

Abstract
Purpose
Complete congenital stationary night blindness (cCSNB) is an incurable inherited retinal disorder characterized by an ON-bipolar cell (ON-BC) defect. GRM6 mutations are the third most prevalent cause of cCSNB. The Grm6−/− mouse model mimics the human phenotype, showing no b-wave in the electroretinogram (ERG) and a loss of mGluR6 and other proteins of the same cascade at the outer plexiform layer (OPL). Our aim was to restore protein localization and function in Grm6−/− adult mice targeting specifically ON-BCs or the whole retina.
Methods
Adeno-associated virus-encoding Grm6 under two different promoters (GRM6-Grm6 and CAG-Grm6) were injected intravitreally in P15 Grm6−/− mice. ERG recordings at 2 and 4 months were performed in Grm6+/+, untreated and treated Grm6−/− mice. Similarly, immunolocalization studies were performed on retinal slices before or after treatment using antibodies against mGluR6, TRPM1, GPR179, RGS7, RGS11, Gβ5, and dystrophin.
Results
Following treatment, mGluR6 was localized to the dendritic tips of ON-BCs when expressed with either promoter. The relocalization efficiency in mGluR6-transduced retinas at the OPL was 2.5% versus 11% when the GRM6-Grm6 and CAG-Grm6 were used, respectively. Albeit no functional rescue was seen in ERGs, relocalization of TRPM1, GPR179, and Gβ5 was also noted using both constructs. The restoration of the localization of RGS7, RGS11, and dystrophin was more obvious in retinas treated with GRM6-Grm6 than in retinas treated with CAG-Grm6.
Conclusions
Our findings show the potential of treating cCSNB with GRM6 mutations; however, it appears that the transduction rate must be improved to restore visual function.
Keywords: CSNB, gene therapy, bipolar cells, mGluR6
Congenital stationary night blindness (CSNB) is a non-progressive retinal disorder.1 Patients suffering from this disease often present an impairment of night vision, delayed light-to-dark adaptation, and difficulty with properly sensing contrasts in dim-light conditions. It is also often associated with high myopia, strabismus, and nystagmus.2 Considering the retinal response to the full-field electroretinogram (ERG), CSNB has been divided in two types: Riggs CSNB, which is associated with a rod-photoreceptor defect, and Schubert–Bornschein CSNB,1 in which the underlying defect relies on the signal transmission from photoreceptors to bipolar cells. The Schubert–Bornschein type of CSNB can be classified as incomplete CSNB or complete CSNB (cCSNB).3 We focused on the latter type in this study. Patients with cCSNB show an electronegative ERG waveform, with normal a-wave amplitudes and severely diminished or absent b-waves under scotopic conditions, thus referring to it as complete. Photopic responses are less altered.2 More specifically, ON-responses are affected but OFF-responses remain globally normal.2 This ERG profile is consistent with a transmission defect between photoreceptors and ON-bipolar cells (ON-BCs). cCSNB is due to mutations in NYX,4,5 TRPM1,6–8 GRM6,9,10 GPR179,11,12 and LRIT3.13 All code for proteins involved in the signaling cascade at the photoreceptor to ON-BC synapse.
Mutations in GRM6 are the third most prevalent cause of cCSNB.2 GRM6 codes for a metabotropic receptor, (mGluR6) that mediates glutamate synaptic transmission between photoreceptors and ON-BCs. In the retina, metabotropic glutamate receptor 6 (mGluR6) is exclusively localized in ON-BCs.14 In the absence of light, glutamate released at the synaptic cleft activates mGluR6, which initiates the ON-BC signaling cascade15 leading to closure of the cation channel transient receptor potential melastatin 1 (TRPM1).16 In contrast, in response to light, less glutamate binds to mGluR6, leading to opening of the TRPM1 channel.17 Subsequently, ON-BCs are depolarized, leading to the b-wave, which is severely reduced or absent in cCSNB. Five mouse models with a Grm6 defect have been described: Grm6tm1Nak (referred to here as Grm6−/−), nob3, nob4, nob7, and nob8.18–22 Of those, four (including Grm6−/−) show a similar ERG phenotype as patients with cCSNB under scotopic conditions—namely, an absent b-wave, whereas the a-wave appears normal. Furthermore, in these four models, mGluR6 is absent.
It has also been suggested that mGluR6 is important for the proper localization of other proteins of the cascade, such as regulator G protein signaling (RGS) proteins (RGS7, RGS11, Gβ5, and R9AP),23,24 and TRPM123 at the dendritic tips of ON-BCs. The absence of TRPM1 at the dendritic tips of ON-BCs renders this channel nonfunctional.23 The interdependence of G protein-coupled receptor 179 (GPR179) on mGluR6, important for the targeting and/or maintenance of RGS proteins,25 was found to be model and condition dependent.26,27 Albeit the gross morphology of the retina in patients and mice lacking mGluR6 is not affected, detailed clinical examinations and morphological studies have detected differences compared to unaffected retinas. Indeed, three patients harboring mutations in GRM6 exhibited reduced retinal thickness in the extrafoveal region, as revealed by spectral-domain optical coherence tomography measurements.28 Furthermore, in mice lacking Grm6, invaginating dendrites of rod BCs are larger and often contain ectopic ribbons, whereas the number of invaginating dendrites of cone ON-BCs and ribbons decreases at the cone pedicles in the Grm6−/− mouse model.29 Tummala and colleagues30 showed that mGluR6 has also a role presynaptically, as several presynaptic matrix-associated proteins such as dystrophin are reduced at the photoreceptor-to-BC synapse in the same Grm6−/− model.
