

Abstract
During the past decade, immunotherapies have made a major impact on the treatment of diverse types of cancer. Inflammatory toxicities are not only a major concern for FDA-approved checkpoint blockade and CAR T cell therapies, but also limit the development and use of combination therapies. Fundamentally, these adverse events highlight the intricate balance of pro- and anti-inflammatory pathways that regulate protective immune responses. Here we discuss the cellular and molecular mechanisms of inflammatory adverse events, current approaches to treatment as well as opportunities for the design of immunotherapies that limit such inflammatory toxicities while preserving anti-tumor efficacy.
I. Introduction
Over the past decade, immunotherapy has had an enormous impact on the treatment of diverse types of cancer, leading to durable remissions in a subset of patients and significantly extending survival for others. The most broadly effective of these therapies are monoclonal antibodies that block the immune checkpoints cytotoxic T lymphocyte antigen (CTLA)-4, Programmed Death (PD)-1 or its ligand PD-L1. Chimeric antigen receptor (CAR) T cells can provide profound efficacy against hematological malignancies. Alongside the tremendous clinical benefit of immunotherapy has come a diverse array of inflammatory toxicities that can affect any organ system in the body. These toxicities are an important cause of morbidity, frequently lead to treatment discontinuation and can have debilitating long-term consequences. For checkpoint blockade in particular, risk factors for predicting these events have not yet emerged; the inability to predict who will develop toxicities that are severe (e.g. myocarditis) or permanent (e.g. autoimmune diabetes) is a challenge where immunotherapy is being developed as an alternative to established treatments (Figure 1). Inflammatory toxicities have also been a substantial barrier to the development of novel immunotherapies such as activating antibodies targeting co-stimulatory receptors and systemically delivered cytokine therapies. Concerns about severe on-target toxicity also substantially limit the choice of antigens targeted by CAR T cells in solid tumors. Here we will discuss the inflammatory toxicities of all current major types of immunotherapies, specifically checkpoint blockade, adoptive T cell therapies and cytokine therapies.
Figure 1. Organs frequently affected by inflammatory toxicities of checkpoint blockade.
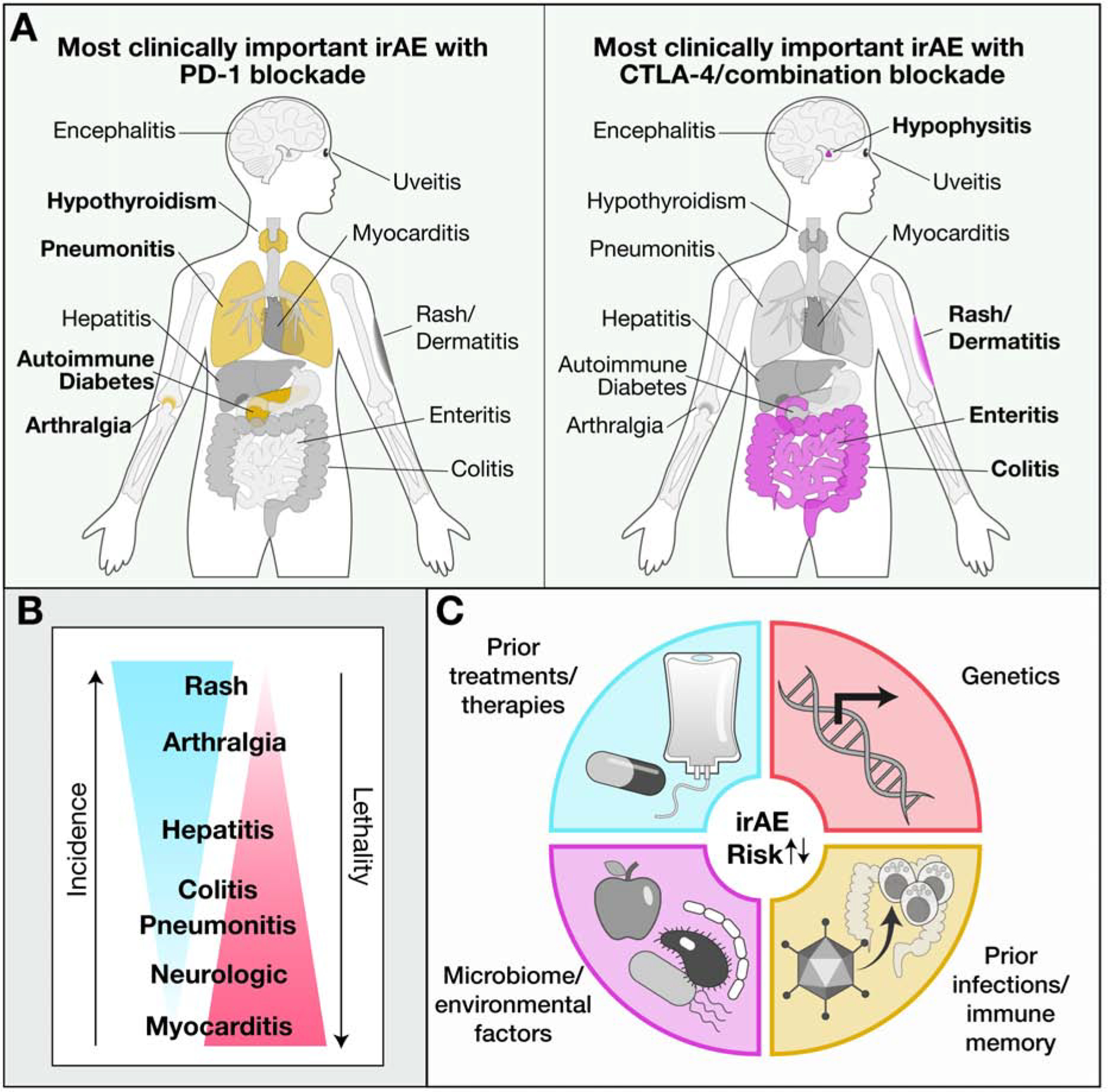
(A) Organs representing the most clinically important sites of inflammatory toxicities induced by PD-1/PD-L1 (left) or CTLA-4/combination (right) blockade.
(B) Relationship between incidence and severity for organs affected by checkpoint inhibitor toxicities.
(C) Potential factors contributing to susceptibility of checkpoint inhibitor toxicities.
II. Adverse events triggered by checkpoint blockade
Function of CTLA-4 and PD-1 inhibitory receptors
Checkpoint blockade immunotherapy has transformed the treatment of multiple malignancies, extending survival, and in some cases producing durable remissions (Ribas and Wolchok, 2018). T cells have a central role in the efficacy of checkpoint blockade based on their ability to recognize MHC-bound tumor cell peptides through the T cell receptor (TCR). Co-stimulatory signals through the CD28 and other receptors contribute to full T cell activation, while the co-inhibitory receptors CTLA-4 and PD-1 attenuate activation (Pauken et al., 2019) (Figure 2). The ligands of the CD28 and CTLA-4 receptors, CD80 and CD86, are expressed by activated antigen presenting cells, and CTLA-4 therefore provides an inhibitory feedback signal during the initiation of T cell responses tissue-draining lymph nodes (Figure 2A). CTLA-4 thus appears to function primarily during early phases of a T cell response but may also inhibit T cell activation by dendritic cells within tumors (Baumeister et al., 2016).
Figure 2. Modulation of T cell function by antibodies targeting the inhibitory CTLA-4 and PD-1 receptors.
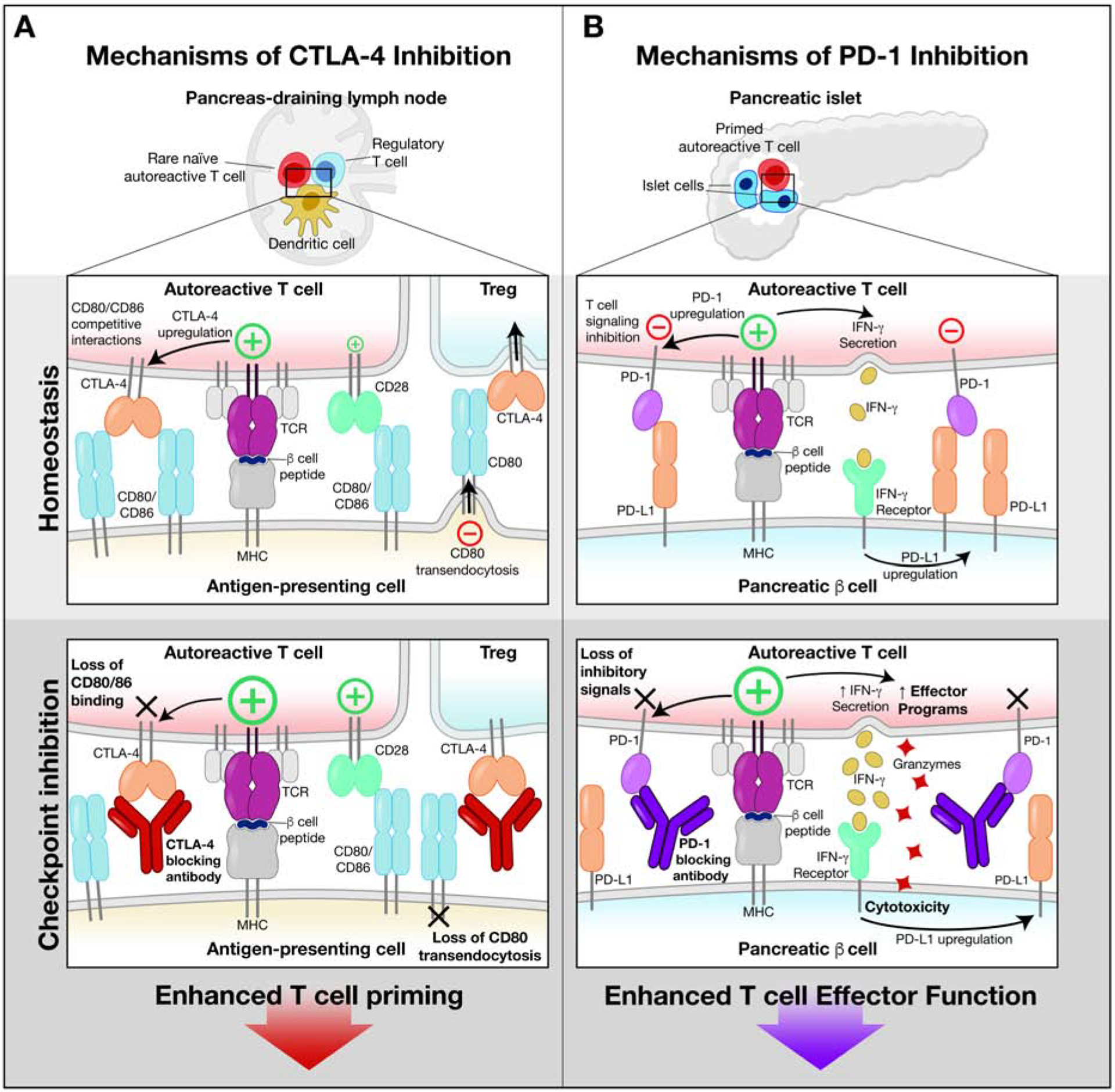
(A) Biology of CTLA-4 receptor. The CTLA-4 inhibitory and CD28 costimulatory receptors bind to the same ligands (CD80/CD86) on antigen presenting cells in lymph nodes. CTLA-4 binds these ligands with higher affinity, thereby reducing ligand availability for CD28. CTLA-4 is stored in an intracellular compartment and transported to the cell surface following initial T cell activation, thereby serving as a negative feedback mechanism. Antibody-mediated inhibition of CTLA-4 function enhances T cell priming by making more CD80/CD86 ligands available for the CD28 costimulatory receptor. Also, the antibody prevents removal of CD80/86 from antigen presenting cells by Tregs.
