

SUMMARY
Aberrant cell proliferation is a hallmark of cancer including glioblastoma (GBM). Here we report that protein arginine methyltransferase (PRMT) 6 activity is required for the proliferation, stem-like properties, and tumorigenicity of glioblastoma stem cells (GSCs), a subpopulation in GBM critical for malignancy. We identified a casein kinase 2 (CK2)-PRMT6-regulator of chromatin condensation 1 (RCC1) signaling axis whose activity is an important contributor to the stem-like properties and tumor biology of GSCs. CK2 phosphorylates and stabilizes PRMT6 through deubiquitylation, which promotes PRMT6 methylation of RCC1, that in turn, is required for RCC1 association with chromatin and activation of RAN. Disruption of this pathway results in defects in mitosis. EPZ020411, a specific small-molecule inhibitor for PRMT6, suppresses RCC1 arginine methylation and improves the cytotoxic activity of radiotherapy against GSC brain tumor xenografts. This study identifies a CK2α-PRMT6-RCC1 signaling axis that can be therapeutically targeted in the treatment of GBM.
Keywords: Mitosis, arginine methylation, glioblastoma (GBM), glioblastoma glioma cell (GSC), protein arginine methyltransferase (PRMT), RCC1, casein kinase 2 (CK2), phosphorylation, tumorigenicity, therapy response
eTOC Blurb
Huang et al. show that PRMT6 methylates RCC1 at arginine 214, which is required for RCC1 association with chromatin and activation of RAN. CK2 phosphorylates and stabilizes PRMT6 through deubiquitylation. Inhibition of PRMT6 reduces tumorigenicity of glioblastoma (GBM) cells and improves the impact of radiotherapy on GBM growth in mice.
Graphical Abstract
INTRODUCTION
GBM is a lethal primary brain tumor with high intra- and inter-tumor heterogeneity containing subpopulations of glioblastoma stem cells (GSCs) that are considered to be responsible for therapy resistance and tumor recurrence (Gimple et al., 2019). A key feature of GBM tumor biology is high mitotic activity (Dunn et al., 2012), suggesting mitosis mediators as a rationale for treating GBM (Dominguez-Brauer et al., 2015). Several approaches have been investigated, including those aimed at microtubules, mitotic kinases, and motor proteins. However, the use of mitotic inhibitors often causes severe side-effects which limit their clinical use (Komlodi-Pasztor et al., 2012). Nonetheless, investigating mitotic processes and mediators continues to reveal therapeutic targets to disrupt this crux of tumor cell biology (Dominguez-Brauer et al., 2015; Haschka et al., 2018).
Regulator of chromosome condensation 1 (RCC1) is a prototype member of the RCC1 super family and the only guanine nucleotide exchange factor (GEF) for RAN GTPase (Hadjebi et al., 2008). RCC1 binds to chromatin through a conformationally diverse loop region containing arginine residues R214 and R217 (McGinty and Tan, 2016; Nemergut et al., 2001), and recruits nuclear RAN-GDP to chromatin for GDP-GTP exchange (Li et al., 2003). Cytoplasmic RAN-GDP and nuclear RAN-GTP establish a gradient that is critical for spindle assembly and spatial coordination of mitosis. RAN GTPase activity promotes cell growth by stimulating mitosis (Clarke and Zhang, 2008). Due to its relationship with RAN GTPase, deficiency of RCC1 or impairment of post-translational modifications that stabilize RCC1, promote cell death due to increased mitotic errors, such as misalignment of chromosomes and formation of multipolar spindles (Chen et al., 2007; Moore et al., 2002). Moreover, RCC1 mutants with defects in phosphorylation or α-N-methylation still bind to the chromatin with reduced affinity (Chen et al., 2007; Hutchins et al., 2004; Li and Zheng, 2004), suggesting that additional mechanisms affect RCC1 association with chromatin and mitosis.
Aberrant methylation of arginine residues is an abundant post-translational protein modification that is increasingly being associated with cancer pathogenesis (Jarrold and Davies, 2019). Post arginine monomethylation (Rme1) as an intermediate, Type II PRMTs generate symmetric arginine dimethylation (sDMA) while Type I PRMTs, including PRMT6, catalyze asymmetric arginine dimethylation (aDMA) (Bedford and Clarke, 2009). PRMT6 is frequently overexpressed in human cancers, and its expression contributes to tumor malignancy (Avasarala et al., 2020; Jarrold and Davies, 2019; Yang and Bedford, 2013). However, the role of PRMT6 in cell mitosis and GBM biology remains largely unknown.
In this study, we report that PRMT6 activity is important for cell mitosis and GSC tumor biology, and specifically with respect to its methylation of RCC1 R214, which promotes RCC1 chromatin association that enhances mitotic activity. CK2α, a ubiquitous protein kinase that is constitutively active in GBM (Nitta et al., 2015), phosphorylates and stabilizes PRMT6. We show that a CK2α-PRMT6-RCC1 signaling axis is critical for GSC cell mitosis, and that inhibiting axis signaling enhances the cytotoxic activity of radiation therapy (RT) in treating GBM preclinical models.
RESULTS
PRMT6 Expression Is Elevated in GSCs and Is a Negative Prognostic Factor for GBM Patients
To identify PRMTs that are involved in glioma tumorigenesis, we evaluated PRMT expression and prognostic significance in GBM, low-grade glioma (LGG), and normal brain (NB) tissue specimens, using RNA-seq datasets from The Cancer Genome Atlas (TCGA), Chinese Glioma Genome Atlas (CGGA), and clinical specimens from Northwestern University (NU). Among the PRMTs, only PRMT1 and PRMT6 show positive association with glioma grade and a worse outcome for glioma patients (Figures 1A–1E, S1A–S1F) in all three datasets. Immunoblot (IB) analysis showed that PRMT6 was undetectable or lowly expressed in NB tissues, normal human astrocytes (NHAs) and neural progenitor cells (NPCs), as compared with glioma tissues, GBM cell lines, and markedly higher in GSCs (Figure 1F and 1G) (Huang et al., 2017; Rohle et al., 2013). PRMT1 did not show appreciable differences in expression among these cell sources (Figure 1G). Multivariate analyses showed an inverse survival association with PRMT6 expression even, when accounting for IDH1 and TP53 mutation status, as well as patient age, and gender (Figure S1G). Elevated PRMT6 expression was also associated with mesenchymal (MES), and classical (CL) compared with proneural (PN) GBM subtypes in the TCGA and CGGA but not NU datasets (Figures S1H–S1J).
Figure 1. PRMT6 Expression Is Elevated in GSCs and Is a Negative Prognostic Factor for GBM Patients.

(A and B), Heatmap and statistical analysis of TCGA (A) and CCGA (B) datasets for expression of PRMT genes in normal brain (NB, A), LGG, and GBM.
(C and D), Kaplan-Meier analysis for PRMT6 expression in the TCGA (C) and CCGA (D) datasets.
(E) Venn diagram of PRMT genes.
(F) IB for PRMT1 and PRMT6 in NB, LGG and GBM of NU glioma cohort.
(G) IB for PRMT1 and PRMT6 in NPCs, NHA, glioma cells, and GSCs.
(H) IB for PRMT6, SOX2, OLIG2, and MYC in GSCs and corresponding differentiated glioma cells (DSCs).
(I) IF of PRMT6 (green) and SOX2 (red), and DAPI (blue for nuclei). Left: images of GBM (n = 5). Right: % of PRMT6+ cells among SOX2+ vs SOX2− cells. Scale bar, 50 μm. Lines, ±SEM.
(H) Pearson correlation between PRMT6 and SOX2 expression in the TCGA GBM dataset. Scale in both axis: log2 (TPM).
*, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, no significance.
Data are representative of two independent experiments with similar results.
We compared PRMT6 expression between GSCs and their corresponding differentiated cells (DSCs), which showed that differentiation is associated with decreased levels of PRMT6 similar to the established stem cell markers SOX2, OLIG2, and MYC (Figure 1H). PRMT6 was preferentially expressed in cells positive for SOX2, OLIG2 and MYC, in GBM patient samples (Figures 1I, S1K and S1L), and PRMT6 mRNA expression showed a direct correlation with SOX2 mRNA levels in GBM samples (Figures 1J and S1M).
PRMT6 Expression Influences GSC Growth, Self-Renewal, and Tumorigenicity
We used short hairpin RNA (shRNA)-mediated knockdown (KD) to suppress and single guide RNA (sgRNA)-mediated knockout (KO) to ablate PRMT6 expression in GSCs (Figure 2A). Both markedly reduced levels of H3R2me2 (Guccione et al., 2007), an established PRMT6 substrate. Moreover, PRMT6 KD and KO suppressed cell growth and sphere-forming frequency, as well as intracranial xenograft growth of cells injected into immunocompromised mice, as indicated by prolonged animal survival (Figures 2B–2D). Re-expression of PRMT6-WT, but not of enzymatically inactive PRMT6 mutant (PRMT6-KLA) (Hyllus et al., 2007), restored H3R2me2, cell growth, and sphere-formation frequency of PRMT6 KD and KO cells (Figures 2E–2G), and increased the intracranial xenograft growth of rescued cells, as indicated by reduced animal survival (Figures 2H and S2). Taken together, these results suggest that PRMT6 expression and activity contribute to the self-renewal and tumorigenicity of GSCs.
Figure 2. PRMT6 Expression Influences Growth, Self-Renewal, and Tumorigenicity of GSCs.

(A) IB for PRMT6 and H3R2me2 in GSC23 and 576 with indicated modifications.
(B and C), Effects of PRMT6 KD or KO on cell proliferation (B), sphere-forming frequency (C) of GSC23 and 576.
(D) BLI of GBM brain xenografts derived from the luciferase-labeled GSC576 with indicated modifications (left). Kaplan-Meier analysis of mice received indicated GSC576 (n=5/group, right).
(E) IB for PRMT6 and H3R2me2 in GSC23/PRMT6 KD and 576/PRMT6 KO cells with indicated modifications. PRMT6-KLA, a catalytically inactive PRMT6.
(F and G) Cell proliferation (F) and sphere-forming frequency (G) of GSC23/PRMT6 KD and 576/PRMT6 KO cells with indicated modifications.
(H) BLI of GBM brain xenografts of GSC576/PRMT6 KO with indicated modifications (left). Kaplan-Meier analysis of mice received indicated GSC576 cells (n = 5, right).
Colored scale bars in (D) and (H) represent photons/s/cm2/steradian. Data in (B, F) are means ± SEM, n=4. **p < 0.01. **p <0.01.
Data are representative of two to three independent experiments with similar results.
See also Figure S2.
PRMT6 Interacts with and Methylates RCC1 at R214
To identify PRMT6 substrates that mediate PRMT6 function in GSCs, we performed mass spectrometry analysis of proteins that co-precipitated with Myc-tagged PRMT6 in HEK293T cells. Among the top candidates, we confirmed PRMT6 association with RCC1 (Hadjebi et al., 2008) (Figure 3A). Compared to the controls, GSCs with PRMT6 KD or KO had lower levels of RAN-GTP (Figure S3A), suggesting that PRMT6 expression is associated with RAN activity. PRMT6 depletion in GSCs also resulted in increase of p21, a cell cycle inhibitor, decrease in histone H3 phosphorylation at S10 (p-S10H3), a mitotic marker (Figure S3A), a G1-phase arrest, and a marked reduction in G2/M phase (Figure S3B). Mapping PRMT6-RCC1 interaction domains revealed that PRMT6 N-terminal sequences (amino acids, AA 1–82) as well as the core domain of RCC1 (AA 200–300) are required for interaction between these proteins (Figures S3C–S3G). Inhibition of PRMT6 increased the frequency of abnormal nuclear morphology at interphase (Figure S3H) and supernumerary spindles at metaphase (Figure S3I), likely due to the disruption of the RCC1-mediated gradient of RAN-GTP formation during interphase and mitosis (Furuta et al., 2016). Additionally, PRMT6 mutant with 1–82 AA deletion and RCC1 mutant with 200–300 AA deletion not only reduced cell growth, sphere-forming frequency of GSCs, but also increased the frequency of abnormal nuclear morphology at interphase and supernumerary spindles at metaphase when compared with their controls, PRMT6 WT and RCC1 WT respectively (Figures S3J–S3M). In silico analysis revealed that high RCC1 expression is associated with glioma progression in TCGA, CGGA, and clinical specimens from NU data, and worse prognosis in TCGA and CGGA glioma (Figures 3B–3C, S3N–3O, and S3Q), and that RCC1 is directly correlated with PRMT6 expression in patient tumors (Figures 3D, S3P, and S3S).
Figure 3. PRMT6 Methylates RCC1 at R214 through Direct Interactions.

