cat. ab246832)29 stainings were performed as described previously. Picrosirius Red staining was visualized using a polarized light microscope (Zeiss Axio Observer) to determine collagen+ areas.
Aortic roots were removed after perfusion with PBS and embedded in OCT (TissueTEK), frozen, and stored at −80°C. Serial sections (6 μm) of roots were obtained by cryosectioning, followed by CD6825 staining with same reagents and protocol described above for aortic arches. Brachiocephalic arteries (BCAs) were collected in 10% formalin, kept overnight at 4°C, and stored in 70% ethanol at 4°C for further processing. BCAs embedded in paraffin were sectioned (5 μm) and every fifth cross section stained using the Movat pentachrome method. The maximal lesion site was determined, and adjacent sections were immunostained using a Mac-2 antibody (Cedarlane Lab, cat. CL8942AP; biotinylated secondary antibody Goat-anti-Rat from Southern Biotech, cat. 3050–08) to detect lesion macrophages, as described previously.30 We used a rat IgG2a isotype control antibody (Cedarlane Lab; cat. CLCR2A00) as a negative control for Mac-2 staining.
ImagePro Plus 7.0 software was used to determine CD68+, Mac-2+ and collagen+ area. ImageJ 1.51r (NIH) was used to determine iNOS+or arginase1+ cells and divided by total cells (DAPI nuclear staining) to get percentage of total cells that were positive for either marker. Acellular areas were measured to determine necrotic core area and fibrous cap was evaluated as none, very thin (<5layers), thin (5–10 layers) and thick (>10 layers) above the necrotic core using Movats stain.
Macrophage phenotyping.
CD68+ cells were selected from atherosclerotic plaques from iLpl−/− and Lplfl/fl mice by laser capture microdissection (LCM) as previously described.31, 32 Briefly, 6μm frozen sections from aortic arches were stained with hematoxylin-eosin and foam cells identified under a microscope and verified by positive CD68 staining. All LCM procedures were performed under RNase-free conditions. After LCM, RNA was isolated using the PicoPure Kit (Molecular Devices, Inc., Sunnyvale, CA), treated with DNase and the quality and quantity determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). RNA was converted to cDNA and amplified using the WT-Ovation Pico RNA Amplification Kit (NuGEN, San Carlos, CA). Data were normalized to HRPT with the ΔΔC(t) method and are presented as relative transcript levels. All primers are listed in Supplemental Table I.
HDL analyses.
Cholesterol efflux assays:
Cholesterol efflux capacity (CEC) was analyzed by methods based on methodology developed by Rothblat et al. as described previously33–35 and as adapted in our laboratory35. Detailed Methodology can be found in the supplementary Materials and Methods. CEC was quantified as the percentage of label in the medium relative to total labelled cholesterol levels in the culture system after subtraction of values obtained without ApoB depleted serum. ABCA1-mediated CEC was determined by analogous method in BHK cells expressing ABCA1 under mifepristone control where the ABCA1-specific CEC represents the difference between CEC of mifepristone stimulated and non-stimulated BHK cells.
HDL particle concentration:
HDL particle number (HDL-P) and particle size distribution was quantified using calibrated differential ion mobility analysis.34, 36 Detailed Methodology can be found in the supplementary Materials and Methods.
HDL proteomics:
Proteome of HDL isolated by two-step sequential density ultracentrifugation (density 1.21–1.063 g/ml) was characterized and quantified using data independent analysis (DIA) mass spectrometry as described previously.35, 37 Detailed Methodology can be found in the supplementary Materials and Methods. Proteomics data were analyzed using Reactome (https://reactome.org).38
Human subjects.
Plasma was obtained from 9 donors with elevated plasma TG levels of whom three were LpL deficient (Characteristics in Supplemental Table VI), and 6 control subjects with normal plasma TG levels. CEC, HDL-P and HDL proteomics analyses were performed as described above.
Statistical analyses.
Doing a standard power analysis, for a 2-fold change in a parameter, with a standard deviation up to 60% of the mean, there is a 94% probability that 10 mice/group will be needed to detect a difference at a 5% level of significance in an unpaired two-tailed student’s t-test or 1-way ANOVA. Thus, we, as many others in the atherosclerosis field, used a group size of 10 in most of the studies. One data point each was excluded for the VLDL-C measurement (Lpl−/− + hCETP), and for the collagen measurement (Lplfl/fl + hCETP).
Data are expressed as mean ± SEM. Data were tested on normality and equal variance (Brown-Forsythe and Bartlett’s) and analyzed by appropriate parametric or non-parametric test as stated in each figure legend. While we adjusted for multiple testing within specific analyses (stated in each figure legend), we did not further adjust for multiple testing. This may be considered a weakness of this study.
P ≤ 0.05 was considered significant (GraphPad Prism 7), and P-values are summarized in supplemental Table VIII.
RESULTS
LpL deficiency increased circulating VLDL-TG and reduced HDL-C, which was further reduced with hCETP expression.
At ~5 weeks of age Ldlr−/− mice were weaned onto WD, which was continued for 16 weeks to promote advanced atherosclerotic lesion formation (baseline group; Figure 1A, Supplemental Figure IA). Plaque-bearing aortic grafts were then transplanted into Lplfl/fl (control) and iLpl−/− mice (HyperTG, induced 3 weeks prior to transplantation) that were normocholesterolemic. Both recipient groups had been maintained on a standard laboratory diet prior to receiving aortic transplants; this diet was continued for 2 weeks to maintain normocholesterolemia and induce plaque regression. A subset of mice was injected with an AAV hCETP (5×1011 genome copies/mouse; Figure 1A, D) to investigate whether the absence of CETP expression in mice was a factor in the effects of HyperTG, especially in light of a previous report that in another model of HyperTG, hCETP expression was associated with the inhibition of the development of early atherosclerotic lesions.39
In contrast to the hypercholesterolemic baseline/donor mice (TC 867 ± 36 mg/dL), both aortic transplant recipient groups were normocholesterolemic (TC 79 ± 10 mg/dL; Figure 1B). Plasma TG levels were normal in Lplfl/fl mice (~67 mg/dL) and, as expected, TG levels were elevated in iLpl−/− mice (~461 mg/dL; Figure 1C). hCETP injection did not affect TC or total TG levels but led to an expected increase in circulating CETP activity (Figure 1D). Body weights of the mice did not change due to the transplant procedure and were similar in all regression groups (Supplemental Figure IB). Body weight was elevated in the baseline group due to WD feeding.