(B) Biology of the PD-1 receptor. PD-1 expression is induced by T cell activation, thereby providing an inhibitory signal that constrains T cell function. IFNγ secreted by activated T cells induces expression of PD-L1 on target cells (such as pancreatic β cells). This pathway inhibits autoimmunity and immunopathology. Antibody-mediated blockade of this pathway enhances T cell activation, resulting in greater cytotoxicity and release of pro-inflammatory cytokines.
CTLA-4 is constitutively expressed by regulatory T cells (Tregs) (Pauken et al., 2019). Although the full function of CTLA-4 in Tregs is incompletely understood, surface CTLA-4 on Tregs can remove CD80/CD86 molecules from the surface of activated dendritic cells, thus reducing the density of these ligands for the CD28 costimulatory receptor (Qureshi et al., 2011) (Figure 2A).
PD-1 functions in T cells through pathways that are largely non-redundant with CTLA-4 (Figure 2B). However, similar to CTLA-4, PD-1 expression on T cells is upregulated by activation, and the highest levels occur in T cells that have been repeatedly stimulated, including tumor-infiltrating T cells (Baumeister et al., 2016; Zhao et al., 2019). The PD-1 ligands, PD-L1 and PD-L2, show distinct expression patterns. PD-L2 is primarily expressed on immune cells, whereas PD-L1 has a much broader distribution. PD-L1 is upregulated by a variety of inflammatory stimuli, including IFNγ secreted by activated T cells, thus serving to limit T cell-mediated inflammation and autoimmunity (Figure 2B). PD-1 recruits SHP-2 phosphatase and thus inhibits early steps in T cell activation (Baumeister et al., 2016; Hui et al., 2017).
Overview of checkpoint blockade toxicities
Checkpoint blockade induces the most diverse array of toxicities of any of the current immunotherapies in widespread clinical use (Pauken et al., 2019) (Figure 1A). These toxicities can present as single organ inflammatory diseases (e.g. dermatitis) or as systemic diseases affecting multiple systems. Interestingly, the precise distribution of toxicities varies widely from person to person, and while most toxicities occur within the first few months of treatment, they can manifest throughout the immunotherapy course (Martins et al., 2019).
Although involvement of nearly every organ system has been reported, checkpoint blockade toxicities most often affect barrier tissues such as the skin, gastrointestinal tract and liver, and the respiratory epithelium, consistent with a fundamental immune regulatory role for CTLA-4 and PD-1/PD-L1 at these barriers, with CTLA-4 having a particularly critical role in the gut (Dougan, 2017) (Figure 1A). Outside of these epithelial barriers, most toxicities of checkpoint blockade occur in endocrine organs (de Filette et al., 2019; Pauken et al., 2019). Joint inflammation is also relatively frequent, affecting ~10% of patients (Cappelli et al., 2017).
Ipilimumab more frequently induces inflammatory toxicities than PD-1/PD-L1 inhibitors, and combination checkpoint blockade with CTLA-4 plus PD-1 antibodies has the highest frequency of toxicities (Pauken et al., 2019) (Figure 1B). GI mucosal, hepatic and pulmonary toxicities are all reasonably common and lead to substantial morbidity, including the need to delay or discontinue immunotherapy, however, fatal adverse events involving these organs are uncommon (Dougan, 2017; Wang et al., 2018).
Cardiac and neurologic toxicities from immunotherapy are rare, but evolve rapidly and account for a substantial fraction of fatal toxicities from checkpoint blockade (Wang et al., 2018) (Figure 1B). Inflammation in endocrine organs is rarely fatal, but often results in permanent organ dysfunction, necessitating lifelong hormonal supplementation which can impact quality of life (de Filette et al., 2019; Wang et al., 2018). Similarly, toxicities involving the joints can persist long after immunotherapy has stopped, causing pain, reducing mobility and requiring extended treatment (Cappelli et al., 2017). Dermatologic toxicities are the most common toxicities seen with checkpoint blockade, but these are often mild, and generally respond to topical treatments (Phillips et al., 2019).
Mechanisms of checkpoint blockade toxicities
Understanding the immune mechanisms driving checkpoint blockade toxicities will have important clinical implications, potentially leading to more targeted therapeutic strategies and methods for identifying high-risk patients prior to treatment initiation. These toxicities also represent an important window into basic immune biology: they are the phenotype of receptor blockade and thus reflect the homeostatic functions of CTLA-4, PD-1 and PD-L1. The ability to study these diseases as they unfold will provide insights into the cell types most directly regulated by these receptor pathways, as well as the downstream inflammatory pathways.
Effector T cell response
Multiple histopathologic analyses across a variety of organs have investigated the cellular infiltrates in checkpoint blockade toxicities, identifying an expanded population of CD8+ T cells and a smaller population of CD4+ cells (Cohen et al., 2020; Johnson et al., 2016; Marthey et al., 2016).
In a detailed immune analysis of colon biopsies from patients who developed colitis following treatment with CTLA-4 or CTLA-4/PD-1 blockade, we identified a large population of proliferating, cytotoxic CD8+ T cells that were present in biopsies from colitis but not control patients (Luoma et al., 2020) (Figure 3). These CD8 T cells expressed IFNγ, granzyme B as well as PD-1 and CTLA-4 receptors. Analysis of the clonal rearrangements of T cell receptor genes, which can serve as genetic barcodes to identify individual T cells and their progeny, indicated that the majority of expanded CD8+ T cells shared the same receptors as cells found within the tissue-resident memory cell population from the same patients (Luoma et al., 2020). This finding strongly suggests that these expanded cells were, at least in part, derived from resident memory cells within the colon that were held in check by the CTLA-4 and/or PD-1 receptors and became reactivated following checkpoint blockade. Both colitis associated CD8+ and CD4+ cells expressed CXCR3, the receptor for chemokines CXCL9 and CXCL10, both of which were highly expressed by colitis-associated myeloid cells. This finding indicates that additional T cells may indeed be entering the colon from the peripheral blood in response to inflammatory chemokine gradients. In this model, initial expansion of resident memory cells reacting to colonic antigens (such as microbial antigens) could induce an inflammatory response, resulting in release of chemokines and cytokines that recruit additional cell populations from the blood into the tissue (Figure 3). Whether similar resident memory cell expansion explains toxicity in other barrier organs such as the skin or lungs remains to be established.
Figure 3. Inflammatory pathways contributing to colon inflammation following checkpoint blockade.