(A) IP-IB and IB of PRMT6 and RCC1 in 293T cells and GSC576.
(B) Box plots of the TCGA for RCC1 gene between NB, LGG, and GBM with indicated median.
(C) Kaplan-Meier analyses of the TCGA LGG+GBM dataset for RCC1 expression.
(D) Pearson correlation between PRMT6 and RCC1 expression in the TCGA LGG+GBM dataset. Scale in both axis: log2 (TPM).
(E) IB of GSC576/PRMT6 KO cells using an anti-aDMA antibody. PRMT6 KO-affected aDMA proteins are indicated with two asterisks and one arrow with a molecular weight (~45 KDa) similar to RCC1.
(F) IP-IB and IB of indicated proteins in GSC23/PRMT6 KD and 576/PRMT6 KO cells, or GSCs with EPZ020411 treatments.
(G) IP-IB and IB of indicated proteins in GSC576/PRMT6 KO with indicated modifications.
(H) AA sequences around R214 residue in RCC1 are conserved across different species. Arrows, serine residues that are conserved across species.
(I) IP-IB and IB of indicated proteins in GSC23 and 576 with indicated modifications.
(J) In vitro methylation assays of PRMT6.
***p < 0.001. Data are representative of two to three independent experiments with similar results.
See also Figure S3.
In GSC576 cells, PRMT6 KO yielded an appreciable decrease in various aDMA proteins, including one protein with a molecular weight similar to RCC1 (Figure 3E). PRMT6 KD, KO or inhibition with a PRMT6 inhibitor, EPZ020411 (EPZ) (Mitchell et al., 2015) markedly reduced aDMA of RCC1 as well as H3R2me2 in GSCs (Figure 3F), and re-expression of PRMT6 WT, but not the KLA mutant, rescued PRMT6-induced aDMA of RCC1 (Figure 3G). Among purified recombinant PRMT1–8 proteins, only PRMT6 induced aDMA of RCC1 (Figure S3T).
RCC1 has been identified as an arginine methylated protein and R214 is the putative methylation site of RCC1 (Larsen et al., 2016). The RCC1 AA sequence surrounding R214 is highly conserved among multiple species (Figure 3H) and matches the consensus methylation motif, RGG/RG of PRMT6 (Thandapani et al., 2013). GSCs with RCC1 KD and subsequent re-expression of RCC1 WT or the R217K mutant, which allows RCC1 to associate with chromatin (England et al., 2010), rescued PRMT6-induced aDMA and R214me2 of RCC1 (Figure 3I). An aDMA 214 blocking-peptide inhibited detection of R214me2 of RCC1 in GSCs (Figure S3U) as well as in clinical GBM specimens when using an anti-RCC1 R214me2 antibody (Figure S3V). In vitro methylation assay showed that PRMT6 methylates RCC1 WT and R217K mutant, but not an R214K mutant (Figure 3J). Lastly, methylation of RCC1 R214 was confirmed by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) (Figure S3W).
RCC1 Methylation and its Association with Chromatin, Mitosis, and Tumorigenicity of GSCs
To determine the role of PRMT6-induced R214me2 in mitosis and GSC biology, we used RCC1 KD cells (Figure S4A) and observed increased spindle defects in mitotic cells, such as supernumerary spindles in metaphase and lagging chromosomes in anaphase (Figure S4B). RAN-GTP was not evident in KD cells (Figure S4C). RCC1 KD markedly suppressed GSC proliferation, sphere-forming frequency, and in vivo tumorigenicity as indicated by prolonged survival of mice injected with RCC1 KD GSCs (Figures S4D–S4F).
We synchronized GSCs in G1 phase with a double-thymidine block, and also arrested cells in metaphase using nocodazole after a double-thymidine block. RCC1 R214me2 was observed in GSC chromatin extracts irrespective of the cell cycle phase from which the extracts were obtained (Figure S4G). Next, we introduced shRNA-resistant RCC1 WT-GFP or R214K-GFP fusion proteins in GSCs with RCC1 KD. Although RCC1 WT- and R214K-GFP protein levels were expressed at similar levels in transduced cells, a marked reduction in chromatin-bound R214K-GFP and RCC1 R214me2 signal was evident in cells modified with R214-GFP, suggesting that methylation of R214 is important for RCC1 chromatin association (Figure 4A). In mitotic GSCs, RCC1 WT-GFP localized predominantly to chromatin, while R214K-GFP was dispersed throughout the cytoplasm (Figures 4B and S4H). The chromatin/cytoplasm intensity ratio of R214K-GFP signals was significantly lower than that of WT-GFP (Figure 4C), indicating that loss of R214me2 decreases RCC1 chromatin association. A significant fraction of GSCs expressing R214K-GFP exhibited multiple mitotic defects including partially condensed or misaligned chromosomes, supernumerary metaphase spindles, and lagging chromosomes in anaphase (Figures 4B, 4D, S4H, S4I). In interphase, both WT-GFP and R214K-GFP were localized in nucleus, but R214K-GFP transduced cells frequently displayed clover-shaped nuclei (Figure S4J) (Furuta et al., 2016). RCC1 relies on chromatin as a scaffold for its nucleotide exchange activity (Zhang et al., 2014). We found that diminishing RCC1-chromatin association by RCC1 KD resulted in a marked reduction of RAN-GTP, which was restored upon rescue of KD cells with RCC1-WT-GFP, and to a much lesser extent by R214K-GFP (Figure S4K). RCC1 KD also induced cell cycle arrest and reduced cell mitotic index, with an associated increase in p21 expression and decreased pS10H3 (Figure S4K). RCC1 KD effects therefore phenocopied effects of PRMT6 inhibition in GSCs (Figure S3A). Re-expression of RCC1-WT increased pS10H3 and decreased p21, as compared to re-expression effects of R214K (Figure S4K). IF and IB showed that RAN was mislocalized to the cytoplasm in RCC1-R214K GSCs, whereas RAN was primarily localized in the nucleus in RCC1-WT GSCs (Figures S4L and S4N). As reduced RAN-GTP levels generally affect nucleocytoplasmic transport of all nuclear localization signal (NLS)-containing proteins and precursor-miRNAs, STAT3 and miR-21 were examined as examples because of their crucial role in tumorigenesis (Feng and Tsao, 2016; Huynh et al., 2019). IF and IB analyses showed nuclear localization of Stat3 in RCC1-WT cells treated with IL6, whereas most STAT3 protein was found to be retained in the cytoplasm of RCC1-RK cells (Figures S4M and S4N). The nuclear pre-miRNAs need Exportin-5/RAN-GTP to be exported to the cytoplasm for miRNA maturation. We observed an increase of nuclear accumulation of the precursor miR-21 in RCC1-RK cells relative to RCC1-WT cells (Figure S4O). These results indicated that reduction of nuclear RAN-GTP levels caused by RCC1 R214K mutation leads to inactivation of the nuclear transport machinery. In relation to RCC1 KD cells rescued with RCC1-WT, GSCs expressing R214K also showed reduced cell growth, sphere formation, and brain tumorigenicity as indicated by animal subject survival (Figures 4E–4G).
Figure 4. R214 Methylation of RCC1 is Required for Its Chromatin Association, Mitotic Process, and GSC Tumorigenicity.

(A and H) IB for whole cell lysates (WCL) and chromatin fractions (Chr) of GSC576 and 83 with indicated modifications. β-actin and histone H3 were loading controls for WCL and Chr, respectively.
(B and I) IF for GSC576 with indicated modifications. GFP, green; tubulin, red; DAPI, blue.
(C and J) Ratios of chromosomal:cytosolic GFP in GSC576 with indicated modifications. Ratios of relative fluorescence intensity at chromosome versus centrosome in 50 mitotic cells for each condition were measured.
(D and K) The frequency of supernumerary spindles in GSC576 with indicated modifications. 50 mitotic cells for each condition were measured.
(E, F, L, M) Cell proliferation (E and L) or sphere-forming frequency (F and M) of GSC576 and 83 with indicated modifications.
(G and N) Left: BLI of GBM brain xenografts of GSC576 with indicated modifications. Right, Kaplan-Meier analysis of animals (n=5/group). Colored scale bars represent photons/s/cm2/steradian.
Scale bars in (B, I), 5 μm.
Data in bar or line graphs are means ± SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Data are representative of two to three independent experiments with similar results.
See also Figure S4.
A decrease in RCC1 chromatin association was observed by disrupting the interaction of RCC1 with RAN (RCC1D182A) (Azuma et al., 1999), and by truncating the N-terminus of RCC1 (RCC1Δ21) (Moore et al., 2002). Notably, combined R214K mutation with D182A or Δ21 resulted in further loss of chromosome association (Figures S4P and S4Q), increased frequency of abnormal nuclear morphology at interphase and supernumerary spindles at metaphase (Figures S4R and S4S), and additional decrease of cell growth and self-renewal of GSCs (Figures S4T and S4U), with robust increase in p21 expression and decreased pS10H3 (Figure S4V), suggesting that multiple mechanisms modulate interactions between RCC1 and chromatin in cells. In addition, R214K mutation had no effect on phosphorylation of RCC1 at its N-terminal tail and the interaction between RCC1 and RAN (Figure S4W). Disruption of phosphorylation and methylation at N-terminal tail of RCC1 by S11A and K4Q mutation respectively, had negligible effects on R214 methylation (Figure S4X).
Next, we expressed RCC1 WT-GFP and R214K-GFP in GSC576 and 83 with PRMT6 KO, respectively, followed by RCC1 KD with a shRNA targeting the 3’UTR of RCC1 mRNA. Subsequently, PRMT6 was re-expressed in these cells. Total RCC1, RCC1 R214me2, and PRMT6 were expressed at comparable levels (Figure 4H). Restoring PRMT6 in RCC1 KD/RCC1-WT-GFP, but not RCC1 KD/R214K-GFP GSCs, restored R214me2 and chromatin-bound RCC1 (Figure 4H). PRMT6 KO did not affect the nuclear localization of RCC1-WT-GFP or R214K-GFP in interphase (data not shown), but caused diffuse distribution of WT-GFP signal, similar to that observed in R214K-GFP mitotic cells (Figure 4I and 4J).
Similar to effects caused by R214K-GFP mutant, PRMT6 KO/RCC1-WT-GFP GSCs frequently exhibited interphase and mitotic defects (Figures 4I, 4K and S4Y). Re-expression of exogenous PRMT6 in PRMT6 KO/RCC1-WT-GFP, but not R214K-GFP GSCs, promoted stable RCC1 association with chromatin as well as decreased defects in mitosis and interphase (Figures 4I, 4K, and S4Y). Rescuing with RCC1-WT-GFP in GSC/PRMT6 KO cells led to increased RAN-GTP and pS10H3, while reducing p21 (Figure S4Z). For PRMT6-KO/PRMT6 GSCs, the expression of RCC1 WT-GFP but not R214K-GFP significantly increased cell proliferation, sphere-forming frequency, and intracranial tumorigenicity as indicated by mouse survival (Figures 4L–4N). However, re-expression of PRMT6 or a control vector could not rescue the impeded cell growth and sphere-forming frequency resulted from the RCC1 KD. These observations corroborate with critical role of RCC1 in cell survival as previously reported (Cekan et al., 2016; Chen et al., 2007; Furuta et al., 2016; Li and Zheng, 2004). Of note, in PRMT6-KO/PRMT6 GSCs, RCC1-R214K-GFP expression also resulted in notable increase in cell proliferation and sphere-forming frequency in vitro, as well as in vivo tumorigenicity, when compared with R214K-GFP expression in PRMT6-KO/vector GSCs, suggesting other PRMT6 substrates may be involved in these processes (Figures 4L–4N).
CK2 Stabilizes PRMT6 Protein through Phosphorylation of PRMT6
We assessed PRMT6 mRNA levels in GSCs and their corresponding DSCs, as well as in non-GSCs. We found no difference in PRMT6 mRNA levels among these cells (Figures S5A and S5B), suggesting that GSC-enriched PRMT6 was not regulated at the transcriptional level. PRMT6 proteins have been shown to undergo proteasome degradation (Singhroy et al., 2013), and we have observed the same in GSCs (Figure S5C). In analyzing post-translational modifications of PRMT6, we observed elevated phospho-PRMT6 (p-PRMT6), rather than elevated arginine methylated PRMT6, in GSCs when compared with DSCs or non-GSCs (Figure 5A). Consistent with the prediction that PRMT6 has extensive post-translational modifications at its N-terminus (Winter et al., 2018), a PRMT6-Δ81 mutant lacking its N-terminus was deficient in p-serine/threonine (p-S/T) compared to PRMT6-WT (Figure 5B). Our mass spectrometry analysis identified S11 and T21 at the N-terminus of PRMT6 protein as phosphorylated residues (p-S and p-T, Figure S5D). When PRMT6-WT, non-phosphorylatable S11A or T21A, or combined S11A/T21A (2A) mutants were expressed individually in HEK293T/PRMT6 KO cells, S11A or T21A mutants showed partial reductions in phosphorylation, in relation to WT, whereas the 2A mutant showed a complete lack of phosphorylation (Figure 5C). When probed with residue-specific phospho-antibodies, S11A or T21A mutants were not detected (Figure 5C). PRMT6 2A had no effect on the nuclear localization (Figure S5E), despite their location in relation to the lysine-rich PRMT6 importin binding motif (AA 3–10) that may function as a nuclear localization signal. Additionally, S11A and T21A single mutations had minor effect on protein stability, but the 2A mutant showed a marked increase in ubiquitylation and protein degradation compared with PRMT6 WT (Figures 5D and S5F). Conversely, a phosphorylation mimic PRMT6 2D mutant, but not S11D or T21D single mutant, showed a marked reduction in ubiquitylation as compared to PRMT6-WT (Figure 5D).