The changes in lipoprotein profile due to LpL deficiency and hCETP expression are shown in Figures 1E–F. The most remarkable lipid change that occurred with hCETP and HyperTG was a marked reduction in HDL-C with smaller changes in other lipoprotein parameters. VLDL-C increased in iLpl−/− compared to Lplfl/fl mice (iLpl−/− 37 ± 7 versus Lplfl/fl 13 ± 3 mg/dL, P = 0.046). As LpL-mediated lipolysis is required for LDL and HDL formation, LpL deficient mice had decreased LDL-C and HDL-C levels; HDL-C was decreased from 29 ± 2 to 17 ± 1 mg/dL (P= 0.004) and even further reduced with hCETP expression to 9 ± 2 mg/dL (P = 3×10−5). As expected, VLDL-TG was increased in iLpl−/− compared to Lplfl/fl mice (iLpl−/− 314 ± 40 vs. Lplfl/fl 60 ± 15 mg/dL, P = 1×10−5, Figure 1F). Neither LDL-TG nor HDL-TG differed between Lplfl/fl and iLpl−/− mice, but HDL-TG significantly increased with hCETP expression to the same extent in mice with and without Lpl expression (average HDL-TG with hCETP 3.3 ± 1.4 mg/dL vs. no hCETP 1.2 ± 0.2 mg/dL). Thus, LpL deficiency induced HyperTG and reduced LDL-C and HDL-C. hCETP reduced HDL-C even further and created TG-enriched HDL as expected from its role in humans in exchanging VLDL-TG for HDL-C.
HyperTG due to global LpL deficiency does not affect atherosclerosis regression in the aortic transplant mouse model.
After two-weeks of plaque exposure to normocholesterolemia (atherosclerosis regression period), lesion areas were similar in all groups (Figure 2A–B). We considered as evidence of atherosclerosis regression decreases in lesion area or macrophage content because the two parameters can be independent of each other (e.g.40) and either is considered to be a beneficial change.
The macrophage content (CD68+ area, Figure 2C) decreased in regression groups compared to baseline, but did not differ between mice with and without LpL/hCETP expression (baseline 21.4% ± 1.3 versus average of 4 regression groups: 11.5% ± 1.4, P = 1.1×10−5). To further evaluate the plaques, we assessed necrotic core, fibrous cap and collagen. Necrotic core (% lesion area) did not significantly differ between groups (Figure 2A, C–D). Fibrous caps (Figure 2E) and collagen (Figure 2G) were increased compared to baseline (P = 0.052), but did not significantly differ between regression groups. Therefore, the favorable remodeling of the cellular composition of the plaques (in particular, reduced macrophage content, an index of the resolution of lesion inflammation) was not impaired by HyperTG and/or hCETP expression. Moreover, we could not correlate lesion area or macrophage content with the degree of HyperTG (Supplemental Figure II).
HyperTG due to non-macrophage LpL deficiency also does not affect atherosclerosis regression in the aortic transplant mouse model.
LpL is also expressed by macrophages and has been shown to promote lipid uptake and foam cell formation.41–43 To exclude possible counterbalancing effects of the putative atherogenic HyperTG and atheroprotective LpL deficiency in macrophages, we performed atherosclerosis regression studies using Lplfl/fl and iLpl−/− transplanted with WT bone marrow (Supplemental Figure III A–B). HyperTG mice with LpL expressing macrophages showed body weights and lipid levels similar to those in total LpL-deficient mice (Supplemental Figure III C–G). Unlike the effects of macrophage LpL to promote atherosclerosis41, 44, the number of macrophages per lesion area (%CD68) during regression was similarly reduced in mice with and without macrophage LpL expression, showing a reduction of >50% compared to baseline (Supplemental Figure III H–I). Thus, knocking out macrophage LpL expression did not alter regression. These data are similar to another study that showed LpL expression does not modify lesional macrophage phenotype in the setting of atherosclerosis regression.20
LpL deficiency mediated HyperTG does not affect atherosclerosis regression in a non-invasive mouse model.
To confirm the results on LpL and atherosclerosis regression in an independent model (Figure 3A), WD-fed Lplfl/fl and iLpl−/− (non-induced) mice were made hypercholesterolemic using LDLR ASO treatment for 16 weeks. At the end of 16 weeks, the ASO treatments stopped and one subset from each genotype was analyzed as the ‘baseline’. The other subset of mice was treated with tamoxifen to induce LpL deletion at week 14. These mice were then treated with LDLR SO at week 16 to rapidly reverse hypercholesterolemia and to induce regression.26 After another 3 weeks, these mice were studied to analyze the regressing plaques. As expected, plasma TC (Figure 3B) decreased in regression groups compared to baseline (baseline 638.5 ± 52 versus Lplfl/fl regression 83.4 ± 9, P = 1×10−14 and baseline 638.5 ± 5 vs iLpl−/− regression 56.2 ± 4, P = 1×10−15), and plasma TG (Figure 3C) increased significantly in iLpl−/− mice (Lplfl/fl regression 146.2 ± 16 versus iLpl−/− regression 480 ± 43, P = 1×10−15).
As with the transplant model, with marked cholesterol reduction there were no statistical differences observed in total plaque areas in the aortic root (Figure 3D). There was a marked reduction in CD68% area (Figure 3E, baseline 24% ± 2.5 versus Lplfl/fl regression 10.8% ± 1.2, P = 1×10−15 and baseline 24% ± 2.5 versus iLpl−/− regression 17.2% ± 1.9, P=0.047), and the presence of HyperTG did not impair this reduction. In parallel, we noted no significant changes in total plaque area in the BCAs (Figure 3F), but as in the root, there was a significant decrease in macrophage area (Figure 3G, baseline 27.5 ± 8% versus Lplfl/fl regression 3.7% ± 0.8, P=0.002 and baseline 27.5% ± 8 versus iLpl−/− regression 5.5% ±1.2, P=0.012). Again, HyperTG did not affect the extent of BCA regression compared to the controls.
Thus, in two independent models of atherosclerosis regression, HyperTG iLpl−/− mice had similar changes as those in the control Lplfl/fl mice after reversal of hypercholesterolemia, and in the LDL ASO model there was no evidence that the changes we observed were specific only to the aortic root.
LpL deficiency-mediated HyperTG with local lipolysis in plaques does not affect atherosclerosis regression in a non-invasive mouse model of regression.