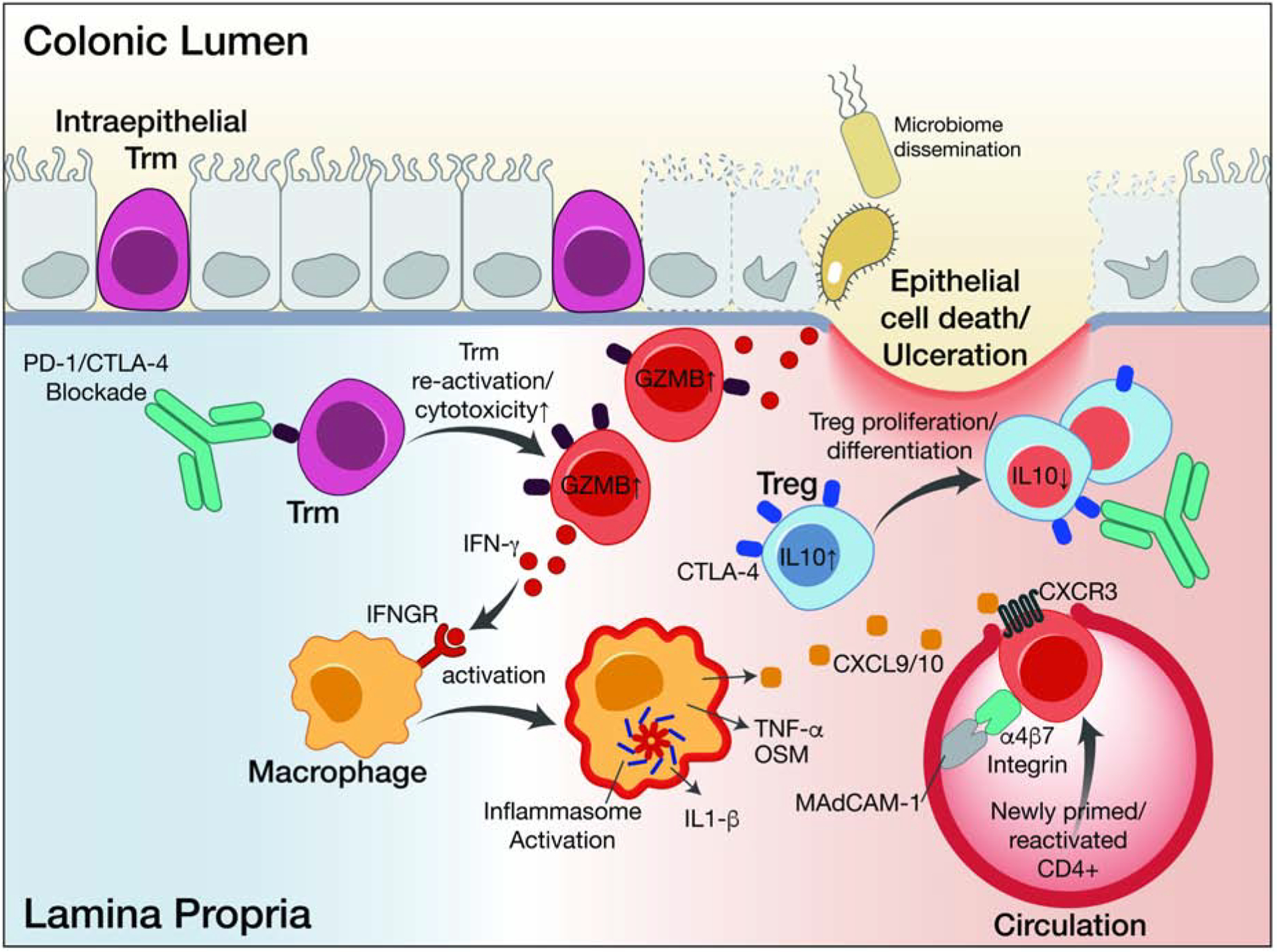
Tissue-resident memory T cells that may be specific for microbial antigens become re-activated following blockade of CTLA-4 and/or PD-1 inhibitory receptors. Re-activated Trm show elevated cytotoxicity, proliferation and inflammatory cytokine (IFNγ) programs. Myeloid cells respond to IFNγ and other cytokines by amplifying the inflammatory response and recruiting T cells from the circulation, thereby overwhelming Treg-mediated suppression. Damage to colon tissue and loss of barrier integrity may result from T cell-mediated cytotoxicity and inflammatory cytokine signaling.
Inhibition of Tregs
In animal models, CTLA-4 targeting antibodies appear to induce antitumor responses through depletion of regulatory T cells (Tregs) which express the highest level of CTLA-4 (Simpson et al., 2013). Whether Treg depletion occurs in humans treated with anti-CTLA-4 antibodies is less clear, and has remained a point of controversy (Pauken et al., 2019). In colitis induced by CTLA-4 blockade, Treg populations were preserved or even expanded, even when biopsies were taken shortly after the onset of symptoms, indicating that Treg depletion is not the driving factor (Luoma et al., 2020) (Figure 3). Transcriptional changes consistent with exposure to IFNγ were found in Tregs from patients with checkpoint colitis (Luoma et al., 2020). Whether the suppressive function of Tregs is impaired in checkpoint colitis remains unknown.
Myeloid cells
Analysis of single-cell RNA-seq (scRNA-seq) data also provides evidence for an important role of myeloid cells in colitis. The transcriptome of myeloid cells was altered in colitis, and myeloid cells showed transcriptional changes induced by IFNγ and TNFα (Luoma et al., 2020) (Figure 3). TNFα is likely to play an important role in driving checkpoint colitis based on clinical data (Mooradian et al., 2020). The scRNA-seq data also provide evidence for an involvement of other inflammatory cytokines, including IL-1β (Luoma et al., 2020).
B cells and antibodies
The role of antibodies in contributing to checkpoint blockade toxicities is not well understood. Key antibody-dependent diseases such as systemic lupus erythematosus (SLE) do not seem to be mimicked by any of the common checkpoint blockade toxicities, nor are antibody-mediated diseases such as pemphigus particularly common in these patients, although some rare toxicities have been reported to respond to B cell-directed therapies (Leonardi et al., 2018; Phillips et al., 2019; Shiuan et al., 2017). Although inflammation of the thyroid gland (thyroiditis) is often observed in patients treated with PD-1/PD-L1 inhibitors, the classic auto-antibodies seen in Hashimoto’s thyroiditis are not universally present (de Filette et al., 2019). Similarly, the classic antibodies in autoimmune hepatitis are frequently absent in patients who develop checkpoint hepatitis, potentially because alternative antigens are targeted (Reynolds et al., 2018; Tahir et al., 2019). One of the most dangerous toxicities from current checkpoint blockade is a neuromuscular inflammatory syndrome that resembles myasthenia gravis (Safa et al., 2019). Some, but not all, of these patients have detectable anti-acetylcholine receptor antibodies suggesting a potential link to classic myasthenia gravis which is defined by these autoantibodies.
Proposed functional subgrouping of inflammatory toxicities
We propose a functional subgrouping of inflammatory toxicities based on the immune repertoire present in organs at the steady state: 1. epithelial organs with a local microbiota and large populations of tissue-resident T cells, 2. sterile internal organs with limited T cell infiltration, including endocrine organs (Figure 4).
Figure 4. Proposed functional subgrouping of inflammatory toxicities.
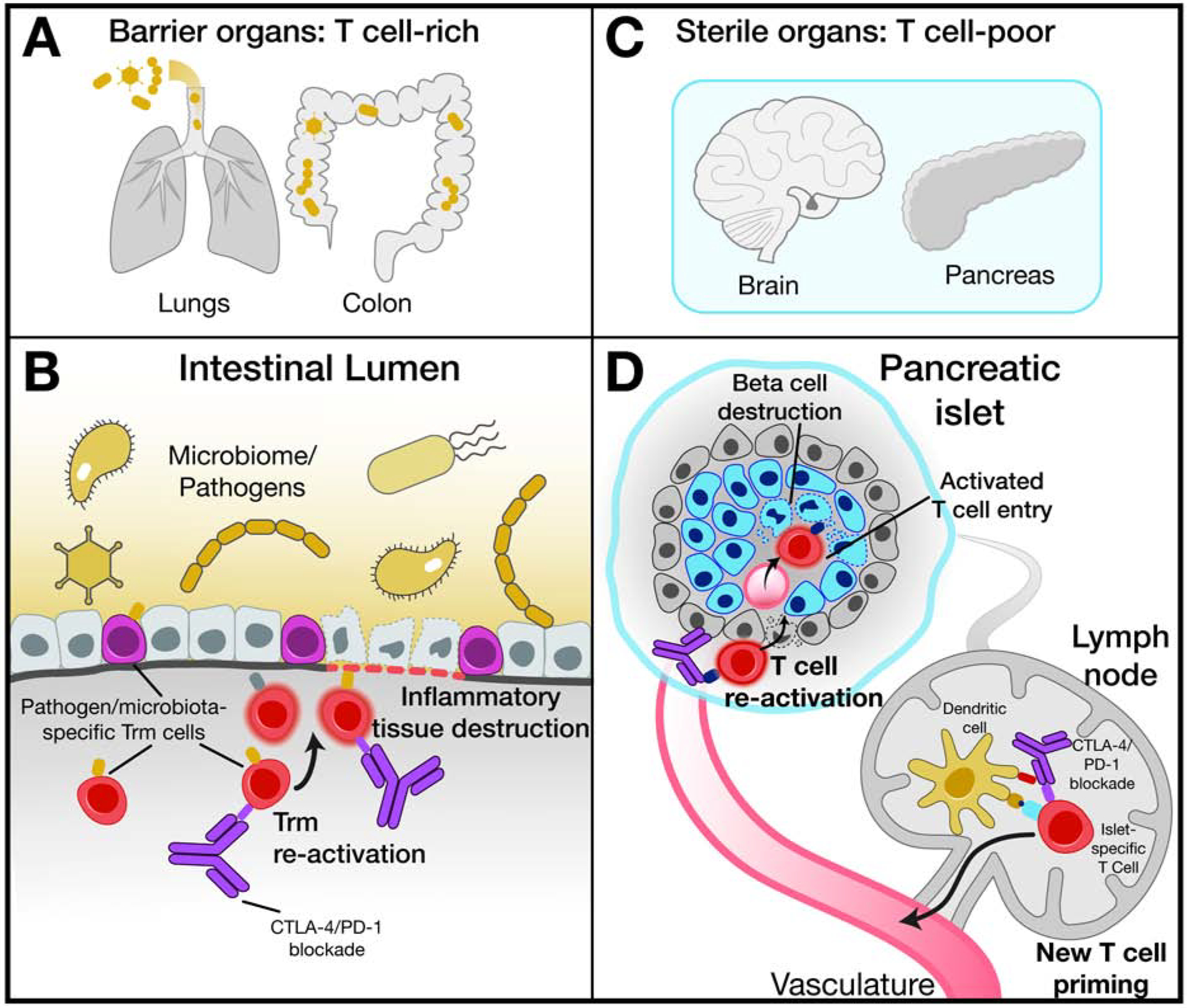
(A) Barrier organs colonized by microbiota harbor an abundant tissue-resident T cell (Trm) population that may recognize microbial antigens.
(B) Re-activation of Trm within barrier organs by checkpoint blockade represents an important step in the development of inflammatory toxicities.
(C) Internal sterile organs tend to have smaller numbers of infiltrating T cells.
(D) Enhanced de novo priming of T cells specific for self-antigens and activation of latent autoreactive T cell clones may represent initial steps in the development of checkpoint blockade induced toxicities in sterile organs, such as endocrine organs or the heart.
