

Abstract
The global spread of SARS-CoV-2/COVID-19 is devastating health systems and economies worldwide. Recombinant or vaccine-induced neutralizing antibodies are used to combat the COVID-19 pandemic. However, the recently emerged SARS-CoV-2 variants B.1.1.7 (UK), B.1.351 (South Africa), and P.1 (Brazil) harbor mutations in the viral spike (S) protein that may alter virus-host cell interactions and confer resistance to inhibitors and antibodies. Here, using pseudoparticles, we show that entry of all variants into human cells is susceptible to blockade by the entry inhibitors soluble ACE2, Camostat, EK-1, and EK-1-C4. In contrast, entry of the B.1.351 and P.1 variant was partially (Casirivimab) or fully (Bamlanivimab) resistant to antibodies used for COVID-19 treatment. Moreover, entry of these variants was less efficiently inhibited by plasma from convalescent COVID-19 patients and sera from BNT162b2-vaccinated individuals. These results suggest that SARS-CoV-2 may escape neutralizing antibody responses, which has important implications for efforts to contain the pandemic.
Keywords: SARS-CoV-2, COVID-19, variants, variants of concern, VOC, B.1.1.7, B.1.351, P.1, spike protein, host cell entry, antibodies, neutralization, escape
Graphical abstract
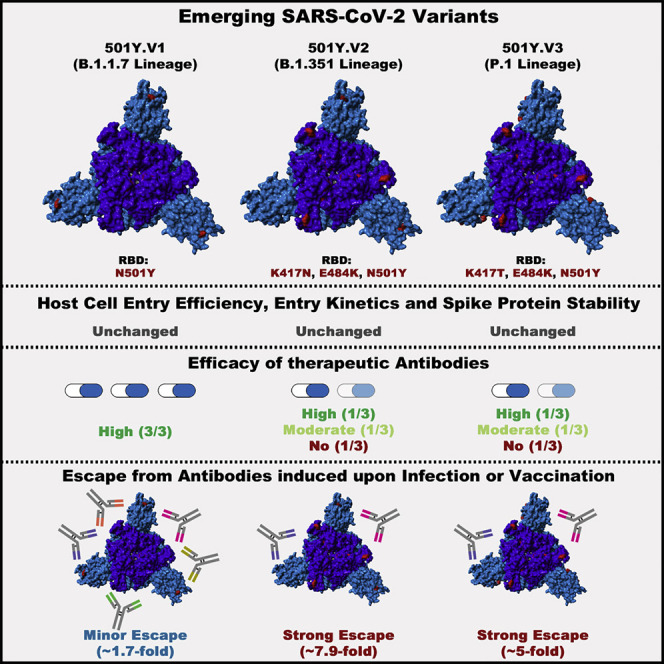
Comparison of the SARS-CoV-2 variants B.1.1.7, B.1.351, and P.1 shows that inhibitors under clinical evaluation are still effective in blocking entry, though the B.1.351 and P.1 variants evade antibody responses induced upon infection as well as vaccination and evade certain therapeutic antibodies.
Introduction
The pandemic spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), is ravaging economies and health systems worldwide and has caused more than 2.3 million deaths (WHO, 2020). SARS-CoV-2, an enveloped, positive-strand RNA virus, uses its envelope protein spike (S) to enter target cells, and the viral and cellular factors involved in cell entry constitute targets for antiviral intervention.
Host cell entry depends on S protein binding to the cellular receptor angiotensin-converting enzyme 2 (ACE2) and S protein priming by the cellular serine protease TMPRSS2 (Hoffmann et al., 2020b; Zhou et al., 2020), and these processes can be disrupted by soluble ACE2 and serine protease inhibitors (Hoffmann et al., 2020b; Monteil et al., 2020; Zhou et al., 2020). The suitability of these agents for COVID-19 treatment is currently being evaluated within clinical trials. Further, the S protein of SARS-CoV-2 is the main target for neutralizing antibodies, and several recombinant neutralizing antibodies have been granted Emergency Use Authorization (EUA) for COVID-19 treatment (Baum et al., 2020a; Chen et al., 2020). Finally, protective mRNA- and vector-based vaccines encoding the SARS-CoV-2 S protein have been approved for human use and are considered key to the containment of the COVID-19 pandemic (Baden et al., 2021; Polack et al., 2020).
The genetic information of SARS-CoV-2 has remained relatively stable after the detection of the first cases in Wuhan, China, in the winter season of 2019. The only exception was a D614G change in the viral S protein that became dominant early in the pandemic and that has been associated with increased transmissibility (Korber et al., 2020; Plante et al., 2020; Volz et al., 2021). In contrast, D614G has only a moderate impact on SARS-CoV-2 neutralization by sera from COVID-19 patients and by sera from vaccinated individuals (Korber et al., 2020; Weissman et al., 2021). In the recent weeks, several SARS-CoV-2 variants emerged that seem to exhibit increased transmissibility. These variants harbor mutations in the viral S protein that may compromise immune control, raising concerns that the rapid spread of these variants might undermine current efforts to control the pandemic.
The SARS-CoV-2 variant B.1.1.7 (UK variant), also termed variant of concern (VOC) 202012/01 or 20I/501Y.V1, emerged in the United Kingdom and was associated with a surge of COVID-19 cases (Kidd et al., 2021; Leung et al., 2021). Subsequently, spread of the B.1.1.7 variant in other countries was reported (Claro et al., 2021; Galloway et al., 2021; Surleac et al., 2021; Umair et al., 2021; Yadav et al., 2021). It harbors nine mutations in the S protein, six of which are located in the surface unit of the S protein, S1, and three are found in the transmembrane unit, S2 (Figure 1 ). Exchange N501Y is located in the receptor-binding domain (RBD), a domain within S1 that interacts with ACE2, and its presence was linked to increased human-human transmissibility (Leung et al., 2021; Zhao et al., 2021). Variants B.1.351 (501Y.V2, also termed South Africa variant; Mwenda et al., 2021) and P.1 (501Y.V3, also termed Brazil variant; Francisco et al., 2021) were also purported to be more transmissible, and these variants harbor 9 and 11 mutations in their S proteins, respectively, including three changes in the RBD, K417N/T, E484K, and N501Y (Figure 1) (CDC, 2021). However, it is largely unclear how the mutations present in the S proteins of B.1.1.7, B.1.351, and P.1 impact host cell interactions and susceptibility to entry inhibitors and antibody-mediated neutralization.
Figure 1.

Schematic overview of the S proteins from the SARS-CoV-2 variants under study
The location of the mutations in the context of S protein domain organization is shown in the upper panel. RBD, receptor-binding domain; TD, transmembrane domain. The location of the mutations in the context of the trimeric S protein is shown in the lower panel. Color code: light blue, S1 subunit with RBD in dark blue; gray, S2 subunit; orange, S1/S2 and S2′ cleavage sites; red, mutated amino acid residues.
Here, we show that the S protein of the B.1.1.7, B.1.351, and P.1 variants mediate robust entry into human cell lines and that entry is blocked by soluble ACE2 (sACE2), protease inhibitors active against TMPRSS2, and membrane fusion inhibitors. In contrast, monoclonal antibodies with EUA for COVID-19 treatment partially or completely failed to inhibit entry driven by the S proteins of the B.1.351 and P.1 variants. Similarly, these variants were less efficiently inhibited by convalescent plasma and sera from individuals vaccinated with BNT162b2. Our results suggest that SARS-CoV-2 variants B.1.351 and P.1 can evade inhibition by neutralizing antibodies.