One possible cause for the postulated atherogenicity of HyperTG is the generation of local lipolysis products.45–47 iLpl−/− mice would be protected from this type of injury due to generalized LpL deficiency. To determine the importance of local lipolysis we assessed atherosclerosis regression in mice with LpL expression in endothelial cells driven by a Tie2-Lpl transgene, described in 48. These mice were allowed to develop atherosclerosis using the LDLR ASO protocol26 and regression was induced by reversing the hypercholesterolemia with SO. Total plasma cholesterol (Supplemental Fig IV A) significantly decreased in regression groups after SO treatment (Baseline versus regression group 1, P=0.001 and regression group 2, P=0.007). Mice with total LpL deficiency and the Tie2-Lpl transgene had mild HyperTG compared to LpL floxed mice expressing the Tie2-Lpl transgene (Supplemental Figure IV B). We quantified aortic root total plaque area (Supplemental Figure IV C) and percentage of plaque that was occupied by macrophages (Figure IV D) along with BCA total plaque area (Supplemental Figure IV E) and percentage of the Mac-2 positive (i.e., macrophage positive) area in BCA plaques (Supplemental Figure IV F). The Mac-2 positive plaque area decreased slightly in iLpl−/− with Tie2-Lpl transgene compared to its baseline, but overall, there were no significant differences attributable to increasing the lipolytic capacity of the endothelial cells. Thus, greater local lipolysis did not impair regression.
Effects of HyperTG on plaque macrophage gene expression.
Given the similarity in the results between the two regression models in terms of changes in plaque macrophage content, we wondered if the phenotype of plaque macrophages was altered by HyperTG. Thus, RNA expression of isolated plaque macrophages and immunofluorescence staining were performed using samples from the transplant studies (Figure 4).
We have previously shown that transcriptomic profiling of atherosclerotic macrophages did not disclose significant differences in metabolism-related genes or pro- or anti-inflammatory genes in LpL-expressing versus LpL-deficient macrophages.20 To determine if hCETP expression affected macrophage phenotype, we isolated CD68+ cells by laser capture microdissection from Lplfl/fl and iLpl−/− mice expressing hCETP. In line with the previous results, we did not observe significant changes in metabolism-related genes (plin2, cd36, glut1, fasn, cpt1a), pro-inflammatory genes (tnfα, nos2, mcp1), or anti-inflammatory genes (mrc1, fizz1, il10) (Figure 4A–C). Staining of arginase-1 (a marker of M2-like macrophages, Figure 4D–E) and iNOS (a marker of M1-like macrophages, Figure 4F–G) in atherosclerotic plaques showed no differences between Lplfl/fl and iLpl−/− mice expressing hCETP, further confirming transcriptomic profiling results.
LpL deficiency-mediated HyperTG reduces HDL-C and HDL-P but does not impair CEC.
We hypothesized that regression was unchanged in our HyperTG mice because although HDL-C was reduced, CEC was maintained. To determine this, we assessed plasma levels of HDL-P and CEC. LpL deficiency leads to lower HDL-C levels in humans49 and in animal models50. We found that LpL deficiency also significantly lowers circulating HDL-P levels (P = 2×10 −8, which were further reduced by hCETP expression (Figure 5A, P = 4.2×10 −4). Dividing total HDL-P into their subclasses according to size showed that the reduction was in small (9.8–10nm) and medium (10.4–11.1nm), but not in large (11.9 – 12.6nm) HDL-P (Figure 5A).
We next measured CEC according the protocol developed by Rothblat and colleagues, in which radiolabeled cAMP-stimulated J774 cells are incubated with ApoB-depleted plasma for 4h, followed by quantification of labelled cholesterol in cells and medium.51 Total CEC and ABCA1 mediated CEC were assessed (Figure 5B). LpL deficiency decreased total CEC (Lplfl/fl 7.5 ± 0.4% vs. iLpl−/− 5.4 ± 0.7%, P = 2×10 −4), but despite the marked reduction in HDL-C, ABCA1-mediated efflux was not reduced. There was no reduction in total CEC or ABCA1-mediated efflux in mice expressing hCETP. When total CEC and ABCA1-mediated efflux on a per particle basis were calculated, there was no significant differences between Lplfl/fl and iLpl−/− mice (Figure 5C). Thus, HyperTG did not produce dysfunctional HDL. hCETP and TG-enrichment of HDL (Figure 1G) increased total CEC per particle (Lplfl/fl + hCETP 1.30 ± 0.06% vs. Lplfl/fl 0.90 ± 0.06%, P = 0.018) and ABCA1-mediated efflux per particle in Lplfl/fl mice (Lplfl/fl + hCETP 8.36 ± 0.62% vs. Lplfl/fl 7.72 ± 0.37%, P = 0.014). Total CEC and ABCA1-mediated CEC per particle showed a trend towards an increase in iLpl−/− mice expressing hCETP. These results show that despite a reduction in HDL-C and HDL-P with HyperTG, the unchanged regression correlated with maintained CEC.
Effects of LpL and CETP on the HDL proteome.
We next determined how LpL deficiency expression altered the HDL proteome. Comparing HDL of Lplfl/fl with iLpl−/− mice, of the 133 detected proteins, 50 proteins were significantly different with 18 being down- and 32 up-regulated (Figure 5D–E, Supplemental Table II–III). Differentially detected proteins included ApoA-I, ApoC-III, and ApoC-IV, and also PLTP (phospholipid transfer protein) and Angptl3 (Angiopoietin-like protein 3; Figure 5E). Thus, the HDL proteome reflects changes in apolipoprotein structural components induced by HyperTG. Further, ApoA-I reduction correlated with reduced HDL-C and HDL-P; this was assessed by HDL proteomics and plasma ApoA-I measurements (Supplemental Figure V).
We next assessed how hCETP expression altered the proteome in a HyperTG environment. Therefore, we compared the HDL proteome from Lplfl/fl + hCETP versus iLpl−/− + hCETP mice. In total, we detected 97 HDL proteins, of which 15 proteins significantly differed (1 was down- and 14 up-regulated in iLpl−/− + hCETP; Figure 5F–G, Supplemental Table IV). Surprisingly, some of the proteins found in the HDL proteome from non-hCETP mice (Figure 5D & E) were not detected in hCETP-expressing mice (Figure 5F & G), such as ApoA-V. In fact, while we detected 133 proteins in non-hCETP mice, we only detected 97 proteins in hCETP expressing mice, suggesting that hCETP activity moves these proteins from HDL to VLDL. Eighty-five of the proteins were present in both groups, and only 4 proteins (1 down- and 3 up-regulated) were significantly different with hCETP expression compared to no hCETP expression. This shows that hCETP has modest effects on the mouse HDL proteome (Supplemental Table V), and mainly leads to a redistribution of proteins between lipoproteins.
Subjects with HyperTG also show decreased HDL-P number, changes in the HDL proteome, and no impairment in total CEC.
