

Summary
The challenge of precision medicine is to model complex interactions among DNA variants, phenotypes, development, environments, and treatments. We address this challenge by expanding the BXD family of mice to 140 fully isogenic strains, creating a uniquely powerful model for precision medicine. This family segregates for 6 million common DNA variants—a level that exceeds many human populations. Because each member can be replicated, heritable traits can be mapped with high power and precision. Current BXD phenomes are unsurpassed in coverage and include much omics data and thousands of quantitative traits. The BXDs can be extended by a single-generation cross to as many as 19,460 isogenic F1 progeny, and this extended BXD family is an effective platform for testing causal modeling and for predictive validation. The BXDs are a unique core resource for the field of experimental precision medicine.
Keywords: Systems genetics, systems biology, gene mapping, complex trait, power calculation, personalized medicine, recombinant inbred strains, GXE
Electronic Table of Contents blurb
Ashbrook et al., have expanded the BXD family to 140 strains, providing a new tool for translational precision and predictive biology, and extended the usefulness of the deep phenome of >100 omics datasets and >7500 classical phenotypes already available. They show increased precision and power by using new genotypes, updated models and more strains.
Introduction
The development of predictive, preventive, personalized, and participatory healthcare requires as prerequisites deep and coherent phenome data. These data must be matched to sophisticated causal models of complex interactions among sequence variants, phenotypes at many levels, and environmental exposures (Auffray et al., 2010; Hood and Friend, 2011; Williams, 2009). For most diseases it is not yet practical to predict risk and best treatment options for the simple reason that each human is unique (Schüssler-Fiorenza Rose et al., 2019). This N-of-1 problem inhibits development and refinement of predictions. However, it is now possible to create optimized experimental platforms that fill this gap in the form of large and fully characterized families of isogenic rodents, such as the BXD.
The BXD family was started fifty years ago by crossing fully inbred female C57BL/6J (B6 or B) to male DBA/2J (D2 or D) mice. The first set of BXD recombinant inbred (RI) strains was used to map Mendelian traits (Taylor et al., 1973, 1999), but the BXDs are now used mainly to map complex traits including cancers, metabolic and cardiovascular disease (Grizzle et al., 2002; Koutnikova et al., 2009; Lee et al., 1995; McGinnis et al., 1993), brain structure (Belknap et al., 1992a; Rosen et al., 2009; Seecharan et al., 2003; Zhou and Williams, 1999), behavior, and pharmacology (Ashbrook et al., 2018a; Belknap et al., 1992b, 1993; Grisel et al., 1997; Jones et al., 2006; Knoll et al., 2018; Palmer et al., 2006; Phillips et al., 1998; Rodriguez et al., 1994; Weimar et al., 1982). We started a second wave of BXDs in the late 1990s (Peirce et al., 2004) using advanced intercross (AI) progeny (Darvasi, 1998) (Figure 1; Figure S1; Table S1). These AI-derived family members incorporate twice as many recombination junctions as do conventional BXDs (Broman, 2005; Crow, 2007; Haldane and Waddington, 1931; Williams et al., 2001) (Figure 1C)—an attribute that improves mapping precision about two-fold.
Figure 1: Production of the BXD family by standard F2 (A) or advanced intercross (B), and recombination differences (C).
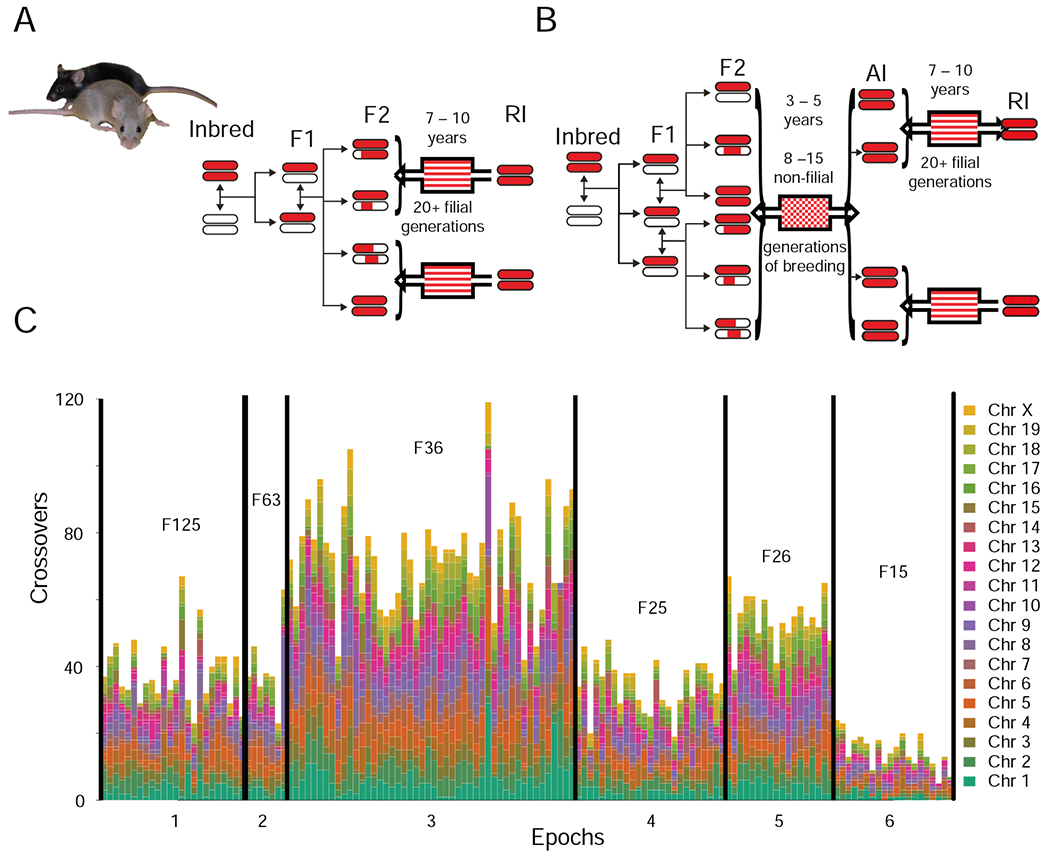
Four epochs of the BXD were derived from F2 (A) (~75 strains; epochs 1,2,4,6 in C). Two epochs were derived from advanced intercross (B) (~65 strains; epochs 3, 5 in C). Red represents regions of the genome coming from C57BL/6J (B6), white represents regions from the DBA/2J (D2). Solid lines represent a single generation. Adapted from Peirce et al., 2004. (C) Genome wide visualization of recombinations in the BXD. Number of recombinations per strain (nRecS), ignoring heterozygous and unknown genotypes plotted using a color gradient. Strain numbers on the X-axis, horizontal lines separate the epochs, epochs are annotated on the X-axis. Mean number of inbreeding generations is shown for each epoch. Epoch 6 appears to have fewer recombinations due to a large number of heterozygous loci at genotyping. Details in Figure S1 and Table S1.
The BXDs are well suited for systems biology (Williams and Williams, 2017) thanks to a well-integrated phenome consisting of over 7500 classical phenotypes and over 100 omics data sets (Williams and Auwerx, 2015; Williams et al., 2001). This phenome extends back to Taylor’s 1973 analysis of cadmium toxicity, through to recent studies of metabolism (Roy et al., 2020; Wang et al., 2013, 2016b; Williams et al., 2016, 2020; Wu et al., 2014), addiction (Dickson et al., 2016, 2019; Mulligan et al., 2013), behavior (Carhuatanta et al., 2014; Graybeal et al., 2014; Mulligan and Williams, 2015; Philip et al., 2010), infectious disease (Hayes et al., 2014; McKnite et al., 2012; Wang et al., 2020), epigenetics (Baker et al., 2019; Sandoval-Sierra et al., 2020), and even indirect genetic effects (Ashbrook et al., 2015a, 2017; Baud et al., 2017). The BXD have been used to test developmental and evolutionary hypotheses (Hager et al., 2012; Oren et al., 2015; Seecharan et al., 2003), to study GXE and consequences of treatments as a function of age, diet, and sex (Fleet et al., 2016; Philip et al., 2010; Roy et al., 2020; Sandoval-Sierra et al., 2020; Williams et al., 2016, 2020), gene pleiotropy (Wang et al., 2016a), and to test behavioral predictions based on differences in brain architecture (Yang et al., 2008).
Here we summarize the current status of this resource with a focus on genetic structure, and on the power and precision of mapping trait variance to loci and genes. We have almost doubled the size of the BXD family, from ~80 (Peirce et al., 2004) to 140 extant strains. Each of the 140 fully inbred and isogenic strains of mice is an immortal genometype that can be resampled at any stage, in both sexes, and under varied but controlled conditions to quantify gene-by-environmental interactions (GXE) and to test the accuracy and errors of genome-to-phenome prediction. Compared to progeny of conventional intercrosses, AIs, or heterogenous stock, the BXDs are advantageous when heritabilities of traits are low or when measurement errors are high because the genetic signal can be boosted by resampling many isogenic cases (Belknap, 1998; Crow, 2007). A further benefit is that each data set adds quadratically to the number of trait-to-trait associations, and even the oldest data becomes more powerful as genetic, genomic, and phenomic contexts are improved and enlarged. The BXD, like other replicable reference populations, are therefore well adapted to extensible multisystems analyses, to modeling, and to quantitative prediction (Andreux et al., 2012; Chesler et al., 2003; Jha et al., 2018b, 2018a; Li et al., 2018; Williams et al., 2016). We have therefore assembled deep companion resources, including full sequence for both parents (Baker et al., 2019; Keane et al., 2011; McKnite et al., 2012; Wang et al., 2016b; Wu et al., 2014). Access to data and statistical tools are available from open-source web services (GeneNetwork.org and Systems-Genetics.org) (Li et al., 2018; Sloan et al., 2016; Williams and Williams, 2017). High-density genetic maps are now combined with linear mixed model mapping to boost power and accuracy.
Results
The BXD family has been expanded to 140 inbred strains
We have approximately doubled the number of extant BXD strains, from ~35 (Taylor et al., 1999), to ~80 (Peirce et al., 2004), to 140 here. We started producing 108 new BXD strains between 2009 and 2013 (BXD104–BXD220). A total of 123 BXD lines are currently distributed by The Jackson Laboratory (JAX, Table S1). Seventeen additional strains are available at UTHSC but will soon be donated to JAX. All strains are available under a standard material transfer agreement.
We have identified three sets of strains that have almost identical genomes. These are marked by the addition of “a” and “b” suffixes. For example, BXD65, BXD65a, and BXD65b are identical by descent across >90% of their genomes. Without correction, the inclusion of three BXD65 substrains will bias results. There is also more subtle kinship among BXD family members that results from drift, backcrossing, and unintended selection. Correcting for both types of kinship when mapping traits can be accomplished using linear mixed models during analyses, for example Genome-wide Efficient Mixed Model Association (GEMMA) (Zhou and Stephens, 2012) or R/qtl2 (Broman et al., 2003, 2019) discussed below.
Improved mapping using updated markers
Affymetrix and Illumina arrays have been used to genotype at more than 100,000 informative markers in 198 BXD strains (Morgan et al., 2015; Peirce et al., 2004; Shifman et al., 2006; Taylor et al., 1999; Zhang et al., 2012). Previously, a subset of 3,830 markers have been used for mapping, but here we have boosted marker numbers nearly two-fold to 7,324. This high-density marker set defines proximal and distal limits of chromosomal intervals that are non-recombinant. Markers are spaced at 0.63 Mb ± 1.0 SE, closely matching the expected asymptotic map resolution of ±1 Mb.
The increase in numbers of markers has significantly improved power and precision of linkage analysis (Figure 2, Figure S2) across all chromosomes (Figure S2). To quantify these improvements in mapping, we computed linkage for the entire BXD phenome—currently 7562 phenotypes—using three different genotype files: the original file from 2001 (Williams et al., 2001), the “classic” genotypes used from 2005 to about 2016, and the updated genotypes. Each of these stage roughly doubled the numbers of markers (from 1,578 to 3,811 to 7,324). We counted the number of traits associated with logarithm of the odds (LOD) linkage scores at three cut-offs: 2.2 (suggestive; a likelihood of one false positive per genome scan), 3.6 (significant; one false positives per 20 scans), and 5.4 (highly significant; one false positives per 100 scans) (Lander and Kruglyak, 1995) (Figure 2C). The largest boost in LOD is achieved for traits with linkage between 3.6 and 5.4 LOD—up 30% compared to older marker sets (Figure 2C and 2D).
Figure 2: Improved, denser, genotypes increase linkage across chromosomes, decades of work, and number of strains.