Results
The S proteins of the SARS-CoV-2 variants mediate robust entry into human cell lines
We first investigated whether the S proteins of SARS-CoV-2 wild type (WT) (Wuhan-1 isolate with D614G exchange), B.1.1.7, B.1.351, and P.1 variants (Figure 1) mediated robust entry into cell lines. For this, we used a vesicular stomatitis virus (VSV)-based vector pseudotyped with the respective S proteins (Figure S1 A). This system faithfully mimics key aspects of SARS-CoV-2 entry into cells, including ACE2 engagement, priming of the S protein by TMPRSS2, and antibody-mediated neutralization (Hoffmann et al., 2020b; Riepler et al., 2020; Schmidt et al., 2020). The following cell lines are frequently used for SARS-CoV-2 research and were employed as target cells in our study: the African green monkey kidney cell line Vero, Vero cells engineered to express TMPRSS2, the human embryonic kidney cell line 293T, 293T cells engineered to express ACE2, the human lung cell line Calu-3, and the human colon cell line Caco-2. All cell lines tested express endogenous ACE2. In addition, Calu-3 and Caco-2 cells express endogenous TMPRSS2 (Böttcher-Friebertshäuser et al., 2011; Kleine-Weber et al., 2018).
Figure S1.

Graphical summary of the generation and use of VSV pseudotype particles bearing SARS-2-S and representative transduction data, related to Figure 2
(A) Schematic illustration of how SARS-2-S-bearing VSV pseudotype particles are generated and used for transduction experiments.
(B) Raw transduction data (cps, counts per second) from a representative experiment. Presented are the data from a single representative experiment conducted with technical quadruplicates. Error bars indicate the SD. Bald pseudotype particles bearing no viral glycoprotein and particles harboring VSV-G served as negative (assay background) and positive controls, respectively.
All S proteins studied were robustly expressed and mediated formation of syncytia in transfected cells (Figure 2 A). Entry into all cell lines was readily detectable (Figure 2B; Figure S1B). Particles bearing the S proteins of the SARS-CoV-2 variants entered 293T (P.1) and 293T-ACE2 (B.1.351 and P.1) cells with slightly reduced efficiency as compared to particles bearing WT S protein, while the reverse observation was made for Calu-3 cells (B.1.351). For the remaining cell lines, no significant differences in entry efficiency were observed (Figure 2B). Collectively, these results indicate that the mutations present in the S proteins of the B.1.1.7, B.1.351, and P.1 variants are compatible with robust entry into human cells.
Figure 2.

S proteins from SARS-CoV-2 variants drive entry into human cell lines
(A) Directed expression of SARS-CoV-2 S proteins (SARS-2-S) in A549-ACE2 cells leads to the formation of syncytia. S protein expression was detected by immunostaining using an antibody directed against a C-terminal HA-epitope tag. Presented are the data from one representative experiment. Similar results were obtained in four biological replicates.
(B) The S proteins of the SARS-CoV-2 variants mediate robust entry into cell lines. The indicated cell lines were inoculated with pseudotyped particles bearing the S proteins of the indicated SARS-CoV-2 variants or wild-type (WT) SARS-CoV-2 S. Transduction efficiency was quantified by measuring virus-encoded luciferase activity in cell lysates at 16–20 h post transduction. Presented are the average (mean) data from six biological replicates (each conducted with technical quadruplicates). Error bars indicate the standard error of the mean (SEM). Statistical significance of differences between WT and variant S proteins was analyzed by one-way analysis of variance (ANOVA) with Dunnett’s posttest (p > 0.05, not significant [not indicated]; p ≤ 0.05, *; p ≤ 0.01, **; p ≤ 0.001, ***). See also Figure S1.
The S proteins of the SARS-CoV-2 variants mediate fusion of human cells
The S protein of SARS-CoV-2 drives cell-cell fusion resulting in the formation of syncytia, and this process might contribute to viral pathogenesis (Buchrieser et al., 2021). We employed a cell-cell fusion assay to determine whether the S proteins of the B.1.1.7, B.1.351, and P.1 variants drive fusion with human cells. For this, the S proteins were expressed in effector cells, which were subsequently mixed with target cells engineered to express ACE2 alone or in conjunction with TMPRSS2. The S protein of SARS-CoV was included as control. The SARS-CoV S protein failed to mediate fusion with target cells expressing only ACE2 but efficiently drove fusion with cells coexpressing ACE2 and TMPRSS2 (Figures 3A and 3B), as expected (Hoffmann et al., 2020a). In contrast, the SARS-CoV-2 S protein mediated efficient membrane fusion in the absence of TMPRSS2 expression in target cells (Figures 3A and 3B), again in keeping with expectations (Hoffmann et al., 2020a). Finally, the S proteins of all SARS-CoV-2 variants tested facilitated cell-cell fusion with similar (B.1.1.7) or slightly reduced (B.1.351 and P.1) efficiency as compared to WT S protein (Figures 3A and 3B).
Figure 3.

The S proteins of the SARS-CoV-2 variants drive robust cell-cell fusion
(A) Quantitative cell-cell fusion assay. S protein-expressing effector cells were mixed with ACE2 or ACE2/TMPRSS2-expressing target cells, and cell-cell fusion was analyzed by measuring luciferase activity in cell lysates. Presented are the average (mean) data from four biological replicates (each performed with technical triplicates). Error bars indicate the SEM. Statistical significance of differences between WT and variant S proteins (or SARS-S) was analyzed by one-way ANOVA with Dunnett’s posttest (p > 0.05, not significant [not indicated]; p ≤ 0.05, *; p ≤ 0.01, **; p ≤ 0.001, ***).
(B) Qualitative fusion assay. A549-ACE2 (left) and A549-ACE2/TMPRSS2 (right) cells were transfected to express the indicated S proteins (or no viral protein) along with DsRed. At 24 h post-transfection, cells were fixed and analyzed for the presence of syncytia by fluorescence microscopy (magnification: 10×). Presented are representative images from a single experiment. Data were confirmed in three additional experiments.
Similar stability and entry kinetics of particles bearing WT and variant S proteins
We next investigated whether the S proteins of the SARS-CoV-2 variants showed altered stability, which may contribute to the alleged increased transmissibility of the viral variants. For this, we incubated S protein-bearing particles for different time intervals at 33°C, a temperature that is present in the nasal cavity, and subsequently assessed their capacity to enter target cells. The efficiency of cell entry markedly decreased upon incubation of particles at 33°C for more than 8 h, but no appreciable differences were observed between particles bearing S proteins from SARS-CoV-2 WT or variants (Figure 4 A).
Figure 4.

Particles bearing the S proteins of SARS-CoV-2 variants exhibit similar stability and entry kinetics
(A) Particles bearing the indicated S proteins were incubated for different time intervals at 33°C, snap frozen, thawed, and inoculated onto Vero cells. Entry of particles that were frozen immediately was set as 100%.
(B) Particles bearing the indicated S proteins were incubated for the indicated time intervals with Vero cells. Subsequently, the cells were washed and luciferase activity determined. Transduction measured for particles incubated with cells for 24 h (maximum incubation time = time of luciferase measurement) was set as 100%.
For both panels, the average (mean) data from three biological replicates (each performed with technical quadruplicates) is presented. Error bars indicate the SEM. Statistical significance of differences between WT and variant S proteins was analyzed by two-way ANOVA with Dunnett’s posttest (p > 0.05, not significant [not indicated]; p ≤ 0.05, *; p ≤ 0.01, **; p ≤ 0.001, ***).
Although the S proteins of the SARS-CoV-2 variants did not differ markedly from WT S protein regarding stability and entry efficiency, they might mediate entry with different kinetics as compared to WT S protein. To investigate this possibility, we incubated target cells with S protein-bearing particles for the indicated time intervals, removed unbound virus by washing, and universally determined entry efficiency at 24 h post inoculation. Entry efficiency increased with the time available for particle adsorption to cells, but no differences were observed between particles bearing WT S protein or S protein from SARS-CoV-2 variants (Figure 4B). Confirmation of the results with lung cells is pending. However, the data available at present indicate that there might be no major differences between WT SARS-CoV-2 and SARS-CoV-2 variants B.1.1.7, B.1.351, and P.1 regarding S protein stability and entry kinetics.