Figure 5. CK2α Stabilizes PRMT6 Protein through Phosphorylation of PRMT6.

(A) IP-IB and IB using indicated antibodies in GSCs, their corresponding differentiated glioma cells (DSCs), and glioma cell lines.
(B) IP-IB and IB using indicated antibodies in GSC576 cells expressing a full-length (FL) or N-terminal deletion mutant (Δ81) of HA-PRMT6.
(C-F) IP-IB and IB for indicated proteins in 293T/PRMT6 KO cells with indicated modifications or treatments.
(G) IP-IB and IB for p-S/T of PRMT6 in GSC576/PRMT6 KO cells that were transduced with indicated plasmids. sh-CK2α targets 3’UTR of CSNK2A1 mRNA, and GSC576 cells were collected after treatment with MG132 for 6 h.
(H) IB for PRMT6 in 293T cells that were transduced with indicated plasmids.
(I) IB for PRMT6 and CK2α in GSC576 cells with indicated modifications.
(J) IB for PRMT6 and CK2α in GSC23/CK2α KD or sh-C cells that were treated with 50 μg/ml cycloheximide (CHX) for the indicated times. Band intensities of PRMT6 proteins were quantified and the results were expressed as PRMT6 levels relative to untreated cells. Error bars, ± SEM, n=3. Two-tailed Student’s t-test. *, p < 0.05;
(K and L) IP-IB and IB for ubiquitination of PRMT6 in 293T P6-KO (K) and GSC576 (L) cells with indicated modifications and treatments.
Data are representative of two to three independent experiments with similar results.
See also Figure S5.
We performed an in silico analysis to identify potential upstream protein kinases of PRMT6 (http://www.cbs.dtu.dk/services/NetPhos/). We also treated HEK293T/PRMT6 KO cells expressing Flag-PRMT6 WT with inhibitors of identified kinases (Figure S5G). As shown in Figure 5E, CX-4945 (CX), a highly selective ATP-competitive inhibitor for CK2α (Nitta et al., 2015; Qiao et al., 2019) markedly reduced p-S11 and p-T21 of PRMT6 compared with vehicle control. The AA sequence surrounding S11 and T21 is highly conserved in PRMT6 among multiple species (Figure S5H) and is comparable to the consensus phosphorylation site for CK2α (Figure S5I), which is not present in other PRMTs (data not shown). CK2α interacted with PRMT6, and unlike a kinase-dead K68M mutant, induced p-S/T of PRMT6 (Figures 5F, 5G, S5J, and S5K). Consistent with previous reports (Nitta et al., 2015; Rowse et al., 2017), we observed elevated expression of CK2α in GSCs relative to DSCs, or non-GSCs, and this elevated expression was correlated with increased p-PRMT6 (Figure S5L). Overexpression of CK2α WT, but not K68M increased cellular PRMT6 (Figure 5H), whereas CK2α KD reduced the level of PRMT6 (Figure 5I and 5J). This reduction was rescued by treatment of cells with proteasome inhibitor MG132, or by expression of exogenous CK2α (Figures 5I and S5M). MG132 treatment also inhibited the ubiquitylation of PRMT6-WT in CK2α-WT cells, but not in cells expressing CK2α-K68M, nor in cells expressing the PRMT6 2A mutant (Figure 5K). Increased PRMT6 ubiquitylation was also found in GSC576 cells with CK2α KD (Figure 5L).
CK2 Phosphorylation of PRMT6 Increases RCC1 Association with Chromatin, Mitosis, and tumorigenicity of GSCs
We examined whether CK2 phosphorylation of PRMT6 is critical for PRMT6-RCC1 association, PRMT6 global enzymatic activity, and RCC1 association with chromatin. Overexpression of CK2α K68M but not WT attenuated PRMT6-RCC1 association (Figure 6A). PRMT6 2A but not WT showed diminished ability to interact with RCC1, caused reductions in cellular RCC1me2, H3R2me2, and reduced chromatin association of RCC1 (Figure 6B). In PRMT6 KO cells, PRMT6 2A, but not WT or 2D mutant, abolished PRMT6-RCC1 interaction and markedly reduced RCC1me2, H3R2me2, the chromatin-bound RCC1 as well as p-PMRT6 and PMRT6-chromatin association (Figure S6A and S6B).
Figure 6. CK2α Phosphorylation of PRMT6 Regulates RCC1 Association with Chromatin, Mitosis, and GSC tumorigenicity.

(A) IP-IB and IB for indicated proteins in 293T/PRMT6 KO cells with indicated modifications.
(B) IP-IB and IB for indicated proteins in 293T cells with indicated modifications.
(C) IP-IB and IB for indicated proteins in GSC83/CK2α KD cells that expressed indicated plasmids.
(D) IP-IB and IB for indicated proteins in GSC576/PRMT6 KO cells with indicated modifications and treatments.
(E) Chromosomal:cytosolic RCC1-GFP in GSC576/PRMT6 KO cells with indicated modifications and treatments. n=50 mitotic cells for each condition.
(F-I) Frequencies of indicated mitotic and interphase defects were determined in GSC576/PRMT6 KO cells with indicated modifications and treatments. n=100 cells for each condition.
(J and K) Cell proliferation (J) or sphere-forming frequency (K) of GSC576/PRMT6 KO cells with indicated modifications and treatments.
(L) BLI of GBM brain xenografts of GSC576/PRMT6 KO cells with indicated modifications (n=5/group).
(M) Kaplan-Meier analyses of animals as indicated in (L, n=5/group). Colored scale bars, photons/s/cm2/steradian.
Data in bar or line graphs are means ± SEM. *, p < 0.05; **, p < 0.01;
Data are representative of two to three independent experiments with similar results.
See also Figure S6.
We also found that both RCC1-WT and the R214K mutant associated with PRMT6-WT, suggesting that RCC1 R214me2 is not involved in the PRMT6-RCC1 association (Figure S6C). In GSC83/CK2α KD cells, CK2α KD or rescuing PRMT6 2A reduced the levels of p-PRMT6, RCC1me2, H3R2me2, and RAN-GTP, as well as the PRMT6-RCC1 interaction (Figure 6C). CK2α KD or rescuing PRMT6 2A also increased diffuse distribution of RCC1-GFP signal, the frequency of mitotic and interphase defects, as well as GSC cell proliferation and sphere forming frequency (Figures S6D–S6F). In GSC576/PRMT6 KO cells rescued by PRMT6-WT, 2A, or 2D expression, CX inhibition of CK2α suppressed exogenous PRMT6-WT phosphorylation, PRMT6 binding with RCC1, and reduced the levels of RCC1me2, H3R2me2, and RAN-GTP (Figure 6D). Targeting CK2α also increased diffuse distribution of RCC1-GFP signal as well as the frequency of mitotic and interphase defects (Figures 6E–6I). Moreover, CX inhibition of CK2α decreased the tumorigenicity of PRMT6 KO/PRMT6 WT GSCs in vitro and in vivo (Figures 6J–6M, S6G–S6I). Observed effects of CK2α inhibition could be partially rescued by PRMT6 2D, but not by the 2A mutant (Figures 6C–6M, S6G–S6I), indicating that in addition to PRMT6, other substrate of CK2α might be involved in these processes. Similar to PRMT6, CK2α and RCC1 also regulated stem-like property, we found that inhibition of CK2α or RCC1 decreased expression of SOX2. In addition, KD or CX inhibition of CK2α, but not KD of RCC1, had effect on PRMT6 global binding with chromatin (Figure S6J).
Inhibiting PRMT6 Attenuates GSC Tumor Initiation and Sensitizes GBM to Ionizing Radiation
We investigated the effects of a selective PRMT6 inhibitor EPZ020411 (EPZ), with established in vivo bioavailability (Mitchell et al., 2015), on GSC tumorigenicity. In a dose- and time-dependent manner, EPZ decreased the level of RCC1 R214me2 and H3R2me2, but not known PRMT1 target H4R3me2 (Blanc and Richard, 2017) (Figure 7A), supporting EPZ specificity for inhibiting PRMT6. In a dose-dependent manner, EPZ reduced the association of RCC1 with chromatin, reduced RAN activation, increased frequency of mitotic and interphase defects, induced cell cycle arrest (p21 induction), and blocked mitotic progression (p-S10H3 reduction) in GSCs (Figure 7B and 7C). EPZ also attenuated GSC sphere-forming frequency and cell viability (Figure 7D and 7E), decreased the expression of GSC markers SOX2 and OLIG2, and increased the expression of a neuron differentiation marker, TUBB3 (Figure S7A). Moreover, chromatin immunoprecipitation (ChIP)-PCR results showed no recruitment of PRMT6 to the transcriptional start site (TSS) of the SOX2 gene promoter regardless the enrichment of PRMT6-targeting genes in this assay (Figure S7B). Additionally, CX and EPZ inhibition did not affect cell viability of NHAs (Figures S7C and S7D), and expression of RCC1 WT or R214K had no effects on cell viability and mitosis of RCC1 KD NHAs (Figures S7E–S7H).
Figure 7. EPZ020411 (EPZ) Inhibition of PRMT6 Attenuates the Tumor-Initiating Ability of GSCs and Sensitizes GSC Tumor Xenografts to Ionizing Radiation (IR).

(A) IB for indicated proteins in GSC576 cells that were treated with the indicated concentrations of EPZ for 12 h (left) or with 20 μM EPZ for the indicated times (right).
(B) Chromosomal:cytosolic RCC-GFP and frequencies of indicated defects in mitotic and interphase in GSC83 cells with the indicated concentrations of EPZ for 12 h. n=50 mitotic cells for each condition.
(C) IP-IB and IB for indicated proteins in GSC576 and 23 were treated with the indicated concentrations of EPZ for 12 hr.
(D) Sphere-forming frequency for GSC576 and 23 with the indicated concentrations of EPZ for 12 h.
(E) Cell viability for GSC576 (left) and 23 (right) cells treated with indicated concentration of EPZ for 5 days.
(F) IB for indicated proteins in GSC576 and 23 treated with DMSO, 20 μM EPZ, IR (5 Gy), or EPZ + IR in 2 days post IR. γH2AX was detected at one hour after IR.
(G and H) Cell viability (G) and sphere-forming frequency (H) for GSC576 and 23 cells at day 5 after indicated treatments.
(I and J) Representative BLI images and Kaplan Meier analysis of GBM brain xenografts of GSC576 (I) and 23 (J) with indicated treatments (n=5/group). Colored scale bars represent photons/s/cm2/steradian.
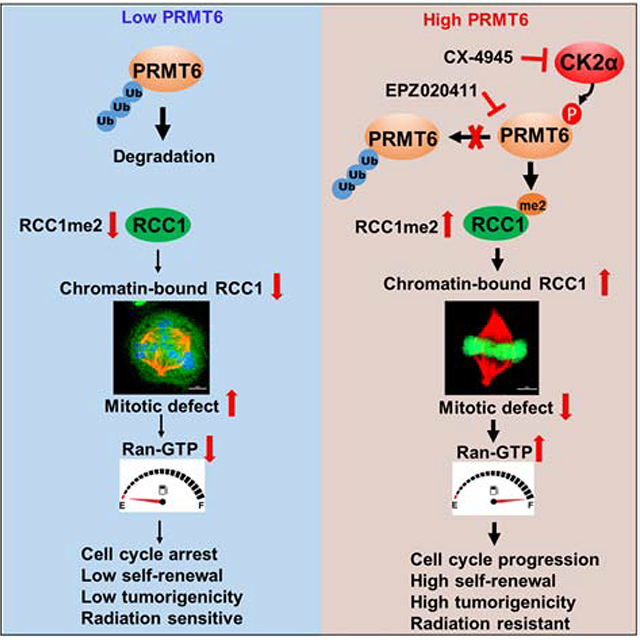
(K) Illustration of the CK2α-PRMT6-RCC1 axis in the regulation of cell mitosis and tumorigenicity of GBM.