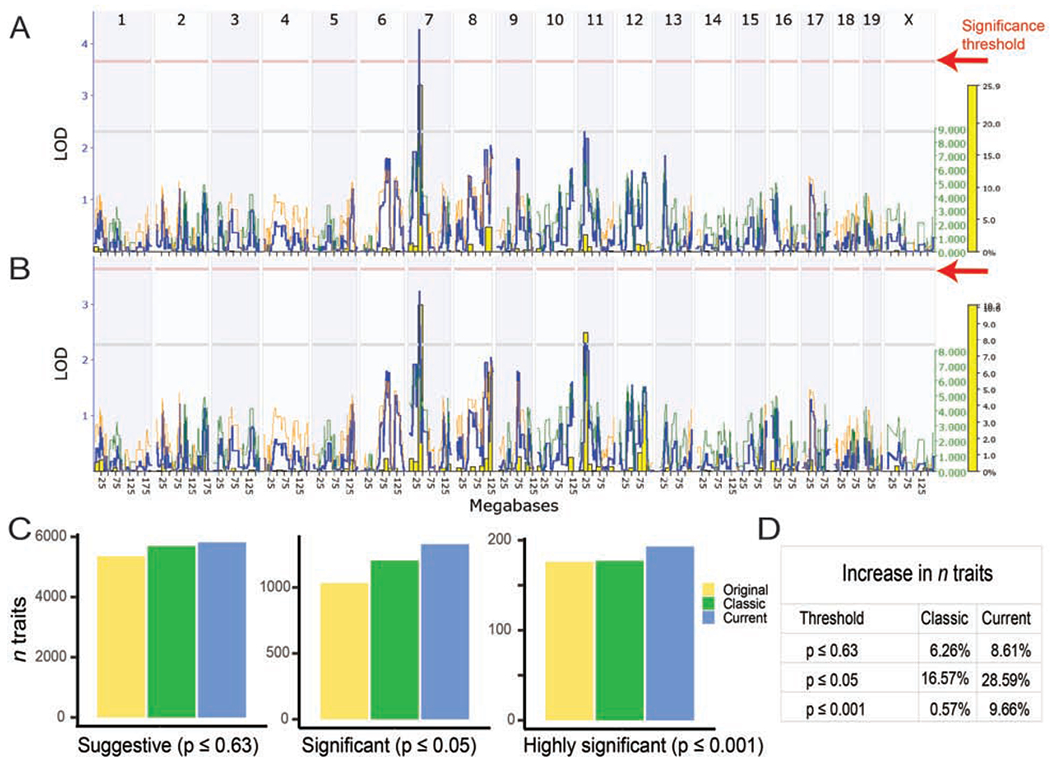
Comparison of current (A) and classic (B) genotypes for BXD_10666, Cytotoxic T-cell (CTL) response (5 x 10^9 PFU AdLacZ iv), measured as interleukin 6 (IL-6) cytokine expression [pg/ml], published by Zhang et al., 2004, measured in 23 strains. A QTL on chromosome 7 is now significant (above the red p = 0.05 significance line) using the current genotypes (A), compared to the classic genotypes (B). Additional examples in Figure S2. (C) Bar chart showing the number of phenotypes with a peak LOD score over the suggestive (LOD ≥ 2.2; p ≤ 0.63), significant (LOD ≥ 3.6; p ≤ 0.05) and highly significant (LOD ≥ 5.4; p ≤ 0.001) thresholds, using original, classic or current marker maps. (D) Percentage increase in the number of traits passing the thresholds in the classic or current genotype map, compared to the original map.
We can further improve power and precision using linear mixed models
Compared to traditionally used Haley-Knott mapping (Haley and Knott, 1992), more recently developed algorithms can correct for variable kinship among genometypes and further improve mapping (Broman et al., 2003, 2019; Sul et al., 2016; Zhou and Stephens, 2012). We calculated linkage for 3300 phenotypes using both the Haley-Knott (H-K) method that does not correct for kinship or using linear mixed model (LMM) mapping that does correct for kinship (Broman et al., 2019). Genome-wide significance was estimated based on 5000 permutations and the improvement is substantial (Figure 3, Table S2). Using H-K, 638 phenotypes reach pgw < 0.05 significance, whereas 763 reach the same criterion using the LMM, a 20% increase. To check that this was not due to phenotypes close to the threshold, we counted the number of phenotypes with a peak LOD ≥ 1 above threshold: 196 for H-K and 262 for LMM, a 33% increase. Note that there is not a substantial average increase in LOD scores when using an LMM compared to H-K (a mean increase of 0.06 and a median of 0.07), nor is there a substantial change in the mean pgw threshold (LMM 3.51 vs H-K 3.55; Table S2), demonstrating that LMM is not simply inflating all values.
Figure 3: Improvement in mapping by using linear mixed models (LMMs) vs Haley-Knott regression (H-K).
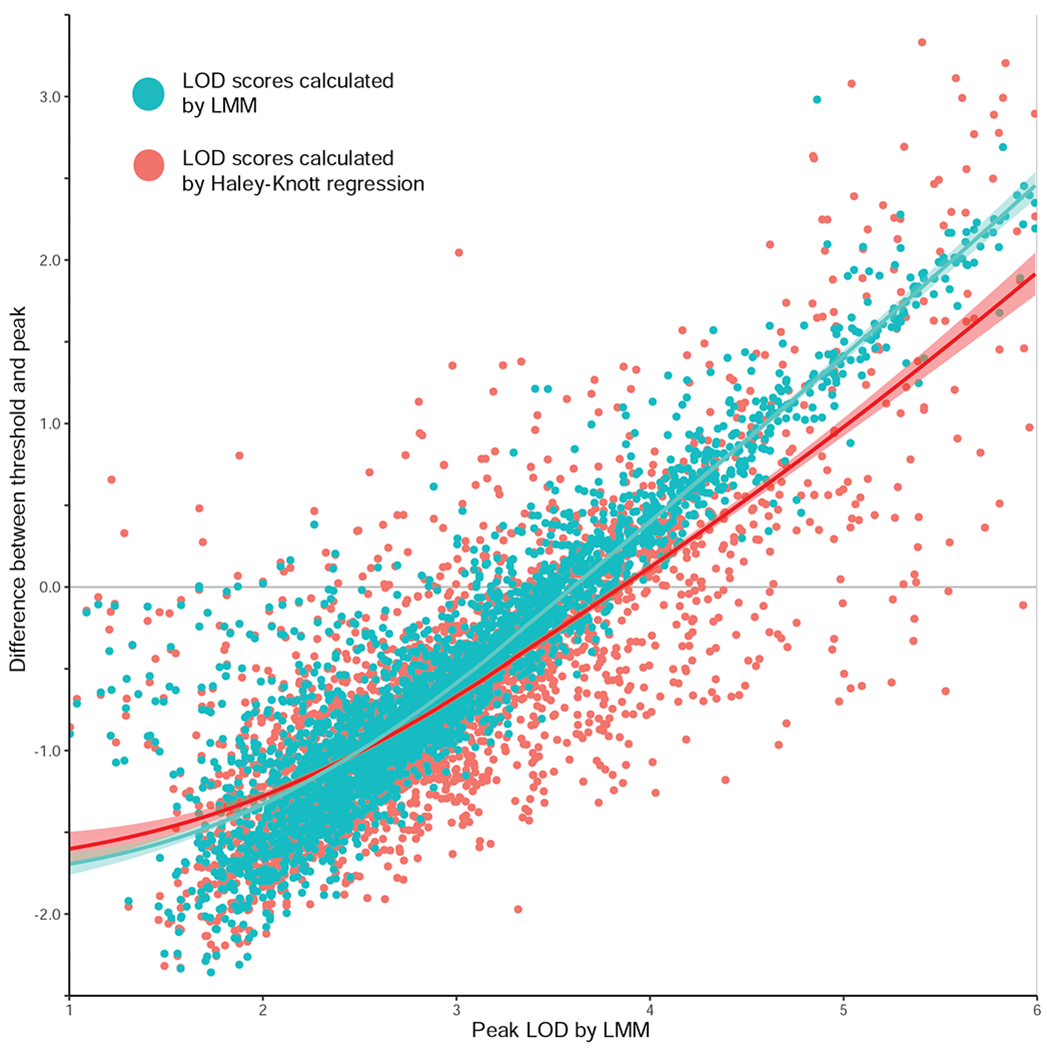
Linkage measured in 3300 phenotypes from GeneNetwork, and mapped using Haley-Knott regression or an LMM in R/qtl2 (Broman et al., 2003, 2019). Y-axis is difference in LOD from the significance threshold (calculated by 5000 permutations) and the peak LOD. X-axis is the peak LOD, calculated by LMM, so that points can be directly compared. Each point represents a phenotype, LMM results plotted turquoise, Haley-Knott plotted red. A smoother was fitted in the same colors for comparison. Underlying data in Table S2.
Empirical mapping precision ranges from 0.25 to 5 Mb
We estimated the empirical precision of linkage maps using ~270,000 cis-acting expression QTLs (eQTLs) that have LOD scores greater than 3.0 and within 10 Mb of the mRNA transcription start site (Figure 4A). These criteria are conservative because an acceptance window of ±10 Mb will include a small number of regional trans-acting variants. However, this approach guards against overly optimistic estimates of precision. Mean precision for 17,805 eQTLs with LODs above 5.0 is about 1.0 Mb using only 60–80 BXD strains. The median precision is about 0.50 Mb. In other words, the offset between the location of a gene and the location of the marker with the highest LOD is generally less than 1 Mb using half the available strains. However, regional variation in precision is high (Figure 4A,B), and intervals with low precision have either few markers or low recombination rates— a problem that is currently being addressed by generating sequence-based infinite marker maps (Ashbrook et al., 2018b). There may also be regions that contain large structural variants—duplication, inversions, insertions—compared to the reference genome of C57BL/6J.
Figure 4: Empirical QTL mapping precision estimated using cis-eQTLs.
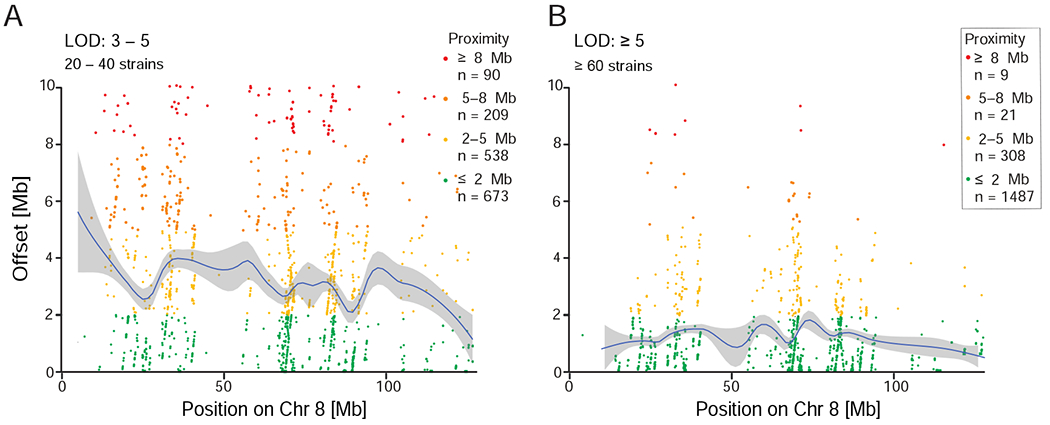
Each point shows the distance between a probe and an eQTL. (A) Precision achieved when mapping modest QTLs with LOD scores of 3-5 and using only 20-40 strains and 2-4 samples per strain. Number of replicates within each strain will affect the precision to a greater extent for QTLs with low heritability and modest LOD scores by increasing the effective heritability (see Belknap 1998). eQTL studies generally use only 1–4 replicates, so precision values are conservative. Mean offset across the genome is 4.67 Mb, median 2.72 Mb. (B) Precision achieved when mapping highly significant QTLs with LOD > 5, using 60–80 strains, with > 2 replicates. Mean offset is 1.38 Mb, median 0.76 Mb.
It is possible to achieve subcentimorgan mapping precision using only half of the full set of BXD strains (Figure 4)—a mean resolution of 500 Kb and a median resolution of 250 Kb for these Mendelian traits. Three factors contribute to this precision: 1. well-balanced distribution of only two haplotype across the genome (minor allele and haplotype frequencies close to 0.5). 2. the ability to boost the effective heritability of traits by resampling (Belknap, 1998); and 3. the high density of recombination junctions captured collectively within the BXD family (Figure 1C). Surprisingly, this level of map precision does not differ appreciably from that typical of the Collaborative Cross, the Diversity Outbred, or heterogeneous stock (HS), mainly because the minor haplotype frequencies (MHF) at loci are about four-fold higher in the BXDs than those typical of crosses made using many parental strains (MHFs of 0.05 to 0.10). In any case, precision much finer than this, while welcome, will often not be critical. The fuzzy functional boundaries of genes and the high density of sequence variants in linkage disequilibrium shifts the burden of proof from pure mapping to functional genomics, comparative analysis of human cohorts, complementary animal models, and direct pharmacological and genetic engineering (Smemo et al., 2014).
Mapping with the BXDs has high power
How many replicates and strains are needed to detect and resolve QTLs? To start with the conclusion—it is almost always better to study small numbers of as many strains as possible (Andreux et al., 2012; Belknap, 1998). Studying as few as 2–4 replicates of each of 100 or more strains may seem counterintuitive to those used to typing knockouts and their wildtype controls, but shallow comprehensive coverage is correct even for traits with low heritability. We have developed an R/shiny application (power.genenetwork.org) that provides guidance on power and resampling for designs using the BXD. When heritability is low, the gain in power by increasing replicates is significant up to an n of 4–6 cases per genometype, but falls off rapidly at heritabilities above 0.5. At heritabilities above 0.5, power is high even using only two replicates (Figure 5). It may still make sense to add replicates for those strains that are in the tails of distributions because these extremes are both potential sampling errors but, if verified, are true research opportunities.
Figure 5: Relations between power, numbers of strains and replicates in BXD, generated using qtlDesign (Sen et al., 2007).