Soluble ACE2, TMPRSS2 inhibitors, and membrane fusion inhibitors block entry
sACE2 blocks SARS-CoV-2 entry into cells and is in clinical development for COVID-19 therapy (Monteil et al., 2020). Similarly, the clinically proven protease inhibitors Camostat and Nafamostat block TMPRSS2-dependent SARS-CoV-2 cell entry, and their potential for COVID-19 treatment is currently being assessed (Hoffmann et al., 2020b, 2020c). Finally, the membrane fusion inhibitor EK-1 and its optimized lipid-conjugated derivative EK-1-C4 block SARS-CoV-2 entry by preventing conformational rearrangements in the S protein that are required for membrane fusion (Xia et al., 2020). We asked whether entry driven by the S proteins of the B.1.1.7, B.1.351, and P.1 variants can be blocked by these inhibitors. All inhibitors were found to be active, although entry mediated by the S proteins of the SARS-CoV-2 variants was slightly less sensitive to blockade by sACE2 as compared to WT S protein (Figure 5 ). In addition, entry driven by the S protein of the P.1 variant was slightly more sensitive to blockade by EK-1 and EK-1-C4 as compared to the other S proteins tested (Figure 5). These results suggest that sACE2, TMPRSS2 inhibitors, and membrane fusion inhibitors will be active against the B.1.1.7, B.1.351, and P.1 variants.
Figure 5.

Entry driven by the S proteins of the SARS-CoV-2 variants can be blocked with soluble ACE2, protease inhibitors targeting TMPRSS2, and a membrane fusion inhibitor
Top row, left panel: S protein-bearing particles were incubated with different concentrations of soluble ACE2 (30 min, 37°C) before being inoculated onto Caco-2 cells. Top row, middle and right panel: Caco-2 target cells were pre-incubated with different concentrations of serine protease inhibitor (Camostat or Nafamostat; 1 h, 37°C) before being inoculated with particles harboring the indicated S proteins. Bottom row, both panels: the peptidic fusion inhibitor EK-1 and its improved lipidated derivate EK-1-C4 were incubated with particles at indicated concentrations (30 min, 37°C) and then added to Caco-2 cells. All panels: transduction efficiency was quantified by measuring virus-encoded luciferase activity in cell lysates at 16–20 h post-transduction. For normalization, SARS-CoV-2 S protein-driven entry in the absence of soluble ACE2 or inhibitor was set as 0% inhibition. Presented are the average (mean) data from three biological replicates (each performed with technical triplicates [EK-1, EK-1-C4] or quadruplicates [soluble ACE2, Camostat, Nafamostat]). Error bars indicate the SEM. Statistical significance of differences between WT and variant S proteins was analyzed by two-way ANOVA with Dunnett’s posttest (p > 0.05, not significant [not indicated]; p ≤ 0.05, *; p ≤ 0.01, **; p ≤ 0.001, ***).
Resistance against antibodies used for COVID-19 treatment
A cocktail of monoclonal antibodies (REGN-COV2, consisting of Casirivimab and Imdevimab) and the monoclonal antibody Bamlanivimab block SARS-CoV-2 WT infection (Figure S2 ) and have received EUA for COVID-19 therapy. We analyzed whether these antibodies can inhibit entry driven by the S proteins of the B.1.1.7, B.1.351, and P.1 variants. Entry driven by the S proteins of all variants was comparably inhibited by Imdevimab, while entry driven by the S proteins of the B.1.351 and P.1 variants was partially resistant against Casirivimab (Figure 6 ). A cocktail of both antibodies (REGN-COV2) efficiently inhibited entry mediated by the S proteins of all variants. Finally, entry mediated by the S proteins of the B.1.351 and P.1 variant was completely resistant to REGN10989 and Bamlanivimab, while the S protein of the B.1.1.7 variant was efficiently blocked by all antibodies tested (Figure 6). Collectively, these data indicate that single antibodies with EUA might provide incomplete (Casirivimab) or no (Bamlanivimab) protection against the B.1.351 and P.1 variants.
Figure S2.

Location of SARS-2-S RBD mutations K417N/T, E484K, and N501Y with respect to the binding interface of the REGN-COV2 antibody cocktail and the monoclonal antibody Bamlanivimab, related to Figure 6
The protein models of the SARS-2-S receptor-binding domain (RBD, blue) in complex with antibodies Casirivimab (orange) and Imdevimab (green) (A) were constructed based on the 6XDG template (Hansen et al., 2020), while the protein models of the SARS-2-S RBD in complex with antibody Bamlanivimab (purple) (B) were based on the 7L3N template (Jones et al., 2020). Residues highlighted in red indicate amino acid variations found in emerging SARS-CoV-2 variants.
Figure 6.

Cell entry mediated by the S proteins of SARS-CoV-2 variants B.1.351 and P.1 is partially or fully resistant to inhibition by monoclonal antibodies with EUA
Pseudotypes bearing the indicated S proteins were incubated (30 min, 37°C) with different concentrations of control antibody (hIgG), four different monoclonal antibodies (Casirivimab, Imdevimab, REGN10989, Bamlanivimab), or a combination of Casirivimab and Imdevimab, as present in the REGN-CoV2 antibody cocktail, before being inoculated onto target Vero cells. Transduction efficiency was quantified by measuring virus-encoded luciferase activity in cell lysates at 16–20 h post-transduction. For normalization, inhibition of S protein-driven entry in samples without antibody was set as 0%. Presented are the data from a single experiment performed with technical triplicates. Data were confirmed in a separate experiment. Error bars indicate standard deviation (SD). See also Figure S2.
Reduced neutralization by plasma from convalescent patients
SARS-CoV-2 infection can induce the production of neutralizing antibodies, and these antibodies are believed to contribute to protection from reinfection (Rodda et al., 2020; Wajnberg et al., 2020). Therefore, it is important to elucidate whether B.1.1.7, B.1.351, and P.1 variants are efficiently neutralized by antibody responses in convalescent COVID-19 patients. We addressed this question using plasma collected from COVID-19 patients undergoing intensive care at Göttingen University Hospital, Germany (Table S1). The plasma samples had been pre-screened for neutralizing activity against WT S protein, and a plasma sample with no neutralizing activity was included as negative control. Spread of SARS-CoV-2 variants in Germany was very limited at the time of sample collection, indicating that serum antibodies were induced in response to SARS-CoV-2 WT infection.
All plasma samples with known neutralizing activity (ID15, 18, 20, 22, 23, 24, 27, 33, 51) efficiently reduced entry driven by WT S protein, while the control plasma (ID16) was inactive (Figure 7 A; Figure S3 ). Blockade of entry driven by the S protein of the B.1.1.7 variant was slightly less efficient (Figure 7A; Figure S3). In contrast, seven out of nine plasma samples inhibited entry driven by the S proteins of the B.1.351 and P.1 variants less efficiently as compared to entry driven by WT S protein. These results suggest that individuals previously infected with WT SARS-CoV-2 might only be partially protected against infection with B.1.351 and P.1 variants of SARS-CoV-2.
Figure 7.

Entry driven by the S proteins of SARS-CoV-2 variants B.1.351 and P.1 shows reduced neutralization by convalescent plasma and sera from BNT162b2-vaccinated individuals
Pseudotypes bearing the indicated S proteins were incubated (30 min, 37°C) with different dilutions of plasma derived from COVID-19 patients (A, see also Table S1) or serum from individuals vaccinated with the Pfizer/BioNTech vaccine BNT162b2 (obtained 13–15 days after the second dose) (B, see also Table S2) and inoculated onto Vero target cells. Transduction efficiency was quantified by measuring virus-encoded luciferase activity in cell lysates at 16–20 h post-transduction (please see Figure S3 for more details) and used to calculate the plasma/serum dilution factor that leads to 50% reduction in S protein-driven cell entry (neutralizing titer 50, NT50). Presented are the average (mean) NT50 from two independent experiments. The lines in the scatterplots indicate the median NT50, while the bars indicate the mean NT50. Identical plasma/serum samples are connected with lines in the bar graphs and the numbers in brackets indicate the average (mean) reduction in neutralization sensitivity for the S proteins of the respective SARS-CoV-2 variants. Statistical significance of differences between WT and variant S proteins was analyzed by paired, two-tailed Student’s t test (p > 0.05, not significant [ns]; p ≤ 0.05, *; p ≤ 0.01, **; p ≤ 0.001, ***).