To date, no treatment is available for cCSNB. A gene replacement strategy might be a suitable approach treating cCSNB, as it represents a stationary nondegenerative disorder, and the genetics of this disorder are well characterized.2 Two different approaches targeting two genes involved in cCSNB, Nyx and Lrit3, resulted in partial restoration of function upon protein relocalization in mouse models.31,32 However, this partial rescue was mainly obtained at a very young age (postnatal day 2 [P2] or 5 [P5]) and only under scotopic conditions. These two strategies aimed at treating cCSNB show the challenges to obtaining functional rescue in adult cCSNB mice. Our study aimed to restore GluR6 localization, along with its missing partners and ERG phenotype, by treating adult P15 Grm6−/−18 mice with an adeno-associated virus (AAV)-mediated intravitreal delivery of the transgene. In order to compare their efficacy, two different promoters were used: (1) a Grm6-200bp/SV40 promoter that specifically targets ON-BCs (referred to here as the GRM6 promoter) and (2) a cytomegalovirus (CMV) enhancer fused to the chicken β-actin promoter (CAG) promoter driving ubiquitous expression of the transgene. Protein localization, and functional rescue were investigated.
Materials and Methods
Ethical Statement
All animal procedures were performed according to Council Directive 2010/63EU of the European Parliament and the Council of September 22, 2010, on the protection of animals used for scientific purposes; National Institutes of Health guidelines; and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The study was approved by the French Minister of National Superior Education and Research (authorization delivered on January 21, 2019; APAFIS: #1132-201812181048827 v2).
Intravitreal Injections
Two different constructs were designed using two different promoters to rescue the phenotype in the Grm6−/− mouse model. To drive the expression of the transgene specifically to ON-BCs, the GRM6 promoter was used.33 To drive ubiquitous expression of the transgene throughout the retina, the CAG promoter was used.34 These two constructs were encapsidated in the AAV2-7m8 serotype34 and are referred to here as GRM6-Grm6 and CAG-Grm6, respectively. Mice were anesthetized by isoflurane inhalation (5% in oxygen for induction and 2% for maintenance). Intravitreal injections were performed at postnatal day 15. Pupils were dilated (0.5% mydriaticum), and a 33-gauge needle was passed through the sclera at the ora serrata level. A total of 1 µL of viral stock solution at maximum concentrations of 1.6 × 1013 viral genomes (vg)/mL (CAG-Grm6 construct) and 5.8 × 1013 vg/mL (GRM6-Grm6 construct) was injected directly in the vitreous cavity of eight Grm6−/− mice for each promoter. Viral vectors were produced as described in Macé et al.33
Immunolocalization Studies
Animals were sacrificed by CO2 inhalation followed by cervical dislocation. Eyes were removed and dissected to keep the posterior part of the eyes, which were then fixed in ice-cold 4% paraformaldehyde for 20 minutes. Subsequently, eyecups were washed in ice-cold PBS and cryoprotected by increasing concentrations of sucrose (ranging from 10% to 30%) in water and 0.24-M phosphate buffer for 1 hour at 4°C for the 10% sucrose and 20% sucrose solutions and overnight at 4°C under agitation for the 30% sucrose solution. The eyecups were then embedded in 7.5% gelatin/10% sucrose; the blocks were frozen at –40°C in isopentane and kept at –80°C until cutting. Sections of 12-µm thickness were generated using a cryostat (Microm HM 560; Thermo Fisher Scientific, Waltham, MA, USA) and mounted on glass slides (Superfrost Plus; Thermo Fisher Scientific). Mouse retinal sections were blocked for 1 hour at room temperature in PBS, 1×; 10% donkey serum (v/v); and 0.3% Triton X-100. Primary antibodies and the dilutions used were sheep anti-TRPM1 (1:500; Cao et al.35), guinea pig anti-mGluR6 (AP20134SU-N, 1:15000; Acris, Herford, Germany), rabbit anti-Gβ5 (C16068, 1:500; Antibodies Online, Limerick, PA, USA), goat anti-RGS11 (sc-9725, 1:300; Santa-Cruz Biotechnology, Dallas, TX, USA), rabbit anti-RGS7 (1:100; Cao et al.36), mouse anti-GPR179 (AB0887-YOM, 1:200; PrimmBiotech, Cambridge, MA, USA), and H4 anti-dystrophin (1:1000; Vacca et al.37). The sections were incubated with primary antibodies diluted in PBS, 1×; 2% donkey serum; and 0.1% Triton X-100 for 1 hour at room temperature. After washes with PBS, 1×, and 0.1% Triton X-100, the sections were incubated with anti-human, anti-guinea pig, anti-goat, anti-rabbit, or anti-mouse secondary antibodies coupled with Alexa Fluor 488, Alexa Fluor 594, or Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) along with 4′,6-diamidino-2-phenylindole, all used at 1:1000, for 0.5 hour at room temperature. Subsequently, the sections were coverslipped with mounting medium (Mowiol; Millipore Sigma, Burlington, MA, USA). Fluorescence images of retinal sections were acquired with a confocal microscope (FV1000; Olympus, Tokyo, Japan). Images for figures were handled with Image J (National Institutes of Health, Bethesda, MD, USA). The percentage of outer plexiform layer (OPL) presenting any mGluR6 staining was automatically calculated using Image J. After selection of the OPL area, the ratio of red staining representing mGluR6 compared to green staining representing PKCα was determined.