Data in bar or line graphs are means ± SEM, *, p < 0.05; **, p < 0.01; ***, p < 0.01;
Data are representative of two to three independent experiments with similar results.
See also Figures S7 and S8.
Radiation therapy is part of the standard care for GBM with limitations on its effects on cell cycle progression (Deckbar et al., 2011). Thus, we investigated the effect of EPZ on GSC response to ionizing irradiation (IR). EPZ enhanced the effects of IR in decreasing GSC viability and sphere-forming frequency when compared to IR treatment alone. EPZ also markedly reduced RCC1me2, with or without IR (Figures 7F to 7H). In vivo co-administration of EPZ to tumor-bearing immunocompromised mice markedly enhanced the cytotoxic activity of IR as indicated by significant increases in animal subject survival in comparison to animals treated with monotherapy (EPZ or IR) (Figures 7I and 7J). Both mono- and combination therapies had negligible effect on the body weight of treated animals (data not shown). EPZ-treated xenografts showed reduced expression of PRMT6 substrates RCC1me2 and H3R2me2 (Figures S7I and S7J), indicating that EPZ is able to penetrate through the blood-brain barrier (BBB) to suppress PRMT6 global enzymatic activity in the xenografts. EPZ and IR decreased Ki67 (cell proliferation), SOX2 (GSC marker), RCC1me2, and p-S10H3 (mitotic index), but increased γH2AX (indicative DNA double-strand breaks), and apoptosis (cleaved caspase-3) in treated tumors. Co-administration of EPZ also enhanced cytotoxic effects of IR on mitosis and tumorigenicity of GSCs when compared with monotherapies (Figures S7I and S7J).
Correlations between CK2α, p-S11/T-21-PRMT6, and RCC1me2 Expression, and Associations with GBM Patient Survival.
We analyzed the expression of CK2α, p-S11, p-T21 of PRMT6, and RCC1me2 in a cohort of 81 patient GBM samples by immunohistochemical (IHC) staining (Figure S8A). The results revealed a positive correlation for the expression of each of these proteins (Figure S8B), and increasing expression of each protein was correlated with worse survival of GBM patients (Figure S8C). The elevated mRNA and protein levels of CK2α, PRMT6 and RCC1 were associated with glioma tumor progression (Figure S8D). Additionally, p-S11, p-T21 of PRMT6, RCC1me2, and RAN-GTP were also associated with glioma malignancy grade (Figures S8E and S8F). Of note, PRMT6 protein levels were lower in normal brain tissue and higher levels in LGG and GBM tissues (Figure 1F). Moreover, the mRNA of PRMT6 showed minimal changes between normal brain tissues and LGG (P>0.05), indicating that PRMT6 expression may be regulated at posttranslational level. Lastly, CSNK2A1, PRMT6 and RCC1 were increased not only in GBM, but also in other types of cancers such as large B-cell lymphoma, lung and skin cancers, further supporting the oncogenic role of the CK2α-PRMT6-RCC1 signaling axis (Figure S8G).
Discussion
This study reveals a CK2α-PRMT6-RCC1 signaling axis that is a key regulator of mitotic process, and whose activity contributes to the tumorigenicity of GSCs. Suppressing the activity of this signaling axis increases GSC sensitivity to IR. Signaling through this axis begins with CK2α phosphorylation of PRMT6 at residues S11 and T21, which protects PRMT6 from ubiquitylation and degradation. This increases PRMT6 methylation of RCC1 at R214, which is necessary for RCC1 to bind to chromatin. Chromatin-bound RCC1 activates RAN-GTPase which is required for mitotic progression and nucleocytoplasmic transport during interphase, thereby promoting GBM tumorigenicity and resistance to IR. Pharmacologic inhibition of this signaling axis with PRMT6 inhibitor EPZ enhances the cytotoxic activity of IR against GBM, as indicated by the extension of animal survival (Figure 7K).
Elevated expression of PRMT6 has been shown in multiple malignancies, including breast (Phalke et al., 2012), prostate (Almeida-Rios et al., 2016), bladder, and lung cancer (Yoshimatsu et al., 2011), which suggests PRMT6 is broadly important in cancer. PRMT6-induced H3R2me2 contributes to global DNA hypomethylation in cancer (Veland et al., 2017), and other diverse PRMT6 functions are required for tumorigenesis in vivo (Avasarala et al., 2020; Bao et al., 2019). Several cancer-associated mechanisms have been proposed for PRMT6 (Guccione and Richard, 2019). For example, PRMT6 transcriptionally represses the expression of tumor suppressors such as p53, p21, and p16 through induction of H3R2me2 (Neault et al., 2012; Phalke et al., 2012; Stein et al., 2012). In this study, we described a stimulatory role for PRMT6 in GSC cell mitosis and tumorigenicity. Elevated expression of PRMT6 and activity are associated with glioma tumor malignancy, patient survival, and GSC tumorigenic properties in vitro and in vivo. PRMT6 methylates RCC1 at R214, promoting RCC1 chromatin association, RAN activation, and mitosis. We also show that CK2 phosphorylation of PRMT6 at p-S11 and p-T21 protects PRMT6 from ubiquitylation, thereby increasing PRMT6 stability and function. Furthermore, our data reveal that the key components of the CK2α-PRMT6-RCC1 signaling pathway are upregulated in glioma cell lines, glioma stem cells, and clinical tumors, which we suggest leads to the specific activation and tumorigenic properties of this pathway. However, the mechanisms controlling the activation of this axis in GBM have yet to be determined. While our epigenomics analyses excluded DNA and histone methylation in regulating the expression of these proteins, it is possible that upregulated transcription factors or other mechanisms are responsible for their dysregulation in tumorigenesis. Another potential regulatory mechanism is the E3 ligases and de-ubiquitinating enzymes that are involved in modulating PRMT6 stability in which CK2α phosphorylation stabilizes the protein.
RCC1 has a core seven bladed propeller structure, with one face for binding with RAN, and the other face interacting with chromatin through an N-terminal tail (Makde et al., 2010). The conformationally diverse loop region of RCC1 includes R214 and R217 residues, which are necessary for a dynamic association between RCC1 and chromatin throughout the cell cycle (Bierbaum and Bastiaens, 2013; England et al., 2010). Here, we provided extensive evidence that indicate a critical role of PRMT6-induced methylation of RCC1 in mitosis. We revealed that PRMT6 specifically methylates RCC1 at R214 which promotes its chromatin association. In RCC1 KD cells, expression of exogenous R214K phenocopied defects in the association of RCC1 with chromatin and RAN activation, leading to increased frequency of mitotic defects and cell cycle arrest. Moreover, the effects of R214K on RCC1 binding to chromatin are additive when R214K is co-expressed with mutants of RCC1Δ21 that lacks its N-terminal or RCC1D182A that is incapable of interacting with RAN, suggesting different mechanisms underlying RCC1 association with chromatin. The AA sequence surrounding R214 in RCC1 matches a PRMT6 consensus methylation RGG/RG motif (Blanc and Richard, 2017), which serves as an interface for protein-protein and RNA-protein interactions (Thandapani et al., 2013). Additionally, our results showing that PRMT6 re-expression was able to partially rescue deficiencies in PRMT6-KO/RCC1 R214K GSCs suggest that in addition to RCC1me2, other PRMT6 substrates such as H3R2me2-mediated recruitment of the chromosomal passenger complex may be involved in PRMT6 regulation of mitosis (Kim et al., 2020).
RAN-GTP has been reported to function as a key player of cell transformation, tumor proliferation and progression (Schnepp et al., 2015). Oncogenes, such as c-Myc, H-Ras, K-Ras, EGFR, mutant BRAF, c-Kit, or Met control or are under the control of the MAPK and PI3K pathways in which RAN is a critical player acting downstream of these oncogenes (Boudhraa et al., 2020). Cancer cells are addicted to RAN-GTP signaling to execute mitosis, and are particularly sensitive to the depletion of RAN (Xia et al., 2008). In this study, we showed that GSCs, but not NHAs, were sensitive to inhibition of CK2α-PRMT6-RCC1 mechanism, suggesting that cancer cells are specifically dependent on the CK2α-PRMT6-RCC1 mechanism. Disrupting PRMT6-induced R214me2 reduced RCC1-chromatin association, impeded RAN activation, and impaired RCC1-mediated mitotic processes and nucleocytoplasmic transport, and in so doing inhibited GBM tumorigenicity and DNA damage response. Our results also reveal a prognostic value of RCC1 R214me2 in predicting clinical outcomes of GBM, indicating the importance of R214me2 in GBM biology.
Targeting mitosis has been an attractive therapeutic approach (Dominguez-Brauer et al., 2015), that has been blunted by severe side effects (Haschka et al., 2018). Our data, together with previous reports, show that suppressing RCC1 function causes defects in mitosis and cell cycle that often lead to cell death (Furuta et al., 2016; Li and Zheng, 2004). In this study, we identified and validated PRMT6 as a key regulator of GSC mitosis through direct arginine methylation of RCC1. Genetic suppression or pharmacologic inhibition of PRMT6 not only reduced RCC1 methylation, as well as the methylation of other established PRMT6 substrates, but also impaired RCC1 chromatin binding and RAN activation, augmenting the defects in mitosis and the cell cycle. Inhibiting PRMT6 also attenuated GSC tumorigenicity in vitro and in vivo.
GSCs are largely responsible for GBM resistance to IR and tumor recurrence (Lathia et al., 2015). High mitotic activity co-opts DNA damage response pathways to preserve genome stability, thereby contributing to radioresistance in cancer cells (Petsalaki and Zachos, 2020). We show that targeting PRMT6 by EPZ enhances the cytotoxic activity of IR and reduces the tumorigenic behaviors of GSCs, relative to individual treatments. This data suggests that EPZ penetrated blood brain barrier and reached GBM brain tumors. Additionally, our data reveal that constitutively active CK2α stimulates PRMT6 by phosphorylating S11 and T21, thereby promoting RCC1-mediated mitosis, cell cycle progression, and GSC tumorigenicity. Taken together, our mechanistic and preclinical results not only elaborate the tumor-promoting roles of PRMT6 (Almeida-Rios et al., 2016; Avasarala et al., 2020; Bao et al., 2019), but also establish PRMT6 as a potential therapeutic target with radiation for GBM.
In conclusion, we have identified a CK2α-PRMT6-RCC1 signaling axis that is important for mitotic activity, malignancy, and therapy resistance in GSCs. Our findings that the PRMT6 inhibitor EPZ sensitizes GSC brain tumor xenografts to IR in animals provide a rationale of targeting PRMT6 to improve the standard treatment of GBM. Additionally, our data support the targeting up-stream regulators of mitosis, namely PRMT6 and CK2α, could bring potential benefits for GBM and other cancers, in which the CK2α-PRMT6-RCC1 signaling axis is active.
Limitations:
This study establishes a CK2α-PRMT6-RCC1 signaling axis whose activity contributes to GBM tumorigenesis. We show that this axis is uniquely activated in gliomas, as compared to normal brain samples, due to CK2α-PRMT6-enhanced RCC1 chromatin binding through R214 methylation and Ran-GTP level during mitosis, it remains uncertain whether this signaling axis drives glioma tumorigenesis or simply sustains tumor growth. Since increasing the fidelity of mitosis is not generally thought to be tumorigenic, it is unclear how the increased expression of these pathway components drives tumorigenesis. However, it is possible that the CK2α-PRMT6-RCC1 axis permits defects in mitosis to be tolerated or perhaps simply makes mitosis more efficient which allows for faster cell proliferation. Another layer of complexity is added as we show that outside of mitotic defects, this signaling axis affects both interphase nuclear shape and nucleocytoplasmic transport, that are thought to generally effect interphase gene expression, and decrease cell proliferation. Though we have started to unravel the role of the CK2α-PRMT6-RCC1 signaling axis in glioma biology, it remains uncertain what the relative contribution of the different roles in mitosis and interphase are to tumorigenesis. While we show that the inhibition of this pathway reduces tumorigenesis, future therapeutic strategies should also test the impact of this pathway in normal cell proliferation as this signaling axis has a potentially important role in normal cell mitosis and nucleocytoplasmic transport.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shi-Yuan Cheng (shiyuan.cheng@northwestern.edu). Alternative Lead Contact, Bo Hu (bo.hu@northwestern.edu)
Materials Availability
All the materials generated in this study are available from the Lead Contact upon completion of a Material Transfer Agreement.
Data and Code Availability
RNA Seq data of Northwestern glioma cohort used in this study has been deposited in the NCBI GEO with accession code GSE147352.