Figure S3.

Representative neutralization data, related to Figure 7
Pseudotypes bearing the indicated S proteins were incubated (30 min, 37°C) with different dilutions of plasma derived from COVID-19 patients (A) or serum from individuals vaccinated with the Pfizer/BioNTech vaccine BNT162b2 (obtained 13-15 days after the second dose) (B) and inoculated onto Vero target cells. Transduction efficiency was quantified by measuring virus-encoded luciferase activity in cell lysates at 16-20 h posttransduction. Presented are the data from a single representative experiment conducted with technical triplicates (results were confirmed in a separate biological replicate). For normalization, inhibition of S protein-driven entry in samples without plasma/serum was set as 0%. Error bars indicate the SD. The data were further used to calculated the plasma/serum dilution that leads to 50% reduction in S protein-driven cell entry (neutralizing titer, NT50, shown in Figure 7). Of note, serum BNT-10 was excluded from further analysis, as its extraordinary high neutralizing activity precluded a reliable NT50 determination.
Reduced neutralization by sera from BNT162b2-vaccinated individuals
The vaccine BNT162b2 is based on an mRNA that encodes for the viral S protein and is highly protective against COVID-19 (Polack et al., 2020). While the S protein harbors T cell epitopes (Grifoni et al., 2020; Peng et al., 2020), efficient protection is believed to require the induction of neutralizing antibodies. We determined the neutralizing activity of sera from 15 donors immunized twice with BNT162b2 (Table S2). All sera efficiently inhibited entry driven by the WT S protein and inhibition of entry driven by the S protein of the B.1.1.7 variant was only slightly reduced (Figure 7B; Figure S3). In contrast, 12 out of 15 sera showed a markedly reduced inhibition of entry driven by the S proteins of the B.1.351 and P.1 variants (Figure 7B), although it should be stated that all sera completely inhibited entry at the lowest dilution tested. In sum, these results suggest that BNT162b2 may offer less robust protection against infection by the B.1.351 and P.1 variants as compared to SARS-CoV-2 WT.
Discussion
The COVID-19 pandemic has taken a major toll on human health and prosperity. Non-pharmaceutic interventions are currently the major instrument to combat the pandemic but are associated with a heavy burden on economies. Protective vaccines became recently available and might become a game changer—it is hoped that efficient vaccine roll out might allow attainment of herd immunity in certain countries in the second half of 2021. The recent emergence of SARS-CoV-2 variants B.1.1.7, B.1.351, and P.1 that harbor mutations in the major target of neutralizing antibodies, the viral S protein, raises the question whether vaccines available at present will protect against infection with these viruses. Similarly, it is largely unclear whether antibody responses in convalescent patients protect against re-infection with the new variants. The results of the present study suggest that SARS-CoV-2 variants B.1.351 and P.1 are partially (Casirivimab) or fully (Bamlanivimab) resistant against antibodies used for COVID-19 treatment and are inhibited less efficiently by convalescent plasma or sera from individuals immunized with the mRNA vaccine BNT162b2. These results suggest that strategies relying on antibody-mediated control of SARS-CoV-2 infection might be compromised by resistance development.
The increased transmissibility postulated for the B.1.1.7, B.1.351, and P.1 variants (Francisco et al., 2021; Leung et al., 2021; Mwenda et al., 2021) raises the possibility that these viruses might exhibit altered host cell interactions or particle stability. The present study demonstrates that S proteins of SARS-CoV-2 WT, B.1.1.7, B.1.351, and P.1 mediate cell-cell and virus-cell fusion with roughly comparable efficiency and entry kinetics. Similarly, particles bearing S protein of WT and variant SARS-CoV-2 did not differ appreciably in stability, although we cannot exclude that examination of more time points might have revealed minor differences. In keeping with these findings, entry driven by the S proteins of SARS-CoV-2 WT and variants was efficiently blocked by inhibitors targeting the cellular factors ACE2 and TMPRSS2, which are critical for lung cell entry. Similarly, membrane fusion inhibitors blocked entry driven by WT S protein and S proteins of the B.1.1.7, B.1.351, and P.1. variants with similar efficiency. These results await confirmation with authentic virus and lung cells, in which entry kinetics might differ. However, the data available at present do not point toward major differences in host cell entry and stability of SARS-CoV-2 and variants B.1.1.7, B.1.351, and P.1.
Although host cell interactions underlying viral entry might not differ markedly between SARS-CoV-2 WT and the B.1.1.7, B.1.351, and P.1 variants, major differences in susceptibility to antibody-mediated neutralization were observed. Entry driven by the S proteins of the B.1.351 and P.1 variants was only partially inhibited by Casirivimab (REGN10933), in keeping with mutations present in the S protein of the B.1.351 and P.1 variants being located at the antibody binding site (Figure S2). Combining Casirivimab and Imdevimab (REGN10987) within an antibody cocktail with EUA (REGN-COV2) restored efficient inhibition, suggesting that REGN-COV2 should be suitable for treatment of patients infected with variant B.1.351 or P.1. In contrast, Bamlanivimab (Baum et al., 2020a, 2020b; Chen et al., 2020; Gottlieb et al., 2021), another antibody with EUA for COVID-19 treatment, failed to block entry driven by the S proteins of B.1.351 and P.1. This finding is in agreement with the E484K mutation being located in the antibody binding site and suggests that Bamlanivimab should not be used for treatment of patients infected with the B.1.351 and P.1 variants.
Vaccination is key to global efforts to contain the COVID-19 pandemic. The mRNA-based vaccine BNT162b2 encodes the viral S protein and is highly efficacious. Closely related vaccines as well as vector-based vaccines followed, and it is believed that these vaccines mainly protect by inducing neutralizing antibody responses. Similarly, neutralizing antibody responses are believed to contribute to protection of convalescent COVID-19 patients against reinfection and disease. The present study showed that entry driven by the S proteins of the B.1.351 and P.1 variants was less susceptible to inhibition by sera/plasma from COVID-19 patients and BNT162b2-vaccinated individuals as compared to entry driven by WT S protein. It should be noted that all plasma and sera tested completely inhibited entry at the lowest dilution tested and that T cell responses will contribute to control of SARS-CoV-2 infection, particularly in re-infected convalescent patients (Grifoni et al., 2020; Peng et al., 2020). Nevertheless, the markedly reduced sensitivity to antibody-mediated neutralization suggests that convalescent and vaccinated individuals might not be fully protected against infection by the B.1.351 and P.1 variants. Such a scenario would be in keeping with preliminary information suggesting that a vaccine based on the S protein might provide less effective protection in South Africa as compared to the United States (Callaway and Mallapaty, 2021). On a more general level, our findings suggest that the interface between the SARS-CoV-2 S protein and ACE2 exhibits high plasticity, favoring emergence of escape variants.
Our finding that entry driven by the S protein of the B.1.1.7 variant can be efficiently inhibited by antibodies induced upon infection and vaccination is in agreement with those of Muik and colleagues, who reported that pseudoparticles bearing the B.1.1.7 S protein are efficiently neutralized by sera from BNT162b2-vaccinated individuals (Muik et al., 2021). Xie and coworkers found that authentic SARS-CoV-2 bearing two mutations present in the S protein of the B.1.1.7 variant (69/70-deletion + N501Y) was still robustly neutralized by antibodies induced by vaccination with BNT162b2. Similarly, neutralization of a virus bearing changes found in the RBD of the B.1.351 and P.1 variants (E484K + N501Y) was moderately reduced (Liu et al., 2021; Wang et al., 2021; Xie et al., 2021), again in keeping with our findings.