To extend our mouse data to humans, we measured HDL-P and CEC in HyperTG patients of whom three were diagnosed with LpL deficiency (full characteristics are shown in Supplemental Table VI). Average plasma TG levels were ~750mg/dL and total cholesterol levels were ~380mg/dL (Figure 6A). HyperTG led to a reduced number of HDL-P (Figure 6B, P = 3×10−6), which was mainly due to reduction in medium and large HDL-P, but not small HDL-P. This differs from the mouse data showing a reduction in predominantly small HDL-P, but not large HDL-P, and is likely due to species differences in HDL metabolism (e.g.,52). As in the humanized mouse data (+hCETP, Figure 5B–C), total CEC did not change, meaning that CEC per HDL-P was significantly increased (P = 0.0044), suggesting an improvement in HDL functionality. This increase was primarily due to an increase in the ABCA1-mediated efflux pathway (Figure 6C–D).
Figure 6. Subjects with HyperTG, incl. LpL deficient subjects show decreased HDL-P, but no impairment in CEC.

(A) Plasma TG and Cholesterol levels (mg/dL). (B) total HDL-P and HDL-P subsets. (C) Total and ABCA1-mediated CEC (%). (D) CEC per HDL-P (E) Volcano-plot of HDL Proteomics showing changes with HyperTG. (F) Main regulated proteins are part of plasma lipoprotein remodeling pathway. White-filled dots refer to HyperTG patients, blue filled dots refer to LpL deficient patients. Proteomics normalized to spiked ApoA-I; complete list of proteins can be found in Supplemental Table VI. N=6 (Control), N=9 (HyperTG). * P < 0.05, ** P<0.01, **** P < 0.0001, students unpaired t-test; (C, D) Mann-Whitney test.
We next assessed HDL proteomics in the human samples. HDL proteomic analysis detected a total of 90 proteins (Figure 6E, Supplemental Table VII). Similar to our humanized mouse data, we found changes in HDL-associated apolipoproteins and PLTP (Figure 6F). Notably, the results of LpL deficient patients are similar to those of patients who had HyperTG for other reasons. Taking the human and mouse results together, HyperTG leads to a reduction in HDL-P without the creation of intrinsically dysfunctional HDL measured as CEC, while proteomic data mainly reflect the observed changes in HDL associated with LpL deficiency/HyperTG, such as reduced ApoA-I.
DISCUSSION
Our study shows that despite reductions in HDL-C and HDL-P, LpL deficiency-mediated HyperTG did not affect atherosclerosis regression as assessed by the macrophage content in plaques in aortic arches, roots and BCAs. Furthermore, LpL-deficiency did not affect macrophage inflammation (as judged by markers of M1/M2 phenotypes) or criteria of plaque stability judged indirectly by composition (necrotic core, collagen and fibrous cap) in aortic arches in mice. These results are surprising, as HDL-C and HDL-P have been shown to be inversely associated with CVD (reviewed in2). However, despite a reduction in HDL-C, HDL-P, and CEC, total CEC per particle did not change in HyperTG mice, and was increased in HyperTG humans. Thus, HDL function (CEC) was maintained in the presence of severe HyperTG. This suggests that the inverse association of HDL-C and CVD is not necessarily due to changes in CEC.
Because mice do not have CETP, the HDL-C and HDL-P reductions were likely due to reduced LpL-mediated lipid and protein transfer from TG-rich lipoproteins to HDL.53 LpL deficiency led to a change in the HDL-P size distribution with reduced levels of small- and medium-sized particles. In the mice, the combination of reduced lipolysis generated HDL components along with more rapid loss in the kidney might have led to this reduction in smaller HDL-P. With expression of CETP, resulting in TG-rich HDL, HDL-C and HDL-P were further reduced. Along with the reduction in HDL-C and HDL-P, ApoA-I was significantly reduced in mice and humans with HyperTG.
Although reduced LpL expression in macrophages decreases atherosclerosis progression in mice, the biology of regression differs. The reduction in lesional macrophage content was similar in the HyperTG/low HDL and control mice despite total body LpL deficiency. Thus, despite increased circulating TG levels and marked reductions in HDL-C, atherosclerosis regression proceeded normally and is evidence that LDL-C reduction overrode changes in macrophage LpL expression and in HDL, as discussed below. Even when macrophage LpL was replenished using bone marrow transplantation and vascular LpL expression was increased with an endothelial cell LpL-expressing transgene, regression was unaffected.
Recent clinical data have implicated HDL function rather than HDL-C as a marker for CVD risk.15–17 Unsuccessful lowering of CVD mortality in HDL-raising trials using CETP inhibition have been explained as a failure to increase reverse cholesterol transport, and perhaps CEC.13 Although HyperTG mouse plasma displayed some lowering of CEC, this reduction was less than what would be expected due to reduced HDL-P. In fact, when corrected for particle number, there was no indication that HDL was dysfunctional. Also notable is that the HyperTG-associated reduction in CEC was lost with hCETP expression, and there was a strong trend towards an increase in CEC per HDL-P.
We provided novel data on the effects of HyperTG on the HDL proteome in mice and humans. These data showed that hyperTG leads to changes in the HDL proteome that are associated with plasma lipoprotein remodeling, e.g. ApoA-I and PLTP were reduced in hyperTG mice and human. The reduction in ApoA-I correlates with HDL-C and HDL-P reduction in hyperTG mice. In line with our data, Pltp-deficient mice show reduced HDL-C, while the effect of HDL-PLTP and its activity on cholesterol efflux capacity remains unclear.54. Our proteomics data also revealed that hCETP likely led to transfer of HDL proteins to VLDL, but had modest effect on proteins that remained on HDL.
In humans, HyperTG is inversely correlated with HDL-C.1 Therefore, HyperTG was expected to lead to reduced CEC. In agreement with our mouse data, however, HyperTG increased CEC per HDL-P in our human samples. It is not entirely clear why the increase in CEC per HDL-P was more robust in humans than in mice, but we can speculate that other apoproteins and their molecular actions could have obscured the effects in the mouse. Differences in CETP activity may be another explanation for the less robust increase in CEC compared to mice expressing hCETP, and also explain the differences in HDL proteomics. Also, mouse ApoA-I does not interact as well as human ApoA-I with CETP.55 Nonetheless, these data confirm that despite the reduction in HDL-P, HDL is still functional in the HyperTG environment. Together, our results broadly indicate that HDL enrichment with TG over cholesterol does not affect function in mice and in people with hyperTG in the 500–700 mg/dL range, as measured by CEC in vitro and by no impairment of atherosclerosis regression. We recognize that this finding cannot automatically be extrapolated to untreated patients with homozygous LpL deficiency, whose plasma TG levels are often more extreme and can exceed 2000 mg/dL.