ERG Recordings
Full-field ERG recordings were performed in accordance with the description in Neuillé et al.38 All scotopic ERG responses were recorded first using six increasing light intensities of flashes ranging from 0.003 to 30.0 cd·s/m2. To ensure saturation of rod photoreceptors and the recording of cone-driven responses, a 10-minute light-adaptation step at 20 cd/m2 was done. All data were analyzed with Prism v.6 (GraphPad Software, La Jolla, CA, USA).
Results
mGluR6 Relocalized at the Dendritic Tips of ON-BCs Following Treatment
In Grm6−/− mice, mGluR6 production was completely absent in the OPL (Fig. 1A)18. Its localization at the dendritic tips of ON-BCs is essential to ensure proper transmission of the visual information between photoreceptors and ON-BCs. Relocalization of mGluR6 at the dendritic tips of ON-BCs 5 months following treatment was investigated through immunolocalization studies. Grm6−/−-GRM6-Grm6 retinas treated at P15 displayed areas where mGluR6 was present in the OPL but which was absent in untreated Grm6−/− retinas (n = 8) (Fig. 1A). We also noticed that ON-BC bodies were sometimes stained (Fig. 1B, asterisks). However, mGluR6 staining was only present in small areas of the retina (∼2.5% of the OPL presented any mGluR6 staining) (Fig. 2C, red arrows). Similarly, Grm6−/−-CAG-Grm6 retinas treated at P15 displayed mGluR6 staining in the OPL (Fig. 1A), which seemed to be stronger and more homogeneously distributed (Figs. 1B, 1C) compared to the Grm6−/−-GRM6-Grm6 treated retinas (∼11% of the OPL presented any mGluR6 staining) (Fig. 1C, red arrows). In addition, for the latter construct, staining was also noted in some areas in the inner plexiform layer (IPL) (Fig. 1B). Furthermore, this mGluR6 staining seemed more diffuse in several areas (Fig. 1B). Using either CAG-Grm6 or GRM6-Grm6 constructs, mGluR6 staining was solely observed at the dendritic tips of rod BCs; no staining was observed at the dendritic tips of cone ON-BCs.
Figure 1.

GRM6 localization after treatment. (A) Representative confocal images of cross-sections centered on the OPL of Grm6+/+, untreated Grm6−/−, Grm6−/−-GRM6-Grm6, and Grm6−/−-CAG-Grm6 retinas stained with an antibody against mGluR6 (red). Scale bar: 10 µm. (B) Specific features of Grm6−/−-GRM6-Grm6 (top row, stars represent putative somas of ON-BCs) and Grm6−/−-CAG-Grm6 (bottom row, diffuse staining) retinas. ONL, outer nuclear layer; INL, inner nuclear layer, IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bar: 10 µm. (C) Representative confocal images of cross-sections of the entire retina of mice treated with Grm6−/−-GRM6-Grm6 or Grm6−/−-CAG-Grm6. Red arrows point to mGluR6 staining. Scale bar: 100 µm.
Figure 2.

Localization of TRPM1 after treatment. Representative confocal images of cross-sections centered on the OPL of Grm6+/+, untreated Grm6−/−, Grm6−/−-GRM6-Grm6, and Grm6−/−-CAG-Grm6 retinas co-stained (yellow, merge) with an antibody against mGluR6 (red) and against TRPM1 (green). Scale bar: 10 µm.
Signaling Partners of mGluR6 Were Relocalized at the Dendritic Tips of ON-BCs
It was previously described that several molecules involved in the signaling cascade of ON-BCs were impacted by the absence of mGluR6.2 This is consistent with our findings, which showed reduced or abolished staining of molecules of the same cascade including TRPM1, RGS7, RGS11, and Gβ5 (Figs. 2, 3, second column). Consistent with several observations regarding proper mGluR6-dependant GPR179 localization,26,27 we observed a severe decrease in the localization of GPR179 at the dendritic tips of ON-BCs in Grm6−/− mice (Fig. 3). Grm6−/−-GRM6-Grm6 and Grm6−/−-CAG-Grm6 retinas revealed restoration of TRPM1, Gβ5, and GPR179 localization at the dendritic tips of presumably rod BCs after treatment (Figs. 2, 3). In addition, a partial restoration of the localization of RGS7 and RGS11 was noted, which was more obvious in retinas treated with GRM6-Grm6 than retinas treated with CAG-Grm6 (Fig. 3). Partial relocalization of the presynaptic protein dystrophin at the presumed rod-to-BC synapse, but not at the cone-to-cone ON-BCs, was obtained in mice retinas treated with GRM6-Grm6 but was almost absent in mice retinas treated with CAG-Grm6 (Supplementary Data S1).
Figure 3.

Localization of proteins of the ON-BC signaling cascade after treatment. Representative confocal images of cross-sections centered on the OPL of Grm6+/+, untreated Grm6−/−, Grm6−/−-GRM6-Grm6, and Grm6−/−-CAG-Grm6 retinas co-stained (yellow, merge) with an antibody against mGluR6 (red) and against GPR179, RGS11, RGS7, or Gβ5 (green). Arrows point out the relocalization at the dendritic tips of the ON-BCs. Scale bar: 10 µm.
mGluR6 Protein Restoration Did Not Induce a Functional Rescue
Due to disruption of the signal transmission between photoreceptors and ON-BCs in the Grm6−/− mouse model, the b-wave was abolished in both scotopic and photopic conditions (Fig. 4). In order to study the functional restoration, ERG recordings were performed on P15-treated mice at two time points: 2 months and 4 months post-injection (n = 8). At both time points, none of the treated animals revealed a restoration of the b-wave under either scotopic or photopic conditions after intravitreal injections of either CAG-Grm6 or GRM6-Grm6 AAVs (Fig. 4).