We present, to the best of our knowledge, the first side-by-side comparison of host cell entry of variants B.1.1.7, B.1.351, and P.1 and its inhibition by small molecules and antibodies. Although our results await confirmation with authentic SARS-CoV-2, they suggest that evasion of antibody responses does not account for the rapid spread of B.1.1.7. In contrast, our findings indicate that the B.1.351 and P.1 variants might be able to spread in convalescent patients or BNT162b2-vaccinated individuals and thus constitute an elevated threat to human health. Containment of these variants by non-pharmaceutic interventions is an important task.
Limitations of the study
Our study has the following limitations: we used VSV pseudotyped with the S proteins of SARS-CoV-2 variants to study SARS-CoV-2 entry and its inhibition. Although this surrogate model is believed to faithfully mimic cell entry of SARS-CoV-2, our results await formal confirmation with authentic virus. Furthermore, host cell entry and its blockade were studied using immortalized cell lines, and these analyses should be extended to primary cell cultures within future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal anti-HA antibody produced in mouse | Sigma-Aldrich | Cat.#: H3663; RRID: AB_262051 |
| Alexa Fluor 568-conjugated anti-mouse antibody | Thermo Fisher Scientific | Cat.#: A-11004; RRID: AB_2534072 |
| Anti-VSV-G antibody (I1, produced from CRL-2700 mouse hybridoma cells) | ATCC | Cat.# CRL-2700; RRID: CVCL_G654 |
| Bacterial and Virus Strains | ||
| One Shot™ OmniMAX™ 2 T1R Chemically Competent E. coli | ThermoFisher Scientific | Cat.#: C854003 |
| VSV∗ΔG-FLuc | Laboratory of Gert Zimmer | N/A |
| Biological Samples | ||
| Patient plasma: ID 15 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 16 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 18 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 20 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 22 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 23 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 24 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 27 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 33 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient plasma: ID 51 | Laboratory of Martin Sebastian Winkler | N/A |
| Patient serum: BNT-01 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-02 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-03 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-04 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-05 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-06 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-07 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-08 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-09 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-10 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-11 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-12 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-13 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-14 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Patient serum: BNT-15 | Ulm University Hospital / Laboratory of Alexander Kleger | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Camostat mesylate | Tocris | Cat.#: 3193 |
| Nafamostat mesylate | Tocris | Cat.#: 3081 |
| EK-1 | Core Facility Functional Peptidomics, Ulm University, Germany | N/A |
| EK-1-C4 | Bachem AG, Bubendorf, Switzerland | N/A |
| hIgG | Laboratory of Hans-Martin Jäck | N/A |
| Casirivimab | Laboratory of Hans-Martin Jäck | N/A |
| Imdevimab | Laboratory of Hans-Martin Jäck | N/A |
| REGN10989 | Laboratory of Hans-Martin Jäck | N/A |
| Bamlanivimab | Lilly, Indianapolis, IN, USA | YLO52IPAMOO |
| Luciferase Cell Culture Lysis 5X Reagent | Promega | Cat.#: E1531 |
| Lipofectamine LTX with Plus Reagent | Thermo Fisher Scientific | Cat.#: 15338100 |
| Critical Commercial Assays | ||
| Beetle-Juice Kit | PJK | Cat.#: 102511 |
| Luciferase Assay System | Promega | Cat.#: E1500 |
| Experimental Models: Cell Lines | ||
| 293T | DSMZ | Cat.#: ACC-635; RRID: CVCL_0063 |
| 293T-ACE2 | This study | N/A |
| A549 | Laboratory of Georg Herrler | ATCC Cat# CRM-CCL-185; RRID: CVCL_0023 |
| A549-ACE2 | This study | N/A |
| A549-ACE2/TMPRSS2 | This study | N/A |
| Caco-2 | Laboratory of Stefan Pöhlmann | ATCC Cat# HTB-37; RRID: CVCL_0025 |
| Calu-3 | Laboratory of Stephan Ludwig | ATCC Cat# HTB-55; RRID: CVCL_0609 |
| Vero | Laboratory of Andrea Maisner | ATCC Cat# CRL-1586; RRID: CVCL_0574 |
| Vero-TMPRSS2 | Laboratory of Stefan Pöhlmann | N/A |
| Oligonucleotides | ||
| pCG1 Seq F (CCTGGGCAACGTGCTGGT) | Sigma-Aldrich | N/A |
| pCG1 Seq R (GTCAGATGCTCAAGGGGCTTCA) | Sigma-Aldrich | N/A |
| SARS-2-S (BamHI) F (AAGGCCGGATCCGCCA CCATGTTCGTGTTTCTGGTGCTGC) |
Sigma-Aldrich | N/A |
| SARS-2-SΔ18 (XbaI) R (AAGGCCTCTAGACT ACTTGCAGCAGCTGCCACAG) |
Sigma-Aldrich | N/A |
| SARS-2-S-HA (XbaI) R (AAGGCCTCTAGATTA CGCATAATCCGGCACATCATACGGATA GGTGTAGTGCAGTTTCACGCCCTTC) |
Sigma-Aldrich | N/A |
| SARS-2-Ssyn Seq-1 (CAAGATCTACAGCAAGCACACC) | Sigma-Aldrich | N/A |
| SARS-2-Ssyn Seq-2 (GTCGGCGGCAACTACAATTAC) | Sigma-Aldrich | N/A |
| SARS-2-Ssyn Seq-3 (CTGTCTGATCGGAGCCGAGCAC) | Sigma-Aldrich | N/A |
| SARS-2-Ssyn Seq-4 (TGAGATGATCGCCCAGTACAC) | Sigma-Aldrich | N/A |
| SARS-2-Ssyn Seq-5 (GCCATCTGCCACGACGGCAAAG) | Sigma-Aldrich | N/A |
| SARS-2-S (D80A) F (AGAGATTCGcCAACCCCGTGC) | Sigma-Aldrich | N/A |
| SARS-2-S (D80A) R (ACGGGGTTGgCGAATCTCTTGGTG) | Sigma-Aldrich | N/A |