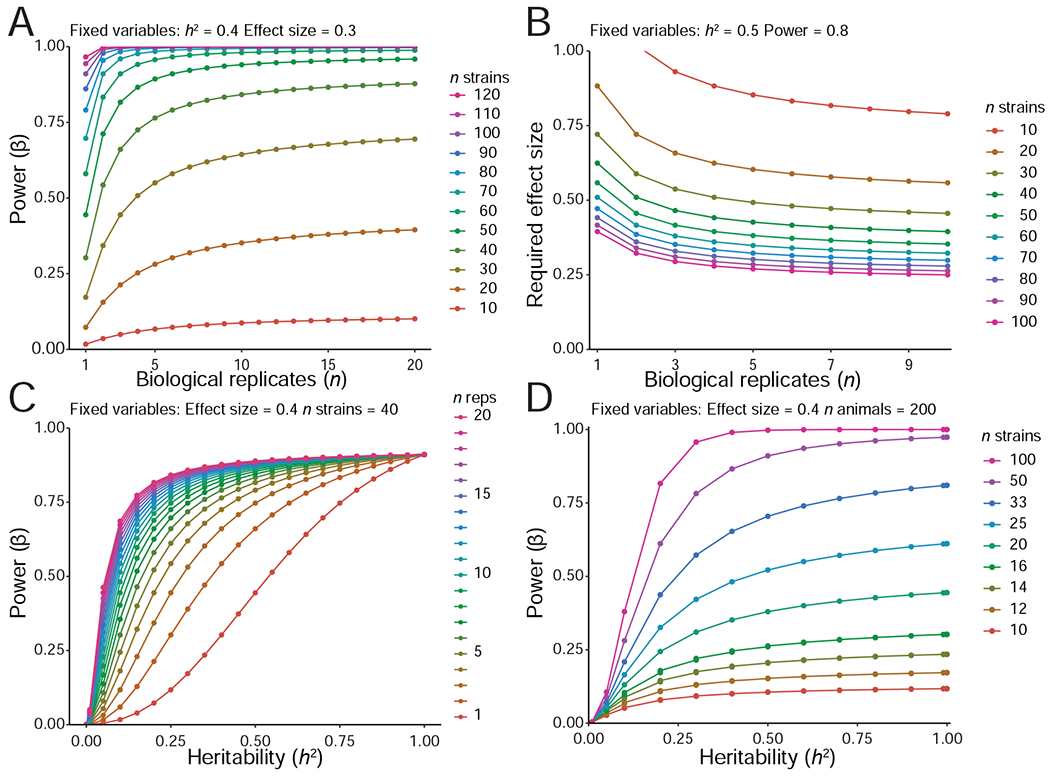
(A) Power to detect a given effect for n replicates and n strains. Locus effect size and heritability constant at 0.3 and 0.4 respectively. More replicates increase power but beyond four replicates gain in power is marginal. (B) Effect size detectable, given n replicates and strains. Heritability and power kept at 0.5 and 0.8. (C) Power to detect a given effect size, dependent upon heritability and n biological replicates. Locus effect size and n strains kept at 0.4 and 40. With very low h2 there is no power, and with lower h2 the gain in power is stronger with increasing number of replicates compared with high h2. Beyond four replicates, the gain in power is marginal. (D) Power to detect a given effect size, dependent upon heritability, with a total of 200 animals. These range from 100 lines with 2 replicates to 10 lines with 20 replicates. In contrast to what one might assume, even for low levels of h2, power always increases with more lines rather than more replicates. Effect size kept at 0.4. Empirical example of power in the BXD can be found in Figure S3.
The effect sizes for loci in BXDs are usually high compared to those of outbred populations and humans. Two factors contribute to higher levels of genetic variance. First, nearly all loci in the BXDs are homozygous. The lack of heterozygotes increases the genetic variance two-fold compared to a matched intercross, and four times as much variance as in a backcross (Belknap, 1998). The drawback is obvious: we cannot detect dominance effects without adding at least a subset of heterozygous loci of the type we can generate easily with sets of F1 progeny and a diallel cross. The second contributor is replicability (Figure 5). When effect size is treated as the proportion of total variance explained by genometypes, this will increase as environmental “noise” is suppressed by replication (Belknap, 1998). To demonstrate this effect we collected all cis-eQTLs (n = 6867) for a brain gene expression data set—55,683 measurements across 129 individuals and 37 BXD strains (GeneNetwork.org ID GN381). We calculated the proportion of variance in expression explained by cis-eQTL markers using either each individual as a unit or each strain mean as a unit. The mean proportion of variance explained is greatly increased by the latter approach (Figure S3), and for variants with a smaller effect sizes (z ≤ 0.2; n = 800) the improvement is 2.5-fold. The average proportion of explained variance is about 1.5-fold higher when using strain means than when using individual values. Across all cis-eQTLs, the effect size using individual as unit is 0.49, whereas using strain means is 0.66—a 56% increase.
Substrains and epochs allow rapid determination of causal variants
Although kinship and epoch structure of the BXDs have been framed as a problem that needs to be solved using linear mixed models, kinship can also provide unique opportunities. In some cases, substrain sets show substantial phenotypic differences and can be used to generate so-called reduced complexity crosses (Bryant et al., 2020). Due to the low genetic variation between substrain sets, causal variants can often be identified rapidly, and these sets can be used as models of disease (Chang et al., 2006; Cook et al., 2006; Rosen et al., 2013).
Each epoch of BXDs has been generated using unique B6 and D2 parents. Although nominally considered identical, these parents will have inherited small numbers of spontaneous mutations (Figure 1A). These de novo sequence variants that accumulated between epochs will lead to expression variation and differences in phenotypes, and due to their very small number, these variants are especially tractable. It becomes straightforward to define the quantitative trait nucleotides causing this higher order variation among epochs using reduced complexity crosses and coisogenic crosses (Heiker et al., 2014; Kirkpatrick and Bryant, 2014; Kumar et al., 2013; Mulligan et al., 2008). Well-defined epoch effect variants include Gpnmb in vision (Lu et al., 2011, 2019); Gabra2 in CNS function (Figure 6; (Hawkins et al., 2020; Mulligan et al., 2019); and Taarl in methamphetamine abuse (Reed et al., 2017; Shi et al., 2016; Stafford et al., 2019).
Figure 6: Impact of epoch-specific mutations in the BXD family.
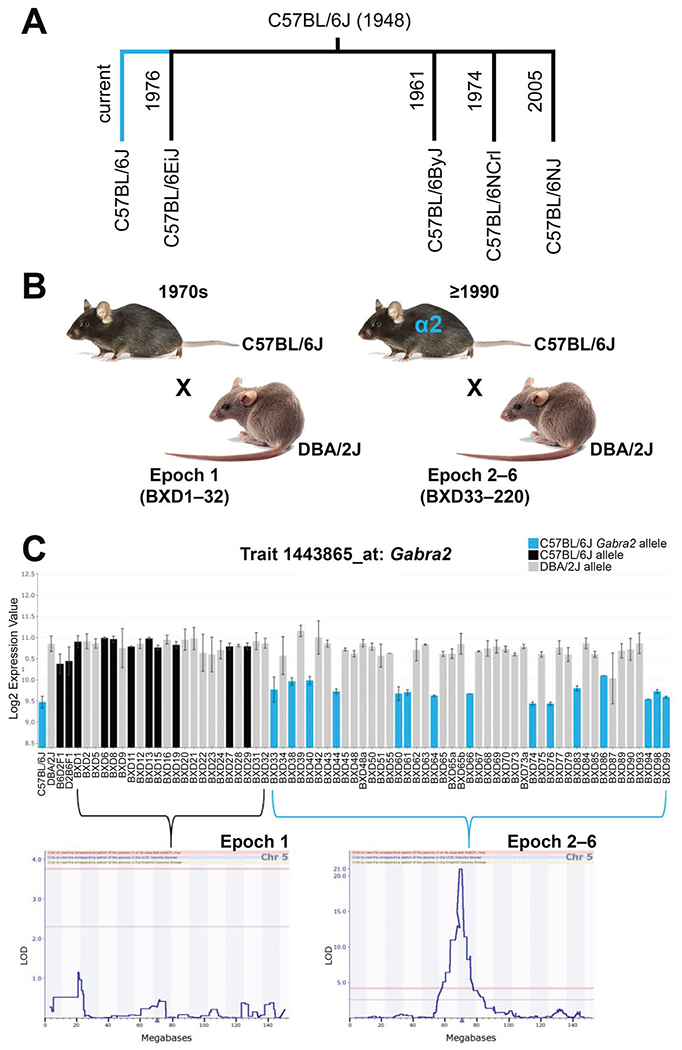
(A) A private mutation (single nucleotide intronic deletion) in the Gamma-aminobutyric acid (GABA-A) receptor, subunit alpha 2 (Gabra2) gene occurred in the C57BL/6J lineage (turquoise line). The spontaneous mutation was fixed in Jackson Laboratory C57BL/6J foundation stock after separation of the C57BL/6EiJ substrain (1976), and (B) after the creation of the first BXD epoch (1970s). All BXD epochs (2–6) created after 1990 are derived from separate crosses between C57BL/6J and DBA/2J mice and segregate for the C57BL/6J Gabra2 mutant allele (shown in turquoise). (C) Inheritance of the C57BL/6J mutant allele (turquoise) results in a reduction of brain Gabra2 expression levels relative to the ancestral C57BL/6J (black) or DBA/2J (gray) allele. Allele color coded based on Gabra2 genotype at rs13478320. Gabra2 mRNA expression values (trait 1443865) are shown for the hippocampus but are replicated in other brain regions and at the protein level. QTL mapping in epoch 1 or later epochs demonstrates how the absence or presence of segregating C57BL/6J mutant Gabra2 alleles controls gene expression.
Discussion
Kinship and genetic drift
The expanded family of BXDs is a well powered resource for both forward and reverse genetic analyses of genome-to-phenome linkage. As this family has grown, relations among individual strains have become complex, requiring the use of linear mixed models (Arends et al., 2010; Sul et al., 2016; Zhou and Stephens, 2014) or nonparametric equivalents such as mixed random forests (Stephan et al., 2015) that account for kinship, epoch, and other cofactors. The family has kinship at several levels. First, there is random fixation and selection of alleles and haplotypes that occurs during inbreeding (Williams et al., 2001). Second, there is strong kinship among AI-derived strains, including BXD43 to BXD102 and BXD160 to BXD186 (Peirce et al., 2004), that has its origins mainly in the 8–14 generations of intercrossing that preceded inbreeding (Darvasi, 1998). Third, there are coisogenic pairs such as BXD29/TyJ and BXD29-Tlr4<lps-2J>/J (Rosen et al., 2013). One member of each pair has at least one highly penetrant spontaneous mutation. Other recombinant congenic strains result from breeding admixture, for example the two BXD48s and the three BXD73s. These small sibships are interesting and useful and have been used to quickly identify causal mutations for immune disfunction in Tlr4 (Cook et al., 2006) and for retinal degeneration in Cep290 (Chang et al., 2006). Finally, there is kinship that results from the epochs of BXD production.
Improved power and precision of mapping BXD phenotypes—a 50 year path of progress
The first set of BXDs were used to map traits to sparse linkage groups, not chromosomes, in the early 1970s (Taylor et al., 1973). While postmillennial mapping resources are obviously far better, there is still much room for improvement. We compared three sequential sets of markers used to map the BXDs—the original genotypes from 2001 (Williams et al., 2001), the set used from 2005 to 2016 (the “classic” set) (Shifman et al., 2006), and the new genotype file released here. This most recent set increases the yield of loci detected at genome-wide significance by about ~25%.
We highlight two key points. First, using the genetic resources we present in this manuscript, it is possible to map quantitative traits with modest LOD scores with good precision, even when using comparatively small numbers of strains (n = 25 to 50; e.g. (Chintalapudi et al., 2017; Houtkooper et al., 2013; Williams et al., 1998). Second, an effective way to transition from QTLs to causal genes and biological processes is to take advantage of complementary resources including other murine mapping resources, efficient in vitro and in vivo screens (Houtkooper et al., 2013; Williams and Auwerx, 2015), and human GWAS data (Ashbrook et al., 2014a, 2015b; Jha et al., 2018b, 2018a; Koutnikova et al., 2009). The brute-force way to improve precision and power is to just phenotype larger numbers of BXD strains. With 140 strains and nearly 20,000 easily made F1 hybrds, traits can be mapped with subcentimorgan precision.
Identification of causal variants is becoming easier thanks to improved genotypes (Rau et al., 2015; Shifman et al., 2006; Simecek et al., 2017; Williams et al., 2001), full genome sequence (Keane et al., 2011; Wang et al., 2016b), powerful omics resources (King et al., 2015; Williams et al., 2016, 2018; Wu et al., 2014), better Bayesian fine-mapping methods (Gonzales and Palmer, 2014; Mulligan et al., 2017; Wakefield, 2008; Wellcome Trust Case Control Consortium et al., 2012; Zhou and Stephens, 2014), and efficient molecular validation methods (Li et al., 2018). The BXD have been used to define specific genes and variants corresponding to well over 20 QTLs, including two tightly linked genes, Iigp2 and Irgb10, for Chlamydia infectivity (Miyairi et al., 2007, 2012), Fmn2 as a master controller of tRNA synthetases in neurons (Mozhui et al., 2008), Ubp1 for blood pressure (Koutnikova et al., 2009), Hc for H5N1 influenza resistance (Boon et al., 2014), Comt as a master controller of neuropharmacological traits (Li et al., 2010), Alpl for hypophosphatasia (Andreux et al., 2012), Mrps5 for longevity (Houtkooper et al., 2013), Bckdhb for maple syrup urine disease, Dhtkd1 for diabetes (Wu et al., 2014), Hp1bp3 for cognitive aging (Neuner et al., 2016), Ahr for locomotor activity (Williams et al., 2014), and Gabra2 and Taar1 for behavioral traits (Mulligan et al., 2019; Reed et al., 2017) .
The BXD family is the largest and the most deeply phenotyped mammalian genetic reference panel
Data sets for the BXD encompass multiple levels—from single molecules to complex behavioral repertoires, and to traits measured with environmental perturbations, including exposure to alcohol and drugs of abuse (Dickson et al., 2019; Mulligan et al., 2018; Théberge et al., 2019; Zhou et al., 2018) , infectious agents (Boon et al., 2014; Chella Krishnan et al., 2016; Russo et al., 2015), dietary modifications (Fleet et al., 2016; Jha et al., 2018b, 2018a; Jones and Jellen, 2017; Reyes Fernandez et al., 2016; Rodrigues et al., 2017), stress (Diessler et al., 2018; Jung et al., 2017) and even as a function of age (Sandoval-Sierra et al., 2020; Williams et al., 2020). This phenome is linked to over 500 publications, including state-of-the-art proteome, metabolome (Williams et al., 2018, 2020), epigenome (Baker et al., 2019; Sandoval-Sierra et al., 2019), and metagenome (Perez-Munoz et al., 2019) data sets available on GeneNetwork.org.
Genetic research is moving rapidly toward causal modeling of health and disease risk and toward the predicted efficacy of prevention and interventions (Hood and Flores, 2012). The size and depth of the BXD phenome makes it a strong foundation for this experimental precision medicine. The genetic architecture of traits can be dissected and causal relations among networks can be explored (Roy et al., 2020). The ability to resample genomes across a stable reference family enables the expansion of data in almost any direction. Improved phenotype ontologies and better access to FAIR data (Wilkinson et al., 2016) enables cross-species translation. This also enables more diverse researcher communities to engage in replicable trans-disciplinary studies of genome-phenome prediction.