In contrast to these results, increasing HDL-P in mice by transgenic overexpression of ApoA-I improves atherosclerosis regression.6, 9 This raises the question why we do not detect impaired regression when HDL-P and ApoA-I are reduced. Perhaps ApoA-I has some function that is exclusive of its actions on HDL to affect CEC. HDL likely has multiple actions in vivo, such as reducing inflammation56 and blood coagulation57. These CEC-independent functions might be improved by marked increases in HDL. Such other actions of HDL rather than its effects on CEC would be consistent with its relationship to CVD and in agreement with clinical data showing similar overall cholesterol balances of humans with high and low HDL-C levels.18 Alternatively, the level of total CEC in the mice with fewer HDL-P particles may have still been sufficient to prevent a greater accumulation of cholesterol in plaque macrophages. For example, in apoE−/− mice we have previously shown that the naturally low level of HDL-P in that model was sufficient to remove the excess free cholesterol from plaques when ACAT was inhibited.58
It should also be considered that the effects on regression by changes in HDL-P were likely attenuated by the preservation of ABCA1-mediated cholesterol efflux that we observed. As Rothblat and colleagues have shown59, as macrophages get progressively loaded by cholesterol, akin to their state in the baseline atherosclerotic plaques, the majority of efflux shifts from the aqueous diffusion pathway to being mediated by ABCA1. Finally, we should also note that despite the decrease in the ABCA1-mediated flux, acceptors other than ApoA-I not captured by our HDL measurements could have contributed to preservation of ABCA1-mediated efflux. These could be, among others, pre-β HDL60, non-HDL ApoA-IV61, ApoE62, or plasminogen63.
LpL genetic variations are a risk factor for CVD, along with hyperchylomicronemia and pancreatitis64. Damaging mutations in LpL lead to higher plasma TG and lower HDL-C65 in most cases, however, a linear association between LpL mutations and CVD has been challenging to establish. There have been reports of LpL-deficient individuals who develop premature CVD66, 67, but these might be the exceptions. Nevertheless, the atherogenic potential of nascent TG-rich lipoproteins is still incompletely known. For that reason, we have focused on whether the reductions in HDL-C and HDL-P due to LpL deficiency alter atherosclerosis regression, which is thought to require reverse cholesterol transport (e.g., 5). We should note that heterozygous carriers of certain damaging mutations in LpL have more coronary artery disease.65 Reduced but residual LpL activity might lead to incomplete lipolysis of TG-rich lipoproteins and accumulation of atherogenic remnants. Also, Ference et al. concluded that TG-lowering LpL polymorphisms (S447X) were associated with lower risk of heart disease per unit difference in plasma ApoB levels.68
Because the risk of CVD with complete LpL deficiency is unclear our animal data are especially germane. Our global LpL-deficient mice had mild-to-moderate plasma TG levels (approx. 500mg/dL). This level, while lower than many LpL-deficient patients, is not dissimilar from that found in LpL-deficient patients on a very restricted diet that might be comparable to a mouse chow diet. However, on a high fat diet, TG levels of iLpl−/− mice increased to >1,000 mg/dL. Thus, the failure to detect differences in regression in these mice is consistent with the hypothesis that nascent TG-rich lipoproteins are not atherogenic.
Regulation of lipolysis and generation of local lipolysis products depends both on LpL activity and circulating TG levels, which provide substrate for the reaction. For this reason, it has been postulated that the combination of partial LpL loss and HyperTG causes high concentrations of liberated toxic lipids or the generation of more atherogenic remnant lipoproteins. Further, our recent published data20 suggested that non-myeloid LpL is needed for recruitment, differentiation and proliferation of macrophages at site of inflammation. Therefore, we also created mice expressing additional LpL in arteries using the Tie2 promoter, which primarily expresses in endothelial cells. Because this transgene was bred onto the LpL knockout background, greater lipolysis along the arterial wall would be expected. This transgene also did not affect atherosclerosis regression. Therefore, neither circulating nascent TG-rich lipoproteins nor increasing the capacity of their hydrolysis along the artery wall prevented vascular repair (i.e., regression), in the setting of a marked reduction in LDL-C.
In addition to the points discussed already, our mouse model of LpL deficiency leads to several changes exclusive of circulating HDL-C and TG that should be considered. Macrophages are the only white blood cells that express significant amounts of LpL and are the major source of LpL in atherosclerotic plaques. Whether LpL in atherosclerotic plaques is pro- or anti-atherogenic is still debated. In vitro, macrophage LpL appears to have atherogenic functions, because it creates atherogenic remnant lipoproteins, which are rapidly internalized by macrophages.69 Macrophage-specific LpL knockout in vivo reduced atherosclerosis41, 42, indicating a role of LpL produced by these cells which is exclusive of changes in circulating lipoproteins.
In contrast to these presumed inflammatory effects of macrophage LpL, fatty acids provided via lipolysis were reported to activate PPARγ70 and convert macrophages to a less inflammatory phenotype.43, 71, 72 Our recent published data20, as well as the present study, however, showed that the transcriptomic profile of regressing atherosclerotic macrophages did not differ between macrophages expressing LpL and LpL-deficient macrophages. Of note, our recent publication also showed that LpL is needed for monocyte recruitment to sites of inflammation and further, that ratio of Ly6Chigh/Ly6Clow monocytes is TG-dependent with an increase in Ly6Chigh monocytes in LpL deficient mice.20 In this study, our comparatively low TG levels (500mg/dl versus 2000mg/dL) did not lead to changes in the Ly6Chigh/Ly6Clow ratio with global LpL deficiency (Supplemental Figure VI A,B). Since macrophage LpL has been implicated in foam cell formation41–43, loss of macrophage LpL could have opposite and counterbalancing effects on atherogenesis. To exclude the possibility that we protected our mouse model from possible atherogenic effects of HyperTG, we created HyperTG mice with macrophages expressing LpL. Again, we observed intact atherosclerosis regression. Therefore, macrophage-LpL-mediated lipolysis in atherosclerotic plaques does not impede regression of atherosclerotic lesions in HyperTG mice.
In conclusion, our study shows that a reduction in HDL-C and HDL-P associated with LpL-deficiency and CETP expression does not affect atherosclerosis regression. Furthermore, we show that the lipoprotein alterations caused by these interventions, including the TG-enrichment of HDL, does not affect CEC per particle. As already noted, perhaps the lack of effects on atherosclerosis is explained by the continued level of sufficient HDL function, especially through the ABCA1 pathway. Furthermore, our data do not support a toxic effect of TG-rich lipoproteins on regression. We expect that these data reflect the human situation in which patients on high doses of LDL-lowering medications show clinical benefit, regardless of their circulating TG levels.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Lipoprotein lipase (LpL) deficiency increases circulating triglycerides and reduces circulating high-density lipoprotein cholesterol (HDL-C), both risk factors for cardiovascular diseases (CVD).
Genetic variations in LpL and its post-translational regulators are also associated with CVD risk.