| SARS-2-S (Δ242-244+R246I) F (AGACACTGCACAtAAGCTACCTGACACC) | Sigma-Aldrich | N/A |
| SARS-2-S (Δ242-244+R246I) R (GTAGCTTaTGTGCAGTGTCTGAAACCGGGTG) | Sigma-Aldrich | N/A |
| SARS-2-S (K417N) F (AGACAGGCA AtATCGCCGACTACAACTACAAG) |
Sigma-Aldrich | N/A |
| SARS-2-S (K417N) R (GTCGGCGATa TTGCCTGTCTGTCCAG) |
Sigma-Aldrich | N/A |
| SARS-2-S (E484K) F (TAACGGCGTGa AAGGCTTCAACTGCTACTTC) |
Sigma-Aldrich | N/A |
| SARS-2-S (E484K) R (TGAAGCCTTt CACGCCGTTACAAGG) |
Sigma-Aldrich | N/A |
| SARS-2-S (N501Y) F (TCAGCCCACAtA TGGCGTGGGCTATC) |
Sigma-Aldrich | N/A |
| SARS-2-S (N501Y) R (CCCACGCCATaTGTGGGCTGAAAGC) | Sigma-Aldrich | N/A |
| SARS-2-S (A701V) F (CTCTGGGCGtCGAGAACAGCGTG) | Sigma-Aldrich | N/A |
| SARS-2-S (A701V) R (CTGTTCTCGaCGCCCAGAGACATTG) | Sigma-Aldrich | N/A |
| SARS-2-S (L18F+T20N+P26S) F (tTtACCAacAGAACCCAGCTGCC TtCAGCCTACACCAACAGC) |
Sigma-Aldrich | N/A |
| SARS-2-S (L18F+T20N+P26S) R (TGaAGGCAGCTGGGTTCTgtTG GTaAaGTTCACACACTGGCTGG) |
Sigma-Aldrich | N/A |
| SARS-2-S (D138Y) F (TTCTGCAACtACCCCTTCCTGG) | Sigma-Aldrich | N/A |
| SARS-2-S (D138Y) R (AGGAAGGGGTaGTTGCAGAACTGG) | Sigma-Aldrich | N/A |
| SARS-2-S (R190S) F (CAAGAACCTGaGCGAGTTCGTGTTC) | Sigma-Aldrich | N/A |
| SARS-2-S (R190S) R (ACACGAACTCGCtCAGGTTCTTGAAG) | Sigma-Aldrich | N/A |
| SARS-2-S (K417T) F (ACAGACAGGCAcGATCGCCGACTACAAC) | Sigma-Aldrich | N/A |
| SARS-2-S (K417T) R (GTCGGCGATCgTGCCTGTCTGTCCAGG) | Sigma-Aldrich | N/A |
| SARS-2-S (E484K+N501Y) F (aAAGGCTTCAACTGCTACTTC CCACTGCAGTCCTACGGCTTT CAGCCCACAtATGGCGTGGGC TATCAGC) |
Sigma-Aldrich | N/A |
| SARS-2-S (E484K+N501Y) R (ATaTGTGGGCTGAAAGCCG TAGGACTGCAGTGGGAAGTAGC AGTTGAAGCCTTtCACGCCGTTA CAAGGGGTGC) |
Sigma-Aldrich | N/A |
| SARS-2-S (H655Y) F (CGGAGCCGAGtACGTGAACAATAGC) | Sigma-Aldrich | N/A |
| SARS-2-S (H655Y) R (TGTTCACGTaCTCGGCTCCGATCAGAC) | Sigma-Aldrich | N/A |
| SARS-2-S (T1027I) F (CTGGCCGCCAtCAAGATGTCTGAGTG) | Sigma-Aldrich | N/A |
| SARS-2-S (T1027I) R (GACATC TTGaTGGCGGCCAGATTGGCAG) |
Sigma-Aldrich | N/A |
| SARS-2-S (V1176F) F (GCCAGCGTCtTcAACATC CAGAAAGAGATCG) |
Sigma-Aldrich | N/A |
| SARS-2-S (V1176F) R (TCTGGATGTTgAaGACGC TGGCATTGATTCC) |
Sigma-Aldrich | N/A |
| SARS-2-S (H69Δ/V70Δ) F (CCACGCCATCTCCGGC ACCAATGGCACCAAG) |
Sigma-Aldrich | N/A |
| SARS-2-S (H69Δ/V70Δ) R (TGGTGCCGGAGATGGC GTGGAACCAGGTCAC) |
Sigma-Aldrich | N/A |
| SARS-2-S (Y144Δ) F (CTGGGCGTC TATCACAAGAACAACAAGAGC) |
Sigma-Aldrich | N/A |
| SARS-2-S (Y144Δ) R (CTTGTGATA GACGCCCAGGAAGGGGTC) |
Sigma-Aldrich | N/A |
| SARS-2-S (A570D) F (CGGGATATCGaCGATACCA CAGACGCCGTTAG) |
Sigma-Aldrich | N/A |
| SARS-2-S (A570D) R (GTGGTATCGt CGATATCCCGGCCAAAC) |
Sigma-Aldrich | N/A |
| SARS-2-S (P681H) F (GACAAACAGC CaCAGACGGGCCAGATCTG) |
Sigma-Aldrich | N/A |
| SARS-2-S (P681H) R (GGCCCGTC TGtGGCTGTTTGTCTGTGTC) |
Sigma-Aldrich | N/A |
| SARS-2-S (T716I) F (CTATCCCCAtC AACTTCACCATCAGC) |
Sigma-Aldrich | N/A |
| SARS-2-S (T716I) R (GTGAAGTTGaTG GGGATAGCGATAGAGTTG) |
Sigma-Aldrich | N/A |
| SARS-2-S (S982A) F (GATATCCTGg cCAGACTGGACAAGGTG) |
Sigma-Aldrich | N/A |
| SARS-2-S (S982A) R (TCCAGTCTGgc CAGGATATCGTTCAGCAC) |
Sigma-Aldrich | N/A |
| SARS-2-S (D1118H) F (ATCACCACC cACAACACCTTCGTGTCTG) |
Sigma-Aldrich | N/A |
| SARS-2-S (D1118H) R (AGGTGTTGTg GGTGGTGATGATCTG) |
Sigma-Aldrich | N/A |
| pQCXIP/BL Seq F (GAGCTCGTTTAGTGAACCG) | Sigma-Aldrich | N/A |
| ACE2 (NotI) F (AAGGCCGCGGCCGCGCCAC CATGTCAAGCTCTTCCTGGCTCC) |
Sigma-Aldrich | N/A |
| ACE2 (PacI) R (AAGGCCTTAATTAAC TAAAAGGAGGTCTGAACATCATC) |
Sigma-Aldrich | N/A |
| ACE2 (PacI) F (AAGGCCTTAATTAAGCCACCAT GTCAAGCTCTTCCTGGCTCC) |
Sigma-Aldrich | N/A |
| solACE2 (SalI) R (AAGGCCGTCGAC AGGGGGCTGGTTAGGAGGTCC) |
Sigma-Aldrich | N/A |
| ACE2 Seq-1 (AGGGCAAAGTTGATGAATGC) | Sigma-Aldrich | N/A |
| ACE2 Seq-2 (TATTACACAAGGACCCTTTACC) | Sigma-Aldrich | N/A |
| pCG1-Fc Seq R (CACAGTGGGCATGTGTGAGG) | Sigma-Aldrich | N/A |
| Recombinant DNA | ||
| Plasmid: pCG1 | Laboratory of Roberto Cattaneo | N/A |
| Plasmid: pCAGGS-DsRed | Laboratory of Stefan Pöhlmann | N/A |
| Plasmid: pCG1-SARS-S | Laboratory of Georg Herrler | N/A |
| Plasmid: pCG1-SARS-2-S | Laboratory of Stefan Pöhlmann | N/A |
| Plasmid: pCG1-SARS-2-S (D614G) | This study | N/A |
| Plasmid: pCG1-SARS-2-S (B.1.1.7) | This study | N/A |
| Plasmid: pCG1-SARS-2-S (B.1.351) | This study | N/A |
| Plasmid: pCG1-SARS-2-S (B.1.1.28) | This study | N/A |
| Plasmid: pQCXIBL-cMYC-hTMPRSS2 | Laboratory of Stefan Pöhlmann | N/A |
| Plasmid: pQCXIP-ACE2 | This study | N/A |
| Plasmid: pCG1-Fc | Laboratory of Georg Herrler | N/A |
| Plasmid: pCG1-sol-ACE2-Fc | This study | N/A |
| Plasmid: Gal4-TurboGFP-Luc | Laboratory of Alexander Hahn | N/A |
| Plasmid: Vp16-Gal4 | Laboratory of Alexander Hahn | N/A |
| Plasmid: pCG1-ACE2 | Laboratory of Georg Herrler | N/A |
| Plasmid: pCAGGS-TMPRSS2 | Laboratory of Stefan Pöhlmann | N/A |
| Software and Algorithms | ||
| Hidex Sense Microplate Reader Software | Hidex Deutschland Vertrieb GmbH | https://www.hidex.de |
| Adobe Photoshop CS5 Extended (version 12.0 × 32) | Adobe | https://www.adobe.com |
| GraphPad Prism (version 8.3.0(538)) | GraphPad Software | https://www.graphpad.com |
| Microsoft Office Standard 2010 (version 14.0.7232.5000) | Microsoft Corporation | https://products.office.com |
| YASARA (version 19.1.27) | YASARA Biosciences GmbH | http://www.yasara.org |
| ZEN imaging software | Carl Zeiss | https://www.zeiss.com |
Resource availability
Lead contact
Requests for material can be directed to Markus Hoffmann (mhoffmann@dpz.eu) and the lead contact, Stefan Pöhlmann (spoehlmann@dpz.eu).