Figure 4.
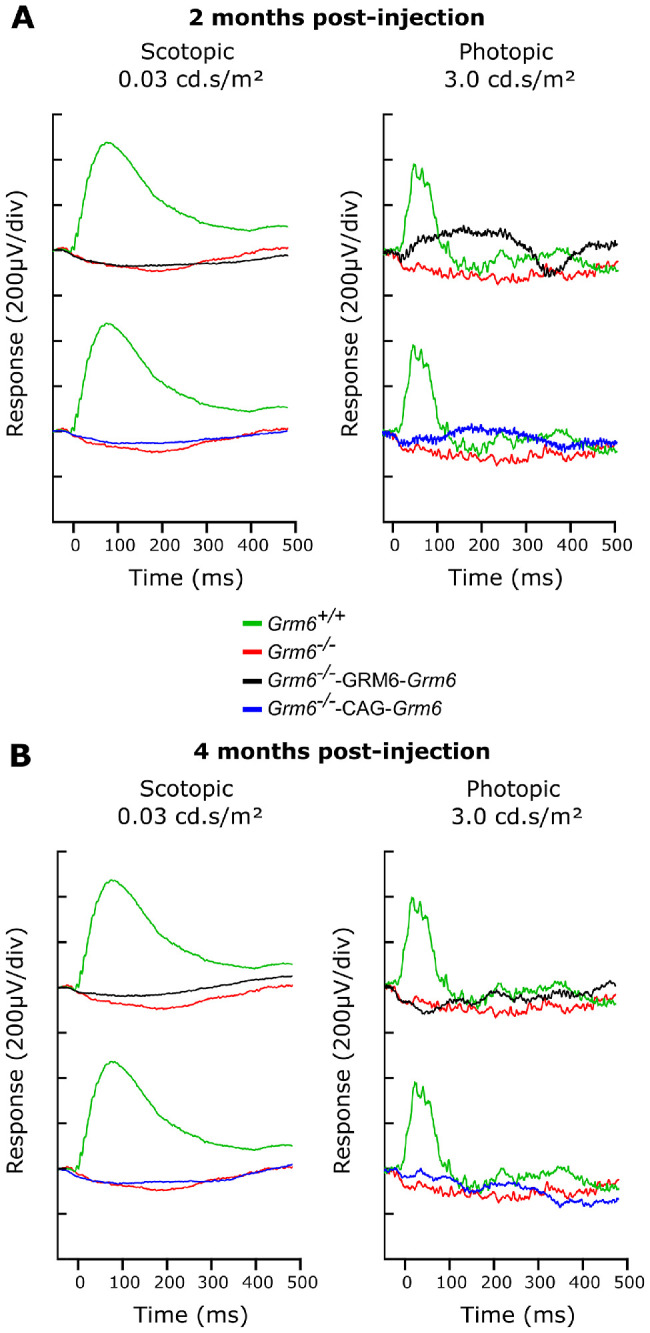
ERG recordings. (A) Representative scotopic ERG traces at 2 months post-injection for Grm6+/+ (green line), untreated Grm6−/− (red line), Grm6−/−-GRM6-Grm6 (black line), and Grm6−/−-CAG-Grm6 (blue line) mice at a flash intensity of 0.03 cd·s/m² under scotopic conditions (left) and 3.0 cd·s/m² under photopic conditions (right). (B) Representative scotopic ERG traces at 4 months post-injection for a flash intensity of 0.03 cd·s/m² (left) and photopic ERG traces for a flash intensity of 3.0 cd·s/m² (right).
Discussion
GRM6 is the first actor of the signaling cascade at the dendritic tips of ON-BCs, responsible for the transmission of the signal from photoreceptors to ON-BCs. When GRM6 is mutated it leads to cCSNB. Patients displaying cCSNB have night blindness, high myopia, nystagmus, and sometimes strabismus, all of which can influence the quality of life during the day and at night. The disease can be correctly diagnosed through full-field ERGs. Patients and mice with this gene defect show loss of mGluR6 function and reveal an electronegative ERG waveform in which the a-wave is preserved but the b-wave is absent.18,39 In addition, mice lacking Grm6 are characterized by the absence of the respective protein, and several other proteins of the same cascade are mislocalized, absent, or reduced at the dendritic tips of ON-BCs. GPR179 and TRPM1 are reduced, and the RGS proteins (Gβ5, RGS7, and RGS11) are absent.24 We investigated the localization of these proteins in Grm6−/− mice and compared our findings with reported studies. We showed that, in Grm6−/− mice, GPR179 localization at the dendritic tips of ON-BCs is dramatically reduced but still present. Ray and co-workers27 reported no difference in GPR179 localization in Grm6−/− mice in immunolocalization studies, but western blot analysis suggested a decrease in GPR179 of ∼50% in Grm6−/− retinas compared to normal retinas. In contrast, Orlandi and co-workers26 reported, as we did, a dramatic reduction of GPR179 at the dendritic tips of ON-BCs in a mouse model lacking Grm6 (nob3). In fact, our study and the study by Orlandi et al.26 used the same antibody, which differed from that of Ray et al.,27 which may explain this discrepancy. Further investigations are needed to explain these different observations. However, GRP179 being reduced but not completely absent at the dendritic tips of ON-BCs in Grm6−/− mice indicates that mGluR6 plays a major but not essential role for the proper localization of GPR179 at the dendritic tips.