It is not known how LpL directly affects CVD risk or how it may influence the regression of atherosclerosis after LDL-C lowering.
What New Information Does This Article Contribute?
LpL deficiency in mice induced hypertriglyceridemia leading to TG-enriched HDL, but does not affect cholesterol efflux capacity (CEC) per HDL particle.
Humans with hypertriglyceridemia have increased CEC per particle, likely mediated by cholesteryl ester transfer protein (CETP).
Despite reductions in HDL-C and HDL-P, LpL deficiency-mediated HyperTG does not affect atherosclerosis regression
Reduced plasma HDL-C and increased triglycerides (TG) have both been implicated in CVD risk. The inverse relationship between plasma TG and HDL-C makes it difficult to assess the separate roles of each factor. However, LpL regulates circulating TG and HDL-C, and its deficiency leads to high TG and low HDL-C in mice and humans alike. GWAS studies have associated variations in LpL gene and that of its regulators to the risk of CVD. However, the direct effects of lowering LDL-C on the ability to promote atherosclerosis regression are not known.
Combining two robust mouse models of studying atherosclerosis regression with an inducible LpL deficient model, we show that after LDL-C lowering, the decrease in macrophage content in plaques was not affected by lack of LpL. Despite a reduction in HDL-C, HDL function, measured as ABCA1-mediated cholesterol efflux capacity (CEC), was preserved in LpL-deficient mice. Excess circulating TG also did not have a negative impact on regression. We anticipate that in humans with hypertriglyceridemia, sufficient LDL-C lowering will lead to cardiovascular benefit, irrespective of circulating TG or HDL-C levels.
ACKNOWLEDGEMENTS
The authors would like to acknowledge Dr. Joseph Witztum for helpful discussions and advice, and thank the Penn Vector Core in the University of Pennsylvania Perelman School of Medicine for the production of AAV vectors used in this study.
SOURCES OF FUDNING
Funding was provided by American Heart Association (Predoctoral Fellowship 18PRE33990436, TJ; Postdoctoral Fellowship 17POST33660283, AHA Career Develpoment Award 20CDA35320109, DB), Hartstichting (2019SB001, BA), NLA Junior Investigator Award (DB), and the NIH (P01 HL092969, P01 HL131481, RO1 HL045095, RO1 HL073029, R01 HL084312, R01 HL129433, P01HL151328, R35HL150754; KEB, EAF, IJG).
Nonstandard Abbreviations and Acronyms:
- ApoA-I
apolipoprotein A1
- CEC
cholesterol efflux capacity
- CETP
cholesterol ester transfer protein
- hCETP
human CETP
- HDL
high-density lipoprotein
- HDL-C
HDL cholesterol
- HDL-P
HDL particle number
- HyperTG
hypertriglyceridemia
- LDLR
low-density lipoprotein receptor
- LpL
lipoprotein lipase
- TG
triglyceride
- VLDL
very low-density lipoprotein
Footnotes
DISCLOSURES
None.
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Dron JS and Hegele RA. Genetics of Triglycerides and the Risk of Atherosclerosis. Curr Atheroscler Rep. 2017;19:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ouimet M, Barrett TJ and Fisher EA. HDL and Reverse Cholesterol Transport. Circ Res. 2019;124:1505–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zilversmit DB. Atherogenesis: a postprandial phenomenon. Circulation. 1979;60:473–85. [DOI] [PubMed] [Google Scholar]
- 4.Ebenbichler CF, Kirchmair R, Egger C and Patsch JR. Postprandial state and atherosclerosis. Curr Opin Lipidol. 1995;6:286–90. [DOI] [PubMed] [Google Scholar]
- 5.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M and Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 2011;108:7166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hewing B, Parathath S, Barrett T, Chung WK, Astudillo YM, Hamada T, Ramkhelawon B, Tallant TC, Yusufishaq MS, Didonato JA, Huang Y, Buffa J, Berisha SZ, Smith JD, Hazen SL and Fisher EA. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2014;34:779–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plump AS, Scott CJ and Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci U S A. 1994;91:9607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG and Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353:265–7. [DOI] [PubMed] [Google Scholar]
- 9.Barrett TJ, Distel E, Murphy AJ, Hu J, Garshick MS, Ogando Y, Liu J, Vaisar T, Heinecke JW, Berger JS, Goldberg IJ and Fisher EA. Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation. Circulation. 2019;140:1170–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC and Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–300. [DOI] [PubMed] [Google Scholar]
- 11.Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, Woollard K, Lyon S, Sviridov D and Dart AM. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–91. [DOI] [PubMed] [Google Scholar]
- 12.Group HTC, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R and Armitage J. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12. [DOI] [PubMed] [Google Scholar]
- 13.Barter PJ, Brewer HB Jr., Chapman MJ, Hennekens CH, Rader DJ and Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–7. [DOI] [PubMed] [Google Scholar]
- 14.Group HTRC, Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, Collins R, Wiviott SD, Cannon CP, Braunwald E, Sammons E and Landray MJ. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N Engl J Med. 2017;377:1217–1227. [DOI] [PubMed] [Google Scholar]
- 15.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA and Shaul PW. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, Mucksavage ML, Luben R, Billheimer J, Kastelein JJ, Boekholdt SM, Khaw KT, Wareham N and Rader DJ. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case-control study. Lancet Diabetes Endocrinol. 2015;3:507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shea S, Stein JH, Jorgensen NW, McClelland RL, Tascau L, Shrager S, Heinecke JW, Yvan-Charvet L and Tall AR. Cholesterol Mass Efflux Capacity, Incident Cardiovascular Disease, and Progression of Carotid Plaque. Arterioscler Thromb Vasc Biol. 2019;39:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blum CB, Dell RB, Palmer RH, Ramakrishnan R, Seplowitz AH and Goodman DS. Relationship of the parameters of body cholesterol metabolism with plasma levels of HDL cholesterol and the major HDL apoproteins. J Lipid Res. 1985;26:1079–88. [PubMed] [Google Scholar]
- 19.Augustus A, Yagyu H, Haemmerle G, Bensadoun A, Vikramadithyan RK, Park SY, Kim JK, Zechner R and Goldberg IJ. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J Biol Chem. 2004;279:25050–7. [DOI] [PubMed] [Google Scholar]