Future Directions: Epistasis, pleiotropy, epigenetics, gene-by-environment interactions
Most work using the BXD family has had the simple goal of defining single gene variants and processes that contribute to heritable differences in disease risk. But we need to tackle a more important problem—the complex interplay among sets of variants, constellations of phenotypes, and different treatments and environments (Ashbrook et al., 2014b; Mulligan and Williams, 2015; Williams and Auwerx, 2015; Williams et al., 2016). The BXDs are now sufficiently large to evaluate predictive models of biological processes of these more complex types (Li et al., 2008; Miyairi et al., 2012; Wang et al., 2016b) and to map epistatic interactions. But power to fit complex models can be greatly amplified by crossing BXD strains to generate a diallel cross of up to 19,460 F1 progeny. All isogenic BXD F1 lines are replicable and have entirely defined genomes. They have the advantage of being non-inbred and heterozygous. A subset of this massive diallel cross (DAX) can be generated efficiently from a central repository for cost-effective cell-based assays, and for matched in vivo predictive validation. Any part of this DAX can be used to study parent-of-origin and sex chromosome effects (Ashbrook and Hager, 2013). It is also practical to cross BXDs to other lines—for example to the humanized 5XFAD line used in Alzheimer’s disease research by Kaczorowski and colleagues (Neuner et al., 2019a, 2019b). By crossing humanized or genetically engineered lines on a single genetic background to a diverse but defined subset of BXDs, variants that modify traits—such as memory loss in the 5XFAD model—can be mapped and can provide valuable information on disease process and outcomes. Kaczorowski and colleagues have also evaluated the efficacy of reverse translation from human to mouse (Neuner et al., 2019a). They generated a polygenic genetic risk score using 21 human genes that increase Alzheimer’s risk and showed that allele dosage was significantly associated with cognitive outcomes in mice. This demonstrates that naturally occurring variation in these networks may have overlapping effects in mouse and humans.
In humans tens-of-thousands of variants have now been mapped to haplotype blocks of under 100 Kb (Huan et al., 2015; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Wood et al., 2014). We need to revamp murine genetic resources to contribute more effectively, and in complementary ways, in an era flooded with GWAS hits. How are rodent resources best repositioned to help deliver on the still unmet and much more integrative promises of predictive genetics and personalized precision health care? The short answer is that we need large genetically complex resources with matched multiscalar and multisystems phenome data. We need resources that define and test progressively more sophisticated computational models as a function of genometype, stage, and exposure. We need replicable populations with the same intrinsic genetic complexity and admixture as humans but without the challenges of clinical research—high cost, marginal compliance and control, confidentiality, and of most importance, ethical constraints on designs and interventions. Rodent populations are ideally positioned to be replicable and extensible testbeds with which to define the power, accuracy, and limits of precision health care.
Conclusions
Systems genetics using rodent models has been revitalized over the last decade thanks to several resources, including the BXD family (Peirce et al., 2004), the Hybrid Mouse Diversity Panel (Bennett et al., 2015; Ghazalpour et al., 2012), and the Collaborative Cross (Churchill et al., 2004; Morgan and Welsh, 2015; Schughart and Williams, 2017; Valdar et al., 2006). The main limitation has been relatively modest mapping power and precision—a simple problem caused by small numbers of strains. With 140 strains now readily available, and expandable to 19,460 isogenic F1s, the extended BXD family has overcome this problem. All phenome data is multiplicatively useful, and like a fine vintage, old data gets better with age.
STAR Methods
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by, the corresponding author David Ashbrook (dashbrook@UTHSC.edu).
Materials Availability
The BXD strains reported in this study are available from the JAX. JAX Stock No (RRIDs) can be found in the Key resources table and in Table S1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| GeneNetwork | Genenetwork.org | RRID:SCR_002388 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BXD1/TyJ | JAX | RRID:000036 |
| Mouse: BXD2/TyJ | JAX | RRID:000075 |
| Mouse: BXD3 | JAX | |
| Mouse: BXD4 | JAX | |
| Mouse: BXD5/TyJ | JAX | RRID:000037 |
| Mouse: BXD6/TyJ | JAX | RRID:000007 |
| Mouse: BXD7 | JAX | |
| Mouse: BXD8/TyJ | JAX | RRID:000084 |
| Mouse: BXD9/TyJ | JAX | RRID:000105 |
| Mouse: BXD10 | JAX | |
| Mouse: BXD11/TyJ | JAX | RRID:000012 |
| Mouse: BXD12/TyJ | JAX | RRID:000045 |
| Mouse: BXD13/TyJ | JAX | RRID:000040 |
| Mouse: BXD14/TyJ | JAX | RRID:000329 |
| Mouse: BXD15/TyJ | JAX | RRID:000095 |
| Mouse: BXD16/TyJ | JAX | RRID:000013 |
| Mouse: BXD17 | JAX | |
| Mouse: BXD18/TyJ | JAX | RRID:000015 |
| Mouse: BXD19/TyJ | JAX | RRID:000010 |
| Mouse: BXD20/TyJ | JAX | RRID:000330 |
| Mouse: BXD21/TyJ | JAX | RRID:000077 |
| Mouse: BXD22/TyJ | JAX | RRID:000043 |
| Mouse: BXD23/TyJ | JAX | RRID:000098 |
| Mouse: BXD24/TyJ-Cep290rd16/J | JAX | RRID:000031 | rd16 |
| Mouse: BXD24/TyJ | JAX | RRID:005243 |
| Mouse: BXD25/TyJ | JAX | RRID:000081 |
| Mouse: BXD26 | JAX | |
| Mouse: BXD27/TyJ | JAX | RRID:000041 |
| Mouse: BXD28/TyJ | JAX | RRID:000047 |
| Mouse: BXD29/Ty | JAX | RRID:010981 |
| Mouse: BXD29-Tlr4lps-2J/J | JAX | RRID:000029 | defective lipopolysaccharide response, 2J |
| Mouse: BXD30/TyJ | JAX | |
| Mouse: BXD31/TyJ | JAX | RRID:000083 |
| Mouse: BXD32/TyJ | JAX | RRID:000078 |
| Mouse: BXD32/TyJ-Galctwi-5J/J | JAX | RRID:003613 | twitcher 5 Jackson |
| Mouse: BXD33/TyJ | JAX | RRID:003222 |
| Mouse: BXD34/TyJ | JAX | RRID:003223 |
| Mouse: BXD35/Ty | JAX | |
| Mouse: BXD36/TyJ | JAX | RRID:003225 |
| Mouse: BXD37/Ty | JAX | |
| Mouse: BXD38/TyJ | JAX | RRID:003227 |
| Mouse: BXD39/TyJ | JAX | RRID:003228 |
| Mouse: BXD40/TyJ | JAX | RRID:003229 |
| Mouse: BXD41/Ty | JAX | |
| Mouse: BXD42/TyJ | JAX | RRID:003230 |
| Mouse: BXD43/RwwJ | JAX | RRID:007093 |
| Mouse: BXD44/RwwJ | JAX | RRID:007094 |
| Mouse: BXD45/RwwJ | JAX | RRID:007096 |
| Mouse: BXD46 | JAX | |
| Mouse: BXD47 | JAX | |
| Mouse: BXD48/RwwJ | JAX | RRID:007097 |
| Mouse: BXD48a/RwwJ | JAX | RRID:007139 |
| Mouse: BXD49/RwwJ | JAX | RRID:007098 |
| Mouse: BXD50/RwwJ | JAX | RRID:007099 |
| Mouse: BXD51/RwwJ | JAX | RRID:007100 |
| Mouse: BXD52 | UTHSC | |
| Mouse: BXD53/2RwwJ | JAX | RRID:017749 |
| Mouse: BXD54 | UTHSC | |
| Mouse: BXD55/RwwJ | JAX | RRID:007103 |
| Mouse: BXD56/RwwJ | JAX | RRID:007104 |
| Mouse: BXD57 | UTHSC | |
| Mouse: BXD58 | UTHSC | |
| Mouse: BXD59 | UTHSC | |
| Mouse: BXD60/RwwJ | JAX | RRID:007105 |
| Mouse: BXD61/RwwJ | JAX | RRID:007106 |
| Mouse: BXD62/RwwJ | JAX | RRID:007107 |
| Mouse: BXD63/RwwJ | JAX | RRID:007108 |
| Mouse: BXD64/RwwJ | JAX | RRID:007109 |
| Mouse: BXD65/RwwJ | JAX | RRID:007110 |
| Mouse: BXD65a/RwwJ | JAX | RRID:007140 |
| Mouse: BXD65b/RwwJ | JAX | RRID:009677 |
| Mouse: BXD66/RwwJ | JAX | RRID:007111 |
| Mouse: BXD67/RwwJ | JAX | RRID:007112 |
| Mouse: BXD68/RwwJ | JAX | RRID:007113 |
| Mouse: BXD69/RwwJ | JAX | RRID:007114 |
| Mouse: BXD70/RwwJ | JAX | RRID:007115 |
| Mouse: BXD71/RwwJ | JAX | RRID:007116 |
| Mouse: BXD72 | UTHSC | |
| Mouse: BXD73/RwwJ | JAX | RRID:007117 |
| Mouse: BXD73a/RwwJ | JAX | RRID:007124 |
| Mouse: BXD73b/RwwJ | JAX | RRID:007146 |
| Mouse: BXD74/RwwJ | JAX | RRID:007118 |
| Mouse: BXD75/RwwJ | JAX | RRID:007119 |
| Mouse: BXD76 | UTHSC | |
| Mouse: BXD77/RwwJ | JAX | RRID:007121 |
| Mouse: BXD78/2RwwJ | JAX | RRID:024029 |
| Mouse: BXD79/RwwJ | JAX | RRID:007123 |
| Mouse: BXD80 | UTHSC | |
| Mouse: BXD81/RwwJ | JAX | RRID:007125 |
| Mouse: BXD82 | UTHSC | |
| Mouse: BXD83/RwwJ | JAX | RRID:007126 |
| Mouse: BXD84/RwwJ | JAX | RRID:007127 |
| Mouse: BXD85/RwwJ | JAX | RRID:007128 |
| Mouse: BXD86/RwwJ | JAX | RRID:007129 |
| Mouse: BXD87/RwwJ | JAX | RRID:007130 |
| Mouse: BXD88/2RwwJ | JAX | RRID:017750 |
| Mouse: BXD89/RwwJ | UTHSC | |
| Mouse: BXD90/RwwJ | JAX | RRID:007133 |
| Mouse: BXD91/2RwwJ | JAX | RRID:017751 |
| Mouse: BXD92 | UTHSC | |
| Mouse: BXD93 | UTHSC | |
| Mouse: BXD94 | UTHSC | |
| Mouse: BXD95/RwwJ | JAX | RRID:007138 |
| Mouse: BXD96 | UTHSC | |
| Mouse: BXD97 | UTHSC | |
| Mouse: BXD98/RwwJ | JAX | RRID:007141 |
| Mouse: BXD99/RwwJ | JAX | RRID:007142 |
| Mouse: BXD100/RwwJ | JAX | RRID:007143 |
| Mouse: BXD101/RwwJ | JAX | RRID:007144 |
| Mouse: BXD102/RwwJ | JAX | RRID:007145 |
| Mouse: BXD103 | UTHSC | |
| Mouse: BXD104 | UTHSC | |
| Mouse: BXD105 | UTHSC | |
| Mouse: BXD106 | UTHSC | |
| Mouse: BXD107 | UTHSC | |
| Mouse: BXD108 | UTHSC | |
| Mouse: BXD109 | UTHSC | |
| Mouse: BXD110 | UTHSC | |
| Mouse: BXD111/RwwJ | JAX | RRID:030872 |
| Mouse: BXD112 | UTHSC | |
| Mouse: BXD113/RwwJ | JAX | RRID:024030 |
| Mouse: BXD114 | ||
| Mouse: BXD115 | UTHSC | |
| Mouse: BXD116 | UTHSC | |
| Mouse: BXD117 | UTHSC | |
| Mouse: BXD118 | UTHSC | |
| Mouse: BXD119 | UTHSC | |
| Mouse: BXD120 | UTHSC | |
| Mouse: BXD121 | UTHSC | |
| Mouse: BXD122/RwwJ | JAX | RRID:030873 |
| Mouse: BXD122a | UTHSC | |
| Mouse: BXD123/RwwJ | JAX | RRID:025974 |
| Mouse: BXD124/RwwJ | JAX | RRID:025975 |
| Mouse: BXD125/RwwJ | JAX | RRID:024031 |
| Mouse: BXD126 | UTHSC | |
| Mouse: BXD127 | ||
| Mouse: BXD128/RwwJ | JAX | RRID:030874 |
| Mouse: BXD128a/RwwJ | JAX | RRID:030875 |
| Mouse: BXD129 | UTHSC | |
| Mouse: BXD130 | UTHSC | |
| Mouse: BXD131 | UTHSC | |
| Mouse: BXD132 | UTHSC | |
| Mouse: BXD133 | UTHSC | |
| Mouse: BXD134 | UTHSC | |
| Mouse: BXD135 | UTHSC | |
| Mouse: BXD136 | UTHSC | |
| Mouse: BXD137 | UTHSC | |
| Mouse: BXD138 | UTHSC | |
| Mouse: BXD139 | UTHSC | |
| Mouse: BXD140 | UTHSC | |
| Mouse: BXD141/RwwJ | JAX | RRID:029865 |
| Mouse: BXD142 | UTHSC | |
| Mouse: BXD143 | UTHSC | |
| Mouse: BXD144/RwwJ | JAX | RRID:025984 |
| Mouse: BXD145 | UTHSC | |
| Mouse: BXD146 | UTHSC | |
| Mouse: BXD147 | UTHSC | |
| Mouse: BXD148 | UTHSC | |
| Mouse: BXD149 | UTHSC | |
| Mouse: BXD150/RwwJ | JAX | RRID:029867 |
| Mouse: BXD151/RwwJ | JAX | RRID:029868 |
| Mouse: BXD152/RwwJ | JAX | RRID:029869 |
| Mouse: BXD153 | UTHSC | |
| Mouse: BXD154/RwwJ | JAX | RRID:025976 |
| Mouse: BXD155 | UTHSC | |
| Mouse: BXD156/RwwJ | JAX | RRID:029871 |
| Mouse: BXD156a | UTHSC | |
| Mouse: BXD157/RwwJ | JAX | RRID:029872 |
| Mouse: BXD158 | UTHSC | |
| Mouse: BXD159 | UTHSC | |
| Mouse: BXD160/RwwJ | JAX | RRID:029873 |
| Mouse: BXD161/RwwJ | JAX | RRID:025985 |
| Mouse: BXD162 | UTHSC | |
| Mouse: BXD163 | UTHSC | |
| Mouse: BXD164 | UTHSC | |
| Mouse: BXD165 | UTHSC | |
| Mouse: BXD166 | UTHSC | |
| Mouse: BXD167 | UTHSC | |
| Mouse: BXD168/RwwJ | JAX | RRID:029874 |
| Mouse: BXD169/RwwJ | JAX | RRID:029876 |
| Mouse: BXD170/RwwJ | JAX | RRID:029878 |
| Mouse: BXD171/RwwJ | JAX | RRID:029879 |
| Mouse: BXD172/RwwJ | JAX | RRID:029880 |
| Mouse: BXD173 | UTHSC | |
| Mouse: BXD174 | UTHSC | |
| Mouse: BXD175 | UTHSC | |
| Mouse: BXD176 | UTHSC | |
| Mouse: BXD177/RwwJ | JAX | RRID:029881 |
| Mouse: BXD178/RwwJ | JAX | RRID:029882 |
| Mouse: BXD179 | UTHSC | |
| Mouse: BXD180/RwwJ | JAX | RRID:029883 |
| Mouse: BXD181 | UTHSC | |
| Mouse: BXD182 | UTHSC | |
| Mouse: BXD183 | UTHSC | |
| Mouse: BXD184/RwwJ | JAX | RRID:029884 |
| Mouse: BXD185 | UTHSC | |
| Mouse: BXD186/RwwJ | JAX | RRID:029885 |
| Mouse: BXD187/RwwJ | JAX | RRID:031974 |
| Mouse: BXD188 | UTHSC | |
| Mouse: BXD189 | UTHSC | |
| Mouse: BXD190/RwwJ | JAX | RRID:030876 |
| Mouse: BXD191/RwwJ | JAX | RRID:030877 |
| Mouse: BXD192 | UTHSC | |
| Mouse: BXD193 | UTHSC | |
| Mouse: BXD194/RwwJ | JAX | RRID:030878 |
| Mouse: BXD195/RwwJ | JAX | RRID:030879 |
| Mouse: BXD196 | UTHSC | |
