

Abstract
All four of the adenosine receptor (AR) subtypes mediate pain and have been targeted by pharmacologists to generate new therapeutics for chronic pain. The vanilloid phytochemicals, which include curcumin, capsaicin, and gingerol, have been shown to alleviate pain. However, there is little to no literature on the interaction of vanilloid phytochemicals with ARs. In this study, photochemical methods were used to generate a novel isomer of curcumin (cis-trans curcumin or CTCUR), and the interactions of both curcumin and CTCUR with the two Gs-linked AR subtypes were studied. Competitive binding assays, docking analysis, and confocal fluorescence microscopy were performed to measure binding affinity; cell survival assays were used to measure toxicity; and cAMP assays were performed to measure receptor activation. Competitive binding results indicated that CTCUR binds to both AR A2A and AR A2B with Ki values of 5 μM and 7 μM, respectively, which is consistent with our docking results. Fluorescence microscopy data also shows binding for A2B and A2A. Cell survival results show that CTCUR and CUR are nontoxic at the tested concentrations in these cell lines. Overall, our results suggest that vanilloid phytochemicals may be slightly modified to increase interaction with Gs-ARs, and thereby can be further explored to provide a novel class of non-opioid antinociceptives.
Keywords: Adenosine, curcumin, Gstimulatory, adenosine receptor A2A, adenosine receptor A2B, chronic pain
Graphical Abstract
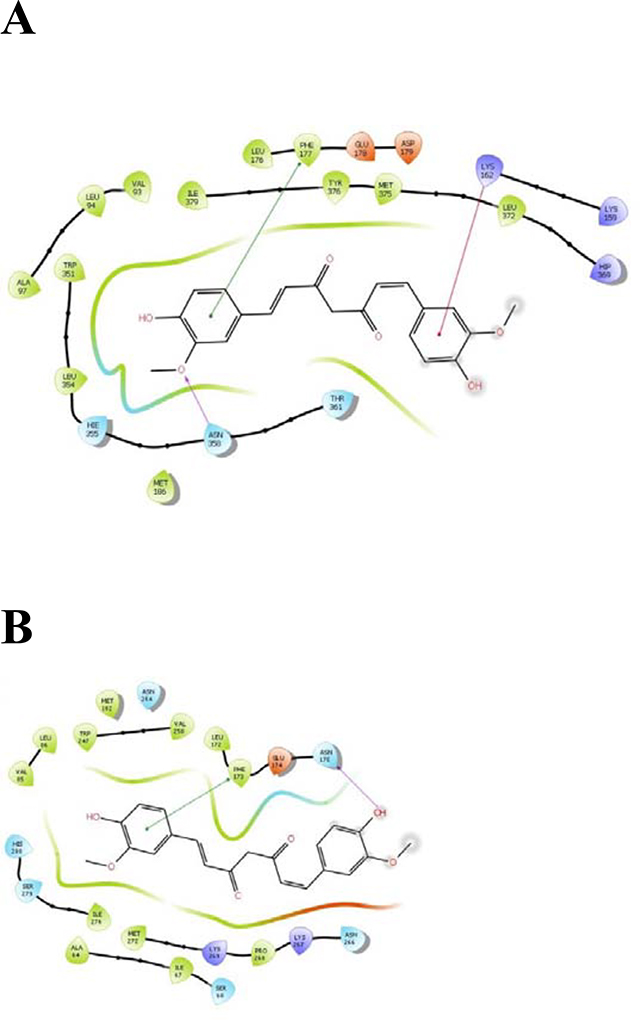
Interaction of CTCUR (keto form) with amino acid residues of A) AR A2A and B) AR A2B.
1.1. Introduction
Chronic pain is estimated to affect 25 million people in the United States alone [1]. From the mid-1990s until recent times, the principal pharmacologic treatment for chronic pain was long term administration of opioid medications [2]. However, between 1999 and 2007, the number of deaths related to opioid drugs rose throughout the world and more than tripled in the USA, causing concern within the biomedical community [3]. In response, studies have been initiated to discover new analgesics with a lower risk of addiction. Among the non-opioid receptor families, one that has shown promise as a target for analgesics are the adenosine receptors [4–9].
Adenosine is a signaling molecule that preserves metabolic homeostasis in humans. Nervous, cardiovascular, immune, respiratory, urinary, and gastrointestinal systems all benefit from the protection afforded by adenosine signaling [10]. The International Union of Pharmacology has described four receptors for adenosine (A1, A2A, A2B, and A3), all of which are G-protein-coupled receptors (GPCRs). Like other GPCRs, each adenosine receptor (AR) is an integral membrane protein whose salient structural feature is seven α helices that span the membrane. The type of G protein to which an AR is coupled varies by subtype. A1 and A3 are coupled to Gi, which depresses cAMP levels, while A2A and A2B are coupled to Gs, which elevates cAMP levels [11]. The four receptor subtypes differ substantially from each other both in their distribution and in their physiological effects [10] and are therefore usually studied separately [12]. To date, only one of the subtypes has a selective ligand that is approved for clinical use, namely A2A, whose agonist regadenoson (Lexiscan) is used in perfusion imaging of the heart. The only other AR ligand approved for clinical use is a non-selective agonist, namely adenosine itself (Adenocard, Adenoscan), which is used for treatment of supraventricular tachyarrythmia [13]. Although there are currently only two AR ligands approved for clinical use, there is much untapped medicinal potential in the ARs, and they have been the subject of hundreds of pharmacological studies [10]. All four AR subtypes have been investigated for their potential as targets for analgesic drugs [6–9]. The work presented here will focus on ligand binding studies with the stimulatory Gα-linked subtypes— A2A and A2B.
A2A ARs are found primarily in the periphery [7], especially on leukocytes and in the cells of blood vessel walls [10]. The majority of A2A ARs that are involved in pain pathways are the peripheral A2A ARs [14]. Indeed, A2A knockout mice show a reduced ability to feel pain, implying that peripheral A2A ARs are pro-nociceptive [14]. This hypothesis has been confirmed through the testing of a synthetic A2A antagonist, which was analgesic [6]. The situation of A2A ARs in the CNS is less clear. For example, sleep deprivation is associated with higher pain sensitivity, and this effect is thought to be partially mediated by A2A ARs within the nucleus accumbens. [15]. However, another study of injections of A2A ligands into the spinal cord found that an A2A agonist relieved pain, implying that A2A ARs in the spinal cord are anti-nociceptive [16]. Since A2A ARs are also found in blood vessel walls, the possibility of cardiovascular side effects must be considered. In fact, A2A agonists have been found to lower blood pressure [17]. It can be inferred from this that A2A antagonists administered as pain medications might raise blood pressure, a concerning side effect.