To date, for most inherited retinal disorders, treatment is unavailable. However, a gene addition approach mediated by AAV for Leber congenital amaurosis was validated by the Food and Drug Administration in the United States 3 years ago, paving the way to treatment of inherited retinal disorders by gene therapy.40–43 CSNB is a rare heterogeneous group of retinal disorders and as yet incurable.2 Two gene therapy approaches for cCSNB have been reported so far, one targeting Nyx31 and the other targeting Lrit3.32 In both strategies, the wild-type copy of the gene was delivered in newborn (P2 or P5) and adult (P30 or P35) CSNB mice. A partial rescue of the scotopic ERG responses was obtained, mainly in newborn treated mice, under scotopic conditions.31,32 To our knowledge, a gene therapy approach to rescue the phenotype due to the GRM6-gene defect is unavailable. Mutations in GRM6 were found to be the third most prevalent gene defect causing cCSNB.2 To rescue the phenotype of mice lacking mGluR6, here we used a combination of the AAV2.7m8 capsid along with either the GRM6-200bp/SV40 promoter to drive the expression of Grm633 (GRM6-Grm6) or a CAG promoter34 (CAG-Grm6). Although functional restoration in the ERG was not observed, intravitreal injections of adult mice revealed relocalization of mGluR6 and their signaling partners. Furthermore, although the specificity of the GRM6-Grm6 construct for ON-BCs was greater than that of the CAG-Grm6 combination, the transduction rate was less than with the non-specific promoter.
Studying the expression of the transgene by RNA in situ hybridization would explain whether the low production of mGluR6 in the treated mice is due to a low transduction efficacy or a problem with ON-BCs producing the protein from the transgene. The lack of functional rescue and relatively low transduction efficiency of our approach may be due to the size and structure of mGluR6. A substantial limitation of AAV vectors is their small packaging capacity, which is generally considered to be <5 kb.44 Our constructs were 3.5 kb and 3.8 kb in size for the GRM6-Grm6 and CAG-Grm6 constructs, respectively (the coding sequence of Grm6 is 2.6 kb). Together, these constructs are in the range of <5 kb, so encapsidation of those should not be an issue. Successful gene therapies resulting in partial functional restoration for nyctalopin31 and LRIT332 (Varin, J, et al., submitted) using similar vectors have focused on even smaller genes (1.4 kb and 2 kb, respectively), encoding smaller and structurally simpler proteins than mGluR6. We speculate it might be more complicated to produce more complex proteins, such as mGluR6, which has multiple transmembrane domains and more disulfide bonds than smaller molecules. In addition, the constructs used may need to be optimized. Of course, using the full-length promoter and intronic regions to enhance and control expression and protein production would most likely improve the efficacy of the treatment.45 However, the size of such a transgene would be too large to be encapsidated by our AAV approach. Oversized AAV vectors encapsidating up to 9 kb of transgene have not been validated for clinical use because gene fragmentation occurs. As an alternative to the oversized AAV approach, various research groups have tested the use of dual and triple AAV vector systems and reported very low levels of protein expression.46
Recently, Lu and co-workers47 tested different modulations of the Grm6 promoter in order to increase its efficiency. They reported two promoters with high specificity and higher expression of the transgene than the one we applied by using the endogenous promoter of Grm6. The modified sequence was only ∼300 bp larger compared to our GRM6-Grm6 construct and thus could be easily used for gene therapy delivery to rescue the mGluR6 phenotype. Compared with the construct we used, the fluorescence intensity of mCherry under the control of this new promoter was ∼5 times higher.47 However, the number of transduced cells was not significantly improved with this promoter compared to ours. It remains questionable if this promoter would be good enough to increase the transduction efficiency to obtain homogeneous protein immunolocalization and functional rescue. Efficient transduction of ON-BCs is still difficult to obtain, and to overcome this issue other improved AAV–promoter combinations should be tested.