| Mouse: BXD197/RwwJ | JAX | RRID:030880 |
| Mouse: BXD198 | UTHSC | |
| Mouse: BXD199/RwwJ | JAX | RRID:030881 |
| Mouse: BXD200 | UTHSC | |
| Mouse: BXD201 | UTHSC | |
| Mouse: BXD202/RwwJ | JAX | RRID:031975 |
| Mouse: BXD203 | UTHSC | |
| Mouse: BXD204 | UTHSC | |
| Mouse: BXD205 | UTHSC | |
| Mouse: BXD206 | UTHSC | |
| Mouse: BXD207 | UTHSC | |
| Mouse: BXD208 | UTHSC | |
| Mouse: BXD209 | UTHSC | |
| Mouse: BXD210/RwwJ | JAX | RRID:031976 |
| Mouse: BXD211 | UTHSC | |
| Mouse: BXD212 | UTHSC | |
| Mouse: BXD213 | UTHSC | |
| Mouse: BXD214/RwwJ | JAX | RRID:030882 |
| Mouse: BXD215/RwwJ | JAX | RRID:031977 |
| Mouse: BXD216/RwwJ | JAX | RRID:031978 |
| Mouse: BXD217 | UTHSC | |
| Mouse: BXD218 | UTHSC | |
| Mouse: BXD219 | UTHSC | |
| Mouse: BXD220 | UTHSC | |
| Mouse: BXD221 | UTHSC | |
| Mouse: BXD222 | UTHSC | |
| Mouse: BXD223 | UTHSC | |
| Mouse: BXD224 | UTHSC | |
| Mouse: BXD225 | UTHSC | |
| Mouse: BXD226 | UTHSC | |
| Mouse: BXD227 | UTHSC | |
| Mouse: BXD228 | UTHSC | |
| Software and Algorithms | ||
| GeneNetwork | Genenetwork.org | RRID:SCR_002388 |
| BXDtools | https://github.com/DannyArends/BXDtools | |
| BXD power calculator app | http://power.genenetwork.org/; this paper | https://github.com/Dashbrook/BXD_power_calculator_app |
| R/qtl2 | Broman et al., 2019 | RRID:SCR_018181; https://kbroman.org/qtl2/ |
Data and Software Availability
This paper analyzes existing, publicly available data. These data sets’ accession numbers are provided in the Key Resource Table, and throughout the manuscript. Genotype files can be found at http://www.genenetwork.org/webqtl/main.py?FormID=sharinginfo&GN_AccessionId=600. GeneNetwork.org original code is publicly available at https://qithub.com/genenetwork/genenetwork2 and https://github.com/genenetwork/genenetwork1.
The scripts used to generate the figures reported in this paper are available in the BXDtools package (https://github.com/DannyArends/BXDtools) or using ggplot2 (version 3.3.2; https://cran.r-project.org/web/packages/ggplot2/index.html) and their use is described in the STAR Methods. Code for computing marker positions and visualization of recombinations is available in the BXDtools package (https://github.com/DannyArends/BXDtools).
Power estimates can be made using http://power.genenetwork.org/, and code is at https://github.com/Dashbrook/BXD_power_calculator_app.
The R/qtl2 software can be found here https://kbroman.org/qtl2/, and its use is described in the STAR Methods.
Experimental Model and Subject Details
Mice
Between 2009 and 2010 we initiated 74 BXD strains (BXD104 to BXD186). We initiated another 34 strains in 2013 (BXD187 to BXD220). We used both conventional F2 intercrosses (n = 88) and AI progeny (n = 20) to make the 108 new lines. BXD160 through BXD186 were derived from unique matings of AI progeny gifted us by Dr. Abraham Palmer at G8 and G9 late in 2010 (Figure 1, Figure S1, Table S1).
In cases of low reproductive fitness, we often attempted to rescue lines by outcrossing young male BXDs to C57BL/6J females followed by three or more sequential backcrosses (N3) to the RI male to produce progeny enriched for the BXD genome. In more recent cases, pairs of at-risk strains were crossed to produce RIX progeny (Tsaih et al., 2005; Williams et al., 2001), which were then inbred by sibling mating. BXD221 to BXD228 are RIX-derived strains of this type (Figure S1, Table S1).
Unless otherwise specified, all animals used in this study were raised in a closed-barrier pathogen-free vivarium at the University of Tennessee Health Science Center. All BXD strains are available under a standard material transfer agreement; the most important limitation being that they cannot be sold or distributed without approval of the Jackson Laboratory or UTHSC. Availability information on all BXD lines can be found in Table S1.
Method details
Genetic map construction and genotype error correction
Individuals from each of the 198 BXD lines were genotyped in late 2011 and in late 2015 using the Neogen/GeneSeek MUGA or GigaMUGA arrays. These were combined with previous genotypes generated using Affymetrix and MUGA platforms. Unknown genotypes were imputed as B (C57BL/6J-like) or D (DBA/2J-like) or were called as H (heterozygous) if the genotype was uncertain.
The full data set contains 37,000 markers and was optimized for mapping efficiency by excluding markers showing identical strain distribution patterns. Markers that flank sites of recombination were retained. As a result, strain distribution patterns are often defined by proximal-distal marker pairs. Whenever possible, we verified and updated genotypes of the original BXD strains (BXD1 - BXD102) to reflect those of stock available from the Jackson Laboratory (Rau et al., 2015) and from Petr Simecek and Gary Churchill; http://cgd.jax.org/datasets/diversityarray.shtml).
Genotypes for all strains were smoothed and curated to remove highly implausible (double) recombination events, e.g. unsupported genotypes (singletons) that introduced two recombination events over less than 100 kb (Williams et al., 2001). In general, we imputed unknown or heterozygous genotypes on the basis of flanking markers. Undefined genotypes between recombinations were coded as heterozygous, and telomere genotypes were imputed using the closest flanking marker.
In the newer BXD strains many regions are still heterozygous. Generated genotypes using standard platforms still show regions of low marker density and a high frequency of recombinations. These regions of low marker density were filled with imputed genotypes using cis-eQTLs of genes in the problematic intervals. Microsatellites and cis-eQTL genotypes were generated by the Williams/Lu laboratory.
The assembled and error-checked genotype file includes 7,324 markers for 191 independent strains, and 7 substrains, has been available since January 2017 at http://www.genenetwork.org/genotypes/BXD.geno and is the default genetic map used when QTL mapping on GeneNetwork (http://www.genenetwork.org).
Genome-wide visualization of recombinations
For each chromosome we compute the number of observed recombinations per strain from the start of the chromosome (nRecS), ignoring the heterozygous and unknown genotypes. We then visualize this by plotting the number of recombinations for each BXD strain per chromosome using a color gradient. Strain numbers are used for the ordering on the X-axis, and horizontal lines are added to visually separate the different epochs and annotate the epochs on the X-axis.
Quantification and Statistical Analysis
Computing centimorgan positions
For each chromosome we compute the number of observed recombinations from one position to the next, ignoring the heterozygous and unknown genotypes (nRecP). We then compute the recombination fraction (R) by dividing the number of recombinations observed (nRecP) by the population size used to generate the genetic map. The recombination fractions (R) are then deflated using the formula:
, which is the inverse of the formula R = 4r/(1+6r) for a recombinant inbred line by sibling mating. We then use the function imf.cf available in the R package R/qtl (Arends et al., 2010; Broman et al., 2003) to convert from centiMorgan using the Carter-Falconer map function (Rédei, 2008a). Positions obtained start at 0 on each chromosome, requiring all positions to be shifted so that the first marker on the map matches the known centiMorgan position of the first marker on the genetic map. The old genetic map and the mouse map converter (http://cgd.jax.org/mousemapconverter/) were used to obtain centiMorgan positions for the first marker on a chromosome, all position on a chromosome are shifted to align the genetic map to the known starting positions of the markers. Code for computing marker positions is available in the BXDtools package (https://github.com/DannyArends/BXDtools), and positions can be recomputed using different map functions when required (e.g. Haldane (Rédei, 2008b), Kosambi (Rédei, 2008c), Morgan (Rédei, 2008d)).
Comparing genotype files
Over the past four decades many genotype files have been used for QTL mapping of the BXDs. The first carefully vetted genome-wide map (which we call the ‘original’) was generated in 2001, and mainly made up of 1,578 microsatellite markers (Wang et al., 2003; Williams et al., 2001). The second ‘classic’ map was used between 2005–2016 and contained 3,811 SNPs from various array platforms (Shifman et al., 2006). The current third-generation file is based on 7,321 informative SNPs described above.
For the 2001 markers, Mouse Genome Informatics was used to update 1419 microsatellites to the current assembly. This left 159 orphaned markers without mm10 positions. We used the NCBI Probe database (www.ncbi.nlm.nih.gov/probe/) to search for these 159 markers, and extracted primer sequence for 107. We ran Primer-BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi) to determine where in the mm10 genome they would amplify. For the remaining 52 markers, we used BLAST to align the target sequence to identify the mm10 position. This left 11 markers which Primer-BLAST either predicted a different chromosome would be amplified, or which did not have any mm10 genomic targets.
These ‘original’ genotypes only exist for the first 36 BXD strains (BXD1–42), and so the remaining strains were imputed (BXD24a and BXD 43+), using the genotype at the microsatellite’s position in the ‘current’ post-2017 genotyping file. To determine our power to detect QTLs, we ran mapping using QTL Reaper for the 7562 phenotypes collected for the BXD, and the number of phenotypes which had a LOD greater than three threshold values: 2.2, 3.6 and 5.4. These correspond to suggestive, significant, and highly significant LOD values. The larger the number of phenotypes above these thresholds, the greater our power to detect QTLs.
Precision and Resolution
To estimate the empirical precision of mapping across the genome we have extracted data from 40 large-scale genetic studies of gene expression of the BXD family. These studies used between 21 and 79 strains with variable numbers of within-strain replicates over 27 tissues. We defined eQTLs that map within ±20 Mb of the gene associated with the transcripťs measurement as cis-acting expression QTLs (cis-eQTLs). Unlike a standard F2 intercross, this ±20 Mb window is roughly equivalent to a recombination distance of ±40 cM in the highly recombinant BXD family, and the statistical association between markers this far apart is generally quite low (r2 less than 0.3).
Precision was estimated using the offset between the location of the gene (using the 5’ end of the probe or probe set as a reference point), and the location of the SNP with the highest LOD score. Again, this is conservative, as the causal variant is unlikely to be exactly at the 5’ end of the gene. There are often two or more neighboring SNPs with equally high LOD scores and we simply take the most proximal marker to compute offset. The light blue local regression (LOESS) smoother curve (Cleveland, 1979) was computed using a window with a size of 0.333% of the genome. Smoothing was carried out and data was plotted using the ggplot2 R package.
QTL mapping
QTL mapping using GeneNetwork has been described in detail elsewhere (Mulligan et al., 2017). However, in brief, quantitative trait loci (QTLs) are segments of the genome affecting a particular phenotype (Falconer and Mackay, 1996). QTL mapping, identifying QTLs to explain the genetic basis of complex traits, relies on being able to make correlations between genetic markers and phenotypic traits in a population. Individuals are scored for their phenotype for a particular trait, and their genotype at a marker. If there is a difference in mean phenotype between those individuals with one genotype compared with the other than we can infer that there is a QTL linked to that marker. If there is no difference between the means we can conclude that the locus does not influence the phenotype in that population (Falconer and Mackay, 1996; Miles and Wayne, 2008).