Materials availability
All materials and reagents will be made available upon installment of a material transfer agreement (MTA).
Data and code availability
The study did not generate unique datasets or code.
Experimental model and subject details
Cell culture
All cell lines were incubated at 37°C in a humidified atmosphere containing 5% CO2. All media were supplemented with 10% fetal bovine serum (FCS, Biochrom or GIBCO), 100 U/mL of penicillin and 0.1 mg/mL of streptomycin (PAN-Biotech). 293T (human, female, kidney; ACC-635, DSMZ, RRID: CVCL_0063), 293T cells stably expressing ACE2 (293T-ACE2), Vero (African green monkey, female, kidney; CRL-1586, ATCC, RRID:CVCL_0574; kindly provided by Andrea Maisner, Institute of Virology, Philipps University Marburg, Marburg, Germany) and Vero-TMPRSS2 cells (Hoffmann et al., 2020b) were cultivated in Dulbecco’s modified Eagle medium (DMEM). Vero-TMPRSS2 cells additionally received puromycin (0.5 μg/mL, Invivogen). A549 (human, male, lung; CRM-CCL-185, ATCC, RRID:CVCL_0023; kindly provided by Georg Herrler), A549-ACE2 and A549-ACE2/TMPRSS2 cells were cultivated in DMEM/F-12 Medium with Nutrient Mix (Thermo Fisher Scientific). A549-ACE2 cells further received 0.5 μg/mL puromycin, while A549-ACE2/TMPRSS2 cells were cultivated in the presence of 0.5 μg/mL puromycin and 1 μg/mL blasticidin. Caco-2 (human, male, intestine; HTB-37, ATCC, RRID:CVCL_0025) and Calu-3 cells (human, male, lung; HTB-55, ATCC, RRID:CVCL_0609; kindly provided by Stephan Ludwig, Institute of Virology, University of Münster, Germany) were cultivated in minimum essential medium supplemented with 1x non-essential amino acid solution (from 100x stock, PAA) and 1 mM sodium pyruvate (Thermo Fisher Scientific). 293T cells that stably express ACE2 were generated by retroviral (murine leukemia virus, MLV) transduction and selection of parental 293T cells with puromycin (4 μg/mL for initial selection and 0.5 μg/mL for sub-culturing). Similarly, we generated A549 cells stably expressing ACE2 (A549-ACE2). A549 cells stably expressing ACE2 and TMPRSS2 (A549-ACE2/TMPRSS2) were obtained by retroviral transduction of A549-ACE2 cells and selection with blasticidin (6 μg/mL for initial selection and 1 μg/mL for sub-culturing). Authentication of cell lines was performed by STR-typing, amplification and sequencing of a fragment of the cytochrome c oxidase gene, microscopic examination and/or according to their growth characteristics. Further, cell lines were routinely tested for contamination by mycoplasma.
Method details
Expression plasmids and transfection of cell lines
Expression plasmids for DsRed (Hoffmann et al., 2020b), vesicular stomatitis virus (VSV, serotype Indiana) glycoprotein (VSV-G) (Brinkmann et al., 2017), SARS-S (derived from the Frankfurt-1 isolate; containing a C-terminal HA epitope tag) (Hoffmann et al., 2020b), SARS-2-S (codon-optimized, based on the Wuhan/Hu-1/2019 isolate; with a C-terminal truncation of 18 amino acid residues or with a C-terminal HA epitope tag) (Hoffmann et al., 2020b), angiotensin-converting enzyme 2 (ACE2) (Hoffmann et al., 2013), TMPRSS2 (Heurich et al., 2014), Gal4-TurboGFP-Luc and Vp16-Gal4 (Hörnich et al., 2021) have been described elsewhere. In order to generate expression vectors for S proteins from emerging SARS-CoV-2 variants, we introduced the required mutations into the parental SARS-2-S sequence by overlap extension PCR. Subsequently, the respective open reading frames were inserted into the pCG1 plasmid (kindly provided by Roberto Cattaneo, Mayo Clinic College of Medicine, Rochester, MN, USA), making use of the unique BamHI and XbaI restriction sites. Further, we cloned the coding sequence for human ACE2 into the pQCXIP plasmid (Brass et al., 2009), yielding pQCXIP_ACE2. For the generation of cell lines stably overexpressing human TMPRSS2 and/or human ACE2 we produced MLV-based transduction vectors using the pQCXIBl_cMYC-hTMPRSS2 (Kleine-Weber et al., 2018) or pQCXIP_ACE2 expression vectors in combination with plasmids coding for VSV-G and MLV-Gag/Pol (Bartosch et al., 2003). In order to obtain the expression vector for soluble human ACE2 harboring the Fc portion of human immunoglobulin G (sol-ACE2-Fc), we PCR amplified the sequence coding for the ACE2 ectodomain (amino acid residues 1-733) and cloned it into the pCG1-Fc plasmid ((Sauer et al., 2014), kindly provided by Georg Herrler, University of Veterinary Medicine, Hannover, Germany). Sequence integrity was verified by sequencing using a commercial sequencing service (Microsynth Seqlab). Specific cloning details (e.g., primer sequences and restriction sites) are available upon request. Transfection of cells was carried out by the calcium-phosphate method or by using polyethylenimin, Lipofectamine LTX (Thermo Fisher Scientific) or Transit LT-1 (Mirus).
Analysis of S protein expression by fluorescence microscopy
A549-ACE2 cells that were grown on coverslips were transfected with plasmids encoding SARS-CoV-2 S protein variants with a C-terminal HA epitope tag or empty expression vector (control). At 24 h posttransfection, cells were fixed with 4% paraformaldehyde solution (30 min, room temperature), washed and incubated (15 min, room temperature) with phosphate-buffered saline (PBS) containing 0.1 M glycine and permeabilized by treatment with 0.2% Triton X-100 solution (in PBS, 15 min). Thereafter, samples were washed and incubated for 1 h at room temperature with primary antibody (anti-HA, mouse, 1:500, Sigma-Aldrich) diluted in PBS containing 1% bovine serum albumin. Next, the samples were washed with PBS and incubated in the dark for 1 h at 4°C with secondary antibody (Alexa Fluor-568-conjugated anti-mouse antibody, 1:750, Thermo Fisher Scientific). Finally, the samples were washed, nuclei were stained with DAPI and coverslips were mounted onto microscopic glass slides with Mowiol/DABCO. Images were taken using a Zeiss LSM800 confocal laser scanning microscope with ZEN imaging software (Zeiss).
Preparation of pseudotyped particles and transduction experiments
Rhabdoviral pseudotype particles were prepared according to a published protocol (Kleine-Weber et al., 2019). For pseudotyping we used a replication-deficient VSV vector that lacks the genetic information for VSV-G and instead codes for two reporter proteins, enhanced green fluorescent protein and firefly luciferase (FLuc), VSV∗ΔG-FLuc (kindly provided by Gert Zimmer, Institute of Virology and Immunology, Mittelhäusern, Switzerland) (Berger Rentsch and Zimmer, 2011). 293T cells transfected to express the desired viral glycoprotein were inoculated with VSV∗ΔG-FLuc and incubated for 1 h at 37°C before the inoculum was removed and cells were washed. Finally, culture medium containing anti-VSV-G antibody (culture supernatant from I1-hybridoma cells; ATCC no. CRL-2700) was added. Following an incubation period of 16-18 h, pseudotype particles were harvested by collecting the culture supernatant, pelleting cellular debris through centrifugation (2,000 x g, 10 min, room temperature) and transferring aliquots of the clarified supernatant into fresh reaction tubes. Samples were stored at −80°C. For transduction experiments, target cells were seeded in 96-well plates, inoculated with the respective pseudotype particles with comparable infectivity and further incubated. At 16-18 h postinoculation, transduction efficiency was analyzed. For this, the culture supernatant was removed and cells were lysed by incubation for 30 min at room temperature with Cell Culture Lysis Reagent (Promega). Next, lysates were transferred into white 96-well plates and FLuc activity was measured using a commercial substrate (Beetle-Juice, PJK; Luciferase Assay System, Promega) and a plate luminometer (Hidex Sense Plate Reader, Hidex or Orion II Microplate Luminometer, Berthold).