A2B ARs are widely distributed through the periphery of the body, including cells of the intestines, lungs, and immune system [10]. The distribution of A2B ARs in the CNS is limited [7], although they are present on glial cells [10]. In mice tested for their pain response to a hot plate, five A2B antagonists all showed an analgesic effect [18]. Thus, A2B ARs are pro-nociceptive, like their peripheral A2A AR counterparts. Not only are A2B ARs pro-nociceptive, evidence suggests that A2B plays a greater role than any of the other four receptor subtypes in the perception of pain related to systemically elevated adenosine. Antagonists of all four subtypes were tested on mice, but only the A2B antagonist reduced the hyperalgesia [19]. Due to the strong evidence of antinociception and relatively low side effect risk, A2Bs rank above A2As in their promise as analgesic drug targets. However, an important limitation of A2Bs is that most of their effect on pain is exerted through immune cells [19]. As a result, A2Bs represent an excellent therapeutic target for inflammatory pain. However, their utility in the treatment of other chronic pain conditions is limited. In addition to their key role in natural adenosine signaling, A2B ARs have been linked to the antinociceptive action of molecules that are structurally dissimilar to adenosine.
Although the vast majority of compounds that have been studied as AR ligands are either derivatives of adenosine itself or derivatives of xanthine [20], not all AR ligands fall into these categories. For example, the antinociception action of diphenyl diselenide is mediated by A2B AR [7]. Previous work in our lab suggested that vanilloids might be an additional group of AR ligands [21]. Desiring to explore this possibility further, we screened vanilloid compounds and found that cis-trans curcumin (CTCUR) is a potential AR ligand.
Curcumin (CUR) is a vanilloid compound (Figure 1) from the herb Curcuma longa, which grows in Asia. CUR has been shown to be effective in autoimmune inflammation [22] and postoperative pain [23]. Previous work has demonstrated that CUR exerts its anti-nociceptive effects in part by acting as a COX pathway inhibitor [24] and a TRPV1 antagonist [25]. CUR is poorly absorbed by the human digestive system and, upon being absorbed, is rapidly metabolized into products such as ferulic acid. These facts limit the distribution of CUR to various tissues, and limit its serum concentration in particular, thus drastically reducing the effectiveness of CUR and its analogs as medications [26]. This limitation has been overcome through a variety of techniques in recent years. Some researchers have complexed curcumin with cyclodextrin and thereby achieved reductions in the size of lung tumors [27]. Others have created micelles that are enriched with curcumin, which can be absorbed via the microfold cells of the intestine [28]. The most attractive solution to the CUR bioavailability problem, however, has been nanoparticles. Over 400 studies have investigated the efficacy of nanoformulations of CUR [29], and one such study successfully used CUR nanoparticles to reduce levels of inflammatory cytokines and reduce fibrosis in a mouse model of liver disease [30]. In short, the concerns about the bioavailability of CUR that have plagued its translation into clinical use are being addressed.
Figure 1:

Structure of curcumin (PubChemCID: 969516)
Since curcumin has gained attention recently due to its multitude of medicinal effects, we explored whether curcumin or its analogs can provide a new class of AR ligands. We tested the hypothesis that isomerization of CUR (CTCUR) would increase binding to A2A AR and/or A2B AR as compared to naturally occurring CUR.
1.2. Methods
1.2.1. Chemical synthesis
CTCUR synthesis was achieved through photochemical cavitand-mediated isomerization. CUR is inherently resistant to photoisomerization and dimerization; however, this was overcome through the cavitand-mediation approach. Approximately 50 mg of CUR was added to 20 mL of water, to which one equivalent of γ-cyclodextrin was added. The mixture was stirred with heating at 70° C for 1h, then the heat was turned off. The cooled solution further stirred for 3h during which a chalky slurry of the inclusion complex resulted. The resulting mixture was irradiated for 48h in a photochemical irradiation chamber with a medium pressure mercury vapor lamp. The irradiated mixture of the complex was de-complexed by adding 50 mL of water to the slurry and stirring the diluted solution with 50 mL of ethyl acetate. The organic layer was isolated and dried over anhydrous magnesium sulfate, then the solvent was removed under vacuum. The mixture was subject to slow flash chromatography on a silica gel column with 0.1% formic acid in dichloromethane. The yellow-colored CTCUR was isolated in 8% yield. The presence of CTCUR was confirmed by proton NMR and GCMS.
1H NMR (CDCl3, 400 MHz): 8.083 ppm (m, 1H); 7.979 ppm (d, 1H); 7.657 ppm (d, 1H); 7.398 ppm (t, 1H); 7.112 (m, 2H); 7.053 (s, 1H); 6.936 ppm (d, 1H); 6.478 ppm (d, 1H); 6.315 ppm (d, 1H); 3.975 (s, 3H); 3.933 (s, 3H).
1.2.2. Cell culture
Two HEK-293 cell lines transfected with human adenosine A2A receptor and human adenosine A2B receptor, respectively, were purchased from Perkin Elmer (Waltham, MA, USA). Cells were cultured in Eagle’s minimum essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS). After two passages to allow recovery from thawing stress, all subsequent passages were cultured with 400 μg/mL geneticin (Thermo Fisher, Waltham, MA, USA) to select for transfected cells.
1.2.3. Cytotoxicity Assays
Cytotoxicity assays were performed using previously described methods [31]. In brief, cells were seeded at 8,000 cells/well in 96-well plates, and after overnight incubation they were treated with different concentrations of CUR or CTCUR for 2h (the same time point used for binding assays). The compounds were dissolved in DMSO at a concentration of 0.1M, and diluted for assays as required. Cell survival was measured using PrestoBlue dye (Thermo Fisher Scientific, Waltham, MA, USA) as per manufacturer’s instructions, and fluorescence was measured at 560/590 nm (ex/em) using a Synergy H1 microplate reader and Gen5 software (BioTek Winooski, VT, USA). Percent change was calculated relative to the average for control cells (no treatment).