Due to the very high density of markers, the mapping algorithm used to map BXD data sets has been modified and is a mixture of simple marker regression, linear interpolation, and standard Haley-Knott interval mapping (Haley and Knott, 1992). When two adjacent markers have identical strain distribution patterns, they will have identical linkage statistics, as will the entire interval between these two markers (assuming complete and error-free haplotype data for all strains). On a physical map the LOD and the additive effect values will, therefore, be constant over this physical interval. Between neighboring markers that have different strain distribution patterns and that are separated by 1 cM or more, we use a conventional interval mapping method (Haley-Knott) combined with a Haldane estimate of genetic distance. When the interval is less than 1 cM, we simply interpolate linearly between markers based on a physical scale between those markers. The result of this mixture mapping algorithm is a linkage map of a trait that has an unusual profile that is particularly striking on a physical (Mb) scale, with many plateaus, abrupt linear transitions between plateaus, and a few regions with the standard graceful curves typical of interval maps.
Prior to the 2017 release of the genotypes described in this manuscript interval mapping in GeneNetwork relied on 3,795 informative SNP markers across all autosomes and the X chromosome (Wang et al., 2003). These markers were generated using the MUGA array in 2011, along with earlier generated genotypes on the Affymetrix and Illumina platforms (Shifman et al., 2006). As described above, loci are identified in GeneNetwork by the computation of a likelihood ratio statistic score (LRS), and significance was determined using at least 5,000 permutations of the phenotype data.
Updated QTL mapping methods, such as R/qtl (Broman et al., 2003, 2019), Multiple QTL mapping (Arends et al., 2010), GEMMA (Zhou and Stephens, 2012) and pyLMM (Sul et al., 2016), have been implemented on the GeneNetwork site (Sloan et al., 2016). The LMM based methods for kinship between BXD strains by computing a genetic kinship matrix given specific strains used in an analysis, allowing for precise kinship correction and improved QTL mapping performance when mapping any of the BXD phenotypes in the GeneNetwork database.
R/qtl2 (Broman et al., 2003, 2019) was also used to carry out QTL mapping, independently, in the R environment. The functions scan1 and scan1perm were used to calculate linkage for all phenotypes, and permutation thresholds. For Haley-Knott mapping no kinship correction was used, and for LMM the calc_kinship function was used, with leave one chromosome out (LOCO).
Power calculation
Power calculations were carried out based on the method of Sen and colleagues (Sen et al., 2007) and implemented in the R program qtlDesign based on the H2RIX defined by Belknap (Belknap, 1998), which we renamed as it is applicable to any isogenic strain, not just recombinant inbreds. The Detectable function was used, to determine the power available with an RI design to detect a locus of a given effect size.
For calculation of the , four variables need to be known: heritability (h2), the number of within-strain replicates (biological replicates), the number of strains used, and the locus effect size (the proportion of total genetic variance explained by the locus). To calculate h2, the genomic variance (gen.var) was kept constant at 1, and the environmental variance (env.var) was varied to produce h2 values ranging between 0 and 1. The number of biological replicates and the number of strains are self-explanatory, and were capped at 10 and 150 respectively, since >10 replicates produced a marginal increase in power, and because there are a maximum of 150 BXD lines.
The locus effect size is the amount of the variance which is due to genetics (gen.var) explained by a single QTL. That is, a value of 0.2 would mean that a QTL explains 20% of the genetic variance, and a value of 1.0 indicates a Mendelian locus (one QTL which explains all of the genetic variance).
The power given is the ability of the experiment to correctly detect a true positive QTL, given the other values above. The BXD power app can be found at http://power.genenetwork.org/ and uses the Shiny R package (https://CRAN.R-proiect.orq/packaqe=shiny).
Supplementary Material
Table S1: Information on the nomenclature, availability, derivation, and genotyping of all BXD strains (BXD1-BXD228), related to Figure 1. Shows alternative names that have been used for BXD family members, details of when breeding was started, when production of these strains began at the Jackson Laboratory (JAX), the stock number (which is also the RRID number) and availability from JAX, the epoch, method of derivation, the origin of the mitochondrial genome, and platforms that each strain has been genotyped on, and the generation number if known.
Table S2: LOD values for 3298 phenotypes, calculated using Haley-Knott (H-K) equations or linear mixed models (LMMs), related to Figure 2. For each phenotype and model combination, the significance threshold is shown (calculated by 5000 permutations), the peak LOD score, and the difference between the threshold and the peak. For each phenotype, the differences in threshold between the H-K and LMM equations are shown, as well as the count of strains in epoch 1, epochs 1 and 2, epoch 3, and epochs higher than 3.
Table S3: List of traits in the BXD phenome on GeneNetwork, related to Figures 1 and S4. An annotated list of BXD publicly available phenotypes, available on GeneNetwork. The table shows the GeneNetwork trait ID, the phenotype category, the full phenotype description, the authors who produced the phenotype, the year of publication (or year of entry into GN if unpublished), the PubMed ID (if published), and the peak LRS score and the peak’s position in the genome.
Highlights.
There are now 140 fully inbred BXD strains available, with high-quality genotypes
More strains, new genotypes, and new models have improved power and precision
We have high power even for traits with low heritability or small effect sizes
A phenome of >100 omics datasets and >7500 classic phenotypes is freely available
Acknowledgments
Thanks to Dr. Abraham Palmer for providing B6D2 advanced intercross progeny (G8 and G9) to make BXD160 through BXD186. We thank Drs. Gerald McClearn and Lisa Tarantino for 60 eighth-generation (G8) AI progeny from Pennsylvania State University contributed to epoch 3. We thank Dr. Benjamin A. Taylor for initiating the BXD and for continued support and encouragement. We thank Dr. Saunak Sen for consultation on power calculations and qtlDesign. We thank members of the EPFL Center for Phenogenomics in particular Drs. Xavier Warot and Emilie Gesina for sharing data from their BXD colony, and Erik Soehnel from Scionics for assistance in obtaining the full breeding statistics.
The UTHSC Center for Integrative and Translational Genomics (CITG) has supported production of the BXD colony at UTHSC and will continue to support this colony for the duration of the grant. The CITG also provides generous support for computer hardware and programming associated with GeneNetwork, and our Galaxy and UCSC Genome Browser instances.
We thank the support of the UT Center for Integrative and Translational Genomics, and funds from the UT-ORNL Governor’s Chair, NIDA grant P30DA044223, NIAAA U01 AA013499 and U01 AA016662 for the work at UTHSC. Work in the Auwerx lab on the BXDs is supported by the Ecole Polytechnique Fédérale de Lausanne, the ERC (AdG-787702), the SNSF (310030B-160318), the AgingX program of the Swiss Initiative for Systems Biology (RTD 2013/153), and the NIH (R01AG043930). The BXD Resource at the Jackson Laboratory is supported by NIH P40 OD011102 awarded to Dr. Cathleen M. Lutz.
The data sets generated and/or analyzed during the current study are available in the GeneNetwork repository, https://www.genenetwork.org/.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Andreux PA, Williams EG, Koutnikova H, Houtkooper RH, Champy M-F, Henry H, Schoonjans K, Williams RW, and Auwerx J (2012). Systems genetics of metabolism: the use of the BXD murine reference panel for multiscalar integration of traits. Cell 150, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arends D, Prins P, Jansen RC, and Broman KW (2010). R/qtl: high-throughput multiple QTL mapping. Bioinformatics 26, 2990–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, and Hager R (2013). Empirical testing of hypotheses about the evolution of genomic imprinting in mammals. Front Neuroanat 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Williams RW, Lu L, Stein JL, Hibar DP, Nichols TE, Medland SE, Thompson PM, and Hager R (2014a). Joint genetic analysis of hippocampal size in mouse and human identifies a novel gene linked to neurodegenerative disease. BMC Genomics 15, 850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Delprato A, Grellmann C, Klein M, Wetzel R, Overall RW, and Badea A (2014b). Transcript co-variance with Nestin in two mouse genetic reference populations identifies Lef1 as a novel candidate regulator of neural precursor cell proliferation in the adult hippocampus. Front Neurosci 8, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Gini B, and Hager R (2015a). Genetic variation in offspring indirectly influences the quality of maternal behaviour in mice. Elife 4, e11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Williams RW, Lu L, and Hager R (2015b). A cross-species genetic analysis identifies candidate genes for mouse anxiety and human bipolar disorder. Front Behav Neurosci 9, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Sharmin N, and Hager R (2017). Offspring genes indirectly influence sibling and maternal behavioural strategies over resource share. Proceedings Biol Sci 284, 20171059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Roy S, Clifford BG, Riede T, Scattoni ML, Heck DH, Lu L, and Williams RW (2018a). Born to cry: A genetic dissection of infant vocalization. Front Behav Neurosci 12, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashbrook DG, Arends D, Mulligan MK, Williams EG, Lutz C, Valenzuela A, Bohl C, Ingels J, McCarty M, Centeno A, et al. (2018b). Sequencing the BXD family, a cohort for experimental systems genetics and precision medicine. In 2018 IBANGS Meeting: The 20TH Annual Genes, Brain & Behavior Meeting, (Rochester, MN USA: International Behavioral and Neural Genetics Society; ), pp. 4–5. [Google Scholar]
- Auffray C, Charron D, and Hood L (2010). Predictive, preventive, personalized and participatory medicine: back to the future. Genome Med 2, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Walker M, Arat S, Ananda G, Petkova P, Powers NR, Tian H, Spruce C, Ji B, Rausch D, et al. (2019). Tissue-specific trans regulation of the mouse epigenome. Genetics 211, 831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud A, Mulligan MK, Casale FP, Ingels JF, Bohl CJ, Callebert J, Launay J-M, Krohn J, Legarra A, Williams RW, et al. (2017). Genetic variation in the social environment contributes to health and disease. PLoS Genet 13, e1006498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belknap JK (1998). Effect of within-strain sample size on QTL detection and mapping using recombinant inbred mouse strains. Behav Genet 28, 29–38. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Phillips TJ, and O’Toole LA (1992a). Quantitative trait loci associated with brain weight in the BXD/Ty recombinant inbred mouse strains. Brain Res Bull 29, 337–344. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Crabbe JC, Plomin R, McClearn GE, Sampson KE, O’Toole LA, and Gora-Maslak G (1992b). Single-locus control of saccharin intake in BXD/Ty recombinant inbred (RI) mice: Some methodological implications for RI strain analysis. Behav Genet 22, 81–100. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Metten P, Helms ML, O’Toole LA, Angeli-Gade S, Crabbe JC, and Phillips TJ (1993). Quantitative trait loci (QTL) applications to substances of abuse: physical dependence studies with nitrous oxide and ethanol in BXD mice. Behav Genet 23, 213–222. [DOI] [PubMed] [Google Scholar]
- Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, Pan C, Packard RRS, Eskin E, Yan M, et al. (2015). Genetic architecture of atherosclerosis in mice: A systems genetics analysis of common inbred strains. PLoS Genet 11, e1005711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon ACM, Williams RW, Sinasac DS, and Webby RJ (2014). A novel genetic locus linked to pro-inflammatory cytokines after virulent H5N1 virus infection in mice. BMC Genomics 15, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW (2005). The genomes of recombinant inbred lines. Genetics 169, 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, and Churchill GA (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890. [DOI] [PubMed] [Google Scholar]
- Broman KW, Gatti DM, Simecek P, Furlotte NA, Prins P, Sen S, Yandell BS, and Churchill GA (2019). R/qtl2: Software for mapping quantitative trait loci with high-dimensional data and multiparent populations. Genetics 211, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Smith DJ, Kantak KM, Nowak TS, Williams RW, Damaj MI, Redei EE, Chen H, and Mulligan MK (2020). Facilitating complex trait analysis via reduced complexity crosses. Trends Genet 36, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhuatanta KAK, Shea CJA, Herman JP, and Jankord R (2014). Unique genetic loci identified for emotional behavior in control and chronic stress conditions. Front Behav Neurosci 8, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, et al. (2006). In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet 15, 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chella Krishnan K, Mukundan S, Alagarsamy J, Hur J, Nookala S, Siemens N, Svensson M, Hyldegaard O, Norrby-Teglund A, and Kotb M (2016). Genetic architecture of group a streptococcal necrotizing soft tissue infections in the mouse. PLoS Pathog 12, e1005732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Wang J, Lu L, Qu Y, Manly KF, and Williams RW (2003). Genetic correlates of gene expression in recombinant inbred strains: a relational model system to explore neurobehavioral phenotypes. Neuroinformatics 1, 343–357. [DOI] [PubMed] [Google Scholar]
- Chintalapudi SR, Maria D, Di Wang X, Bailey JNC, NEIGHBORHOOD consortium, International Glaucoma Genetics consortium, Hysi PG, Wiggs JL, Williams RW, and Jablonski MM (2017). Systems genetics identifies a role for Cacna2d1 regulation in elevated intraocular pressure and glaucoma susceptibility. Nat Commun 8, 1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. (2004). The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36, 1133–1137. [DOI] [PubMed] [Google Scholar]