Original data for figures in this study are accessible upon request and deposited to Mendeley Data: https://data.mendeley.com/datasets/942×54gf5t/draft?a=d807130b-bb51–4a30-b84b-bb72335f682b.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
For Brain tumor intracranial models, immunocompromised Ncr nu/nu mice at 6–8 weeks of age were obtained from Taconic Farms. Five mice were grouped in each cage. All experiments using animals were conducted under the Institutional Animal Care and Use Committee (IACUC)-approved protocols at Northwestern University in accordance with NIH and institutional guidelines.
Xenograft Studies
For the tumorigenicity studies, male and female mice in equal number were intracranially injected with 1 or 5 X105 GSCs with indicated expression constructs. For in vivo therapeutic experiments, the mice stereotactically transplanted with GSC 576 or 23 cells were randomized into four treatment groups: 1) control (saline, pH 6.5), single-treatment groups 2) EPZ020411 (EPZ), 3) irradiation, or 4) concurrent EPZ and irradiation, The EPZ treatment groups received EPZ at a dose of 10 mg/kg by subcutaneous administration daily for three weeks. The irradiation groups received irradiation at 2 Gy for five consecutive days. The EPZ treatment was started one day after brain transplantation and irradiation treatment was started one week after brain transplantation. Brain GBM xenograft-bearing mice were injected 300 mg/kg of D-luciferin (potassium salt, Gold Biotechnology) before isoflurane anesthesia. Bioluminescence imaging (BLI) was carried out to monitor in vivo brain tumor growth using an IVIS Lumina imaging station (Caliper Life Sciences). The mice were maintained until pathological symptoms from tumor burden developed or 70 days post brain transplantation.
GBM Specimens
De-identified and paraffin-embedded human GBM (WHO grade III and IV) specimens were obtained from year 2001 to 2013 at Saitama Medical University, Saitama, Japan. These clinical GBM specimens were examined and diagnosed by board-certified pathologists at Saitama Medical University. Tumor collections with informed consents and analyses were approved by the Institutional Review Board (IRB) at Saitama Medical University, Saitama, Japan. Fresh and snap-frozen tissue fragments were collected from surgical samples of glioma patients with surgical resections at Northwestern Memorial Hospital through the Northwestern Nervous System Tissue Bank (NSTB) directed by Dr. C. Horbinski. In addition, normal brain (NB) tissues were obtained from the NeuroBioBank at NIH (https://neurobiobank.nih.gov/). Written informed consent was obtained from all patients in these three sources for the use of their samples. The receipt and analyses of these glioma and NB specimens were under a current IRB protocol approved by the Institutional Review Board at Northwestern University in accordance with guidelines by the Declaration of Helsinki, NIH, and institutional Ethics Committee.
Cell Lines and Cell Culture
HEK293T cells and GBM cells used in this study were obtained from ATCC (U87, T98G) or Dr. EG Van Meir (LN443, LN444) (Ishii et al., 1999) and described previously (Feng et al., 2014). These cell lines were cultured in DMEM (Invitrogen) with 10% heat-inactivated FBS and 1% penicillin and streptomycin. Normal human astrocytes (NHAs, LONZA, ThermoFisher) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% bovine calf serum (HyClone). Patient-derived glioma stem-like cells (GSCs) TS 576 was kindly provided by Dr. Cameron Brennan, MSKCC (Rohle et al., 2013); GSC23 were acquired from Dr. Erik P. Sulman, MD Anderson (Bhat et al., 2013); GSC19, 84, 157 and 83 were obtained from Dr. Ichiro Nakano, University of Alabama at Birmingham (Mao et al., 2013); GSC46 was kindly provided by from Dr. John A. Kessler, Northwestern University (Srikanth et al., 2013). GSCs were cultured as non-adherent spheroids in serum-free defined medium containing DMEM/F12 (Invitrogen), supplemented with B27 (2%, Invitrogen), penicillin and streptomycin (1%, Invitrogen), Heparin (5 μg/ml, Sigma-Aldrich), EGF (20 ng/ml), and bFGF (20 ng/ml, Peprotech). Human NPC lines derived from fetal brains (Lonza) were cultured in Neurobasal media (Invitrogen) supplemented with B27 (Invitrogen), EGF, and bFGF (20 ng/ml each; R&D Systems) and maintained according to the vendor’s instruction. For screening the upstream kinase for PRMT6, the 293T cells were separately treated for 4 h with GSK inhibitor SB 216763 (20 μM), CSNK1a1/CK1α inhibitor D4476 (10 μM), CaMKII inhibitor KN-93 (10 μM), PKA inhibitor H-89 (20 μM), CDK1 inhibitor RO-3306 (10 μM), CSNK2a1/CK2α inhibitor CX-4945 (10 μM), p38 MAPK inhibitor SB-202190 (10 μM) or CDK5 inhibitor Roscovitine (20 μM), respectively.
METHOD DETAILS
Bioinformatic Analyses of PRMT6 Expression
To comprehensively study the expression profiles and prognostic implications of PRMT family members and RCC1 genes in gliomas, analyzed RNA-seq data with FPKM values for each gene and corresponding clinical data were downloaded from TCGA through GDC Data Portal (https://portal.gdc.cancer.gov), Firebrowse (http://firebrowse.org/), and CGGA (http://www.cgga.org.cn) (Tables S1 and S2). RNA-seq data and corresponding clinical data of PRMT family genes in clinical glioma specimens from Northwestern NSTB and normal brain tissue specimens form NIH NeuroBiobank were included in Table S3. NU RNA-seq data has been deposited in NBCI GEO with accession code GSE147352. The expression of PRMT family members from RNA-seq data were clustered and visualized with ClustVis online tool (https://biit.cs.ut.ee/clustvis/), using correlation distance and Ward linkage. Significance levels were determined using one-way ANOVA followed by Dunnett’s multiple comparisons test.
REMBRANDT GBM RNA-seq data of PRMT6 and SOX2 were downloaded from Gliovis website (http://gliovis.bioinfo.cnio.es/) (Table S4).
The Mann-Whitney U-test was performed to determine whether PRMTs and RCC1 were differentially expressed between NB, LGG, and GBM.
Kaplan-Meier analyses were used to assess the relevance and importance of indicated genes or proteins in human glioma tumors.
In the analyses of these datasets, the upper median samples were defined as PRMTs- or RCC1-high, and the rest of glioma samples were considered as PRMTs- or RCC1-low. The numerical data were presented as mean ± standard deviation (SD) of at least three determinations.
Plasmid Construction
Lentiviral shRNAs for PRMT6 were purchased from Thermo Fisher; shRNAs for RCC1 were obtained from Dharmacon; shRNA plasmids and pcDNA3-HA-CSNK2A1 wild-type and mutant vectors were provided by Dr. Zhimin Lu, at MD Anderson Cancer Center, University of Texas. pFN21K-Halo-tagged PRMT1–8 were from Dr. Wei Xu’s laboratory at University of Wisconsin-Madison, Madison, Wisconsin. pGFP-RCC1, pCI3-Flag-RCC1 and pDEST-Myc-RCC1 plasmids were kindly provided from Dr. Evelyne Manet at Centre International de Recherche en Infectiologie, France. Open reading frames (ORFs) of human PRMT6 or RCC1 cDNAs were subcloned into the pCDH-EF1-MCS-IRES-copGFP vector to generate pCDH-PRMT6 or RCC1. ORFs of PRMT6 cDNAs were inserted into the pCMV6 vector to generate a pCMV6-PRMT6 with 3XFlag or Myc tag. ORFs of CSNK2A1 cDNAs were inserted into the pLVX vector to generate pLVX-CSNK2A1. pGFP-RCC1 was subcloned into the pLVX vector to generate pLVX- GFP-RCC1. pGFP-PRMT6 WT, or 2A was subcloned into the pLVX vector to generate pLVX-GFP-PRMT6 WT, or 2A. ORFs for PRMT6 and RCC1 cDNAs and its variants were cloned by PCR and then sub-cloned into pET-28a for expression in E. coli. Site-directed mutagenesis was conducted with a QuikChange mutagenesis kit or Q5® site-directed mutagenesis kit, according to the manufacturer’s instructions. All the resulted cDNA constructs and mutants were confirmed for accuracy by DNA sequencing at the NUSeq Core facility at Northwestern University Feinberg School of Medicine (FSM).
Lentiviral Production and Infection
Lentiviral constructs expressing shRNAs, or full-length CSNK2A1, PRMT6, RCC1 or their mutant cDNAs were transfected into HEK293T using Lipofectamine 2000 (Life Technologies) in accordance to the manufacturer’s instructions. For the gene rescue experiments, shRNA targeting 3’UTR of CSNK2A1, PRMT6 or RCC1 was used for knockdown. The supernatants containing lentivirus were collected at 48 and 72 h after transfection and used to infect target cells with polybrene (6.0 μg/ml, Sigma). After 72 h of transduction, were subjected to 1 μg/mL puromycin selection for four days.
CRISPR-mediated PRMT6 Knockout
The gRNAs for PRMT6 knockout were designed using the MIT online tool. The forward and reverse primers with 20 bp target sequence and inserted into the lentiCRISPRv2GFP or pSpCas9(BB)-2A-GFP (PX458) using BsmBI or BbsI sticky ends, respectively. HEK293T cells were seeded in 10 cm plate and transfected with 10 μg lentiCRISPRv2GFP-PRMT6-KO or lentiCRISPRv2 control plasmids, 5 μg psPAX2 and 2.5 μg pVSV-G plasmids using Lipofectamine 2000 to produce CRISPR-lentivirus. Forty-eight to 72 h after transfection, the supernatants containing lentivirus were harvested, filtered, and used to infect GSCs. For generation of HEK293T-PRMT6-KO, HEK293T cells were seeded in 10 cm plate and transfected with 10 μg PX458-PRMT6-KO or PX458 control plasmids, using Lipofectamine 2000. For generation of GSCs-PRMT6-KO single clone without GFP background, GSC cells at a final density of 1 × 107 cells/ml were transfected with 10 μg PX458 constructs through Neon transfection system (ThermoFisher Scientific) following the manufacturer’s directions. Four to 5 days after lentivirus infection, or 7 to 15 days post PX458 construct transfection, 293T and GSC cells were collected to determine the knockout efficiency through genomic DNA sequencing at the NUSeq Core facility at Northwestern University FSM and IB analysis.
Cell Proliferation, Viability and Cell Cycle Assays
GSCs were dissociated into single cells with StemPro Accutase, and were plated into a 24-well plate at a density of 5000 cells per well for cell proliferation assay, or in a 96-well plate at 2000 cells per well for cell viability assay (n=4 in each condition or point in both assays). The number of living cells was counted at seeding or different time points by counting viable (Trypan Blue negative) cells using a hemotocytometer. Cell viabilities were also evaluated using CellTiter-Glow 2.0 Assay (Promega) according to the manufacturer’s instructions. For cell cycle analyses, cultured cells were pulsed with VybrantTM DyeCycleTM Violet Stain for 30 minutes following by processing for FACS
Limited Dilution Assays/Sphere Forming Frequency for GSC
Limiting dilution assays/sphere forming frequency was performed as described previously (Huang et al., 2017). Briefly, dissociated GSCs seeded in 96-well plates at density of 1, 5, 10, 20 or 50 (n=24 in each dilution) for GSC 576, 83, and 23 cells. After 7 day, each well was examined for formation of tumor spheres. Extreme limiting dilution assays/sphere forming frequency were analyzed using a software available at http://bioinf.wehi.edu.au/software/elda
Immunoprecipitation (IP)
Cells were lysed with IP lysis buffer (ThermoFisher Scientific) supplemented with proteinase inhibitor and phosphatase inhibitor cocktail (Sigma-Aldrich), incubated on ice for 15 min, and cleared by centrifugation at 16,000 x g at 4°C for 15 min. After pre-clearing step with protein G- agarose beads (Sigma-Aldrich), cell lysate (5 mg) was subjected to IP with the indicated antibodies for overnight at 4°C. Then, immune complexes were washed three times in cold lysis buffer. The input and output samples were resolved by SDS-PAGE and detected by immunoblot analysis with indicated antibodies.
Immunoblot Analysis (IB)
Cells were lysed with a RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM Sodium chloride,1% NP-40,0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 2mM EDTA) supplemented with proteinase inhibitor and phosphatase inhibitor cocktail (Sigma-Aldrich). Protein samples were quantified using the Bradford assay reagent (Bio-Rad) in accordance with the manufacturer instructions. Protein samples were resolved by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were incubated with appropriate antibodies for overnight at 4°C. Following triple washing with TBS-T buffer (TBS containing 0.1% Tween-20), the blot was further incubated with corresponding peroxidase-labeled secondary antibodies (1:200) and developed with enhanced chemiluminescence (ECL, Amersham Bioscience) reaction according to manufacturer instructions.