1.2.4. Competitive binding assays
Cells were seeded at 30,000 cells per well onto a black-walled 96-well plate and allowed to incubate for approximately two days. Once confluence was achieved, wells were treated with either A) media only, B) media + test compound at 10−5 M, C) media + fluorescent compound at 60 nM, or D) media + fluorescent compound at 60 nM + test compound at varying concentrations. The test compound was either CUR or CTCUR, and the fluorescent compound was CA200623 CellAura fluorescent adenosine agonist, a NECA derivative (HelloBio, Princeton, NJ, USA). After a 2h incubation, wells were washed twice with PBS. An additional 100 μL of PBS per well was added at the end to prevent desiccation during reading. The plate was read with a Synergy H1 microplate reader (BioTek). To generate a binding curve from competitive binding assay (CBA) data, the following calculations were used-
| Equation 1. |
| Equation 2. |
| Equation 3. |
1.2.6. Docking Studies
1.2.6.1. Preparation of protein structures—
The X-ray crystal structures of AR A2A (PDB id: 5NM4) [32] was retrieved from the RCSB Protein Data Bank. The protein was prepared in MOE[33], and the side chain atoms and ligand atoms were optimized, first with the main chain atoms fixed in order to reduce steric repulsion and at the same time to minimize the perturbation to the main chain structure. After that, the whole structure was optimized using AMBER force field. There was no crystal structure of AR A2B. Thus, a homology model for A2B was built using the Homology Model module in MOE, using human adenosine A2B receptor protein sequence (GenBank: AAA51598.1) as the query sequence. The template structure to build the homology model was 5NM4. After the homology model was built, structural alignment to the template 5NM4 was carried out in MOE to evaluate the quality of the model proteins. The model proteins were optimized first with main chain atom fixed and then further optimized with all atoms being relaxed. The homology model of A2B was aligned to 5NM4, and the ligand in 5NM4 was adopted to the homology model proteins to help identify the binding pocket for docking studies.
The MOE optimized proteins (AR A2A and AR A2B) were imported to Maestro 12.4 and were subsequently prepared using the Protein Preparation Wizard in the Schrödinger software suite 2020.2[34]. During the Protein Preparation Wizard procedure, the side-chain structures of Gln and Asn were allowed to flip to maximize H-bond interactions. Both proteins were then subjected to 500 iterations of energy minimization with backbone atoms being restrained using the OPLS force field in the MacroModel module in the Schrödinger software[34].
1.2.6.2. Preparation of ligand structures—
To help study the binding selectivity between AR subtypes A2A and A2B, compounds A, B, and KW3902 (NAX) were built based on literature, along with CTCUR, which contains one cis and one trans on the alkene double bonds of otherwise all-trans CUR. This CTCUR can be represented as EZ-keto isomer (Figure 2). All ligands were built in MOE[33] and were optimized using MMFF94 force field using default settings. The optimized ligands then were exported to Maestro for docking studies. All imported ligands in Maestro were further minimized using the OPLS force field in the MacroModel module in the Schrödinger software[34].
Figure 2.

Structures of Compound A, B, KW3902 (NAX), which were used as controls, and CTCUR in its EZ-keto form.
1.2.6.3. Glide docking procedures—
Grid files, for A2A and A2B respectively, were generated using the Glide Grid Generation protocol with the bound ligands as centroids. Ligands were docked to each of the two grid files, which defined the binding pockets of respective proteins. During the docking process, the scaling factor for receptor van der Waals for the nonpolar atoms was set to 0.8 to allow for some flexibility of the receptor, and an extra-precision method was used to calculate the docking scores. All other parameters were used as defaults. The binding affinity was expressed in terms of docking scores. The more negative the docking score, the more favorable the interaction of the complex.
1.2.7. Confocal fluorescence microscopy
Cells were seeded at 700,000 cells per well onto 12-well glass-bottom plates (Cellvis, Mountain View, CA, USA) then incubated until confluent. On the day images were collected, cells were treated with either A) media only, B) CTCUR at 1 μM, C) CA200623 at 60 nM, or D) CA200623 at 60 nM + CTCUR at 1 μM. Our microscopy protocol was a modification of a previously published protocol [35]. Cells were incubated for 1.5–2.0h, then washed twice with PBS, immersed in clear DMEM, and viewed with a 60× oil immersion objective and an Olympus FV3000 laser scanning confocal microscope (Olympus, Tokyo, Japan). Images were acquired at a resolution of 512 by 512 pixels. For the media-only and CTCUR-only treatments, a single representative image was obtained. For the CA200623-only and the CA200623/CTCUR combination treatments, three representative locations within the well were selected. At each location, a stack of images at 0.5 μm intervals in the z axis were acquired. To minimize bias in image selection, we invariably selected images that were 62% of the way through each stack and used those for analysis. All images were processed using CellSens Dimension Desktop V1.18 (Olympus).
Quantitative analysis was done with ImageJ [36] using a modification of a protocol from the Queensland Brain Institute [37]. The background fluorescence level was determined by measuring the mean gray value of the media-only image. For each selected image from the CA200623 only and the CA200623/CTCUR combination treatments, nine cells were selected to be analyzed. The corrected total cell fluorescence (CTCF) was then calculated according to the formula CTCF= Integrated density – (Area of selected cell) x (Mean gray value of background fluorescence). The CTCF was then divided by the area to yield the corrected mean gray value (CMGV).
1.2.8. Cyclic AMP assays
Cells were seeded at 50,000 cells/well in a 96-well plate and allowed to incubate overnight. After incubation, cells were treated with test compounds or control solutions and allowed to incubate for 2h. The rest of the experiment was performed according to the manufacturer’s protocol (ab138880 cAMP Direct Immunoassay Kit, Abcam, Cambridge, UK). The plate was read with a Synergy H1 microplate reader (BioTek, Winooski, VT, USA).
1.2.9. Statistical Analysis
For each CBA dataset, Ki was calculated according to the Cheng-Prusoff equation [38] (eq. 4).
Equation 4. Ki is the inhibitory constant. IC50 is the test compound concentration when half of binding is inhibited. [L] is the concentration of the fluorescent compound. Kd is the dissociation constant of the fluorescent ligand, equal to .
Treatment groups were compared via ANOVA with Tukey’s post hoc tests for binding assays. For fluorescence microscopy, the CMGVs from the CA200623-only and the CA200623/CTCUR combination treatments were compared with t-tests. T-tests, ANOVAs, Ki calculations, and tests to assess the normality and homoscedasticity of datasets, were all performed with Prism 8.4.3 (GraphPad, San Diego, CA, USA). For all analyses, p<0.05 was considered significant.