- Cleveland WS (1979). Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc 74, 829–836. [Google Scholar]
- Cook DN, Whitehead GS, Burch LH, Berman KG, Kapadia Z, Wohlford-Lenane C, and Schwartz DA (2006). Spontaneous mutations in recombinant inbred mice: mutant toll-like receptor 4 (Tlr4) in BXD29 mice. Genetics 172, 1751–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF (2007). Haldane, Bailey, Taylor and recombinant-inbred lines. Genetics 176, 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvasi A (1998). Experimental strategies for the genetic dissection of complex traits in animal models. Nat Genet 18, 19–24. [DOI] [PubMed] [Google Scholar]
- Dickson PE, Miller MM, Calton MA, Bubier JA, Cook MN, Goldowitz D, Chesler EJ, and Mittleman G (2016). Systems genetics of intravenous cocaine self-administration in the BXD recombinant inbred mouse panel. Psychopharmacology (Berl) 233, 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson PE, Roy TA, McNaughton KA, Wilcox TD, Kumar P, and Chesler EJ (2019). Systems genetics of sensation seeking. Genes Brain Behav 18, e12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diessler S, Jan M, Emmenegger Y, Guex N, Middleton B, Skene DJ, Ibberson M, Burdet F, Götz L, Pagni M, et al. (2018). A systems genetics resource and analysis of sleep regulation in the mouse. PLoS Biol 16, e2005750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS, and Mackay TFC (1996). Introduction to quantitative genetics (Burnt Mill, England: Longman; ). [Google Scholar]
- Fleet JC, Replogle RA, Reyes-Fernandez P, Wang L, Zhang M, Clinkenbeard EL, and White KE (2016). Gene-by-Diet interactions affect serum 1,25-Dihydroxyvitamin D levels in male BXD recombinant inbred mice. Endocrinology 157, 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A, Rau CD, Farber CR, Bennett BJ, Orozco LD, van Nas A, Pan C, Allayee H, Beaven SW, Civelek M, et al. (2012). Hybrid mouse diversity panel: a panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm Genome 23, 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales NM, and Palmer AA (2014). Fine-mapping QTLs in advanced intercross lines and other outbred populations. Mamm Genome 25, 271–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybeal C, Bachu M, Mozhui K, Saksida LM, Bussey TJ, Sagalyn E, Williams RW, and Holmes A (2014). Strains and stressors: an analysis of touchscreen learning in genetically diverse mouse strains. PLoS One 9, e87745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisel JE, Belknap JK, O’Toole LA, Helms ML, Wenger CD, and Crabbe JC (1997). Quantitative trait loci affecting methamphetamine responses in BXD recombinant inbred mouse strains. J Neurosci 17, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizzle WE, Mountz JD, Yang P-A, Xu X, Sun S, Van Zant GE, Williams RW, Hsu H-C, and Zhang H-G (2002). BXD recombinant inbred mice represent a novel T cell-mediated immune response tumor model. Int J Cancer 101, 270–279. [DOI] [PubMed] [Google Scholar]
- Hager R, Lu L, Rosen GD, and Williams RW (2012). Genetic architecture supports mosaic brain evolution and independent brain-body size regulation. Nat Commun 3, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JB, and Waddington CH (1931). Inbreeding and linkage. Genetics 16, 357–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley CS, and Knott SA (1992). A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity (Edinb) 69, 315–324. [DOI] [PubMed] [Google Scholar]
- Hawkins NA, Nomura T, Duarte S, Williams RW, Homanics GE, Mulligan MK, Contractor A, and Kearney JA (2020). Gabra2 is a genetic modifier of Dravet syndrome in mice. BioRxiv 2020.04.19.048546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes KS, Hager R, and Grencis RK (2014). Sex-dependent genetic effects on immune responses to a parasitic nematode. BMC Genomics 15, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiker JT, Kunath A, Kosacka J, Flehmig G, Knigge A, Kern M, Stumvoll M, Kovacs P, BlQher M, and Kloting N (2014). Identification of genetic loci associated with different responses to high-fat diet-induced obesity in C57BL/6N and C57BL/6J substrains. Physiol Genomics 46, 377–384. [DOI] [PubMed] [Google Scholar]
- Hood L, and Flores M (2012). A personal view on systems medicine and the emergence of proactive P4 medicine: predictive, preventive, personalized and participatory. N Biotechnol 29, 613–624. [DOI] [PubMed] [Google Scholar]
- Hood L, and Friend SH (2011). Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat Rev Clin Oncol 8, 184–187. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, and Auwerx J (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T, Meng Q, Saleh MA, Norlander AE, Joehanes R, Zhu J, Chen BH, Zhang B, Johnson AD, Ying S, et al. (2015). Integrative network analysis reveals molecular mechanisms of blood pressure regulation. Mol Syst Biol 11, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha P, McDevitt MT, Gupta R, Quiros PM, Williams EG, Gariani K, Sleiman MB, Diserens L, Jochem A, Ulbrich A, et al. (2018a). Systems analyses reveal physiological roles and genetic regulators of liver lipid species. Cell Syst 6, 722–733.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha P, McDevitt MT, Halilbasic E, Williams EG, Quiros PM, Gariani K, Sleiman MB, Gupta R, Ulbrich A, Jochem A, et al. (2018b). Genetic regulation of plasma lipid species and their association with metabolic phenotypes. Cell Syst 6, 709–721.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BC, and Jellen LC (2017). Systems genetics analysis of iron and its regulation in brain and periphery. Methods Mol Biol 1488, 467–480. [DOI] [PubMed] [Google Scholar]
- Jones LC, McCarthy KA, Beard JL, Keen CL, and Jones BC (2006). Quantitative genetic analysis of brain copper and zinc in BXD recombinant inbred mice. Nutr Neurosci 9, 81–92. [DOI] [PubMed] [Google Scholar]
- Jung SH, Brownlow ML, Pellegrini M, and Jankord R (2017). Divergence in Morris Water Maze-based cognitive performance under chronic stress Is associated with the hippocampal whole transcriptomic modification in mice. Front Mol Neurosci 10, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R, Lu L, Williams RW, and Geisert EE (2015). Transcriptome networks in the mouse retina: An exon level BXD RI database. Mol Vis 21, 1235–1251. [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick SL, and Bryant CD (2014). Behavioral architecture of opioid reward and aversion in C57BL/6 substrains. Front Behav Neurosci 8, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Jiang K, and Levitt P (2018). Quantitative trait locus mapping and analysis of heritable variation in affiliative social behavior and co-occurring traits. Genes Brain Behav 17, e12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutnikova H, Laakso M, Lu L, Combe R, Paananen J, Kuulasmaa T, Kuusisto J, Haring HU, Hansen T, Pedersen O, et al. (2009). Identification of the UBP1 locus as a critical blood pressure determinant using a combination of mouse and human genetics. PLoS Genet 5, e1000591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Kim K, Joseph C, Kourrich S, Yoo S-H, Huang HC, Vitaterna MH, de Villena FP-M, Churchill G, Bonci A, et al. (2013). C57BL/6N mutation in cytoplasmic FMRP interacting protein 2 regulates cocaine response. Science 342, 1508–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E, and Kruglyak L (1995). Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11, 241–247. [DOI] [PubMed] [Google Scholar]
- Lee GH, Bennett LM, Carabeo RA, and Drinkwater NR (1995). Identification of hepatocarcinogen-resistance genes in DBA/2 mice. Genetics 139, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang X, Rukina D, Huang Q, Lin T, Sorrentino V, Zhang H, Bou Sleiman M, Arends D, McDaid A, et al. (2018). An integrated systems genetics and omics toolkit to probe gene function. Cell Syst 6, 90–102.e4. [DOI] [PubMed] [Google Scholar]
- Li R, Svenson KL, Donahue LRB, Peters LL, and Churchill GA (2008). Relationships of dietary fat, body composition, and bone mineral density in inbred mouse strain panels. Physiol Genomics 33, 26–32. [DOI] [PubMed] [Google Scholar]
- Li Z, Mulligan MK, Wang X, Miles MF, Lu L, and Williams RW (2010). A transposon in Comt generates mRNA variants and causes widespread expression and behavioral differences among mice. PLoS One 5, e12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wang X, Pullen M, Guan H, Chen H, Sahu S, Zhang B, Chen H, Williams RW, Geisert EE, et al. (2011). Genetic dissection of the Gpnmb network in the eye. Invest Ophthalmol Vis Sci 52, 4132–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhou D, Lu H, Xu F, Yue J, Tong J, and Lu L (2019). Investigating a downstream gene of Gpnmb using the systems genetics method. Mol Vis 25, 222–236. [PMC free article] [PubMed] [Google Scholar]
- McGinnis JF, Lerious V, Pazik J, and Elliott RW (1993). Chromosomal assignment of the recoverin gene and cancer-associated retinopathy. Mamm Genome 4, 43–45. [DOI] [PubMed] [Google Scholar]
- McKnite AM, Perez-Munoz ME, Lu L, Williams EG, Brewer S, Andreux PA, Bastiaansen JWM, Wang X, Kachman SD, Auwerx J, et al. (2012). Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS One 7, e39191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles CM, and Wayne M (2008). Quantitative trait locus (QTL) analysis. Nat Educ 1, 208. [Google Scholar]
- Miyairi I, Tatireddigari VRRA, Mahdi OS, Rose LA, Belland RJ, Lu L, Williams RW, and Byrne GI (2007). The p47 GTPases Iigp2 and Irgb10 regulate innate immunity and inflammation to murine Chlamydia psittaci infection. J Immunol 179, 1814–1824. [DOI] [PubMed] [Google Scholar]
- Miyairi I, Ziebarth J, Laxton JD, Wang X, van Rooijen N, Williams RW, Lu L, Byrne GI, and Cui Y (2012). Host genetics and Chlamydia disease: prediction and validation of disease severity mechanisms. PLoS One 7, e33781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, and Welsh CE (2015). Informatics resources for the Collaborative Cross and related mouse populations. Mamm Genome 26, 521–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, Fu C-P, Kao C-Y, Welsh CE, Didion JP, Yadgary L, Hyacinth L, Ferris MT, Bell TA, Miller DR, et al. (2015). The mouse universal genotyping array: From substrains to subspecies. G3 (Bethesda) 6, 263–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozhui K, Ciobanu DC, Schikorski T, Wang X, Lu L, and Williams RW (2008). Dissection of a QTL hotspot on mouse distal chromosome 1 that modulates neurobehavioral phenotypes and gene expression. PLoS Genet 4, e1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, and Williams RW (2015). Systems genetics of behavior: a prelude. Curr Opin Behav Sci 2, 108–115. [Google Scholar]
- Mulligan MK, Ponomarev I, Boehm SL, Owen JA, Levin PS, Berman AE, Blednov YA, Crabbe JC, Williams RW, Miles MF, et al. (2008). Alcohol trait and transcriptional genomic analysis of C57BL/6 substrains. Genes Brain Behav 7, 677–689. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, Dubose C, Yue J, Miles MF, Lu L, and Hamre KM (2013). Expression, covariation, and genetic regulation of miRNA Biogenesis genes in brain supports their role in addiction, psychiatric disorders, and disease. Front Genet 4, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Mozhui K, Prins P, and Williams RW (2017). GeneNetwork: A Toolbox for Systems Genetics. Methods Mol Biol 1488, 75–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Zhao W, Dickerson M, Arends D, Prins P, Cavigelli SA, Terenina E, Mormede P, Lu L, and Jones BC (2018). Genetic contribution to initial and progressive alcohol intake among recombinant inbred strains of mice. Front Genet 9, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Abreo T, Neuner SM, Parks C, Watkins CE, Houseal MT, Shapaker TM, Hook M, Tan H, Wang X, et al. (2019). Identification of a functional non-coding variant in the GABAA Receptor α2 subunit of the C57BL/6J mouse reference genome: Major implications for neuroscience research. Front Genet 10, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner SM, Garfinkel BP, Wilmott LA, Ignatowska-Jankowska BM, Citri A, Orly J, Lu L, Overall RW, Mulligan MK, Kempermann G, et al. (2016). Systems genetics identifies Hp1bp3 as a novel modulator of cognitive aging. Neurobiol Aging 46, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner SM, Heuer SE, Huentelman MJ, O’Connell KMS, and Kaczorowski CC (2019a). Harnessing genetic complexity to enhance translatability of Alzheimer’s disease mouse models: A path toward precision medicine. Neuron 101, 399–411.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner SM, Heuer SE, Zhang J-G, Philip VM, and Kaczorowski CC (2019b). Identification of pre-symptomatic gene signatures that predict resilience to cognitive decline in the genetically diverse AD-BXD model. Front Genet 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren Y, Nachshon A, Frishberg A, Wilentzik R, and Gat-Viks I (2015). Linking traits based on their shared molecular mechanisms. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AA, Lessov-Schlaggar CN, Ponder CA, McKinnon CS, and Phillips TJ (2006). Sensitivity to the locomotor-stimulant effects of ethanol and allopregnanolone: a quantitative trait locus study of common genetic influence. Genes Brain Behav 5, 506–517. [DOI] [PubMed] [Google Scholar]
- Peirce JL, Lu L, Gu J, Silver LM, and Williams RW (2004). A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Munoz ME, McKnite AM, Williams EG, Auwerx J, Williams RW, Peterson DA, and Ciobanu DC (2019). Diet modulates cecum bacterial diversity and physiological phenotypes across the BXD mouse genetic reference population. PLoS One 14, e0224100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip VM, Duvvuru S, Gomero B, Ansah TA, Blaha CD, Cook MN, Hamre KM, Lariviere WR, Matthews DB, Mittleman G, et al. (2010). High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes Brain Behav 9, 129–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips TJ, Belknap JK, Buck KJ, and Cunningham CL (1998). Genes on mouse chromosomes 2 and 9 determine variation in ethanol consumption. Mamm Genome 9, 936–941. [DOI] [PubMed] [Google Scholar]
- Rau CD, Parks B, Wang Y, Eskin E, Simecek P, Churchill GA, and Lusis AJ (2015). High-density genotypes of inbred mouse strains: Improved power and precision of association mapping. G3 (Bethesda) 5, 2021–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rédei GP (2008a). Carter-Falconer Mapping Function. In Encyclopedia of Genetics, Genomics, Proteomics and Informatics, (Dordrecht: Springer Netherlands; ), pp. 276–276. [Google Scholar]
- Rédei GP (2008b). Haldane’s Mapping Function. In Encyclopedia of Genetics, Genomics, Proteomics and Informatics, (Dordrecht: Springer Netherlands; ), pp. 836–836. [Google Scholar]