Epithelial tissues are colonized by a diverse array of microorganisms and consequently are infiltrated by large populations of tissue-resident T cells (Trm) (Masopust and Soerens, 2019) (Figure 4A). The single cell data from patients with checkpoint colitis demonstrate that the majority of colitis-associated CD8 T cells with highly proliferative and cytotoxic states originated from Trm based on use of T cell receptors as molecular barcodes (Luoma et al., 2020). Tissue-resident memory T cells are an abundant T cell population in the gut mucosa where they act as first responders and form an integral part of an immune sensing network. They can exert cytotoxic function against infected cells and release cytokines/chemokines that rapidly activate surrounding immune cells and recruit additional immune cell populations from the blood (Masopust and Soerens, 2019; Rosato et al., 2020). We hypothesize that the CTLA-4 and PD-1 receptors keep Trm populations in check, and that loss of these inhibitory signal can result in rapid clonal expansion and conversion to a highly cytotoxic state (Figure 4B).
Sterile internal organs have a limited repertoire of tissue-resident memory T cells, although such populations can be present due to prior infections at these sites (Figure 4C). Two different mechanisms could account for the development of these inflammatory toxicities: 1. activation of previously expanded self-reactive lymphocytes, and 2. priming of a de novo T cell response (Figure 4D). Not all self-reactive T cells escape negative selection in the thymus, but the frequency of such cells tends to be very low (Wucherpfennig and Sethi, 2011). Blockade of the PD-1 and/or CTLA-4 inhibitory receptors on T cells is likely to facilitate the activation and expansion of these rare self-reactive T cells. One of the major cytokines released by activated T cells is IFNγ which induces PD-L1 expression on target cells (Alspach et al., 2019). This negative feedback loop is abrogated when cancer patients receive PD-1/PD-L1 blocking antibodies. Genetic studies in murine models have shown that PD-L1 expression on pancreatic islet cells inhibits tissue destruction by T cells, pointing to an important role of PD-L1 in the protection of parenchymal cells against immune attack (Keir et al., 2006).
Relationship between checkpoint blockade induced inflammatory toxicities and spontaneous autoimmune diseases
We will consider the relationship between checkpoint blockade toxicities and spontaneous autoimmunity by comparing ulcerative colitis (UC) to checkpoint colitis (example of epithelial tissue infiltrated by tissue-resident memory cells) and autoimmune type 1 diabetes to checkpoint diabetes (example of a sterile organ with limited immune cell infiltration). UC and checkpoint colitis resemble each other clinically, although checkpoint colitis typically has a more rapid onset. Endoscopically, the diseases are indistinguishable, and while histopathologic characteristics of checkpoint colitis have been identified, they are not consistent enough to readily distinguish it from UC. Single-cell RNA-seq datasets were recently reported for both ulcerative colitis (UC) and checkpoint colitis (Luoma et al., 2020; Smillie et al., 2019). In ulcerative colitis, an increase in IL17+ CD8 T cells was observed, whereas IL17 expression was unchanged in checkpoint colitis. The IL-23 receptor is essential for differentiation of IL-17 producing T cells, and polymorphisms in the IL23 gene are associated with susceptibility to ulcerative colitis (Neurath, 2019). Increased expression of IL-17 by immune cells in ulcerative colitis has also been demonstrated by analysis of biopsy specimens and serum samples. In contrast, expression of IFNγ by T cells and IFNγ inducible genes by myeloid cells were key aspects of the immune microenvironment in checkpoint colitis (Luoma et al., 2020).
In both spontaneous type 1 diabetes (T1D) and checkpoint diabetes, the insulin-producing β cells are destroyed by an immune attack, necessitating insulin replacement therapy (Pugliese, 2017). T1D occurs most commonly in childhood and progresses slowly, commonly overs years, from prediabetes to clinically manifest disease, with residual islet function frequently still detectable at disease onset (van Belle et al., 2011). In contrast, in checkpoint diabetes, insulin-producing β cells are rapidly lost and patients typically present with hypoglycemia, ketoacidosis and no detectable β cell function (Stamatouli et al., 2018). All patients who developed checkpoint diabetes had been treated with a PD-1/PD-L1 blocking antibodies or a combination of PD-1 plus CTLA-4 antibodies (Stamatouli et al., 2018; Tsang et al., 2019). Patients with an early onset of checkpoint diabetes may have already harbored previously expanded populations of islet-reactive T cells with checkpoint blockade enabling rapid reactivation and further expansion of such T cells; this may also be the case in other rapidly progressive toxicities such as myocarditis.
T1D and checkpoint diabetes also differ in immunological parameters and immunogenetics. While >95% of spontaneous T1D patients have autoantibodies against at least one islet antigen at diagnosis, autoantibodies were detected in only 40% of checkpoint diabetes patients (Stamatouli et al., 2018). Susceptibility to spontaneous T1D is strongly associated with particular alleles of HLA-DR and HLA-DQ genes (van Belle et al., 2011). The number of investigated checkpoint diabetes patients is currently too small for a definite assessment, but 3 of 10 patients in one case series had HLA alleles known to provide protection from spontaneous T1D (Tsang et al., 2019).Taken together these findings establish important differences in kinetics and immunological parameters between these diseases.
Approaches to treatment of checkpoint blockade toxicities
Treatment for checkpoint blockade toxicities must be managed in light of the patient’s cancer diagnosis, and optimal treatment strategies should mitigate the toxicity while preserving anti-tumor activity (Figure 5). Optimal treatment strategies established by randomized, controlled trials are not available for any toxicity at present, and current practice guidelines are all based on retrospective analyses, experience from cancer treatment trials, and anecdotal experience (Pauken et al., 2019). Nearly all toxicities from checkpoint blockade respond to high-dose systemic glucocorticoids, with the exception of dermatologic toxicities for which topical agents are generally sufficient, and endocrine toxicities which are typically identified after loss of function by the target organ (Pauken et al., 2019). In most cases, toxicities that resolved following immune suppression will not recur if immune suppression can be successfully tapered, unless patients are re-challenged with checkpoint blockade targeting the same pathway (Pauken et al., 2019). How immunosuppressive glucocorticoids impact antitumor responses requires further investigation. Animal models suggest that glucocorticoids may inhibit antitumor T cell responses, and glucocorticoids are well known to induce apoptosis of activated T cells (Pauken et al., 2019). Findings from retrospective studies of patients on checkpoint blockade who received systemic glucocorticoids have been mixed, though overall suggestive that they have deleterious effects on optimal antitumor responses, particularly when systemic glucocorticoids are used at initiation of immunotherapy (Arbour et al., 2018; Faje et al., 2018; Horvat et al., 2015). Nevertheless, systemic glucocorticoids are first line therapy for most severe toxicities where a rapid response is necessary to prevent significant morbidity and mortality.
Figure 5. Potential therapeutic targets for inflammatory toxicities induced by checkpoint blockade.
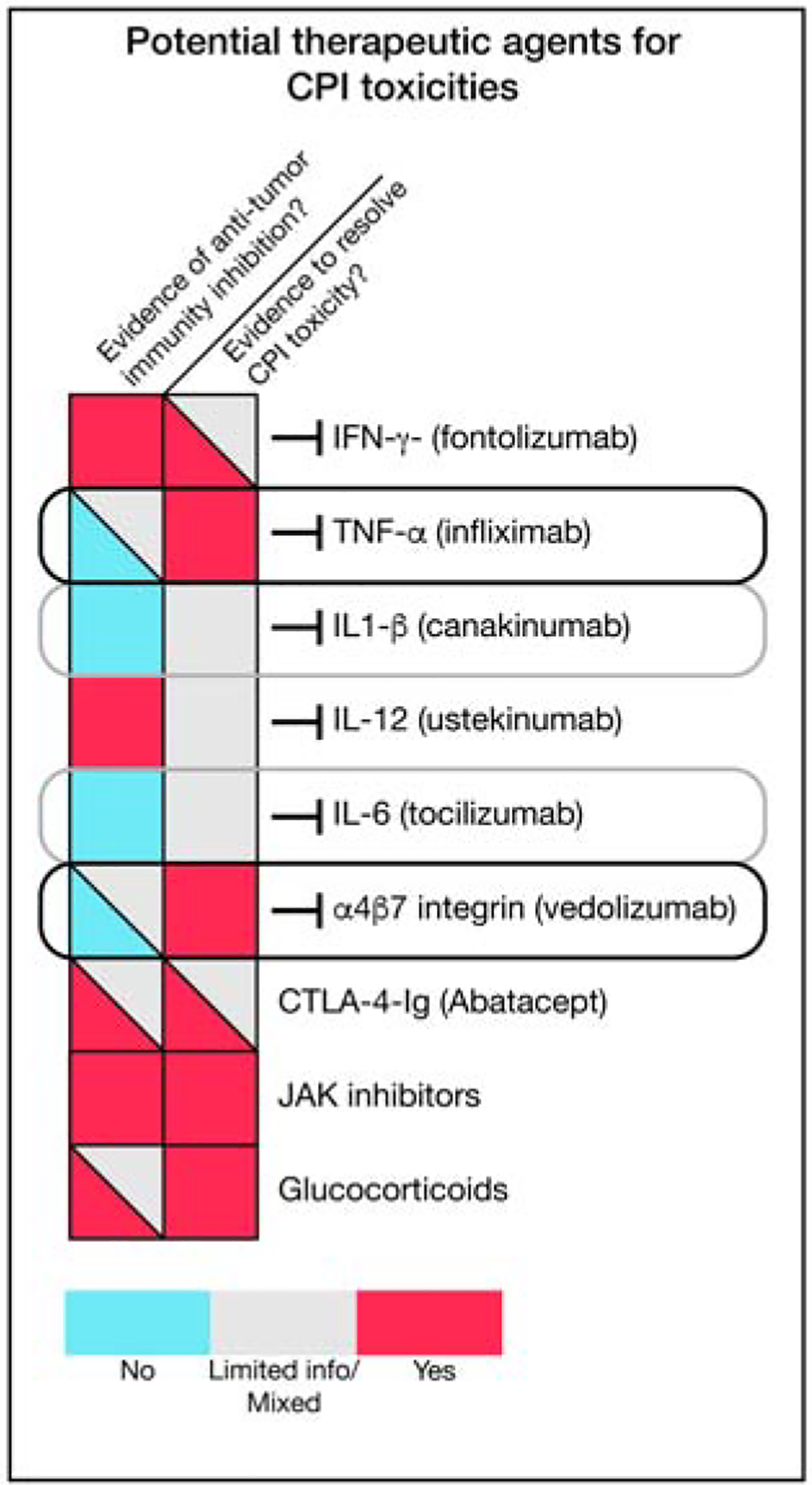
Potential therapeutic agents for checkpoint inhibitor (CPI) toxicities are shown along with their potential impact on tumor immunity. Black outlines represent therapeutic agents with preliminary evidence of clinical activity that may not impair anti-tumor immunity. Grey outlines highlight promising therapeutic targets based on available knowledge on inflammatory toxicities and mechanisms of tumor immunity.