1.3. Results
Treatment with CTCUR for 2h in both A2A and A2B cells caused no significant change in cell survival from 10−8 to 10−4 M concentration of the test compound (Fig. 3A and 3B respectively). There was a 25% decrease in survival at the 10−4 M CTCUR in the A2A cells, however this concentration is much higher than what was used for binding studies.
Figure 3:

Cell survival of A2A cells (A) and A2B cells (B) in presence of varying concentrations of CT-CUR (log M) treated for 2h (n=3).
Isomerization from CUR to CTCUR led to substantial increases in affinity for AR A2A and AR A2B (fig. 4). The curve for the binding of CUR to AR A2A is sufficiently flat that a meaningful Ki cannot be calculated. By contrast, the binding data for CTCUR at AR A2A indicate binding in the 1 μM to 100 μM range with a Ki of 5.1X10−6 M (fig. 4, A and B). Unlike A2A, the binding of CUR to A2B gave a Ki, equal to 5.7X10−3 M. As with A2A, the pattern observed for CTCUR binding to AR A2B was substantially different from the pattern observed for CUR, with binding in the 1 μM to 100 μM range and a Ki of 6.7X10−6 M (fig. 4, C and D).
Figure 4.

Binding curves of CUR to AR A2A and A2B (A and C) and CTCUR to AR A2A and A2B (B and D). Treatment was performed for 2h. Competitive binding analysis was performed in presence of a known adenosine receptor fluorescent ligand, CA200623 (n=3).
From the docking study calculations, the average absolute value of the error between expected ΔG values and obtained ΔG values for the control compounds (A, B, and NAX) was 1.85 kcal/mol. This low error value demonstrated that the methodology being used was valid. For A2A, CTCUR (keto form) binds to the cytoplasmic ends of transmembrane helices 5, 6, and 7 [32] (fig. 5A). For A2B, CTCUR (keto form) binds to transmembrane helices 6 and 7 and to the cytoplasmic linker region near transmembrane helix 5 [39] (fig. 5B). For A2B, the docking results are in close agreement with the in vitro studies, with a percent error ΔG of only 3% (Table 1). For A2A the docking analysis indicated that CTCUR binds substantially more strongly than what was observed in vitro, with a percent error ΔG of 25% (Table 1).
Figure 5.

Interaction of CTCUR (keto form) with amino acids within A) A2A and B) A2B, according to docking analysis.
Table 1.
Quantitative results of docking analysis of CTCUR.
| Subtype | Ki (nM) | ΔGexperimental (kcal/mol) | ΔGdocking (kcal/mol) | % error ΔG (kcal/mol) |
|---|---|---|---|---|
| A2A | 5107 | −7.2 | −9.6 | 25 |
| A2B | 6722 | −7.1 | −7.3 | 3 |
Analysis of binding using confocal microscopy revealed considerable binding of the fluorescent compound CA200623 at both A2A and A2B receptors (Figure 6). Qualitative assessment of the images indicated that CTCUR was effective in displacing the fluorescent compound in A2B cells but that displacement was not as evident in the A2A cells. Quantitative analysis corroborated this finding, i.e. CTCUR outcompeted the fluorescent compound and thereby lowered the CMGV. For A2A cells the difference was not significant (p>0.99) (Figure 6A), but for A2B cells it was significant (p=0.04) (Figure 6B).
Figure 6.

Images and data obtained from confocal microscopy of A) A2A and B) A2B HEK cells. White ovals highlight outlines of individual cells. Each point in the graph represents one individual cell. Bright red outlines of the cells indicate binding of the fluorescent compound at the receptor (n=3, *p <0.05).
To determine receptor activity, agonism and antagonism of CTCUR binding to both the receptors was studied. Since A2A and A2B are Gs (stimulatory receptors), direct cAMP production associated with CTCUR binding was studied for agonism. Interestingly, CTCUR treatment did not show any change in cAMP production compared to media alone in the test for agonism for both the receptors (Figure 7 A and B). In order to test for antagonism, competitive inhibition of cAMP production was performed using CTCUR against a known AR agonist, NECA. With A2A receptor cells, a Kruskal-Wallis test indicated that there was significant variation between groups in general (p<0.01), but a follow-up multiple comparisons test indicated that no specific pair of groups was significantly different from each other (P>0.05) (Figure 7C). With A2B receptor cells, all of the treatments that included NECA were significantly higher than the media control, as expected (p<0.01). Although none of the combination treatments (NECA + 10−x M CTCUR) was significantly different from the NECA-only treatment (p>0.05), the NECA+ 10−5 M CTUCR treatment group had more significantly elevated cAMP levels (p<0.0001 compared to media) relative to the NECA+10−6 M CTUCR and NECA+10−7 M CTCUR treatment groups (p<0.01 compared to media) (Figure 7D).
Figure 7.

Effect of CTCUR on cAMP production: agonist action in A2A and A2B cells (A and B), and antagonist action in A2A and A2B cells (C and D) (n=3, *p <0.01 compared to media control, **p <0.0001 compared to media control).
1.4. Discussion
CTCUR binds to both AR A2A and AR A2B, as demonstrated by both binding studies in vitro and docking studies in silico. In the case of AR A2B, this conclusion is strengthened by fluorescence microscopy data, while for AR A2A the microscopy data are not conclusive. Regarding receptor activation, cAMP studies paradoxically failed to show either consistent agonistic activity or antagonistic activity.
Cytotoxicity assays show that CTCUR is not toxic to these cells except at very high (100 micromolar) concentrations. Clinical trials in patients with knee osteoarthritis [40,41] and major depressive disorder [42] have shown that CUR is safe for humans at a dose of 1 g per day. Based on the data, we predict that CTCUR as an AR ligand would likewise be safe for human consumption if taken at a comparable dose.