Immunofluorescent (IF) Staining
For IF analyses of cultured cells, GSCs were grown on chamber slides precoated with poly (L-lysine). Cells were fixed with cold methanol, permeabilized with PBS containing 0.1% Triton X-100, and blocked with AquaBlock (East Coast Bio, North Berwick, ME). Cells were probed with following primary antibodies indicated in the related figures. After three times of washing with PBS-T, cells were incubated with 594 labeled secondary antibodies (1:200) and DAPI-containing mounting solution Vectashield (Vector Laboratories). The slices were visualized by using a Nikon inverted microscope Eclipse Ti-U equipped with a digital camera.
For IF analyses of GBM xenograft tissue sections, frozen brain tissue sections with GBM xenografts were dried at room temperature for 30 min, and then fixed in PBS with 2% paraformaldehyde for 5 minutes. For IF analyses of clinical glioma specimens tissue sections were deparaffinized, rehydrated through an alcohol series followed by antigen retrieval with sodium citrate buffer. GBM xenograft tumor or clinical glioma tissue sections were blocked with AquaBlock (East Coast Bio, North Berwick, ME) for one hour and then incubated with appropriate primary antibodies at 4°C for overnight, followed by staining with Alexa 488 or 594 labeled secondary antibodies (1:200) and DAPI-containing mounting solution Vectashield (Vector Laboratories). The images were acquired using a Nikon inverted microscope Eclipse Ti-U equipped with a digital camera.
In vitro Kinase Assay
The pcDNA3-HA-CSNK2A1 (CK2α)-WT and –K68M were transfected into HEK293T cells by using lipofectamine 2000 (ThermoFisher Scientific). Three days after transfection, cells were lysed with lysis buffer (20 mM Tris-HCl (pH 8.0), 0.5% Nonidet P-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM DTT, 0.5 mM phenylmethylsulfonyfluoride (PMSF), 20 mM beta-glycerolphosphate, 1 mM sodium orthovanadate and 1 μg/ml leupeptin. HA-CK2α proteins were immunoprecipitated by using a mouse anti-HA.11 epitope Tag antibody (BioLegend, Cat # 901516) and protein G-agarose beads (Sigma-Aldrich) by rotation at 4°C overnight. The precipitated CK2α proteins were re-suspended in 40 μl of 1x kinase buffer (Cell Signaling, Cat #9802) supplemented with 200 μM ATP (Cell Signaling) and purified His-PRMT6 WT, S11A, T21A or 2A protein, which was expressed in pET-28a vector in E. coli. The kinase reaction was conducted at 30 °C for 30 min, and was stopped by addition of 20 μl 3x SDS sample buffer. Each sample was then boiled for 10 min at 100 °C, followed by IB analysis with indicated antibodies.
Arginine Methylation Assay
For in vivo arginine methylation, PRMT6-KO HEK293T cells were co-transfected with Flag-RCC1 and Halo-PRMT1–8 plasmids. Forty-eight h later, the cells were lysed with IP lysis buffer (ThermoFisher Scientific) and IP by using an anti-Flag antibody was performed to pull down the Flag-RCC1 protein. The input and eluted protein lysates were resolved by SDS-PAGE and analyzed by IB with indicated antibodies. The level of asymmetric dimethylated arginine (aDMA) in Flag-RCC1 was assessed by using an anti-aDMA antibody (1:1000, EMD Millipore). For in vitro arginine methylation assay, halo-PRMT6 and Flag-RCC1, proteins were incubated in 15 μl of 5 mM MgCl2, 20 mM HEPES, pH 7.9, 1 mM ethylenediaminetetraacetic acid, 1 mM DTT and 10% glycerol containing 100 μM unlabeled S-(5’-Adenosyl)-L-methionine iodide (Sigma-Aldrich) or 1μ mCi of S-adenosyl-L-methionine {methyl-3H, 3H-SAM} (Amersham) for 1 h. The reaction mix was resolved by SDS-PAGE and fixed in methanol:acetic acid (50:10%) containing Coomassie brilliant blue for 1 h followed by de-staining in methanol:acetic acid (40:10%). The radioactive signals from RCC1 were visualized by fluorography with En3Hance (Perkin-Elmer) according to manufacturer’s instructions.
Proteomics Analysis of PRMT6 Phosphorylation and RCC1 Arginine Methylation
For identification for PRMT6 phosphorylation, Flag-PRMT6 was immunoprecipitated from GSC 576 cells that stably overexpress exogenous HA-CK2α and Flag-PRMT6. The immune complexes were resolved by SDS-PAGE and stained with Coomassie brilliant blue. For RCC1 arginine methylation, RCC1 was incubated with PRMT6 protein in the system of in vitro arginine methylation assays with unlabeled S-(5’-Adenosyl)-L-methionine iodide, and then the reaction mix was separated on SDS-PAGE and visualized by Coomassie brilliant blue. The gel pieces containing PRMT6 and RCC1 proteins were dehydrated in acetonitrile, dried in a speed vacuum, and digested with trypsin. The peptides were extracted from the polyacrylamide and were evaporated for liquid chromatography mass spectrometry (LC-MS) analysis at the Proteomics Core at the Northwestern University Feinberg School of Medicine (http://proteomics.northwestern.edu).
Subcellular Fractionation
Cytoplasmic and nuclear fractions were prepared using a NE-PER™ Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific) according to the manufacturer’s instructions. Briefly, cells were washed with ice-cold PBS buffer and centrifuged at 500xg for 5 min in microcentrifuge tubes and then incubated in ice-cold CER I buffer. Then, ice-cold CER II buffer was added, vortexed and centrifuged. Supernatant was collected and used as the cytosolic fraction. The remaining pellet was suspended in ice-cold NER buffer and vigorously vortexed for 40 min with 15-s breaks every 10 min. Finally, the resulting homogenates were centrifuged for 15 min at 16,000 x g and 4°C to obtain the supernatant as the nuclear fraction.
GSCs expressing PRMT6 WT, 2A, 2D or a vector control were lysed in the lysis buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, 150 mM KCl, 2 mM MgCl2, 0.1% NP40; 1 mM dithiothreitol/protease inhibitor). After 30 min on ice, the soluble fractions (S) were collected by centrifugation (10 min, 16,000 x g, 4°C), and chromatin fractions were isolated from the cells using a chromatin extraction kit according to the manufacturer’s instructions (Abcam). Whole-cell (W), chromatin (C), and soluble (S) fractions were analyzed by 5–20% SDS–PAGE.
Cell Synchronization
For G1-S phase cell synchronization experiments, GSCs were cultured in a complete medium and then washed twice with PBS, followed by double thymidine block (17 h incubation with 2 mM thymidine, 9 h release and then 17 h of incubation with 2 mM thymidine). Following the second block, cells were released into complete medium and assayed.
After the double-thymidine block, GSCs were washed with PBS for three time, and released in complete medium for 6 h, followed by the treatment with 20 ng/ml nocodazole for the indicated time in the figures.
Ubiquitination Assay
Cells were treated with the proteasome inhibitor MG132 (20 μM) for 6 h, and then lysed using RIPA lysis buffer {(50 mM Tris-base pH 6.8, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 10 mM NaF, 10 mM dithiothreitol (DTT), 0.2 mM Na3VO4,1% cocktail protease inhibitors, 1 mM phenylmethylsulfonyl fluoride (PMSF)}. Cell lysates were immunoprecipitated using the indicated antibodies as described in the previous section. To exclude nonspecific ubiquitin-modified species from the PRMT6 complex, The precipitated proteins were washed with a ubiquitylation wash buffer (50 mM Tris- base pH 6.8, 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 1M urea, 1 mM N- ethylmaleimide (NEM), and protease inhibitors) for three times. Then, the protein samples were resolved by SDS-PAGE and then subjected to IB with the indicated antibodies.
Chromosome Immunoprecipitation (ChIP)
GSCs were cross-linked with 1% formaldehyde for 10 min at 37°C and harvested followed by washing twice with cold PBS. The chromatin of GSCs were extracted as described above. The chromatin was sonicated and immunoprecipitated with antibodies against PRMT6, with IgG antibody as the negative control. The immunoprecipitated DNA was used for quantitative real-time PCR (qPCR).
Quantitative Real-time PCR (qPCR)
Total RNA was extracted using Trizol reagent (Invitrogen) and cDNA synthesized using the PrimeScript cDNA synthesis kits (Takara Bio USA, Cat#ab117152) according to the manufacturer’s instructions. The reverse-transcribed cDNA products were used for qPCR analysis using SYBR Select Master Mix (Life Technologies). Supplementary Table 5 includes detailed information about the sequence of the used primers.
RAN Activation Assay
Measurement of RAN activity was conducted using the RAN Activation Assay kit (Cell Biolabs) according to the manufacturer instructions. Briefly, 1 mg of cell lysates was incubated for one h at 4°C with agarose beads conjugated to RAN-binding protein 1 (RANBP1), which specifically binds RAN-GTP rather than RAN-GDP. Beads were pelleted, washed, and resuspended in SDS-PAGE buffer, followed by IB with an anti-RAN antibody and β-actin as a negative control.
Antibody Production
Rabbit polyclonal anti-p-S11-PRMT6 and p-T21-PRMT6 antibodies were commercially produced at ABclonal Science, Inc. Woburn, MA by immunizing animals with a synthetic phosphor- peptide corresponding to residues surrounding S11, T21 of human PRMT6, respectively.
A rabbit polyclonal anti-R124me2-RCC1 antibody was commercially produced at Genemed Synthesis, Inc., San Antonio, TX by immunizing animals with a synthetic asymmetrically dimethylated- peptide corresponding to residues surrounding R214 of human RCC1.
These antibodies were then affinity purified and characterized for their specificity using IB, IP and IHC with or without corresponding modified and unmodified peptides. Nonspecific IgGs were used as a negative control in these assays.
IHC Analysis of Clinical Glioma Specimens.
All human tissue research in this study was conducted according to protocols approved by the Northwestern University Institutional Review Board (IRB) in Chicago, IL, USA. The formalin-fixed paraffin-embedded glioma patient tumors were described in the “GBM specimens” section above. The GBM tissue sections were stained with antibodies against CK2α (1:50, Santa Cruz Biotechnology), p-S11-PRMT6 (1:50), p-T21-PRMT6 (1:50) and RCC1me2 (1:50).
IHC analyses were performed, and percentage of positively stained cells was quantified and statistically analyzed as previously described (Huang et al., 2017). Briefly, IHC staining was quantified as follows: 3+, positive signals in ~50% tumor cells; 2+, positive signals in ~25% tumor cells; 1+, positive signals in ~5% to 25% tumor cells; ±, low or no positive signals in <1% tumor cells; –, no detectable signals in all tumor cells (0%). Tumors with – or ± staining were considered as low expression, and tumors with 1+ to 3+ scores were considered as high expression. Due to the variations on the sensitivity of each antibody that was used for IHC analyses and the inherent tumor heterogeneity between individual glioma tumor samples, even the cut-off/definition for the staining scores described above was applied, numbers of glioma samples with high or low expression of these four individual proteins are different in each graph.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Microsoft Excel 2018 and GraphPad Prism version 6.0 for Windows. For comparing two groups, the two-tailed Student t-tests were used unless otherwise stated. The Mann–Whitney test was conducted to determine whether PRMT family genes and other indicated genes were differentially expressed among normal brain, LGG, and GBM samples. Kaplan-Meier survival data were analyzed using the log-rank test. All grouped data are presented as mean ± SEM unless otherwise stated.
Supplementary Material
Table S1. Expression of PRMT family members and corresponding clinical data of TCGA-LGG and GBM RNA seq dataset, related to Figure 1.
Table S2. Expression of PRMT family members and corresponding clinical data of CGGA RNA-seq dataset, related to Figure 1.
Table S3. Expression of PRMT family members and corresponding clinical data of Northwestern glioma RNA-seq dataset, related to Figure 1.
Table S4. REMBRANDT GBM RNA-seq data for PRMT6 and SOX2 correlation analysis, related to Figure 1.