To test the efficiency of those different promoters, fluorescent proteins such as green fluorescent protein or mCherry are often used33,47. However, these promoters used in combination with these genes will localize the respective protein in ON-BCs but not specifically at the dendritic tips of ON-BCs where mGluR6 should be localized for functional rescue33,47. Thus, the transduction efficiency of those vectors may not indicate if trafficking to the dendritic tips of ON-BCs can be achieved. The targeting of transmembrane proteins to neuronal dendrites was previously reported to be myosin dependent.48 We tried to improve this trafficking to the dendritic tips by adding a myosin-binding domain to Grm6; however, this did not result in further improvement in transduction efficiency nor phenotypic rescue (data not shown). In addition, we waited several months after injection to record ERGs and perform immunolocalization studies, as the peak of transgene expression was shown to be at a maximum 6 to 8 weeks after injection.49 However, even 4 months after treatment, the phenotype of treated animals could not be rescued. As the full-field ERG reflects the global response of the retina, it is also possible that the few ON-BCs expressing mGluR6 after treatment were able to depolarize in response to light but, because their numbers were very low, these responses were not detected by ERG. Multi-electrode array recordings or patch-clamp recordings, as described by Scalabrino et al.,31 on treated retinas or ON-BC expressing mGluR6 could have addressed this issue, although a major problem is still the transduction rate. Furthermore, optomotor responses could have been used to investigate the functional rescue similarly to the maze test for Leber congenital amaurosis patients treated with AAV-RPE65,41 but a range of 2.5% to 11% of ON-BCs expressing mGluR6 most likely would not have improved these responses. A subretinal route of delivery could also be considered to enhance the transduction rate of the AAV.50
As previously described, proteins involved in cCSNB also have a role in synapse formation and maintenance. Indeed, in Lrit3−/− mice, a decreased number of invaginating contacts made by cone ON-BCs at the cone pedicle and a striking decrease in the number of triads51 were observed. Normal cone pedicles were also noted, indicating that LRIT3 is not essential for the normal development of the cone-to-cone BC synapse but does play a major role in this process.51 The same observation was made in Gβ5−/− mice in which the number of triads in the OPL was significantly reduced.52 Finally, mGluR6 was shown to play a role at the photoreceptor-to-BC synapse. Indeed, invaginating dendrites of rod BCs are larger and often contain ectopic ribbons, whereas the number of invaginating dendrites of cone ON-BCs and ribbons decreases at the cone pedicles in the Grm6−/− mouse model,29,53 and the reduced localization at the OPL of presynaptic proteins such as dystrophin has been described.30
In conclusion, there are strong indications that proteins localized at the synapse between photoreceptors and ON-BCs also play a structural role. Interestingly, structural alterations have also been noted in patients28. Structural synaptic alterations, or failure to efficiently relocalize presynaptic proteins, could therefore explain the lack of functional rescue despite restoration of mGluR6 in our Grm6−/− mice, which were treated at an adult age when the synapses are fully formed. In addition, mGluR6 was also detectable in the rat retina at P6 to P8, diffusely distributed in the somata and dendrites of the INL cells.54 This finding may indicate that the presence of mGluR6 may be essential as early as P6 to ensure correct synaptogenesis and functional restoration later. However, with regard to the potential plasticity of the photoreceptors to BC synapses, Wang and colleagues55 recently studied the plasticity of the synapses between rods and rod–BCs in a mouse model of retinitis pigmentosa. They showed that in 4-week-old mice a certain plasticity of the synapses could be noticed as treated mice rescued triads of invaginating BC dendrites, and there was more robust and distributed mGluR6 staining in the treated animals. On the other hand, Shen et al.56 showed a steep maturational decline of cone-to-cone BC homeostatic plasticity by comparing this plasticity in mice whose cones begin to degenerate at P10 or at P30. Therefore, at least in the case of rod-to-rod BC synapses and potentially in the case of cone-to-cone BC synapses, it appears that P15 retinas are able to remodel the synapse, which could lead to functional rescue. It would be interesting to document if such a plasticity could be obtained in treated cCSNB models through electron microscopy.
Finally, regulatory proteins missing at the dendritic tips of ON-BCs (RGS7, RGS11, and Gβ5) when mGluR6 was absent were detected when mGluR6 was present in the OPL after delivery of the GRM6-Grm6 construct. This was less obvious after delivery of the CAG-Grm6 construct, which might partially explain why, even when the transduction rate was increased using a CAG promoter, there was still no functional rescue observed by ERG recordings in these treated mice, as RGS11 and RGS7 are involved in the sensitivity and time course of light-evoked responses36.
We showed here for the first time, to our knowledge, the restoration of mGluR6 localization at the dendritic tips of ON-BCs following a gene addition strategy in P15-treated Grm6−/− mice. This restoration led to relocalization of other partners of the cascade, such as TRPM1, GPR179, RGS7, RGS11, and Gβ5; however, functional rescue after treatment as measured by ERG failed to appear. Further studies are needed to improve vector–promoter combinations and potentially the mode of administration (and therefore the efficiency of ON-BC transduction), but our findings are encouraging and will serve as a base to develop further gene therapy trials for cCSNB due to mutations in GRM6.
Supplementary Material
Acknowledgments
The authors are grateful for the animal housing and imaging provided at the Institut de la Vision and, more specifically, for the assistance of Julie Degradin, Manuel Simonutti, Quenol Cesar, Marie-Laure Niepon, and Stéphane Fouquet. We thank Kirill Martemyanov, Yan Cao, and Anna Verschueren for the fruitful discussions. We also thank Cyrille Vaillend for his help regarding the dystrophin experiment.
Supported by Retina France (CZ); the French Muscular Dystrophy Association (AFM-Téléthon; CZ); UNADEV-Aviesan call 2015 (CZ); Fondation Voir et Entendre (CZ); Prix Dalloz for “la recherche en ophtalmologie” (CZ); Fondation pour la Recherche Médicale (FRM DVS20131228918) in partnership with Fondation Roland Bailly (CZ); and Ville de Paris and Région Ile de France, LABEX LIFESENSES (reference ANR-10-LABX-65), supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d'Avenir programme (ANR11-IDEX-0004-0), with the support of the Programme Investissements d'Avenir IHU FOReSIGHT (ANR-18-IAHU-01) and the Ministère de l'enseignement supérieur et de la recherché (JV). The funders had no role in the study design; data collection, analysis, and interpretation; decision to publish, or preparation of the manuscript.