The primary result of this work is the demonstration that isomerizing CUR into CTCUR drastically increases the affinity of the molecule for both of the Gs-linked ARs. The affinity of CTCUR for ARs is generally stronger than the affinity of caffeine and theophylline (xanthine ligands) but weaker than NECA, regadenoson, and adenosine itself (nucleoside ligands). Unlike the three nucleoside ligands, which are selective for A2A to varying extents, CTCUR is not selective between the two A2 subtypes (Table 2). The fact that the pattern of evidence for competition with NECA is somewhat stronger for AR A2B compared to A2A (only in A2B was competition confirmed by confocal microscopy) is consistent with the binding patterns of NECA and CTCUR. Experiments with AR A2B mutants have demonstrated that Trp247 and Val250, both within the 6th transmembrane helix of AR A2B, are important for the binding of NECA [39]. Our docking studies of AR A2B show that Trp247 and Val250 are probably also involved in the binding of CTCUR (Figure 5B), meaning that CTCUR directly interferes with the binding of NECA. For A2A, by contrast, it is less clear which amino acids are involved in the competition between NECA and CTCUR. Also, for A2B, there is near-perfect agreement between our in vitro assessment of binding affinity and the affinity predicted by docking, with only 3% error between the two. The error for A2A is eight-fold higher. Overall, the data suggest that CTCUR binds to both A2A and A2B, but the data for A2B binding are more definitive than the A2A data.
Table 2.
Binding affinities of adenosine (endogenous agonist, prescription drug), NECA (agonist used in AR research), regadenoson (agonist, prescription drug), caffeine (antagonist, widely used over-the-counter drug), theophylline (antagonist used in AR research). Values are Ki (nM).
Our microscopy data must be interpreted with caution because the cells themselves and CTCUR itself showed substantial fluorescence in the range at which they were read. An Olympus representative whom we contacted about this issue indicated that this autofluorescence was most likely seen because we made our detection range too broad, thereby admitting some wavelengths that should have been filtered out. Even though most of this background noise was subtracted out in the quantitative analysis, it still constitutes a serious confounding variable. However, it must be noted that the fact that CTCUR displays fluorescence within the range being studied only strengthens the case that binding is occurring. In the case of A2B, the wells treated with both CA200623 and CTCUR had significantly lower fluorescence despite the fact that both compounds fluoresce, which suggests that each of the two was interfering with the ability of the other to bind to A2B.
The failure of the receptor activation studies to demonstrate whether CTCUR acts as an agonist or antagonist is another confounding factor. The relatively high cAMP levels that resulted in the 10−5 M treatment groups within the assays for antagonism suggest that CTCUR may in fact be an agonist, and that the non-significant results we obtained from our tests for agonism may be an artifact from diluting our samples too severely. Further tests for agonism using undiluted samples, along with assays of Gs protein levels in response to CTCUR, will be needed in order to reach a conclusion about CTCUR’s action. Future studies should focus on discovering whether CTCUR acts as an agonist or antagonist at ARs.
Curcumin’s vanilloid moiety is a component of at least four other phytochemicals that have been shown to have antinociceptive properties, namely capsaicin [47], incarvillateine [48], 6-gingerol [49], and 6-shogaol [50]. At least in the cases of curcumin [25] and capsaisin [47], it has been demonstrated that the antinociceptive effect is mediated by TRPV1 receptors. It is generally accepted that the vanillyl moiety is important for the binding of these compounds to TRPV1 [51], and docking studies have confirmed that the vanillyl moiety plays a key role in the binding of capsaicin to TRPV1 [52]. Accordingly, others have found it useful to think of the vanilloid phytochemicals as a group when investigating their pharmacology [53,54]. Under this approach, it is worthwhile to compare the binding patterns of CTCUR with other vanilloids. For example, the interaction of capsaicin with TRPV1 has an EC50 of around 712 nM [55]. While EC50 values cannot be compared directly to Ki values, we can say that the interaction of CTCUR with ARs is about one order of magnitude weaker than that of capsaicin with TRPV1. Interpretation of the pharmacological meaning of such comparisons must be done with caution, because whether it is desirable for vanilloid compounds to act as TRPV1 agonists [47] TRPV1 antagonists [25], AR agonists, or AR antagonists depends on the specific type of chronic pain being studied and the specific AR subtypes involved.
Although we have focused on the benefits of modulating Gs-linked ARs in the context of relieving pain, modulation of these receptors has medicinal applications across several organ systems, which have been extensively reviewed elsewhere [10]. For example, A2A agonism is beneficial in the heart, where A2A AR activity exerts an anti-inflammatory effect. In hypertensive rats with experimentally induced myocardial infarctions, the administration of the A2A agonist LASSBio-294 has been shown to prevent fibrosis and tumor necrosis factor α secretion [56]. A2A antagonism is beneficial in the brain, where A2A ARs are concentrated in the striatum, a region important for motor function and mood regulation [57]. The A2A AR antagonist istradefylline has already been approved in Japan for the treatment of Parkinson’s disease, and experiments in rats show that istradefylline can also reverse depression [58]. A2A ARs are also present to a lesser extent in the hippocampus of the brain, and mouse experiments have shown that treatment with the A2A AR antagonist SCH58261 can rescue hippocampal synaptic plasticity and thereby rescue short term memory in Alzheimer’s disease [59]. A2B agonism is beneficial in the pancreas and liver, where A2B AR activation helps to maintain a healthy response to insulin. In mice that were fed a high fat diet to induce metabolic dysfunction, administration of the A2B agonist BAY 60–6583 lowered plasma glucose and plasma insulin levels, improved insulin sensitivity, and restored proper levels of insulin receptor substrate 2. This evidence suggests that A2B agonists could be used to treat type 2 diabetes [60]. Finally, A2B antagonism is beneficial for combatting cancer, because A2B malfunction in is a substantial contributor to immune suppression, angiogenesis, and other factors that facilitate tumor growth [61]. In mouse models of melanoma and mammary adenocarcinoma, administration of the A2B antagonist PSB1115 has been shown to decrease metastasis [62]. As this brief review of AR pharmacology demonstrates, an AR ligand can have vastly different effects depending on which organ system it is present in. Finding ways to improve localized drug delivery is therefore an important consideration in the effort to translate AR ligands into effective medications.
Given that the vanilloid phytochemicals are known to have a therapeutic effect on pain and given that ARs are known to mediate pain, it is unfortunate that almost no literature exists on the interaction of vanilloid phytochemicals with ARs. The important role of TRPV1 in the pain-reducing effect of these phytochemicals is established, but it is unknown whether AR interactions might enhance or dampen the antinociception. A more detailed understanding of the mechanism of action of these phytochemicals may lead to novel insight on how they can be used to help those who suffer with chronic pain.
1.5. Acknowledgements
Funding: This work was supported by the Great Plains IDeA CTR program (NIGMS # 1U54GM115458-01, SC and MP) and the American Chemical Society Petroleum Research Fund (# 54862-UR4, MP). Cell culture facility and fluorescence microscopy facilities at UNK were supported by grants from the National Institute for General Medical Science (NIGMS) (5P20GM103427), a component of the National Institutes of Health (NIH), as well as Nebraska Research Initiative.