Table S5. The primer sequences used for quantitative real-time PCR, related to the STAR Methods.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-β-actin | Santa Cruz Biotechnology | Cat# sc-47778; RRID: AB_626632 |
| anti-SOX2 (For IB) | Santa Cruz Biotechnology | Cat# sc-365823; RRID AB_10842165 |
| anti-PRMT6 (For IF) | Santa Cruz Biotechnology | Cat# sc-271744; RRID:AB_10715087 |
| anti-PRMT1 | Santa Cruz Biotechnology | Cat# sc-59648; RRID: AB_785301 |
| anti-CK2α | Santa Cruz Biotechnology | Cat# sc-12738, RRID:AB_2276843 |
| anti-Ran | Santa Cruz Biotechnology | Cat# sc-271376; RRID:AB_10610890 |
| anti-p21 | Santa Cruz Biotechnology | Cat# sc-6246; RRID:AB_628073 |
| anti-PRMT6 (For IB and IP) | Cell Signaling Technology | Cat# 14641; RRID:AB_2798552 |
| anti-S139-H2A.X | Cell Signaling Technology | Cat# 9718S; RRID:AB_2118009 |
| anti-H3 | Cell Signaling Technology | Cat# 9715; RRID:AB_331563 |
| anti-pS10H3 | Cell Signaling Technology | Cat# 9701; RRID:AB_331535) |
| anti-SOX2 (For IF) | Cell Signaling Technology | Cat# 3579S; RRID:AB_2195767 |
| anti-OLIG2 | R&D Systems | Cat# AF2418-SP; |
| anti-c-Myc | Cell Signaling Technology | Cat# 18583; |
| anti-Ran (For IF) | Novus Biologicals | Cat# NBP2-61832 |
| anti-Stat3 | Cell Signaling Technology | Cat#12640; RRID:AB_2629499 |
| anti-cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661S; RRID:AB_2341188 |
| anti-HaloTag | Promega | Cat# G9211; RRID:AB_2688011 |
| anti-H4R3me2a | Active Motif | Cat# 39705; RRID:AB_2793313 |
| anti-HA.11 | BioLegend | Cat# 901515; RRID:AB_2565334 |
| anti-H3R2me2 | GeneTex | Cat# GTX54134; RRID:AB_2752252 |
| Anti-p-RCC1 (Ser11) | Cell Signaling Technology | Cat# 5500; RRID:AB_2797613 |
| anti-PRMT6 (for IHC and IF) | Abcam | Cat# ab72205; RRID:AB_1270032 |
| anti-RCC1 | Abmart | Cat# ab36760; RRID:AB_2179587 |
| anti-Flag | Sigma-Aldrich | Cat# F3165;RRID:AB_259529 Cat# F7425; RRID:AB_439687 |
| anti-α-Tubulin | Sigma-Aldrich | Cat# T9026; RRID:AB_477593 |
| anti-p-Ser/Thr | BD Biosciences | Cat# 612549; RRID:AB_399844 |
| anti-RCC1 R214me2 | Genemed Synthesis, | this manuscript |
| anti-p-S11-PRMT6 | ABclonal Science | this manuscript |
| anti-p-T21-PRMT6 | ABclonal Science | this manuscript |
| Peroxidase-IgG Fraction Monoclonal Mouse Anti-Rabbit IgG, Light Chain Specific | Jackson ImmunoResearch Laboratories | Cat# 211-032-171; RRID:AB_2339149 |
| anti-Ki-67 | EMD Millipore | Cat# AB9260; RRID:AB_2142366 |
| anti-αDMA | EMD Millipore | Cat# 07-414; RRID:AB_310596 |
| Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11008; RRID:AB_143165 |
| Alexa Fluor 594 | Thermo Fisher Scientific | Cat# R37117; RRID:AB_2556545 |
| Horseradish Peroxidase Streptavidin antibody | Vector Laboratories | Cat# SA-5004; RRID:AB_2336509 |
| Anti-Mouse IgG (H+L), rat adsorbed, made in horse antibody | Vector Laboratories | Cat# BA-2001; RRID:AB_2336180 |
| Biotinylated Goat Anti-Rabbit IgG antibody | Vector Laboratories | Cat# BA-1000; RRID:AB_2313606 |
| anti-Rabbit Immunoglobulins | DAKO | Cat# P0217 |
| anti-Mouse Immunoglobulins | DAKO | Cat# P0260 |
| anti-Goat Immunoglobulins | DAKO | Cat# P0449 |
| Biological Samples | ||
| Paraffin-emended glioma tissues | Saitama Medical University | Saitama, Japan |
| snap-frozen glioma tissue | Northwestern University | Chicago, USA |
| normal brain tissues | NeuroBioBank, NIH | Bethesda, MD, USA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| aDMA of R214me2 RCC1 peptide | Genemed Synthesis Inc. | this manuscript |
| Unmodified R214 RCC1 peptide | Genemed Synthesis Inc. | this manuscript |
| p-T21 PRMT6 peptide | ABclonal Science Inc | this manuscript |
| Unmodified T21 PRMT6 peptide | ABclonal Science Inc | this manuscript |
| p-S11 PRMT6 peptide | ABclonal Science Inc | this manuscript |
| Unmodified S11 PRMT6 peptide | ABclonal Science Inc | this manuscript |
| SB 216763 | Sigma-Aldrich | Cat# S3442 |
| D4476 | Sigma-Aldrich | Cat# 218696 |
| KN-93 | Sigma-Aldrich | Cat# K1385 |
| H-89 | Sigma-Aldrich | Cat# B1427 |
| RO-3306 | Sigma-Aldrich | Cat# SML0569 |
| SB 202190 | Sigma-Aldrich | Cat# S7067 |
| Roscovitine | Sigma-Aldrich | Cat# R7772 |
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| SAM | Sigma-Aldrich | Cat# A4377 |
| Thymidine | Sigma-Aldrich | Cat# T9250 |
| Nocodazole | Sigma-Aldrich | Cat# M1404 |
| CX-4945 | AdooQ BioScience | Cat# A11060 |
| EPZ020411 | AdooQ BioScience | Cat# A16377 |
| Vectashield mounting medium with DAPI | Vector Laboratories | Cat# H-1200 |
| MG-132 | Selleckchem | Cat# S2619 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Kinase buffer | Cell Signaling Technology | Cat# 9802 |
| ATP | Cell Signaling Technology | Cat# 9804 |
| D-Luciferin | Gold Bio | Cat# PP82 |
| AquaBlock | EastCoast Bio | Cat# 2144 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11836153001 |
| Phosphatase inhibitor | Sigma-Aldrich | Cat# 4906837001 |
| Protein G Agarose | Sigma-Aldrich | Cat# 16-266 |
| 2% paraformaldehyde/2.5% glutaraldehyde | Polysciences | Cat# 22872 |
| IP Lysis Buffer | Thermo Fisher Scientific | Cat# 87788 |
| O.C.T Compound | Thermo Fisher Scientific | Cat# 23-730-571 |
| Critical Commercial Assays | ||
| QuikChange mutagenesis kit | Agilent Technologies | Cat# 200524 |
| CellTiter Glo 2.0 | Promega | Cat# 200515-5 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat# 78833 |
| QIAquick Gel Extraction kit | Qiagen | Cat#59104 |
| SYBR Select Master Mix | Life Technologies | Cat#4472913 |
| RAN Activation Assay kit | Cell Biolabs | Cat# STA-409 |
| Q5® Site-Directed Mutagenesis Kit | New England Biolabs | Cat# E0554S |
| Chromatin Extraction Kit | Abcam | Cat# ab117152 |
| Deposited Data | ||
| RNA Seq data of Northwestern glioma cohort | NCBI GEO | GSE147352 |
| Original data that associates with this study | Mendeley Data | https://data.mendeley.com/datasets/942x54gf5t/draft?a=d807130b-bb51-4a30-b84b-bb72335f682b |
| Experimental Models: Cell Lines | ||
| U87 | ATCC | Cat# HTB-14 |
| T98G | ATCC | Cat# CRL-1690 |
| LN443 | (Ishii et al., 1999) | N/A |
| LN444 | (Ishii et al., 1999) | N/A |
| GSC19 | (Mao et al., 2013) | N/A |
| GSC84 | (Mao et al., 2013) | N/A |
| GSC157 | (Mao et al., 2013) | N/A |
| GSC83 | (Mao et al., 2013) | N/A |
| HEK293T | ATCC | Cat# CRL-3216 |
| GSC46 | (Srikanth et al., 2013) | N/A |
| GSC23 | (Bhat et al., 2013) | N/A |
| GSC576 | (Rohle et al., 2013) | N/A |
| NPC or NHNP | LONZA | Cat# PT-2599 |
| NHA | LONZA | Cat# CC-2565 |
| Experimental Models: Organisms/Strains | ||
| Athymic (Ncr nu/nu) mice | Taconic Farms | NCRNU-F and NCRNU-M |
| Oligonucleotides | ||
| PRMT6-KO construction primers on lentiCRISPv2GFP or PX458 | Table S5 | N/A |
| PRMT6 qPCR primers | Table S5 | N/A |
| SOX2 qPCR primers | Table S5 | N/A |
| OLIG2 qPCR primers | Table S5 | N/A |
| TUBB3 qPCR primers | Table S5 | N/A |
| ACTB qPCR primers | Table S5 | N/A |
| Recombinant DNA | ||
| TRC shRNA Control | Dharmacon | Cat#RHS6848 |
| GIPZ shRNA Control | Dharmacon | Cat#RHS4346 |
| PRMT6 shRNA #3 | Dharmacon | V2LHS_155923 |
| PRMT6 shRNA #4 | Dharmacon | TRCN0000034685 |
| RCC1 shRNA #2 | Dharmacon | TRCN0000152440 |
| RCC1 shRNA #3 | Dharmacon | TRCN0000157236 |
| CK2α shRNA #1 | Dharmacon | TRCN0000000606 |
| CK2α shRNA #2 | (Ji et al., 2009) | N/A |
| CK2α shRNA#3 | (Ji et al., 2009) | N/A |
| pcDNA3 CK2α and mutants | this manuscript | N/A |
| CD530-PRMT6-WT | this manuscript | N/A |
| CD530-PRMT6-KLA | this manuscript | N/A |
| CD530-PRMT6-S11A | this manuscript | N/A |
| CD530-PRMT6-S11D | this manuscript | N/A |
| CD530-PRMT6-T21A | this manuscript | N/A |
| CD530-PRMT6-T21D | this manuscript | N/A |
| CD530-PRMT6-2A | this manuscript | N/A |
| CD530-PRMT6-2D | this manuscript | N/A |
| CD530-PRMT6 domain deleted | this manuscript | N/A |
| pLvx-PRMT6-Myc | this manuscript | N/A |
| pCMV6-myc-WT | this manuscript | N/A |
| CD530-RCC1-WT | this manuscript | N/A |
| CD530-RCC1-R214K | this manuscript | N/A |
| CD530-RCC1-R217K | this manuscript | N/A |
| CD530-PRMT6-S11D | this manuscript | N/A |
| CD530-RCC1 domain deleted | this manuscript | N/A |
| pLVX-EGFP-RCC1 mutants | this manuscript | N/A |
| Halo-PRMT1-8 | (Wang et al., 2014) | N/A |
| pET28a –PRMT6 WT and mutants | this manuscript | N/A |
| pET28a –RCC1 WT | this manuscript | N/A |
| psPAX2 | Addgene | Cat#12260; RRID:Addgene_12260 |
| pCMV-VSV-G | Addgene | Cat#8454; RRID:Addgene_8454 |
| Software and Algorithms | ||
| Firebrowse | N/A | http://firebrowse.org/ |
| Gliovis | N/A | http://gliovis.bioinfo.cnio.es/ |
| gRNA design MIT online tool | N/A | https://zlab.bio/guide-design-resources |
| GraphPad Prism 6.0 | GraphPad | https://www.graphpad.com/ |
| Extreme Limiting Dilution Analysis (ELDA) | (Hu and Smyth, 2009) | http://bioinf.wehi.edu.au/software/elda/ |
Highlights.
PRMT6 regulates the growth, sphere formation, and tumorigenicity of GBM cells.
PRMT6 methylation of RCC1 regulates RCC1 chromatin binding and RAN activation.
CK2α phosphorylates and stabilizes PRMT6 through deubiquitylation.
Inhibiting PRMT6 enhances the anti-tumor effects of radiotherapy in GBM PDX models.
ACKNOWLEDGMENTS:
All Shared Resources at the Robert H. Lurie Comprehensive Cancer Center, Northwestern University Feinberg School of Medicine that contributed to this study were supported by an NIH NCI Cancer Center grant P30CA060553. The Northwestern Nervous System Tumor Bank was supported by NIH P50CA221747. This work was also supported by US NIH grants NS093843, NS095634, NS115403 (S.Y.C.); CA209345 (W.Z., S.Y.C), CA213293, CA236356 (W.X.), CA201402, NS107071 (I.N.), NS095642 (C.D.J.), AG054429 (J.A.K.), NS102669 and NS117104 (C.H.), and The Lou and Jean Malnati Brain Tumor Institute at Northwestern Medcine (S.Y.C., B.H.). N.S. and D.T. were supported by NIH under award number F31CA232630 (N.S) and K00CA234799 (D.T.). S.Y.C. is a Zell Scholar at Northwestern University.