Strategies for management of toxicities that do not respond to glucocorticoids are derived from experience in treating autoimmunity and transplant rejection. These strategies fall into several mechanistic categories that will likely have different effects on antitumor responses.
Cytokine blockade
The acute inflammatory cytokines TNFα, interleukin (IL)-6 and IL-1β play important roles in numerous autoimmune diseases, making them attractive targets for the treatment of checkpoint blockade toxicities. Of these, TNFα inhibitors have been the most directly studied. Based on several retrospective studies as well as limited numbers of patients from cancer immunotherapy trials, blockade of TNFα appears to be highly effective for treatment of glucocorticoid-refractory enterocolitis induced by either CTLA-4 or PD-1/PD-L1 inhibitors (Dougan et al., 2020; Mooradian et al., 2020). This is consistent with the established efficacy of TNFα inhibitors in inflammatory bowel disease, and indicates an important role for this cytokine in perpetuating inflammation in the gastrointestinal tract (Dougan et al., 2020).
The role of TNFα in antitumor responses is unclear. In animal models, inhibition of TNFα synergizes with checkpoint blockade, leading to enhanced CD8 T cell immunity (Bertrand et al., 2017). Whether this is the case in humans is unknown; small retrospective studies of patients with colitis have shown no negative effect on overall survival, but a prospective cohort study from the Netherlands found a correlation between the use of TNFα blockers and decreased cancer survival after immunotherapy (Mooradian et al., 2020; Verheijden et al., 2020). This cohort study lacked information on the indication for TNFα blockade, and precise data were not available on the clinical context (Verheijden et al., 2020). Since most patients who receive TNFα blockade also receive extended courses of high-dose glucocorticoids, this correlation may reflect high-dose glucocorticoid use rather than TNFα blockade itself.
Although inhibition of IL-1β and IL-6 have not yet been adequately explored as treatments for checkpoint blockade toxicities, their success in treatment of autoimmunity suggests potential therapeutic benefit (Esfahani et al., 2020). This possibility is particularly intriguing for IL-1β, which has already been implicated in the promotion of lung cancer in humans (Ridker et al., 2017). In a secondary analysis of a phase 3 trial of the anti-IL-1β monoclonal antibody canakinumab designed to test secondary prevention of cardiovascular events in patients who had had a myocardial infarction, canakinumab treatment was associated with a dose-dependent reduction in incident lung cancer and lung cancer mortality (Ridker et al., 2017). The ability of IL-1β inhibition to synergize with PD-1 blockade is currently undergoing direct testing in clinical trials. IL-6 inhibition has shown efficacy in a limited number of immunotherapy toxicities (Esfahani et al., 2020), and, elevated IL-6 correlates with worse tumor outcomes in patients treated with checkpoint blockade, potentially indicating a deleterious role for this cytokine in antitumor immunity (Keegan et al., 2020). This is consistent with several mouse models that have shown synergy between checkpoint blockade and IL-6 inhibition (Esfahani et al., 2020). This strategy is now being testing in a multicenter trial in patients with metastatic melanoma.
Inhibition of T cell migration
Integrin inhibitors can suppress inflammatory responses with organ selectivity, providing an attractive target for immunotherapy toxicities. Vedolizumab, an inhibitor of α4β7 integrin that prevents immune cell trafficking to the gastrointestinal mucosa and is approved for the treatment of inflammatory bowel disease has shown efficacy comparable to TNFα blockade in retrospective analyses of patients with steroid-refractory enterocolitis (Abu-Sbeih et al., 2018).
Inhibition of costimulation
CTLA-4-Ig is effective in several autoimmune diseases and is the therapy of choice for patients with CTLA-4 haploinsufficiency (Kuehn et al., 2014). CTLA-4-Ig binds to the CD80/CD86 costimulatory proteins and prevents their association with CD28 (Esfahani et al., 2020; Kuehn et al., 2014; Pauken et al., 2019). CTLA-4-Ig has been used in patients with refractory checkpoint blockade toxicities with some apparent success, though the number of treated patients has been very small (Salem et al., 2019). How this therapy would impact antitumor responses requires further investigation.
Checkpoint Blockade Toxicities in High Risk Patients
Currently we have minimal information about the risk factors that predispose patients to develop specific toxicities of checkpoint blockade (Figure 1C). A limited number of reports have investigated the risk of adverse events in patients with underlying autoimmune disease (Abu-Sbeih et al., 2019; Leonardi et al., 2018; Menzies et al., 2017). These retrospective studies have had mixed results, and indicate that the risk of toxicity is probably dependent on the nature of the underlying autoimmune disease.
Patients who have developed a treatment-limiting toxicity from immunotherapy represent another important high-risk population. Depending on the specific toxicity, a substantial fraction of patients who develop a toxicity are likely to have recurrence, though this risk drops if the immunotherapy target is changed (e.g. combination checkpoint blockade changed to single agent PD-1 blockade) (Dolladille et al., 2020; Pollack et al., 2018). An alternative approach is to treat the toxicity with a concurrent therapy that is not expected to inhibit antitumor immunity when immunotherapy is restarted. This concurrent treatment approach is the standard for endocrine toxicities where hormone replacement is used, and is often effective for dermatologic toxicities that can be treated topically (de Filette et al., 2019; Phillips et al., 2019). For checkpoint enterocolitis, TNFα blockade has been used alongside checkpoint blockade to prevent recurrence, as have local steroids (Badran et al., 2019; Hughes et al., 2019).
III. Adverse events induced by adoptive T cell therapies
A major advance in the cancer immunotherapy field is the development of cellular therapies in which T cells are engineered to express a tumor-specific antigen receptor. Discovery of the TCR and costimulatory receptors enabled the development of chimeric antigen receptors (CARs) composed of a tumor-directed antibody domain and cytoplasmic signaling domains from the TCR (ζ chain) and costimulatory receptors (commonly CD28 or 41BB). Such second-generation CARs engage both TCR and costimulatory signaling pathways required for full T cell activation (Eshhar et al., 2014; Kalos and June, 2013; Sadelain et al., 2013). CAR T cells that recognize the CD19 antigen have shown striking efficacy against relapsed, treatment-refractory disease in multiple B cell malignancies, including acute lymphoblastic leukemia (ALL) and B cell lymphomas. CD19 CAR T cells have now been approved by the FDA for the treatment of ALL in children and young adults as well as B cell lymphomas (Maude et al., 2018; Neelapu et al., 2017; Park et al., 2018). Also, CAR T cells specific for B cell maturation antigen (BCMA) have shown promising results for the treatment of multiple myeloma (Raje et al., 2019). Serious toxicities remain a major limitation of current CAR T cell therapies, and fall into 3 categories: 1. Cytokine release syndrome (CRS), 2. Neurotoxicity, 3. On-target and off-target killing of healthy cells (Figure 6). These toxicities are also relevant for cellular therapies based on tumor-specific TCRs, and to a lesser extent for bispecific antibodies that target a tumor surface antigen and activate T cells by CD3 binding.
Figure 6. Inflammatory toxicities induced by CAR T cell therapies.