- 20.Chang HR, Josefs T, Scerbo D, Gumaste N, Hu Y, Huggins LA, Barrett TJ, Chiang SS, Grossman J, Bagdasarov S, Fisher EA and Goldberg IJ. Role of LpL (Lipoprotein Lipase) in Macrophage Polarization In Vitro and In Vivo. Arterioscler Thromb Vasc Biol. 2019;39:1967–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanigawa H, Billheimer JT, Tohyama J, Zhang Y, Rothblat G and Rader DJ. Expression of cholesteryl ester transfer protein in mice promotes macrophage reverse cholesterol transport. Circulation. 2007;116:1267–73. [DOI] [PubMed] [Google Scholar]
- 22.Reis ED, Li J, Fayad ZA, Rong JX, Hansoty D, Aguinaldo JG, Fallon JT and Fisher EA. Dramatic remodeling of advanced atherosclerotic plaques of the apolipoprotein E-deficient mouse in a novel transplantation model. J Vasc Surg. 2001;34:541–7. [DOI] [PubMed] [Google Scholar]
- 23.Chereshnev I, Trogan E, Omerhodzic S, Itskovich V, Aguinaldo JG, Fayad ZA, Fisher EA and Reis ED. Mouse model of heterotopic aortic arch transplantation. J Surg Res. 2003;111:171–6. [DOI] [PubMed] [Google Scholar]
- 24.Trogan E, Fayad ZA, Itskovich VV, Aguinaldo JG, Mani V, Fallon JT, Chereshnev I and Fisher EA. Serial studies of mouse atherosclerosis by in vivo magnetic resonance imaging detect lesion regression after correction of dyslipidemia. Arterioscler Thromb Vasc Biol. 2004;24:1714–9. [DOI] [PubMed] [Google Scholar]
- 25.Rahman K, Vengrenyuk Y, Ramsey SA, Vila NR, Girgis NM, Liu J, Gusarova V, Gromada J, Weinstock A, Moore KJ, Loke P and Fisher EA. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J Clin Invest. 2017;127:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basu D, Hu Y, Huggins LA, Mullick AE, Graham MJ, Wietecha T, Barnhart S, Mogul A, Pfeiffer K, Zirlik A, Fisher EA, Bornfeldt KE, Willecke F and Goldberg IJ. Novel Reversible Model of Atherosclerosis and Regression Using Oligonucleotide Regulation of the LDL Receptor. Circ Res. 2018;122:560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, Amengual J and Fisher EA. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanter JE, Kramer F, Barnhart S, Duggan JM, Shimizu-Albergine M, Kothari V, Chait A, Bouman SD, Hamerman JA, Hansen BF, Olsen GS and Bornfeldt KE. A novel strategy to prevent advanced atherosclerosis and lower blood glucose in a mouse model of metabolic syndrome. Diabetes. 2018;67:946–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan C, Hu J, Parathath S, Grauer L, Cassella CB, Bagdasarov S, Goldberg IJ, Ramasamy R and Fisher EA. Human Aldose Reductase Expression Prevents Atherosclerosis Regression in Diabetic Mice. Diabetes. 2018;67:1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johansson F, Kramer F, Barnhart S, Kanter JE, Vaisar T, Merrill RD, Geng L, Oka K, Chan L, Chait A, Heinecke JW and Bornfeldt KE. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc Natl Acad Sci U S A. 2008;105:2082–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trogan E and Fisher EA. Laser capture microdissection for analysis of macrophage gene expression from atherosclerotic lesions. Methods Mol Biol. 2005;293:221–31. [DOI] [PubMed] [Google Scholar]
- 32.Feig JE and Fisher EA. Laser Capture Microdissection for Analysis of Macrophage Gene Expression from Atherosclerotic Lesions. Lipoproteins and Cardiovascular Disease: Methods and Protocols. 2013:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rubinow KB, Vaisar T, Chao JH, Heinecke JW and Page ST. Sex steroids mediate discrete effects on HDL cholesterol efflux capacity and particle concentration in healthy men. J Clin Lipidol. 2018;12:1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ronsein GE, Hutchins PM, Isquith D, Vaisar T, Zhao XQ and Heinecke JW. Niacin Therapy Increases High-Density Lipoprotein Particles and Total Cholesterol Efflux Capacity But Not ABCA1-Specific Cholesterol Efflux in Statin-Treated Subjects. Arterioscler Thromb Vasc Biol. 2016;36:404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaisar T, Kanter JE, Wimberger J, Irwin AD, Gauthier J, Wolfson E, Bahnam V, Wu IH, Shah H, Keenan HA, Greenbaum CJ, King GL, Heinecke JW and Bornfeldt KE. High Concentration of Medium-Sized HDL Particles and Enrichment in HDL Paraoxonase 1 Associate With Protection From Vascular Complications in People With Long-standing Type 1 Diabetes. Diabetes Care. 2020;43:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutchins PM, Ronsein GE, Monette JS, Pamir N, Wimberger J, He Y, Anantharamaiah GM, Kim DS, Ranchalis JE, Jarvik GP, Vaisar T and Heinecke JW. Quantification of HDL particle concentration by calibrated ion mobility analysis. Clin Chem. 2014;60:1393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vaisar T, Tang C, Babenko I, Hutchins P, Wimberger J, Suffredini AF and Heinecke JW. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J Lipid Res. 2015;56:1519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H and D’Eustachio P. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018;46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayek T, Masucci-Magoulas L, Jiang X, Walsh A, Rubin E, Breslow JL and Tall AR. Decreased early atherosclerotic lesions in hypertriglyceridemic mice expressing cholesteryl ester transfer protein transgene. J Clin Invest. 1995;96:2071–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ and Fisher EA. Diabetes adversely affects macrophages during atherosclerotic plaque regression in mice. Diabetes. 2011;60:1759–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Babaev VR, Fazio S, Gleaves LA, Carter KJ, Semenkovich CF and Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in vivo. J Clin Invest. 1999;103:1697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Eck M, Zimmermann R, Groot PH, Zechner R and Van Berkel TJ. Role of macrophage-derived lipoprotein lipase in lipoprotein metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:E53–62. [DOI] [PubMed] [Google Scholar]
- 43.Lindqvist P, Ostlund-Lindqvist AM, Witztum JL, Steinberg D and Little JA. The role of lipoprotein lipase in the metabolism of triglyceride-rich lipoproteins by macrophages. J Biol Chem. 1983;258:9086–92. [PubMed] [Google Scholar]
- 44.Takahashi M, Yagyu H, Tazoe F, Nagashima S, Ohshiro T, Okada K, Osuga J, Goldberg IJ and Ishibashi S. Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. J Lipid Res. 2013;54:1124–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW and Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009;50:204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zilversmit DB. Role of triglyceride-rich lipoproteins in atherogenesis. Ann N Y Acad Sci. 1976;275:138–44. [DOI] [PubMed] [Google Scholar]