Disclosure: J. Varin, None; N. Bouzidi, None; M.M.D.S. Dias, None; T. Pugliese, None; C. Michiels, None; C. Robert, None; M. Desrosiers, None; J.-A. Sahel, None; I. Audo, None; D. Dalkara, Adverum Biotechnologies (P); C. Zeitz, None
References
- 1. Schubert G, Bornschein H. Analysis of the human electroretinogram. Ophthalmologica. 1952; 123(6): 396–413. [DOI] [PubMed] [Google Scholar]
- 2. Zeitz C, Robson AG, Audo I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res. 2015; 45: 58–110. [DOI] [PubMed] [Google Scholar]
- 3. Miyake Y, Yagasaki K, Horiguchi M, Kawase Y, Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch Ophthalmol. 1986; 104(7): 1013–1020. [DOI] [PubMed] [Google Scholar]
- 4. Pusch CM, Zeitz C, Brandau O, et al.. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet. 2000; 26(3): 324–327. [DOI] [PubMed] [Google Scholar]
- 5. Bech-Hansen NT, Naylor MJ, Maybaum TA, et al.. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet. 2000; 26(3): 319–323. [DOI] [PubMed] [Google Scholar]
- 6. Audo I, Kohl S, Leroy BP, et al.. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2009; 85(5): 720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Genderen MM, Bijveld MM, Claassen YB, et al.. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009; 85(5): 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Z, Sergouniotis PI, Michaelides M, et al.. Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet. 2009; 85(5): 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeitz C, van Genderen M, Neidhardt J, et al.. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci. 2005; 46(11): 4328–4335. [DOI] [PubMed] [Google Scholar]
- 10. Dryja TP, McGee TL, Berson EL, et al.. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci USA. 2005; 102(13): 4884–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Audo I, Bujakowska K, Orhan E, et al.. Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2012; 90(2): 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peachey NS, Ray TA, Florijn R, et al.. GPR179 is required for depolarizing bipolar cell function and is mutated in autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2012; 90(2): 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeitz C, Jacobson SG, Hamel CP, et al.. Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2013; 92(1): 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakajima Y, Iwakabe H, Akazawa C, et al.. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for L-2-amino-4-phosphonobutyrate. J Biol Chem. 1993; 268(16): 11868–11873. [PubMed] [Google Scholar]
- 15. Nawy S. The metabotropic receptor mGluR6 may signal through G(o), but not phosphodiesterase, in retinal bipolar cells. J Neurosci. 1999; 19(8): 2938–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morgans CW, Brown RL, Duvoisin RM. TRPM1: the endpoint of the mGluR6 signal transduction cascade in retinal ON-bipolar cells. BioEssays. 2010; 32(7): 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morgans CW, Zhang J, Jeffrey BG, et al.. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc Natl Acad Sci USA. 2009; 106(45): 19174–19178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masu M, Iwakabe H, Tagawa Y, et al.. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell. 1995; 80(5): 757–765. [DOI] [PubMed] [Google Scholar]
- 19. Maddox DM, Vessey KA, Yarbrough GL, et al.. . Allelic variance between GRM6 mutants, Grm6nob3 and Grm6nob4 results in differences in retinal ganglion cell visual responses. J Physiol. 2008; 586(pt 18): 4409–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinto LH, Vitaterna MH, Shimomura K, et al.. Generation, identification and functional characterization of the nob4 mutation of Grm6 in the mouse. Vis Neurosci. 2007; 24(1): 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qian H, Ji R, Gregg RG, Peachey NS. Identification of a new mutant allele, Grm6nob7, for complete congenital stationary night blindness. Vis Neurosci. 2015; 32: E004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peachey NS, Hasan N, FitzMaurice B, et al.. A missense mutation in Grm6 reduces but does not eliminate mGluR6 expression or rod depolarizing bipolar cell function. J Neurophysiol. 2017; 118(2): 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu Y, Dhingra A, Fina ME, Koike C, Furukawa T, Vardi N. mGluR6 deletion renders the TRPM1 channel in retina inactive. J Neurophysiol. 2012; 107(3): 948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gregg RG, Ray TA, Hasan N, McCall MA; Peachey NS. Interdependence among members of the mGluR6 G-protein mediated signalplex of retinal depolarizing bipolar cells. In: Martemyanov KA, Sampath AP, eds. G Protein Signaling Mechanisms in the Retina. New York: Springer; 2014: 67–79 [Google Scholar]
- 25. Orlandi C, Posokhova E, Masuho I, et al.. GPR158/179 regulate G protein signaling by controlling localization and activity of the RGS7 complexes. J Cell Biol. 2012; 197(6): 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orlandi C, Cao Y, Martemyanov KA. Orphan receptor GPR179 forms macromolecular complexes with components of metabotropic signaling cascade in retina ON-bipolar neurons. Invest Ophthalmol Vis Sci. 2013; 54(10): 7153–7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ray TA, Heath KM, Hasan N, et al.. GPR179 is required for high sensitivity of the mGluR6 signaling cascade in depolarizing bipolar cells. J Neurosci. 2014; 34(18): 6334–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Godara P, Cooper RF, Sergouniotis PI, et al.. Assessing retinal structure in complete congenital stationary night blindness and Oguchi disease. Am J Ophthalmol. 2012; 154(6): 987–1001.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsukamoto Y, Omi N. Effects of mGluR6-deficiency on photoreceptor ribbon synapse formation: comparison of electron microscopic analysis of serial sections with random sections. Vis Neurosci. 2014; 31(1): 39–46. [DOI] [PubMed] [Google Scholar]