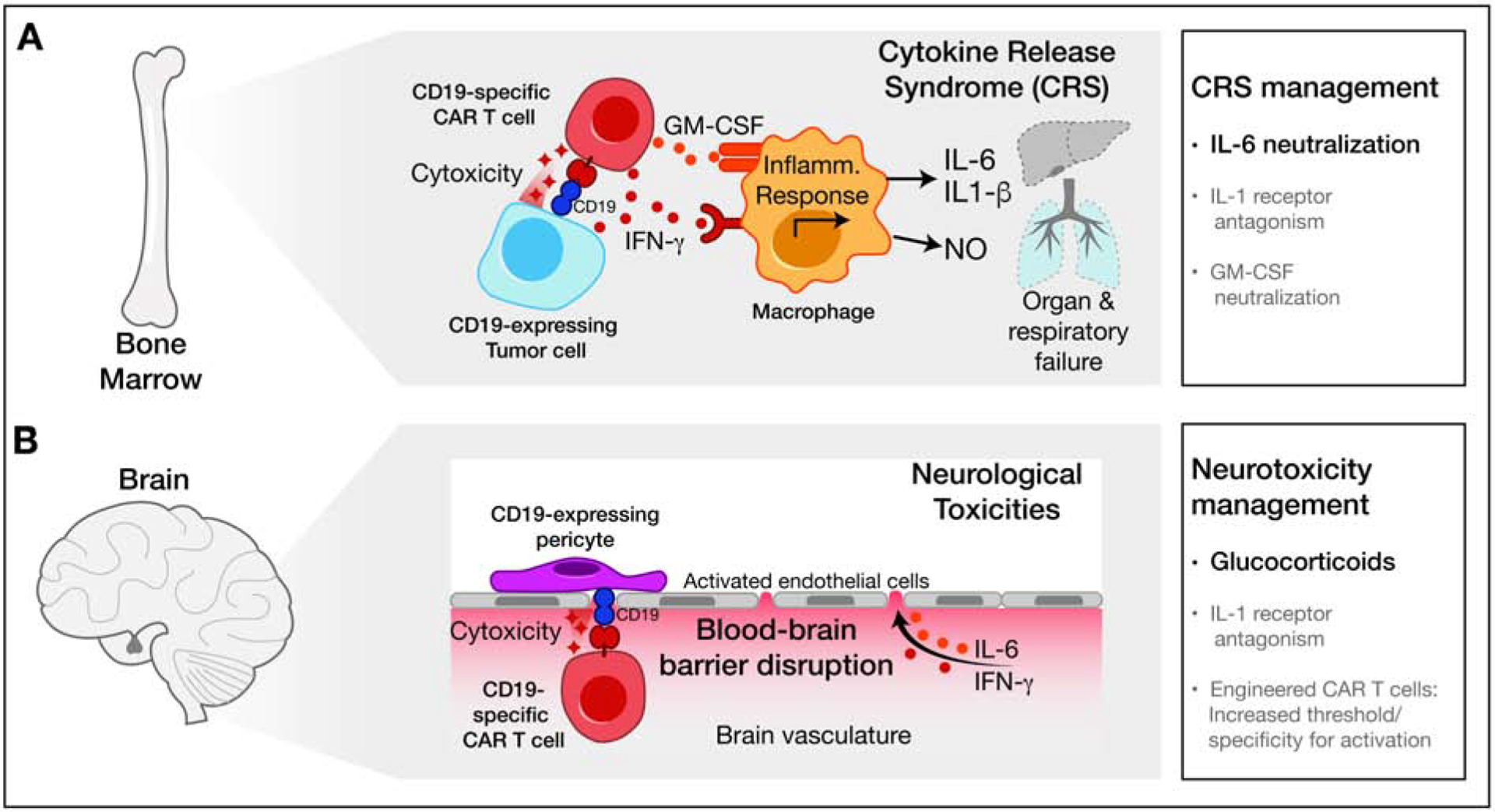
(A) CAR T cells transduced with a CD19-specific receptor eradicate CD19-expressing leukemia cells while inducing potent myeloid cell activation and release of inflammatory mediators, resulting in cytokine release syndrome (CRS). Indicated CRS therapies are colored according to current clinical use (black) or promising pre-clinical data (grey).
(B) CD19-specific T cells can recognize CD19 expressed by perivascular cells at the blood brain barrier. Direct cytotoxicity and indirect cytokine-mediated damage disrupt the blood-brain barrier, resulting in neurological toxicities. Therapies for neurotoxicity are shown colored according to current clinical use (black) or promising pre-clinical data (grey).
Cytokine release syndrome
In hematological malignancies, CAR T cells activation and expansion can result in high-level secretion of cytokines/chemokines. Cytokine release syndrome (CRS) can be a severe and potentially life-threatening toxicity of CAR T cell therapy (Teachey et al., 2016). For the two FDA-approved CD19 CAR T cell products, CRS occurs in 74–94% of patients, with grade 3 or higher toxicity in 13–49% (KYMRIAH, 2017; YESCARTA, 2017). The first presenting symptom of CRS is typically fever, but patients can rapidly progress to develop hypotension and hypoxemia along with impaired function of vital organs. Peak levels of 24 cytokines and soluble cytokine receptors, including IFNγ, IL-6 and GM-CSF are strongly associated with severe CRS (Teachey et al., 2016). The IL-6 pathway plays a prominent role in CRS pathogenesis: the IL-6 receptor blocking antibody tocilizumab is widely used to treat severe CRS and can induce rapid clinical improvement (Maude et al., 2014). Glucocorticoids are also used to treat severe CRS but could negatively impact CAR T cell function. Contributing factors for development of severe CRS include a high burden of malignant cells, a large dose of transferred CAR T cells and the choice of costimulatory signaling domain in the CAR construct. Knowledge of these factors has enabled a risk-adapted approach, including transfer of smaller numbers of CAR T cells to patients with higher burden of malignant cells (Brudno and Kochenderfer, 2019).
Early clinical, laboratory and cytokine data pointed to significant similarities between CRS and macrophage activation syndrome, a potentially life-threatening complication of systemic inflammatory disorders (Crayne et al., 2019; Teachey et al., 2016). A humanized mouse model of CRS demonstrated that activated CAR T cells induced massive recruitment of monocytes/macrophages that produced high levels of IL-6, IL-1 and nitric oxide (Giavridis et al., 2018) (Figure 6A). IL-1β may play an important role in the pathogenesis of CRS and could be targeted with FDA-approved IL-1β inhibitors (Cavalli and Dinarello, 2015; Giavridis et al., 2018; Teachey et al., 2016).
Neurotoxicity
Neurotoxicity represents a second major complication of CAR T cell therapy that can be fatal. For the two FDA-approved CD19 CAR T cell products, neurotoxicity is observed in 72–87% of patients, with grade 3 or higher neurotoxicity in 18–31% (KYMRIAH, 2017; YESCARTA, 2017). A wide range of clinical symptoms are observed including headache, confusion, language disturbances, seizures and cerebral edema (Gust et al., 2018). Neurotoxicity is associated with CRS, and cytokines such as IFNγ and IL-6 are elevated not only in the blood but also the cerebrospinal fluid (CSF). Increased blood brain barrier permeability and local cytokine production by CAR T cells are thought to account for elevated CSF cytokine levels (Figure 6B). Neuropathological investigation demonstrated perivascular infiltration by CAR T cells in the CNS, macrophage infiltration as well as widespread white matter and neuronal injury. Tocilizumab has limited efficacy for the treatment of neurotoxicity, potentially due to low antibody penetration into the CNS. Therefore, high-dose glucocorticoids are currently used for the treatment of neurotoxicity (Gust et al., 2018). In a humanized model of neurotoxicity, monocytes were identified as the major source of IL-1 and IL-6, and the IL-1 receptor antagonist anakinra, which has a molecular weight of 17 kDa, was shown to inhibit both CRS and neurotoxicity (Norelli et al., 2018).
Interestingly, rates of neurotoxicity appear to be lower for CAR T cell therapies that do not target the CD19 antigen, although neurotoxicity has also been observed with CAR T cells specific for other antigens (Gust et al., 2018). A recent study indicated that neurotoxicity may not only be mediated by cytokines, but also on-target activity of CD19 CAR T cells against CNS-resident cells. CD19 was thought to have a B cell restricted expression pattern, but analysis of human brain scRNA-seq data demonstrated CD19 mRNA in perivascular cells critical for blood brain barrier integrity (Parker et al., 2020) (Figure 6B). Targeting of cells by CD19 CAR T cells could result in endothelial activation, increased blood brain barrier permeability and myeloid cell recruitment (Parker et al., 2020).
On-target and off-target toxicity of adoptive T cell therapies
In adoptive T cell therapies, large numbers of activated cells that express a tumor-reactive CAR or TCR are transferred to patients. The expression patterns of potential target antigens are therefore a critical consideration in the design of adoptive T cell therapies. CAR T cells show two types of on-target activity against healthy cells: 1. Engineered CD19 CAR T cells eliminate not only malignant but also healthy B cells (Deya-Martinez et al., 2020). Similarly, BCMA CAR T cells eliminate malignant and healthy plasma cells which provide long-term protection from infectious agents by high-level antibody production (Raje et al., 2019). Loss of B cells or plasma cells can be managed clinically by injection of polyclonal IgG from healthy donors. T cell-mediated elimination of transformed cells and the corresponding healthy cell type represents a major limitation of CAR T cell therapy. This issue has become highly relevant for CAR T cell therapy of solid tumors, many of which originate from essential cell types, such as epithelial cells. 2. Even though a target antigen may appear to have a restricted expression pattern, CAR T cells can also target other healthy cell types expressing the antigen at a lower level, such as brain perivascular cells that express CD19 (Parker et al., 2020). For example, a CAR specific for ERBB2 (Her-2/neu) that was based on the FDA approved antibody trastuzumab (Herceptin) induced fatal pulmonary edema in a patient with colon cancer following transfer of a large dose of engineered T cells. Further investigation revealed low level expression of ERBB2 by lung epithelial cells (Morgan et al., 2010). This example illustrates that a CAR may not be safe even when it is derived from a widely utilized therapeutic antibody, a finding explained by the highly activated state of T cells induced by CAR signaling.
A number of engineering approaches are being developed to protect healthy cell types from on-target attack by CAR T cells. The general concept is to control CAR T cell activation by combinatorial logic based on differential expression of two antigens by healthy versus transformed cells. For example, T cells can be engineered to express activating and inhibitory CARs directed against different surface antigens. Healthy cells that express the ligand for the inhibitory CAR can thus be protected, even if they also express the antigen for the activating CAR (equivalent to TCR + PD-1 signaling in normal T cells) (Fedorov et al., 2013). The SynNotch approach also increases the selectivity of T cell activation by requiring expression of two antigens by tumor cells. SynNotch receptors take advantage of Notch receptor biology: mechanical force exerted by ligand binding induces proteolytic cleavage of the cytoplasmic domain which then acts as a transcription factor. A SynNotch receptor recognizing antigen A induces a transcriptional response, resulting in expression of a CAR specific for antigen B. Tumor cells that express antigens A + B are effectively targeted, but healthy cells that express only antigens A or B are spared (Roybal et al., 2016; Srivastava et al., 2019). The transcription factor activated in the SynNotch system can also be used to further engineer the functional program, including release of cytokines or other therapeutic proteins (Morsut et al., 2016).