Footnotes
DECLARATION of INTEREST:
The authors, and our immedaite family members, are not members of the Molecular Cell Advisory board. We have no further financial or competing interests to declare.
SUPPLEMENTAL INFORMATION
Supplemental Information includes eight figures and five tables and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Almeida-Rios D, Graca I, Vieira FQ, Ramalho-Carvalho J, Pereira-Silva E, Martins AT, Oliveira J, Goncalves CS, Costa BM, Henrique R, et al. (2016). Histone methyltransferase PRMT6 plays an oncogenic role of in prostate cancer. Oncotarget 7, 53018–53028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avasarala S, Wu PY, Khan SQ, Yanlin S, Van Scoyk M, Bao J, Di Lorenzo A, David O, Bedford MT, Gupta V, et al. (2020). PRMT6 Promotes Lung Tumor Progression via the Alternate Activation of Tumor-Associated Macrophages. Mol Cancer Res 18, 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma Y, Renault L, Garcia-Ranea JA, Valencia A, Nishimoto T, and Wittinghofer A (1999). Model of the ran-RCC1 interaction using biochemical and docking experiments. J Mol Biol 289, 1119–1130. [DOI] [PubMed] [Google Scholar]
- Bao J, Di Lorenzo A, Lin K, Lu Y, Zhong Y, Sebastian MM, Muller WJ, Yang Y, and Bedford MT (2019). Mouse Models of Overexpression Reveal Distinct Oncogenic Roles for Different Type I Protein Arginine Methyltransferases. Cancer Res 79, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, and Clarke SG (2009). Protein arginine methylation in mammals: who, what, and why. Mol Cell 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Wani K, Heathcock L, James JD, Goodman LD, et al. (2013). Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 24, 331–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbaum M, and Bastiaens PI (2013). Cell cycle-dependent binding modes of the ran exchange factor RCC1 to chromatin. Biophys J 104, 1642–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc RS, and Richard S (2017). Arginine Methylation: The Coming of Age. Mol Cell 65, 8–24. [DOI] [PubMed] [Google Scholar]
- Boudhraa Z, Carmona E, Provencher D, and Mes-Masson AM (2020). Ran GTPase: A Key Player in Tumor Progression and Metastasis. Front Cell Dev Biol 8, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekan P, Hasegawa K, Pan Y, Tubman E, Odde D, Chen JQ, Herrmann MA, Kumar S, and Kalab P (2016). RCC1-dependent activation of Ran accelerates cell cycle and DNA repair, inhibiting DNA damage-induced cell senescence. Mol Biol Cell 27, 1346–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Muratore TL, Schaner-Tooley CE, Shabanowitz J, Hunt DF, and Macara IG (2007). N-terminal alpha-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat Cell Biol 9, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PR, and Zhang C (2008). Spatial and temporal coordination of mitosis by Ran GTPase. Nat Rev Mol Cell Biol 9, 464–477. [DOI] [PubMed] [Google Scholar]
- Deckbar D, Jeggo PA, and Lobrich M (2011). Understanding the limitations of radiation-induced cell cycle checkpoints. Crit Rev Biochem Mol Biol 46, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, and Mak TW (2015). Targeting Mitosis in Cancer: Emerging Strategies. Mol Cell 60, 524–536. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 26, 756–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England JR, Huang J, Jennings MJ, Makde RD, and Tan S (2010). RCC1 uses a conformationally diverse loop region to interact with the nucleosome: a model for the RCC1-nucleosome complex. J Mol Biol 398, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Lopez GY, Kim CK, Alvarez A, Duncan CG, Nishikawa R, Nagane M, Su AJ, Auron PE, Hedberg ML, et al. (2014). EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates AKT-promoted tumorigenesis. J Clin Invest 124, 3741–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng YH, and Tsao CJ (2016). Emerging role of microRNA-21 in cancer. Biomed Rep 5, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta M, Hori T, and Fukagawa T (2016). Chromatin binding of RCC1 during mitosis is important for its nuclear localization in interphase. Mol Biol Cell 27, 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimple RC, Bhargava S, Dixit D, and Rich JN (2019). Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev 33, 591–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guccione E, Bassi C, Casadio F, Martinato F, Cesaroni M, Schuchlautz H, Luscher B, and Amati B (2007). Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 449, 933–937. [DOI] [PubMed] [Google Scholar]
- Guccione E, and Richard S (2019). The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol 20, 642–657. [DOI] [PubMed] [Google Scholar]
- Hadjebi O, Casas-Terradellas E, Garcia-Gonzalo FR, and Rosa JL (2008). The RCC1 superfamily: from genes, to function, to disease. Biochim Biophys Acta 1783, 1467–1479. [DOI] [PubMed] [Google Scholar]
- Haschka M, Karbon G, Fava LL, and Villunger A (2018). Perturbing mitosis for anti-cancer therapy: is cell death the only answer? EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X, Shi T, Yang Y, Sastry N, Horbinski CM, et al. (2017). MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 32, 840–855 e848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins JR, Moore WJ, Hood FE, Wilson JS, Andrews PD, Swedlow JR, and Clarke PR (2004). Phosphorylation regulates the dynamic interaction of RCC1 with chromosomes during mitosis. Curr Biol 14, 1099–1104. [DOI] [PubMed] [Google Scholar]
- Huynh J, Chand A, Gough D, and Ernst M (2019). Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer 19, 82–96. [DOI] [PubMed] [Google Scholar]
- Hyllus D, Stein C, Schnabel K, Schiltz E, Imhof A, Dou Y, Hsieh J, and Bauer UM (2007). PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev 21, 3369–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, and Van Meir EG (1999). Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9, 469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrold J, and Davies CC (2019). PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol Med 25, 993–1009. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim NH, Park JE, Hwang JW, Myung N, Hwang KT, Kim YA, Jang CY, and Kim YK (2020). PRMT6-mediated H3R2me2a guides Aurora B to chromosome arms for proper chromosome segregation. Nat Commun 11, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett DL, and Fojo AT (2012). Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res 18, 51–63. [DOI] [PubMed] [Google Scholar]
- Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, Daniel JA, Jensen LJ, and Nielsen ML (2016). Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signal 9, rs9. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, and Rich JN (2015). Cancer stem cells in glioblastoma. Genes Dev 29, 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, Wirtz D, and Zheng Y (2003). A mechanism of coupling RCC1 mobility to RanGTP production on the chromatin in vivo. J Cell Biol 160, 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, and Zheng Y (2004). Phosphorylation of RCC1 in mitosis is essential for producing a high RanGTP concentration on chromosomes and for spindle assembly in mammalian cells. Genes Dev 18, 512–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makde RD, England JR, Yennawar HP, and Tan S (2010). Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, et al. (2013). Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proceedings of the National Academy of Sciences of the United States of America 110, 8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, and Tan S (2016). Recognition of the nucleosome by chromatin factors and enzymes. Curr Opin Struct Biol 37, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell LH, Drew AE, Ribich SA, Rioux N, Swinger KK, Jacques SL, Lingaraj T, Boriack-Sjodin PA, Waters NJ, Wigle TJ, et al. (2015). Aryl Pyrazoles as Potent Inhibitors of Arginine Methyltransferases: Identification of the First PRMT6 Tool Compound. ACS Med Chem Lett 6, 655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore W, Zhang C, and Clarke PR (2002). Targeting of RCC1 to chromosomes is required for proper mitotic spindle assembly in human cells. Curr Biol 12, 1442–1447. [DOI] [PubMed] [Google Scholar]
- Neault M, Mallette FA, Vogel G, Michaud-Levesque J, and Richard S (2012). Ablation of PRMT6 reveals a role as a negative transcriptional regulator of the p53 tumor suppressor. Nucleic Acids Res 40, 9513–9521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemergut ME, Mizzen CA, Stukenberg T, Allis CD, and Macara IG (2001). Chromatin docking and exchange activity enhancement of RCC1 by histones H2A and H2B. Science 292, 1540–1543. [DOI] [PubMed] [Google Scholar]
- Nitta RT, Gholamin S, Feroze AH, Agarwal M, Cheshier SH, Mitra SS, and Li G (2015). Casein kinase 2alpha regulates glioblastoma brain tumor-initiating cell growth through the beta-catenin pathway. Oncogene 34, 3688–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petsalaki E, and Zachos G (2020). DNA damage response proteins regulating mitotic cell division: double agents preserving genome stability. FEBS J 287, 1700–1721. [DOI] [PubMed] [Google Scholar]
- Phalke S, Mzoughi S, Bezzi M, Jennifer N, Mok WC, Low DH, Thike AA, Kuznetsov VA, Tan PH, Voorhoeve PM, et al. (2012). p53-Independent regulation of p21Waf1/Cip1 expression and senescence by PRMT6. Nucleic Acids Res 40, 9534–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Chen T, Yang H, Chen Y, Lin H, Qu W, Feng F, Liu W, Guo Q, Liu Z, et al. (2019). Small molecule modulators targeting protein kinase CK1 and CK2. Eur J Med Chem 181, 111581. [DOI] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. (2013). An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowse AL, Gibson SA, Meares GP, Rajbhandari R, Nozell SE, Dees KJ, Hjelmeland AB, McFarland BC, and Benveniste EN (2017). Protein kinase CK2 is important for the function of glioblastoma brain tumor initiating cells. J Neurooncol 132, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp RW, Khurana P, Attiyeh EF, Raman P, Chodosh SE, Oldridge DA, Gagliardi ME, Conkrite KL, Asgharzadeh S, Seeger RC, et al. (2015). A LIN28B-RAN-AURKA Signaling Network Promotes Neuroblastoma Tumorigenesis. Cancer Cell 28, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhroy DN, Mesplede T, Sabbah A, Quashie PK, Falgueyret JP, and Wainberg MA (2013). Automethylation of protein arginine methyltransferase 6 (PRMT6) regulates its stability and its anti-HIV-1 activity. Retrovirology 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth M, Das S, Berns EJ, Kim J, Stupp SI, and Kessler JA (2013). Nanofiber-mediated inhibition of focal adhesion kinase sensitizes glioma stemlike cells to epidermal growth factor receptor inhibition. Neuro-oncology 15, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Riedl S, Ruthnick D, Notzold RR, and Bauer UM (2012). The arginine methyltransferase PRMT6 regulates cell proliferation and senescence through transcriptional repression of tumor suppressor genes. Nucleic Acids Res 40, 9522–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thandapani P, O’Connor TR, Bailey TL, and Richard S (2013). Defining the RGG/RG motif. Mol Cell 50, 613–623. [DOI] [PubMed] [Google Scholar]
- Veland N, Hardikar S, Zhong Y, Gayatri S, Dan J, Strahl BD, Rothbart SB, Bedford MT, and Chen T (2017). The Arginine Methyltransferase PRMT6 Regulates DNA Methylation and Contributes to Global DNA Hypomethylation in Cancer. Cell Rep 21, 3390–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter DL, Hart-Smith G, and Wilkins MR (2018). Characterization of Protein Methyltransferases Rkm1, Rkm4, Efm4, Efm7, Set5 and Hmt1 Reveals Extensive Post-Translational Modification. J Mol Biol 430, 102–118. [DOI] [PubMed] [Google Scholar]
- Xia F, Lee CW, and Altieri DC (2008). Tumor cell dependence on Ran-GTP-directed mitosis. Cancer Res 68, 1826–1833. [DOI] [PubMed] [Google Scholar]
- Yang Y, and Bedford MT (2013). Protein arginine methyltransferases and cancer. Nat Rev Cancer 13, 37–50. [DOI] [PubMed] [Google Scholar]
- Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, Kelly JD, Neal DE, Maehara Y, Ponder BA, et al. (2011). Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer 128, 562–573. [DOI] [PubMed] [Google Scholar]
- Zhang MS, Arnaoutov A, and Dasso M (2014). RanBP1 governs spindle assembly by defining mitotic Ran-GTP production. Dev Cell 31, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Expression of PRMT family members and corresponding clinical data of TCGA-LGG and GBM RNA seq dataset, related to Figure 1.
Table S2. Expression of PRMT family members and corresponding clinical data of CGGA RNA-seq dataset, related to Figure 1.
Table S3. Expression of PRMT family members and corresponding clinical data of Northwestern glioma RNA-seq dataset, related to Figure 1.
Table S4. REMBRANDT GBM RNA-seq data for PRMT6 and SOX2 correlation analysis, related to Figure 1.
Table S5. The primer sequences used for quantitative real-time PCR, related to the STAR Methods.
Data Availability Statement
RNA Seq data of Northwestern glioma cohort used in this study has been deposited in the NCBI GEO with accession code GSE147352.
Original data for figures in this study are accessible upon request and deposited to Mendeley Data: https://data.mendeley.com/datasets/942×54gf5t/draft?a=d807130b-bb51–4a30-b84b-bb72335f682b.