Abbreviations
- AR
adenosine receptor
- cAMP
cyclic adenosine monophosphate
- CBA
competitive binding assay
- CNS
central nervous system
- CUR
curcumin
- CTCUR
cis-trans curcumin
- NECA
5’-N-ethylcarboxamidoadenosine
- HEK
human embryonic kidney
- MOE
molecular operating environment
Footnotes
Declarations of interest: none
Chemical Compounds
Chemical compounds studied in this article: Curcumin (PubChem CID: 969516), cis-trans curcumin (PubChem CID: 969516), 5’-N-Ethylcarboxamidoadenosine (PubChem CID: 4400)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
1.6 Literature Cited
- [1].Nahin RL, Estimates of Pain Prevalence and Severity in Adults: United States, 2012, J. Pain 16 (2015) 769–780. 10.1016/j.jpain.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kolodny A, Courtwright DT, Hwang CS, Kreiner P, Eadie JL, Clark TW, Alexander GC, The Prescription Opioid and Heroin Crisis : A Public Health Approach to an Epidemic of Addiction, Annu. Rev. Public Health 36 (2015) 559–574. 10.1146/annurev-publhealth-031914-122957. [DOI] [PubMed] [Google Scholar]
- [3].Dhalla IA, Persaud N, Juurlink DN, Facing up to the prescription opioid crisis, BMJ. 343 (2011) d5142. 10.1136/bmj.d5142. [DOI] [PubMed] [Google Scholar]
- [4].Zylka MJ, Pain-relieving prospects for adenosine receptors and ectonucleotidases, Trends Mol. Med 17 (2011) 188–196. 10.1016/j.molmed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen Z, Janes K, Chen C, Doyle T, Bryant L, Tosh DK, Jacobson KA, Salvemini D, Controlling murine and rat chronic pain through A3 adenosine receptor activation, FASEB J. 26 (2012) 1855–1865. 10.1096/fj.11-201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Godfrey L, Yan L, Clarke GD, Ledent C, Kitchen I, Hourani SMO, Modulation of paracetamol antinoiception by caffeine and by selective adenosine A 2 receptor antagonists in mice, Eur. J. Pharmacol 531 (2006) 80–86. 10.1016/j.ejphar.2005.12.004. [DOI] [PubMed] [Google Scholar]
- [7].Savegnago L, Jesse CR, Nogueira CW, Caffeine and a selective adenosine A 2B receptor antagonist but not imidazoline receptor antagonists modulate antinociception induced by diphenyl diselenide in mice, Neurosci. Lett 436 (2008) 120–123. 10.1016/j.neulet.2008.03.003. [DOI] [PubMed] [Google Scholar]
- [8].Goldman N, Chen M, Fujita T, Xu Q, Peng W, Liu W, Jensen TK, Pei Y, Wang F, Han X, Chen J-F, Schnermann J, Takano T, Bekar L, Tieu K, Nedergaard M, Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture, 13 (2010) 883–888. 10.1038/nn.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Little JW, Ford A, Symons-liguori AM, Chen Z, Janes K, Doyle T, Xie J, Luongo L, Tosh DK, Maione S, Bannister K, Dickenson AH, Vanderah TW, Porreca F, Jacobson KA, Salvemini D, Endogenous adenosine A 3 receptor activation selectively alleviates persistent pain states, Brain. 138 (2015) 28–35. 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K, Pharmacology of adenosine receptors: the state of the art, Physiol. Rev 98 (2018) 1591–1625. 10.1152/physrev.00049.2017. [DOI] [PubMed] [Google Scholar]
- [11].Fredholm BB, Ijzerman AP, Jacobson KA, Klotz K-N, Linden J, International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors, Pharmacol. Rev 53 (2001) 527–552. [PMC free article] [PubMed] [Google Scholar]
- [12].Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE, International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors — An Update, Pharmacol. Rev 63 (2011) 1–34. 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen J, Eltzschig HK, Fredholm BB, Adenosine receptors as drug targets — what are the challenges?, Nat. Rev. Drug Discov 12 (2013) 265–286. 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bailey A, Ledent C, Kelly M, Hourani SMO, Kitchen I, Changes in Spinal Delta and Kappa Opioid Systems in Mice Deficient in the A 2A Receptor Gene, J. Neurosci 22 (2002) 9210–9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sardi N, Tobaldini G, Morais R, Fischer L, Nucleus accumbens mediates the pronociceptive effect of sleep deprivation, Pain. 159 (2018) 75–84. [DOI] [PubMed] [Google Scholar]
- [16].Kwilasz AJ, Ellis A, Wieseler J, Loram L, Favret J, Mcfadden A, Springer K, Falci S, Rieger J, Maier SF, Watkins LR, Sustained reversal of central neuropathic pain induced by a single intrathecal injection of adenosine A 2A receptor agonists, Brain Behav. Immun 69 (2018) 470–479. 10.1016/j.bbi.2018.01.005. [DOI] [PubMed] [Google Scholar]
- [17].Schindler CW, Karcz-kubicha M, Thorndike EB, Mu CE, Goldberg SR, Tella SR, Ferre S, Role of central and peripheral adenosine receptors in the cardiovascular responses to intraperitoneal injections of adenosine A 1 and A 2A subtype receptor agonists, (2005) 642–650. 10.1038/sj.bjp.0706043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Abo-salem OM, Hayallah AM, Bilkei-gorzo A, Filipek B, Zimmer A, Mu CE, Antinociceptive Effects of Novel A2B Adenosine Receptor Antagonists Antinociceptive Effects of Novel A 2B Adenosine Receptor Antagonists, (2004). 10.1124/jpet.103.056036. [DOI] [PubMed] [Google Scholar]