The field has recently seen a resurgence of interest in adoptive T cell therapies based on TCRs which perform surveillance of the cellular proteome in the form of short peptides bound to MHC proteins. TCRs thus offer more opportunities for tumor antigen discovery. A substantial effort is underway to identify the antigen specificity of tumor-infiltrating T cells in patients responding to checkpoint blockade. TCRs have a high degree of specificity for the relevant MHC-bound peptide but can cross-react with peptides that meet the sequence requirements for MHC binding and TCR recognition (Wucherpfennig and Sethi, 2011). T cells undergo a rigorous selection process in the thymus that eliminates those cells with high affinity for self-antigens, including highly cross-reactive T cells. This natural selection process is sidestepped when TCRs are selected in vitro from recombinant libraries or subjected to significant mutagenesis. The affinity of TCRs isolated from cancer patients is frequently suboptimal, but mutagenesis to enhance TCR affinity can introduce undesirable cross-reactivity, as illustrated by a TCR specific for a MAGE-A3 peptide presented by HLA-A1. The first two patients who received MAGE-A3 specific T cells died of cardiogenic shock several days following T cell transfer. In both patients, severe myocardial damage with substantial T cell infiltration was observed, but MAGE-A3 expression could not be detected in the heart. Rather, this TCR cross-reacted with titin, a striated muscle specific protein (Linette et al., 2013). It is therefore important to systematically examine the cross-reactivity of engineered TCRs prior to initiation of clinical trials.
Currently, the toxicities of CAR T cell therapies represent a significant challenge, but further engineering of these cells provides an opportunity to significantly improve their safety profile while maintaining anti-tumor activity.
IV. Adverse events induced by cytokine therapies
Cytokines are natural immune modulatory agents (Dranoff, 2004). Often acting in autocrine or paracrine fashion, cytokines typically mediate responses in a spatially restricted context. Nevertheless, some cytokines act systemically on distant organs such as the hypothalamus, liver and bone marrow to augment systemic immunity. Recombinant cytokines known to be important for the effector T cell response to cancer have been administered to cancer patients, often with severe treatment-limiting side effects (Dranoff, 2004). Local delivery strategies that more closely mimic the natural production of cytokines have shown promising results in preclinical models and serve to limit the toxicities associated with systemic delivery. Such strategies include delivery by direct intratumoral injection, oncolytic viruses, nanoparticles, and antibody or nanobody conjugates (Dougan and Dougan, 2017). In general, to understand the toxicity associated with a cytokine, one must understand the full array of cell types that express cognate receptors and induced cellular responses. Advances in protein engineering have also allowed the design of cytokines with improved targeting capabilities and reduced signaling by immunosuppressive cells or cell types responsible for systemic toxicities.
IL-2 provides an excellent example for design strategies that mitigate severe systemic toxicities and simultaneously enhance antitumor activity. IL-2 serves as a critical growth factor for activated T cells, and considerable efforts have been made to harness it for therapeutic use. High-dose IL-2 is curative in a small fraction of patients with renal cell carcinoma and melanoma, although determining maximum dosing is difficult, and fatal capillary leak syndrome has been observed due to expression of CD25 by pulmonary endothelial cells. (Dranoff, 2004; Letourneau et al., 2010; Zhu et al., 2015). These severe toxicities have greatly limited clinical use of high-dose IL-2.
The IL-2 receptor is composed of IL2Rβ (CD122) and the gamma common chain (γc), which together form an intermediate-affinity receptor for IL-2 (Leonard et al., 2019). The intermediate-affinity receptor is expressed by all T cells and NK cells, and IL-2 is an important growth factor for these lymphocytes. IL-2Rα (CD25) associates with the IL2Rβ and γc to form the trimeric high-affinity IL-2 receptor complex. CD25 is expressed at the highest level by Foxp3+ CD4 Tregs, which preferentially expand when IL-2 is limiting. Given that neither expansion of Tregs nor pulmonary edema are desirable outcomes, several strategies to reduce binding of IL-2 to CD25 while preserving binding to the intermediate-affinity IL-2 receptor have been developed. Using yeast display and targeted evolution, a variant of IL-2 (Super-2) that lacks binding to CD25 was developed, as has a computationally designed de novo protein that activates the intermediate-affinity IL-2 receptor but lacks CD25 binding (Levin et al., 2012; Silva et al., 2019). Alternatively, attachment of polyethylene glycol (PEG) chains to IL-2 increases its serum half-life and inhibits binding to the high-affinity IL-2 receptor. Slow turnover of the PEG chains releases active versions of IL-2, and pegylated IL-2 (Bempegaldesleukin) is currently in clinical trials in combination with PD-1 blockade (Charych et al., 2016).
Strategies that mimic the localized nature of cytokine signaling are likely to be both less toxic and more effective as cancer therapeutics. Synthetic cytokines and cytokine mimetics also open the possibility of further engineered variants with altered binding properties.
V. General considerations and conclusions
Activated T cells serve as the major effector cells of all current cancer immunotherapies. Targeting of negative feedback mechanisms (checkpoint blockade) or engineering of synthetic receptors (CAR T cells) can enable effective T cell-mediated tumor immunity but also unleash severe inflammatory toxicities. Defining the cellular and molecular pathways responsible for major inflammatory toxicities will be important in order to develop therapeutic approaches compatible with sustained antitumor immunity. Available data suggest that recruitment and activation of myeloid cells by T cells can result in local and/or systemic release of proinflammatory cytokines (including IL-6, TNFα and IL-1β) that play a central role in these inflammatory toxicities, as illustrated by the significant therapeutic activity of IL-6 receptor blockade in cytokine release syndrome induced by CAR T cells. Targeting of such inflammatory pathways may enable treatment or even prevention of inflammatory toxicities while preserving anti-tumor immunity.
Acknowledgements
Funding: This work was supported by supported by a Team Award from the American Cancer Society MRAT-18-113-01 and the Melanoma Research Alliance 597698 (to K.W.W., S.K.D., M.D.), NIH grants R01 CA238039, R01CA251599, P01 CA163222 (to K.W.W.) and T32CA207021 (to A.L. and K.W.W.), a National Institutes of Health Mentored Clinical Scientist Development Award 1K08DK114563-01 (M.D.) and the American Gastroenterological Association Research Scholars Award (M.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
K.W.W. serves on the scientific advisory board of TCR2 Therapeutics, T-Scan Therapeutics, SQZ Biotech and Nextechinvest and receives sponsored research funding from Novartis. He is a scientific co-founder of Immunitas Therapeutics. M.D. is a consultant for Tillotts Pharma, Partner Therapeutics, and Genentech-Roche, receives research funding from Novartis, and is on the Scientific Advisory Board for Neoleukin Therapeutics. S.K.D. receives research funding from Novartis, Bristol-Myers Squibb, and Eli Lilly and is a scientific co-founder of Kojin.
References
- Abu-Sbeih H, Ali FS, Alsaadi D, Jennings J, Luo W, Gong Z, Richards DM, Charabaty A, and Wang Y (2018). Outcomes of vedolizumab therapy in patients with immune checkpoint inhibitor-induced colitis: a multi-center study. J Immunother Cancer 6, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Sbeih H, Faleck DM, Ricciuti B, Mendelsohn RB, Naqash AR, Cohen JV, Sellers MC, Balaji A, Ben-Betzalel G, Hajir I, et al. (2019). Immune Checkpoint Inhibitor Therapy in Patients With Preexisting Inflammatory Bowel Disease. J Clin Oncol, JCO1901674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alspach E, Lussier DM, and Schreiber RD (2019). Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harbor perspectives in biology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour KC, Mezquita L, Long N, Rizvi H, Auclin E, Ni A, Martinez-Bernal G, Ferrara R, Lai WV, Hendriks LEL, et al. (2018). Impact of Baseline Steroids on Efficacy of Programmed Cell Death-1 and Programmed Death-Ligand 1 Blockade in Patients With Non-Small-Cell Lung Cancer. J Clin Oncol 36, 2872–2878. [DOI] [PubMed] [Google Scholar]
- Badran YR, Cohen JV, Brastianos PK, Parikh AR, Hong TS, and Dougan M (2019). Concurrent therapy with immune checkpoint inhibitors and TNFalpha blockade in patients with gastrointestinal immune-related adverse events. J Immunother Cancer 7, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister SH, Freeman GJ, Dranoff G, and Sharpe AH (2016). Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol 34, 539–573. [DOI] [PubMed] [Google Scholar]
- Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, Rochaix P, Andrieu-Abadie N, Levade T, Meyer N, et al. (2017). TNFalpha blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun 8, 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno JN, and Kochenderfer JN (2019). Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev 34, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelli LC, Gutierrez AK, Bingham CO 3rd, and Shah AA (2017). Rheumatic and Musculoskeletal Immune-Related Adverse Events Due to Immune Checkpoint Inhibitors: A Systematic Review of the Literature. Arthritis Care Res (Hoboken) 69, 1751–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli G, and Dinarello CA (2015). Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology (Oxford) 54, 2134–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, et al. (2016). NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 680–690. [DOI] [PubMed] [Google Scholar]
- Cohen JV, Dougan M, Zubiri L, Reynolds KL, Sullivan RJ, and Misdraji J (2020). Liver biopsy findings in patients on immune checkpoint inhibitors. Mod Pathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crayne CB, Albeituni S, Nichols KE, and Cron RQ (2019). The Immunology of Macrophage Activation Syndrome. Front Immunol 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Filette J, Andreescu CE, Cools F, Bravenboer B, and Velkeniers B (2019). A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated with Immune Checkpoint Inhibitors. Horm Metab Res 51, 145–156. [DOI] [PubMed] [Google Scholar]
- Deya-Martinez A, Alonso-Saladrigues A, Garcia AP, Faura A, Torrebadell M, Vlagea A, Catala A, Esteve-Sole A, Juan M, Rives S, et al. (2020). Kinetics of humoral deficiency in CART19-treated children and young adults with acute lymphoblastic leukaemia. Bone Marrow Transplant. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolladille C, Ederhy S, Sassier M, Cautela J, Thuny F, Cohen AA, Fedrizzi S, Chretien B, Da-Silva A, Plane AF, et al. (2020). Immune Checkpoint Inhibitor Rechallenge After Immune-Related Adverse Events in Patients With Cancer. JAMA Oncol 6, 865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M (2017). Checkpoint Blockade Toxicity and Immune Homeostasis in the Gastrointestinal Tract. Front Immunol 8, 1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan M, and Dougan SK (2017). Targeting Immunotherapy to the Tumor Microenvironment. J Cell Biochem 118, 3049–3054. [DOI] [PubMed] [Google Scholar]
- Dougan M, Wang Y, Rubio-Tapia A, and Lim JK (2020). AGA Clinical Practice Update on Diagnosis and Management of Immune Checkpoint Inhibitor (ICI) Colitis and Hepatitis: Expert Review. Gastroenterology. [DOI] [PubMed] [Google Scholar]
- Dranoff G (2004). Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 4, 11–22. [DOI] [PubMed] [Google Scholar]
- Esfahani K, Elkrief A, Calabrese C, Lapointe R, Hudson M, Routy B, Miller WH Jr., and Calabrese L (2020). Moving towards personalized treatments of immune-related adverse events. Nat Rev Clin Oncol 17, 504–515. [DOI] [PubMed] [Google Scholar]
- Eshhar Z, Waks T, and Gross G (2014). The emergence of T-bodies/CAR T cells. Cancer J 20, 123–126. [DOI] [PubMed] [Google Scholar]
- Faje AT, Lawrence D, Flaherty K, Freedman C, Fadden R, Rubin K, Cohen J, and Sullivan RJ (2018). High-dose glucocorticoids for the treatment of ipilimumab-induced hypophysitis is associated with reduced survival in patients with melanoma. Cancer. [DOI] [PubMed] [Google Scholar]
- Fedorov VD, Themeli M, and Sadelain M (2013). PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Science translational medicine 5, 215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, and Sadelain M (2018). CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med 24, 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust J, Taraseviciute A, and Turtle CJ (2018). Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 32, 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvat TZ, Adel NG, Dang TO, Momtaz P, Postow MA, Callahan MK, Carvajal RD, Dickson MA, D’Angelo SP, Woo KM, et al. (2015). Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol 33, 3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MS, Molina GE, Chen ST, Zheng H, Deshpande V, Fadden R, Sullivan RJ, and Dougan M (2019). Budesonide treatment for microscopic colitis from immune checkpoint inhibitors. J Immunother Cancer 7, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, et al. (2017). T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355, 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, et al. (2016). Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med 375, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, and June CH (2013). Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 39, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan A, Ricciuti B, Garden P, Cohen L, Nishihara R, Adeni A, Paweletz C, Supplee J, Janne PA, Severgnini M, et al. (2020). Plasma IL-6 changes correlate to PD-1 inhibitor responses in NSCLC. J Immunother Cancer 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, and Sharpe AH (2006). Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 203, 883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, et al. (2014). Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345, 1623–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KYMRIAH (2017). FDA approval. https://wwwfdagov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel.