- 30. Tummala SR, Dhingra A, Fina ME, Li JJ, Ramakrishnan H, Vardi N. Lack of mGluR6-related cascade elements leads to retrograde trans-synaptic effects on rod photoreceptor synapses via matrix-associated proteins. Eur J Neurosci. 2016; 43(11): 1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scalabrino ML, Boye SL, Fransen KM, et al.. Intravitreal delivery of a novel AAV vector targets ON bipolar cells and restores visual function in a mouse model of complete congenital stationary night blindness. Hum Mol Genet. 2015; 24(21): 6229–6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hasan N, Pangeni G, Cobb CA, et al.. Presynaptic expression of LRIT3 transsynaptically organizes the postsynaptic glutamate signaling complex containing TRPM1. Cell Rep. 2019; 27(11): 3107–3116.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Macé E, Caplette R, Marre O, et al.. Targeting channelrhodopsin-2 to ON-bipolar cells with vitreally administered AAV Restores ON and OFF visual responses in blind mice. Mol Ther. 2015; 23(1): 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dalkara D, Byrne LC, Klimczak RR, et al.. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013; 5(189): 189ra176. [DOI] [PubMed] [Google Scholar]
- 35. Cao Y, Posokhova E, Martemyanov KA. TRPM1 forms complexes with nyctalopin in vivo and accumulates in postsynaptic compartment of ON-bipolar neurons in mGluR6-dependent manner. J Neurosci. 2011; 31(32): 11521–11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cao Y, Pahlberg J, Sarria I, Kamasawa N, Sampath AP, Martemyanov KA. Regulators of G protein signaling RGS7 and RGS11 determine the onset of the light response in ON bipolar neurons. Proc Natl Acad Sci USA. 2012; 109(20): 7905–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vacca O, Charles-Messance H, El Mathari B, Sene A, Barbe P, Fouquet S, Aragón J, Darche M, Giocanti-Aurégan A, Paques M, Sahel J-A, Tadayoni R, Montañez C, Dalkara D, Rendon A. AAV-mediated gene therapy in Dystrophin-Dp71 deficient mouse leads to blood-retinal barrier restoration and oedema reabsorption. Hum Mol Genet. 2016; 25(14): 3070–3079. [DOI] [PubMed] [Google Scholar]
- 38. Neuille M, El Shamieh S, Orhan E, et al.. Lrit3 deficient mouse (nob6): a novel model of complete congenital stationary night blindness (cCSNB). PLoS One. 2014; 9(3): e90342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zeitz C, Forster U, Neidhardt J, et al.. Night blindness-associated mutations in the ligand-binding, cysteine-rich, and intracellular domains of the metabotropic glutamate receptor 6 abolish protein trafficking. Hum Mutat. 2007; 28(8): 771–780. [DOI] [PubMed] [Google Scholar]
- 40. Leber T. Über retinitis pigmentosa und angeborene amaurose [in German]. Archiv für Ophthalmologie. 1869; 15: 1–25. [Google Scholar]
- 41. Bainbridge JW, Smith AJ, Barker SS, et al.. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008; 358(21): 2231–2239. [DOI] [PubMed] [Google Scholar]
- 42. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008; 358(21): 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cideciyan AV, Aleman TS, Boye SL, et al.. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA. 2008; 105(39): 15112–15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010; 18(1): 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Powell SK, Rivera-Soto R, Gray SJ. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov Med. 2015; 19(102): 49–57. [PMC free article] [PubMed] [Google Scholar]
- 46. French LS, Mellough CB, Chen FK, Carvalho LS. A review of gene, drug and cell-based therapies for Usher syndrome. Front Cell Neurosci. 2020; 14: 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lu Q, Ganjawala TH, Ivanova E, Cheng JG, Troilo D, Pan ZH. AAV-mediated transduction and targeting of retinal bipolar cells with improved mGluR6 promoters in rodents and primates. Gene Ther. 2016; 23(8-9): 680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lewis TL Jr, Mao T, Svoboda K, Arnold DB. Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat Neurosci. 2009; 12(5): 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Natkunarajah M, Trittibach P, McIntosh J, et al.. Assessment of ocular transduction using single-stranded and self-complementary recombinant adeno-associated virus serotype 2/8. Gene Ther. 2008; 15(6): 463–467. [DOI] [PubMed] [Google Scholar]
- 50. Trapani I, Auricchio A. Seeing the light after 25 years of retinal gene therapy. Trends Mol Med. 2018; 24(8): 669–681. [DOI] [PubMed] [Google Scholar]
- 51. Neuille M, Cao Y, Caplette R, et al.. LRIT3 differentially affects connectivity and synaptic transmission of cones to ON- and OFF-bipolar cells. Invest Ophthalmol Vis Sci. 2017; 58(3): 1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rao A, Dallman R, Henderson S, Chen CK. Gβ5 is required for normal light responses and morphology of retinal ON-bipolar cells. J Neurosci. 2007; 27(51): 14199–14204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ishii M, Morigiwa K, Takao M, et al.. Ectopic synaptic ribbons in dendrites of mouse retinal ON- and OFF-bipolar cells. Cell Tissue Res. 2009; 338(3): 355–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nomura A, Shigemoto R, Nakamura Y, Okamoto N, Mizuno N, Nakanishi S. Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell. 1994; 77(3): 361–369. [DOI] [PubMed] [Google Scholar]
- 55. Wang T, Pahlberg J, Cafaro J, et al.. Activation of rod input in a model of retinal degeneration reverses retinal remodeling and induces formation of functional synapses and recovery of visual signaling in the adult retina. J Neurosci. 2019; 39(34): 6798–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shen N, Wang B, Soto F, Kerschensteiner D. Homeostatic plasticity shapes the retinal response to photoreceptor degeneration. Curr Biol. 2020; 30(10): 1916–1926.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.