- [19].Hu X, Adebiyi MG, Luo J, Sun K, Le T-TT, Zhang Y, Wu H, Zhao S, Karmouty-quintana H, Liu H, Huang A, Etslzchig HK, Blackburn MR, Walters ET, Huang D, Sustained elevated adenosine via ADORA2B promotes chronic pain through neuro-immune interaction, Cell Rep. 16 (2017) 106–119. 10.1016/j.celrep.2016.05.080.Sustained. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Müller CE, Jacobson KA, Recent developments in adenosine receptor ligands and their potential as novel drugs ☆, Biochim. Biophys. Acta 1808 (2011) 1290–1308. 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Priebe A, Hunke M, Tonello R, Sonawane Y, Berta T, Natarajan A, Bhuvanesh N, Pattabiraman M, Chandra S, Ferulic acid dimer as a non-opioid therapeutic for acute pain, J. Pain Res 11 (2018) 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ahmadi M, Hajialilo M, Dolati S, Fard SE, The effects of nanocurcumin on Treg cell responses and treatment of ankylosing spondylitis patients : A randomized, double - blind, placebo - controlled clinical trial, J. Cell. Biochem 121 (2020) 103–110. 10.1002/jcb.28901. [DOI] [PubMed] [Google Scholar]
- [23].Agarwal KA, Tripathi CD, Agarwal BB, Saluja S, Efficacy of turmeric (curcumin) in pain and postoperative fatigue after laparoscopic cholecystectomy : a double-blind, randomized placebo-controlled study, Surg. Endosc 25 (2011) 3805–3810. 10.1007/s00464-011-1793-z. [DOI] [PubMed] [Google Scholar]
- [24].Zhu X, Li Q, Chang R, Yang D, Song Z, Guo Q, Curcumin Alleviates Neuropathic Pain by Inhibiting p300 / CBP Histone Acetyltransferase Activity-Regulated Expression of BDNF and Cox-2 in a Rat Model, PLoS One. 9 (2014) e91303. 10.1371/journal.pone.0091303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yeon KY, Kim SA, Kim YH, Lee MK, Ahn DK, Kim HJ, Kim JS, Jung SJ, Oh SB, Curcumin Produces an Antihyperalgesic Effect via Antagonism of TRPV1, J. Dent. Res 89 (2009) 170–174. 10.1177/0022034509356169. [DOI] [PubMed] [Google Scholar]
- [26].Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB, Bioavailability of Curcumin : Problems and Promises, Mol. Pharm 4 (2007) 807–818. 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- [27].Rocks N, Bekaert S, Coia I, Paulissen G, Gueders M, Evrard B, Van Heugen J, Chiap P, Foidart J, Noel A, Cataldo D, Curcumin – cyclodextrin complexes potentiate gemcitabine effects in an orthotopic mouse model of lung cancer, Br. J. Cancer 107 (2012) 1083–1092. 10.1038/bjc.2012.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chuang E, Lin K, Huang T, Chen H, Miao Y, Lin P, Chen C, Juang J, Sung H, An Intestinal “ Transformers ” -like Nanocarrier System for Enhancing the Oral Bioavailability of Poorly Water-Soluble Drugs, ACS Nano. 12 (2018) 6389–6397. 10.1021/acsnano.8b00470. [DOI] [PubMed] [Google Scholar]
- [29].Yallapu MM, Nagesh PKB, Jaggi M, Chauhan SC, Review Article Therapeutic Applications of Curcumin Nanoformulations, AAPS J. 17 (2015) 1341–1356. 10.1208/s12248-015-9811-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bisht S, Khan MA, Bekhit M, Bai H, Cornish T, Mizuma M, Rudek MA, Zhao M, Maitra A, Ray B, Lahiri D, Maitra A, Anders RA, A polymeric nanoparticle formulation of curcumin ( NanoCurc t ) ameliorates CCl 4 -induced hepatic injury and fibrosis through reduction of pro-inflammatory cytokines and stellate cell activation, Lab. Investig 91 (2011) 1383–1395. 10.1038/labinvest.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hunke M, Martinez W, Kashyap A, Bokoskie T, Pattabiraman M, Chandra S, Antineoplastic Actions of Cinnamic Acids and Their Dimers in Breast Cancer Cells: A Comparative Study, Anticancer Res. 38 (2018) 4469–4474. 10.21873/anticanres.12749. [DOI] [PubMed] [Google Scholar]
- [32].Weinert T, Olieric N, Cheng R, Brünle S, James D, Ozerov D, Gashi D, Vera L, Marsh M, Jaeger K, Dworkowski F, Panepucci E, Basu S, Skopintsev P, Doré AS, Geng T, Cooke RM, Liang M, Prota AE, Panneels V, Nogly P, Ermler U, Schertler G, Hennig M, Steinmetz MO, Wang M, Standfuss J, Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons, Nat. Commun 8 (2017) article number 542. 10.1038/s41467-017-00630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].The Molecular Operating Environment (MOE), 2020.
- [34].The Protein Preparation Wizard, Maestro, MacroModel, and Glide, Schrodinger Release 2020-2, 2020. [Google Scholar]
- [35].Nabb DL, Song S, Kluthe KE, Daubert TA, Luedtke BE, Nuxoll AS, Polymicrobial Interactions Induce Multidrug Tolerance in Staphylococcus aureus Through Energy Depletion, Front. Microbiol 10 (2019) 2803. 10.3389/fmicb.2019.02803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rasband W, Image J, Natl. Institutes Heal Bethesda, Maryland, USA. (1997). https://imagej.nih.gov/ij/. [Google Scholar]
- [37].Hammond L, Measuring cell fluorescence using ImageJ, Open Lab B. (2014). https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html. [Google Scholar]
- [38].Stoddart LA, White CW, Nguyen K, Hill SJ, Pfleger KDG, Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding, Br. J. Pharmacol 173 (2016) 3028–3037. 10.1111/bph.13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Thimm D, Schiedel AC, Sherbiny FF, Hinz S, Hochheiser K, Bertarelli DCG, Maaß A, Mu CE, Ligand-Specific Binding and Activation of the Human Adenosine A2B receptor, Biochemistry. 52 (2013) 726–740. 10.1021/bi3012065. [DOI] [PubMed] [Google Scholar]
- [40].Haroyan A, Mukuchyan V, Mkrtchyan N, Minasyan N, Gasparyan S, Sargsyan A, Narimanyan M, Hovhannisyan A, Efficacy and safety of curcumin and its combination with boswellic acid in osteoarthritis : a comparative, randomized, double-blind, placebo-controlled study, BMC Complement. Altern. Med 18 (2018) 7. 10.1186/s12906-017-2062-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shep D, Khanwelkar C, Gade P, Karad S, Safety and efficacy of curcumin versus diclofenac in knee osteoarthritis : a randomized open-label parallel-arm study, Trials. 20 (2019) 214. 10.1186/s13063-019-3327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sanmukhani J, Satodia V, Trivedi J, Patel T, Tiwari D, Panchal B, Goel A, Tripathi CB, Efficacy and Safety of Curcumin in Major Depressive Disorder : A Randomized Controlled Trial, Phyther. Res 28 (2014) 579–585. 10.1002/ptr.5025. [DOI] [PubMed] [Google Scholar]