- Leonard WJ, Lin JX, and O’Shea JJ (2019). The gammac Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 50, 832–850. [DOI] [PubMed] [Google Scholar]
- Leonardi GC, Gainor JF, Altan M, Kravets S, Dahlberg SE, Gedmintas L, Azimi R, Rizvi H, Riess JW, Hellmann MD, et al. (2018). Safety of Programmed Death-1 Pathway Inhibitors Among Patients With Non-Small-Cell Lung Cancer and Preexisting Autoimmune Disorders. J Clin Oncol 36, 1905–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneau S, van Leeuwen EM, Krieg C, Martin C, Pantaleo G, Sprent J, Surh CD, and Boyman O (2010). IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proceedings of the National Academy of Sciences of the United States of America 107, 2171–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, et al. (2012). Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 484, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. (2013). Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma AM, Suo S, Williams HL, Sharova T, Sullivan K, Manos M, Bowling P, Hodi FS, Rahma O, Sullivan RJ, et al. (2020). Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 182, 655–671 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthey L, Mateus C, Mussini C, Nachury M, Nancey S, Grange F, Zallot C, Peyrin-Biroulet L, Rahier JF, Bourdier de Beauregard M, et al. (2016). Cancer Immunotherapy with Anti-CTLA-4 Monoclonal Antibodies Induces an Inflammatory Bowel Disease. J Crohns Colitis 10, 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, Shabafrouz K, Ribi C, Cairoli A, Guex-Crosier Y, et al. (2019). Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol 16, 563–580. [DOI] [PubMed] [Google Scholar]
- Masopust D, and Soerens AG (2019). Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol 37, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Barrett D, Teachey DT, and Grupp SA (2014). Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J 20, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong ANM, Park JJ, McQuade JL, Shoushtari AN, Tsai KK, Eroglu Z, et al. (2017). Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol 28, 368–376. [DOI] [PubMed] [Google Scholar]
- Mooradian MJ, Wang DY, Coromilas A, Lumish M, Chen T, Giobbie-Hurder A, Johnson DB, Sullivan RJ, and Dougan M (2020). Mucosal inflammation predicts response to systemic steroids in immune checkpoint inhibitor colitis. J Immunother Cancer 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, and Lim WA (2016). Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. (2017). Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377, 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF (2019). IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev 45, 1–8. [DOI] [PubMed] [Google Scholar]
- Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. (2018). Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 24, 739–748. [DOI] [PubMed] [Google Scholar]
- Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et al. (2018). Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 378, 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, Haris M, Wilson NE, Liu F, Gabunia K, et al. (2020). Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 183, 126–142 e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Dougan M, Rose NR, Lichtman AH, and Sharpe AH (2019). Adverse Events Following Cancer Immunotherapy: Obstacles and Opportunities. Trends in immunology 40, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GS, Wu J, Hellmann MD, Postow MA, Rizvi NA, Freites-Martinez A, Chan D, Dusza S, Motzer RJ, Rosenberg JE, et al. (2019). Treatment Outcomes of Immune-Related Cutaneous Adverse Events. J Clin Oncol 37, 2746–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack MH, Betof A, Dearden H, Rapazzo K, Valentine I, Brohl AS, Ancell KK, Long GV, Menzies AM, Eroglu Z, et al. (2018). Safety of resuming anti-PD-1 in patients with immune-related adverse events (irAEs) during combined anti-CTLA-4 and anti-PD1 in metastatic melanoma. Ann Oncol 29, 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese A (2017). Autoreactive T cells in type 1 diabetes. J Clin Invest 127, 2881–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, et al. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, Liedtke M, Rosenblatt J, Maus MV, Turka A, et al. (2019). Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med 380, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds K, Thomas M, and Dougan M (2018). Diagnosis and Management of Hepatitis in Patients on Checkpoint Blockade. Oncologist 23, 991–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, and Group CT (2017). Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842. [DOI] [PubMed] [Google Scholar]
- Rosato PC, Wijeyesinghe S, Stolley JM, and Masopust D (2020). Integrating resident memory into T cell differentiation models. Curr Opin Immunol 63, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, and Lim WA (2016). Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 419–432 e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R, and Riviere I (2013). The basic principles of chimeric antigen receptor design. Cancer discovery 3, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safa H, Johnson DH, Trinh VA, Rodgers TE, Lin H, Suarez-Almazor ME, Fa’ak F, Saberian C, Yee C, Davies MA, et al. (2019). Immune checkpoint inhibitor related myasthenia gravis: single center experience and systematic review of the literature. J Immunother Cancer 7, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem JE, Allenbach Y, Vozy A, Brechot N, Johnson DB, Moslehi JJ, and Kerneis M (2019). Abatacept for Severe Immune Checkpoint Inhibitor-Associated Myocarditis. N Engl J Med 380, 2377–2379. [DOI] [PubMed] [Google Scholar]
- Shiuan E, Beckermann KE, Ozgun A, Kelly C, McKean M, McQuade J, Thompson MA, Puzanov I, Greer JP, Rapisuwon S, et al. (2017). Thrombocytopenia in patients with melanoma receiving immune checkpoint inhibitor therapy. J Immunother Cancer 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labao-Almeida C, Ali LR, Quijano-Rubio A, Ruterbusch M, Leung I, et al. (2019). De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210, 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 178, 714–730 e722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, Smythe KS, Dudakov JA, Pierce RH, Rader C, et al. (2019). Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer cell 35, 489–503 e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, Gettinger S, Sznol M, Young A, Rushakoff R, et al. (2018). Collateral Damage: Insulin-Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes 67, 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir SA, Gao J, Miura Y, Blando J, Tidwell RSS, Zhao H, Subudhi SK, Tawbi H, Keung E, Wargo J, et al. (2019). Autoimmune antibodies correlate with immune checkpoint therapy-induced toxicities. Proc Natl Acad Sci U S A 116, 22246–22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, et al. (2016). Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer discovery 6, 664–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang VHM, McGrath RT, Clifton-Bligh RJ, Scolyer RA, Jakrot V, Guminski AD, Long GV, and Menzies AM (2019). Checkpoint Inhibitor-Associated Autoimmune Diabetes Is Distinct From Type 1 Diabetes. J Clin Endocrinol Metab 104, 5499–5506. [DOI] [PubMed] [Google Scholar]
- van Belle TL, Coppieters KT, and von Herrath MG (2011). Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91, 79–118. [DOI] [PubMed] [Google Scholar]
- Verheijden RJ, May AM, Blank CU, Aarts MJB, van den Berkmortel F, van den Eertwegh AJM, de Groot JWB, Boers-Sonderen MJ, van der Hoeven JJM, Hospers GA, et al. (2020). Association of Anti-TNF with Decreased Survival in Steroid Refractory Ipilimumab and Anti-PD1-Treated Patients in the Dutch Melanoma Treatment Registry. Clin Cancer Res 26, 2268–2274. [DOI] [PubMed] [Google Scholar]
- Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, Zhao S, Das S, Beckermann KE, Ha L, et al. (2018). Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol 4, 1721–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig KW, and Sethi D (2011). T cell receptor recognition of self and foreign antigens in the induction of autoimmunity. Semin Immunol 23, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YESCARTA (2017). FDA approval. https://wwwfdagov/media/108377/download.
- Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, Xiao C, Bonorino C, Lu LF, Bui JD, et al. (2019). PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 51, 1059–1073 e1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu EF, Gai SA, Opel CF, Kwan BH, Surana R, Mihm MC, Kauke MJ, Moynihan KD, Angelini A, Williams RT, et al. (2015). Synergistic innate and adaptive immune response to combination immunotherapy with anti-tumor antigen antibodies and extended serum half-life IL-2. Cancer cell 27, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]