- Rédei GP (2008c). Kosambi’s Function. In Encyclopedia of Genetics, Genomics, Proteomics and Informatics, (Dordrecht: Springer Netherlands; ), pp. 1066–1066. [Google Scholar]
- Rédei GP (2008d). Morgan. In Encyclopedia of Genetics, Genomics, Proteomics and Informatics, (Dordrecht: Springer Netherlands; ), pp. 1255–1255. [Google Scholar]
- Reed C, Baba H, Zhu Z, Erk J, Mootz JR, Varra NM, Williams RW, and Phillips TJ (2017). A spontaneous mutation in Taar1 impacts methamphetamine-related traits exclusively in DBA/2 mice from a single vendor. Front Pharmacol 8, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes Fernandez PC, Replogle RA, Wang L, Zhang M, and Fleet JC (2016). Novel genetic loci control calcium absorption and femur bone mass as well as their response to low calcium intake in male BXD recombinant inbred mice. J Bone Miner Res 31, 994–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues B. de A., Munoz VR, Kuga GK, Gaspar RC, Nakandakari SCBR, Crisol BM, Botezelli JD, Pauli LSS, da Silva ASR, de Moura LP., et al. (2017). Obesity increases mitogen-activated protein kinase phosphatase-3 levels in the hypothalamus of mice. Front Cell Neurosci 11, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez LA, Plomin R, Blizard DA, Jones BC, and McClearn GE (1994). Alcohol acceptance, preference, and sensitivity in mice. I. Quantitative genetic analysis using BXD recombinant inbred strains. Alcohol Clin Exp Res 18, 1416–1422. [DOI] [PubMed] [Google Scholar]
- Rosen GD, Pung CJ, Owens CB, Caplow J, Kim H, Mozhui K, Lu L, and Williams RW (2009). Genetic modulation of striatal volume by loci on Chrs 6 and 17 in BXD recombinant inbred mice. Genes Brain Behav 8, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GD, Azoulay NG, Griffin EG, Newbury A, Koganti L, Fujisaki N, Takahashi E, Grant PE, Truong DT, Fitch RH, et al. (2013). Bilateral subcortical heterotopia with partial callosal agenesis in a mouse mutant. Cereb Cortex 23, 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Sleiman MB, Jha P, Williams EG, Ingels JF, Chapman CJ, McCarty MS, Hook M, Sun A, Zhao W, et al. (2020). Gene-by-environmental modulation of longevity and weight gain in the murine BXD family. BioRxiv 776559. [Google Scholar]
- Russo LM, Abdeltawab NF, O’Brien AD, Kotb M, and Melton-Celsa AR (2015). Mapping of genetic loci that modulate differential colonization by Escherichia coli O157:H7 TUV86-2 in advanced recombinant inbred BXD mice. BMC Genomics 16, 947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval-Sierra JV, Helbing AHB, Williams EG, Ashbrook DG, Roy S, Williams RW, and Mozhui K (2019). Influence of body weight at young adulthood on the epigenetic clock and lifespan in the BXD murine family. BioRxiv 791582. [Google Scholar]
- Sandoval-Sierra JV, Helbing AHB, Williams EG, Ashbrook DG, Roy S, Williams RW, and Mozhui K (2020). Body weight and high-fat diet are associated with epigenetic aging in female members of the BXD murine family. Aging Cell e13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schughart K, and Williams RW (2017). Systems Genetics (New York, NY: Springer New York; ). [Google Scholar]
- Schüssler-Fiorenza Rose SM, Contrepois K, Moneghetti KJ, Zhou W, Mishra T, Mataraso S, Dagan-Rosenfeld O, Ganz AB, Dunn J, Hornburg D, et al. (2019). A longitudinal big data approach for precision health. Nat Med 25, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seecharan DJ, Kulkarni AL, Lu L, Rosen GD, and Williams RW (2003). Genetic control of interconnected neuronal populations in the mouse primary visual system. J Neurosci 23, 11178–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen Ś, Satagopan JM, Broman KW, and Churchill GA (2007). R/qtlDesign: Inbred line cross experimental design. Mamm Genome 18, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Walter NAR, Harkness JH, Neve KA, Williams RW, Lu L, Belknap JK, Eshleman AJ, Phillips TJ, and Janowsky A (2016). Genetic Polymorphisms Affect Mouse and Human Trace Amine-Associated Receptor 1 Function. PLoS One 11, e0152581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifman S, Bell JT, Copley RR, Taylor MS, Williams RW, Mott R, and Flint J (2006). A high-resolution single nucleotide polymorphism genetic map of the mouse genome. PLoS Biol 4, 2227–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simecek P, Forejt J, Williams RW, Shiroishi T, Takada T, Lu L, Johnson TE, Bennett B, Deschepper CF, Scott-Boyer M-P, et al. (2017). High-resolution maps of mouse reference populations. G3 (Bethesda) 7, 3427–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan Z, Arends D, Broman, K. W, Centeno A, Furlotte N, Nijveen H, Yan L, Zhou X W. Williams R, and Prins P (2016). GeneNetwork: framework for web-based genetics. J Open Source Softw 1, 25. [Google Scholar]
- Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gomez-Marin C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. (2014). Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford AM, Reed C, Baba H, Walter NA, Mootz JR, Williams RW, Neve KA, Fedorov LM, Janowsky AJ, and Phillips TJ (2019). Taar1 gene variants have a causal role in methamphetamine intake and response and interact with Oprm1. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan J, Stegle O, and Beyer A (2015). A random forest approach to capture genetic effects in the presence of population structure. Nat Commun 6, 7432. [DOI] [PubMed] [Google Scholar]
- Sul JH, Bilow M, Yang W-Y, Kostem E, Furlotte N, He D, and Eskin E (2016). Accounting for population structure in gene-by-environment interactions in genome-wide association studies using mixed model. PLoS Genet 12, e1005849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BA, Heiniger HJ, and Meier H (1973). Genetic analysis of resistance to cadmium-induced testicular damage in mice. Proc Soc Exp Biol Med 143, 629–633. [DOI] [PubMed] [Google Scholar]
- Taylor BA, Wnek C, Kotlus BS, Roemer N, MacTaggart T, and Phillips SJ (1999). Genotyping new BXD recombinant inbred mouse strains and comparison of BXD and consensus maps. Mamm Genome 10, 335–348. [DOI] [PubMed] [Google Scholar]
- Théberge ET, Baker JA, Dubose C, Boyle JK, Balce K, Goldowitz D, and Hamre KM (2019). Genetic influences on the amount of cell death in the neural tube of BXD mice exposed to acute ethanol at midgestation. Alcohol Clin Exp Res 43, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaih S-W, Lu L, Airey DC, Williams RW, and Churchill GA (2005). Quantitative trait mapping in a diallel cross of recombinant inbred lines. Mamm Genome 16, 344–355. [DOI] [PubMed] [Google Scholar]
- Valdar W, Flint J, and Mott R (2006). Simulating the collaborative cross: power of quantitative trait loci detection and mapping resolution in large sets of recombinant inbred strains of mice. Genetics 172, 1783–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J (2008). Reporting and interpretation in genome-wide association studies. Int J Epidemiol 37, 641–653. [DOI] [PubMed] [Google Scholar]
- Wang J, Williams RW, and Manly KF (2003). WebQTL: web-based complex trait analysis. Neuroinformatics 1, 299–308. [DOI] [PubMed] [Google Scholar]
- Wang J, Yoon TW, Read R, Yi A-K, Williams RW, and Fitzpatrick EA (2020). Genetic variability of T cell responses in hypersensitivity pneumonitis identified using the BXD genetic reference panel. Am J Physiol Lung Cell Mol Physiol 318, L631–L643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jiao Y, Wang Y, Zhang M, and Gu W (2016a). Self-Confirmation and Ascertainment of the Candidate Genomic Regions of Complex Trait Loci - A None-Experimental Solution. PLoS One 11, e0153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LS, Jiao Y, Huang Y, Liu XY, Gibson G, Bennett B, Hamre KM, Li DW, Zhao HY, Gelernter J, et al. (2013). Critical evaluation of transcription factor Atf2 as a candidate modulator of alcohol preference in mouse and human populations. Genet Mol Res 12, 5992–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pandey AK, Mulligan MK, Williams EG, Mozhui K, Li Z, Jovaisaite V, Quarles LD, Xiao Z, Huang J, et al. (2016b). Joint mouse-human phenome-wide association to test gene function and disease risk. Nat Commun 7, 10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimar WR, Lane PW, and Sidman RL (1982). Vibrator (vb): a spinocerebellar system degeneration with autosomal recessive inheritance in mice. Brain Res 251, 357–364. [DOI] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium, Maller JB, McVean G, Byrnes J, Vukcevic D, Palin K, Su Z, Howson JMM, Auton A, Myers S, et al. (2012). Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet 44, 1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MD, Dumontier M, Aalbersberg, Ij JJJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, et al. (2016). The FAIR Guiding Principles for scientific data management and stewardship. Sci Data 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW (2009). Herding cats: the sociology of data integration. Front Neurosci 3, 154–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EG, and Auwerx J (2015). The convergence of systems and reductionist approaches in complex trait analysis. Cell 162, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW, and Williams EG (2017). Resources for Systems Genetics. In System Genetics: Methods and Protocols, Schughart K, and Williams RW, eds. (New York, NY: Springer New York; ), pp. 3–29. [Google Scholar]
- Williams EG, Mouchiroud L, Frochaux M, Pandey A, Andreux PA, Deplancke B, and Auwerx J (2014). An evolutionarily conserved role for the aryl hydrocarbon receptor in the regulation of movement. PLoS Genet 10, e1004673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EG, Wu Y, Jha P, Dubuis S, Blattmann P, Argmann CA, Houten SM, Amariuta T, Wolski W, Zamboni N, et al. (2016). Systems proteomics of liver mitochondria function. Science 352, aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EG, Wu Y, Wolski W, Kim JY, Lan J, Hasan M, Halter C, Jha P, Ryu D, Auwerx J, et al. (2018). Quantifying and localizing the mitochondrial proteome across five tissues in a mouse population. Mol Cell Proteomics 17, 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EG, Roy S, Statzer C, Ingels J, Bohl C, Hasan M, Cuklina J, Lu L, Ewald CY, Williams RW, et al. (2020). The Molecular Landscape of the Aging Mouse Liver. BioRxiv Syst Biol 2020.08.20.222968. [Google Scholar]
- Williams RW, Strom RC, and Goldowitz D (1998). Natural variation in neuron number in mice is linked to a major quantitative trait locus on Chr 11. J Neurosci 18, 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW, Gu J, Qi S, and Lu L (2001). The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol 2, RESEARCH0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan J, Kutalik Z, et al. (2014). Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46, 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Williams EG, Dubuis S, Mottis A, Jovaisaite V, Houten SM, Argmann CA, Faridi P, Wolski W, Kutalik Z, et al. (2014). Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 158, 1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RJ, Mozhui K, Karlsson R-M, Cameron HA, Williams RW, and Holmes A (2008). Variation in mouse basolateral amygdala volume is associated with differences in stress reactivity and fear learning. Neuropsychopharmacology 33, 2595–2604. [DOI] [PubMed] [Google Scholar]
- Zhang W, Korstanje R, Thaisz J, Staedtler F, Harttman N, Xu L, Feng M, Yanas L, Yang H, Valdar W, et al. (2012). Genome-wide association mapping of quantitative traits in outbred mice. G3 (Bethesda) 2, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, and Williams RW (1999). Eye1 and Eye2: gene loci that modulate eye size, lens weight, and retinal area in the mouse. Invest Ophthalmol Vis Sci 40, 817–825. [PubMed] [Google Scholar]
- Zhou X, and Stephens M (2012). Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, and Stephens M (2014). Efficient multivariate linear mixed model algorithms for genomewide association studies. Nat Methods 11, 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Zhao Y, Hook M, Zhao W, Starlard-Davenport A, Cook MN, Jones BC, Hamre KM, and Lu L (2018). Ethanol’s effect on Coq7 expression in the hippocampus of mice. Front Genet 9, 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Information on the nomenclature, availability, derivation, and genotyping of all BXD strains (BXD1-BXD228), related to Figure 1. Shows alternative names that have been used for BXD family members, details of when breeding was started, when production of these strains began at the Jackson Laboratory (JAX), the stock number (which is also the RRID number) and availability from JAX, the epoch, method of derivation, the origin of the mitochondrial genome, and platforms that each strain has been genotyped on, and the generation number if known.
Table S2: LOD values for 3298 phenotypes, calculated using Haley-Knott (H-K) equations or linear mixed models (LMMs), related to Figure 2. For each phenotype and model combination, the significance threshold is shown (calculated by 5000 permutations), the peak LOD score, and the difference between the threshold and the peak. For each phenotype, the differences in threshold between the H-K and LMM equations are shown, as well as the count of strains in epoch 1, epochs 1 and 2, epoch 3, and epochs higher than 3.
Table S3: List of traits in the BXD phenome on GeneNetwork, related to Figures 1 and S4. An annotated list of BXD publicly available phenotypes, available on GeneNetwork. The table shows the GeneNetwork trait ID, the phenotype category, the full phenotype description, the authors who produced the phenotype, the year of publication (or year of entry into GN if unpublished), the PubMed ID (if published), and the peak LRS score and the peak’s position in the genome.
Data Availability Statement
This paper analyzes existing, publicly available data. These data sets’ accession numbers are provided in the Key Resource Table, and throughout the manuscript. Genotype files can be found at http://www.genenetwork.org/webqtl/main.py?FormID=sharinginfo&GN_AccessionId=600. GeneNetwork.org original code is publicly available at https://qithub.com/genenetwork/genenetwork2 and https://github.com/genenetwork/genenetwork1.
The scripts used to generate the figures reported in this paper are available in the BXDtools package (https://github.com/DannyArends/BXDtools) or using ggplot2 (version 3.3.2; https://cran.r-project.org/web/packages/ggplot2/index.html) and their use is described in the STAR Methods. Code for computing marker positions and visualization of recombinations is available in the BXDtools package (https://github.com/DannyArends/BXDtools).
Power estimates can be made using http://power.genenetwork.org/, and code is at https://github.com/Dashbrook/BXD_power_calculator_app.
The R/qtl2 software can be found here https://kbroman.org/qtl2/, and its use is described in the STAR Methods.