- [43].Klotz K-N, Adenosine receptors and their ligands, Nauyn-Schmiedeberg’s Arch. Pharmacol 362 (2000) 382–391. 10.1007/s002100000315. [DOI] [PubMed] [Google Scholar]
- [44].Gao Z, Li Z, Baker SP, Lasley RD, Meyer S, Elzein E, Palle V, Zablocki JA, Blackburn B, Belardinelli L, Novel Short-Acting A 2A Adenosine Receptor Agonists for Coronary Vasodilation : Inverse Relationship between Affinity and Duration of Action of A 2A Agonists, J. Pharmacol. Exp. Ther 298 (2001) 209–218. [PubMed] [Google Scholar]
- [45].Bertarelli DCG, Diekmann M, Hayallah AM, Rüsing D, Iqbal J, Preiss B, Verspohl EJ, Müller CE, Characterization of human and rodent native and recombinant adenosine A 2B receptors by radioligand binding studies, Purinergic Signal. 2 (2006) 559–571. 10.1007/s11302-006-9012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jacobson KA, IJzerman AP, Linden J, 1, 3-Dialkylxanthine Derivatives Having High Potency as Antagonists at Human A 2B Adenosine Receptors, Drug Dev. Res 47 (1999) 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Starowicz K, Maione S, Cristino L, Palazzo E, Marabese I, Rossi F, De Novellis V, Di Marzo V, Tonic Endovanilloid Facilitation of Glutamate Release in Brainstem Descending Antinociceptive Pathways, J. Neurosci 27 (2007) 13739–13749. 10.1523/JNEUROSCI.3258-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakamura M, Chi Y, Yan W, Nakasugi Y, Yoshizawa T, Irino N, Hashimoto F, Kinjo J, Nohara T, Sakurada S, Strong Antinociceptive Effect of Incarvillateine, a Novel Monoterpene Alkaloid from Incarvillea sinensis, J. Nat. Prod (1999) 1293–1294. 10.1021/np990041c. [DOI] [PubMed] [Google Scholar]
- [49].Young H, Luo Y, Cheng H, Hsieh W, Liao J-C, Peng W-H, Analgesic and anti-inflammatory activities of [ 6 ] -gingerol, J. Ethnopharmacol 96 (2005) 207–210. 10.1016/j.jep.2004.09.009. [DOI] [PubMed] [Google Scholar]
- [50].Hitomi S, Ono K, Terawaki K, Matsumoto C, Mizuno K, Yamaguchi K, Imai R, Omiya Y, Hattori T, Kase Y, Inenaga K, [6]-gingerol and [6]-shogaol, active ingredients of the traditional Japanese medicine hangeshashinto, relief oral ulcerative mucositis-induced pain via action on Na + channels, Pharmacol. Res 117 (2017) 288–302. 10.1016/j.phrs.2016.12.026. [DOI] [PubMed] [Google Scholar]
- [51].Premkumar LS, Transient Receptor Potential Channels as Targets for Phytochemicals, ACS Chem. Neurosci 5 (2014) 1117–1130. 10.1021/cn500094a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yang F, Xiao X, Cheng W, Yang W, Yu P, Song Z, Zheng J, Structural mechanism underlying capsaicin binding and activation of TRPV1 ion channel, Nat. Chem. Biol 11 (2015) 518–524. 10.1038/nchembio.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hail N, Mechanisms of vanilloid-induced apoptosis, Apoptosis. 8 (2003) 251–262. [DOI] [PubMed] [Google Scholar]
- [54].Imm J, Zhang G, Chan L, Nitteranon V, Parkin KL, [6]-Dehydroshogaol, a minor component in ginger rhizome, exhibits quinone reductase inducing and anti-inflammatory activities that rival those of curcumin, Food Res. Int 43 (2010) 2208–2213. 10.1016/j.foodres.2010.07.028. [DOI] [Google Scholar]
- [55].Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D, The capsaicin receptor : a heat-activated ion channel in the pain pathway, Nature. 389 (1997) 816–824. [DOI] [PubMed] [Google Scholar]
- [56].da Silva JS, Gabriel-Costa D, Sudo RT, Wang H, Groban L, Ferraz EB, Nascimento JHM, Fraga CAM, Barriero EJ, Zapata-Sudo G, Adenosine A 2A receptor agonist prevents cardiac remodeling and dysfunction in spontaneously hypertensive male rats after myocardial infarction, Drug Des. Dev. Ther 11 (2017) 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Schwarzschild MA, Agnati L, Fuxe K, Chen J, Morelli M, Targeting adenosine A2A receptors in Parkinson’s disease, Trends Neurosci. 29 (2006) 647–654. 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- [58].Yamada K, Kobayashi M, Shiozaki S, Ohta T, Mori A, Jenner P, Tomoyuki K, Antidepressant activity of the adenosine A2A receptor antagonist, istradefylline (KW-6002) on learned helplessness in rats, Psychopharmacology (Berl). 231 (2014) 2839–2849. 10.1007/s00213-014-3454-0. [DOI] [PubMed] [Google Scholar]
- [59].da Silva SV, Haberl MG, Zhang P, Bethge P, Lemos C, Goncalves N, Gorlewicz A, Malezieux M, Goncalves FQ, Grosjean N, Blanchet C, Frick A, Nagerl UV, Cunha RA, Mulle C, Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors, Nat. Commun 7 (2016) 11915. 10.1038/ncomms11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Johnston-Cox H, Koupenova M, Yang D, Corkey B, Gokce N, Farb MG, LeBrasseur N, Ravid K, The A2b Adenosine Receptor Modulates Glucose Homeostasis and Obesity, PLoS One. 7 (2012) e40584. 10.1371/journal.pone.0040584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gao Z-G, Jacobson KA, A2B Adenosine Receptor and Cancer, Int. J. Mol. Sci 20 (2019) 5139. 10.3390/ijms20205139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, Caramia F, Haibe-Kains B, Stagg J, Khanna KK, Loi S, Smyth MJ, Adenosine 2B Receptor Expression on Cancer Cells Promotes Metastasis, Cancer Res. 76 (2016) 4372–4383. 10.1158/0008-5472.CAN-16-0544. [DOI] [PubMed] [Google Scholar]