- 47.Goldberg IJ, Eckel RH and McPherson R. Triglycerides and heart disease: still a hypothesis? Arterioscler Thromb Vasc Biol. 2011;31:1716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi M, Hiyama Y, Yokoyama M, Yu S, Hu Y, Melford K, Bensadoun A and Goldberg IJ. In vivo arterial lipoprotein lipase expression augments inflammatory responses and impairs vascular dilatation. Arterioscler Thromb Vasc Biol. 2008;28:455–62. [DOI] [PubMed] [Google Scholar]
- 49.Babirak SP, Iverius PH, Fujimoto WY and Brunzell JD. Detection and characterization of the heterozygote state for lipoprotein lipase deficiency. Arteriosclerosis. 1989;9:326–34. [DOI] [PubMed] [Google Scholar]
- 50.Goldberg IJ, Blaner WS, Vanni TM, Moukides M and Ramakrishnan R. Role of lipoprotein lipase in the regulation of high density lipoprotein apolipoprotein metabolism. Studies in normal and lipoprotein lipase-inhibited monkeys. J Clin Invest. 1990;86:463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH and Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walsh A, Ito Y and Breslow JL. High levels of human apolipoprotein A-I in transgenic mice result in increased plasma levels of small high density lipoprotein (HDL) particles comparable to human HDL3. J Biol Chem. 1989;264:6488–94. [PubMed] [Google Scholar]
- 53.Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, Shulman GI, Blaner WS and Goldberg IJ. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem. 2010;285:37976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang XC, Jin W and Hussain MM. The impact of phospholipid transfer protein (PLTP) on lipoprotein metabolism. Nutr Metab (Lond). 2012;9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayek T, Chajek-Shaul T, Walsh A, Agellon LB, Moulin P, Tall AR and Breslow JL. An interaction between the human cholesteryl ester transfer protein (CETP) and apolipoprotein A-I genes in transgenic mice results in a profound CETP-mediated depression of high density lipoprotein cholesterol levels. J Clin Invest. 1992;90:505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cockerill GW, Rye KA, Gamble JR, Vadas MA and Barter PJ. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:1987–94. [DOI] [PubMed] [Google Scholar]
- 57.Mineo C, Deguchi H, Griffin JH and Shaul PW. Endothelial and antithrombotic actions of HDL. Circ Res. 2006;98:1352–64. [DOI] [PubMed] [Google Scholar]
- 58.Rong JX, Blachford C, Feig JE, Bander I, Mayne J, Kusunoki J, Miller C, Davis M, Wilson M, Dehn S, Thorp E, Tabas I, Taubman MB, Rudel LL and Fisher EA. ACAT inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arterioscler Thromb Vasc Biol. 2013;33:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC and Rothblat GH. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. 2007;48:2453–62. [DOI] [PubMed] [Google Scholar]
- 60.Favari E, Lee M, Calabresi L, Franceschini G, Zimetti F, Bernini F and Kovanen PT. Depletion of pre-beta-high density lipoprotein by human chymase impairs ATP-binding cassette transporter A1- but not scavenger receptor class B type I-mediated lipid efflux to high density lipoprotein. J Biol Chem. 2004;279:9930–6. [DOI] [PubMed] [Google Scholar]
- 61.Remaley AT, Stonik JA, Demosky SJ, Neufeld EB, Bocharov AV, Vishnyakova TG, Eggerman TL, Patterson AP, Duverger NJ, Santamarina-Fojo S and Brewer HB Jr. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem Biophys Res Commun. 2001;280:818–23. [DOI] [PubMed] [Google Scholar]
- 62.Rhainds D and Tardif JC. From HDL-cholesterol to HDL-function: cholesterol efflux capacity determinants. Curr Opin Lipidol. 2019;30:101–107. [DOI] [PubMed] [Google Scholar]
- 63.Pamir N, Hutchins PM, Ronsein GE, Wei H, Tang C, Das R, Vaisar T, Plow E, Schuster V, Koschinsky ML, Reardon CA, Weinberg R, Dichek DA, Marcovina S, Getz GS and Heinecke JW. Plasminogen promotes cholesterol efflux by the ABCA1 pathway. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agrawal N, Freitas Corradi P, Gumaste N and Goldberg IJ. Triglyceride Treatment in the Age of Cholesterol Reduction. Progress in Cardiovascular Diseases. 2016;59:107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khera AV, Won HH, Peloso GM, O’Dushlaine C, Liu D, Stitziel NO, Natarajan P, Nomura A, Emdin CA, Gupta N, Borecki IB, Asselta R, Duga S, Merlini PA, Correa A, Kessler T, Wilson JG, Bown MJ, Hall AS, Braund PS, Carey DJ, Murray MF, Kirchner HL, Leader JB, Lavage DR, Manus JN, Hartzel DN, Samani NJ, Schunkert H, Marrugat J, Elosua R, McPherson R, Farrall M, Watkins H, Lander ES, Rader DJ, Danesh J, Ardissino D, Gabriel S, Willer C, Abecasis GR, Saleheen D, Dewey FE, Kathiresan S, Myocardial Infarction Genetics Consortium DSGCEC and Global Lipids Genetics C. Association of Rare and Common Variation in the Lipoprotein Lipase Gene With Coronary Artery Disease. JAMA. 2017;317:937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ebara T, Okubo M, Horinishi A, Adachi M, Murase T and Hirano T. No evidence of accelerated atherosclerosis in a 66-yr-old chylomicronemia patient homozygous for the nonsense mutation (Tyr61-->stop) in the lipoprotein lipase gene. Atherosclerosis. 2001;159:375–9. [DOI] [PubMed] [Google Scholar]
- 67.Kawashiri MA, Higashikata T, Mizuno M, Takata M, Katsuda S, Miwa K, Nozue T, Nohara A, Inazu A, Kobayashi J, Koizumi J and Mabuchi H. Long-term course of lipoprotein lipase (LPL) deficiency due to homozygous LPL(Arita) in a patient with recurrent pancreatitis, retained glucose tolerance, and atherosclerosis. J Clin Endocrinol Metab. 2005;90:6541–4. [DOI] [PubMed] [Google Scholar]
- 68.Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, Laufs U, Oliver-Williams C, Wood AM, Butterworth AS, Di Angelantonio E, Danesh J, Nicholls SJ, Bhatt DL, Sabatine MS and Catapano AL. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321:364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mead JR, Irvine SA and Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl). 2002;80:753–69. [DOI] [PubMed] [Google Scholar]
- 70.Orasanu G, Ziouzenkova O, Devchand PR, Nehra V, Hamdy O, Horton ES and Plutzky J. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone represses inflammation in a peroxisome proliferator-activated receptor-alpha-dependent manner in vitro and in vivo in mice. J Am Coll Cardiol. 2008;52:869–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, Yan C, Du H, Abumrad NA, Urban JF Jr., Artyomov MN, Pearce EL and Pearce EJ. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ and Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article and its online supplementary files.