Depending on the experimental set-up target cells or pseudotype particles were pre-incubated with different compounds. Target cells were incubated with different concentrations of serine protease inhibitor (Camostat or Nafamostat, Caco-2, 1 h at 37°C). Alternatively, pseudotype particles were pre-incubated with different concentrations of either sol-ACE2-Fc, fusion inhibitor (EK-1 or EK-1-C4), monoclonal antibodies (Casirivimab, Imdevimab, REGN-COV2 [Casirivimab and Imdevimab], REGN10989, Bamlanivimab), or plasma/sera from COVID-19 patients or vaccinated (Pfizer/BioNTech vaccine BNT162b2) individuals (30 min at 37°C). S protein stability was analyzed as follows. Pseudotype particles were incubated for different time intervals at 33°C, then snap-frozen and stored at −80°C until all samples were collected. Thereafter, samples were thawed, inoculated onto target cells, and the cells incubated as described above. For the investigation of the entry speed of S protein-bearing pseudotypes, the respective particles were inoculated onto target cells and adsorbed for different time intervals before the inoculum was removed and cells were washed and incubated with fresh medium.
Production of soluble ACE2 (sol-ACE2-Fc)
293T cells were grown in a T-75 flask and transfected with 20 μg of sol-ACE2-Fc expression plasmid. At 10 h posttransfection, the medium was replaced and cells were further incubated for 38 h before the culture supernatant was collected and centrifuged (2,000 x g, 10 min, 4°C). Next, the clarified supernatant was loaded onto Vivaspin protein concentrator columns with a molecular weight cut-off of 30 kDa (Sartorius) and centrifuged at 4,000 x g, 4°C until the sample was concentrated by a factor of 20. The concentrated sol-ACE2-Fc was aliquoted and stored at −80°C until further use.
Collection of serum and plasma samples
Sera from individuals vaccinated with BioNTech/Pfizer vaccine BNT162b2 were obtained 13-15 days after the second dose. The study was approved by the Ethic committee of Ulm university (vote 31/21 – FSt/Sta). Collection of plasma samples from COVID-19 patients treated at the intensive care unit was approved by the Ethic committee of the University Medicine Göttingen (SeptImmun Study 25/4/19 Ü). For collection of plasma, Cell Preparation Tube (CPT) vacutainers with sodium citrate were used and plasma was collected as supernatant over the PBMC layer. For vaccinated patients, blood was collected in S-Monovette® Serum Gel tubes (Sarstedt). Subsequently, the plasma and serum samples were incubated at 56°C for 30 min to inactivate putative infectious virus. For convalescent plasma, pre-screening for detection of neutralizing activity was performed on Vero76 cells using SARS-2-S- and VSV-G bearing pseudotypes as control, normalized for equal infectivity.
Qualitative cell-cell fusion assay
A549-ACE2 or A549-ACE2/TMPRSS2 cells were transfected with DsRed expression plasmid along with either expression vector for wildtype or mutant SARS-2-S, SARS-S or empty plasmid. At 24 h posttransfection, cells were fixed with 4% paraformaldehyde solution (30 min, room temperature), washed and nuclei were stained with DAPI. Next, cells were washed again with PBS and images were taken using a Zeiss LSM800 confocal laser scanning microscope with ZEN imaging software (Zeiss).
Quantitative cell-cell fusion assay
293T target-cells were seeded in a 48-well plate at 50.000 cells/well and transfected with Gal4-TurboGFP-Luciferase reporter plasmid (Gal4-TurboGFP-Luc) as well as expression plasmid for ACE2 alone or in combination with TMPRSS2 (7:1 ratio). 293T effector-cells were seeded in a 6-well dish at 70%–80% confluency and transfected with the Vp16-Gal4 expression plasmid as well as expression plasmid for WT or mutant SARS-2-S, SARS-S or empty plasmid. At 24 h posttransfection, effector-cells were detached by resuspending them in culture medium and added to the target-cells in a 1:1 ratio. After 24 h luciferase activity was analyzed using the Beetle-Juice Luciferase Assay according to manufacturer’s instructions and a Biotek Synergy 2 plate reader.
Sequence analysis and protein models
S protein sequences of emerging SARS-CoV-2 S variants B.1.1.7 (GISAID: EPI_ISL_601443), B.1.351 (GISAID: EPI_ISL_700428) and P1 (GISAID: EPI_ISL_792683) were retrieved from the GISAID (global initiative on sharing all influenza data) database (https://www.gisaid.org/). Protein models are based on PDB: 6XDG (Hansen et al., 2020) or PDB: 7L3N (Jones et al., 2020), or a template generated by modeling the SARS-2-S sequence on a published crystal structure (PDB: 6XR8,(Cai et al., 2020)), using the SWISS-MODEL online tool (https://swissmodel.expasy.org/), and were generated using the YASARA software (http://www.yasara.org/index.html).
Quantification and statistical analysis
The presented data either show (i) results from single representative experiment (conducted with technical triplicates or quadruplicates) that were confirmed in at least one additional biological replicate or (ii) average (mean) data from three or four biological replicates (conducted with technical triplicates or quadruplicates).
Data analysis was performed using Microsoft Excel as part of the Microsoft Office software package (version 2019, Microsoft Corporation) and GraphPad Prism 8 version 8.4.3 (GraphPad Software). Data normalization was done as follows: (i) To compare efficiency of cell entry driven by the different S protein variants under study, transduction was normalized against SARS-CoV-2 S WT (set as 100%); (ii) For experiments investigating inhibitory effects, transduction was normalized against a reference sample (e.g., control-treated cells or pseudotypes, set as 0% inhibition). Serum dilutions that cause a 50% reduction of transduction efficiency (neutralizing titer 50, NT50) were calculated using a non-linear regression model (inhibitor versus normalized response, variable slope). Statistical significance was tested by one- or two-way analysis of variance (ANOVA) with Dunnett’s post hoc test, or by paired student`s t test. Only P values of 0.05 or lower were considered statistically significant (p > 0.05, not significant [ns]; P ≤ 0.05, ∗; P ≤ 0.01, ∗∗; P ≤ 0.001, ∗∗∗). Specific details on the statistical test and the error bars (standard deviation, SD; standard error of the mean, SEM) are indicated in the figure legends.
Acknowledgments
J.M. acknowledges funding by a Collaborative Research Centre grant of the German Research Foundation (316249678 – SFB 1279), the European Union’s Horizon 2020 research and innovation program under grant agreement no. 101003555 (Fight-nCoV) and the Federal Ministry of Economics, Germany (Combi-CoV-2). J.M. and A.K. acknowledge funding from the Ministry for Science, Research and the Arts of Baden-Württemberg, Germany and the German Research Foundation (Fokus-Förderung COVID-19). A.K. further acknowledges funding from the German Research Foundation through the DFG Heisenberg Programm (KL 2544/6-1). R.G. and A.S. are part of the International Graduate School in Molecular Medicine Ulm. S.P. acknowledges funding by BMBF (RAPID Consortium, 01KI1723D and 01KI2006D; RENACO, 01KI20328A; SARS_S1S2 01KI20396; COVIM consortium, 01KX2021), the county of Lower Saxony and the German Research Foundation (PO 716/11-1, PO 716/14-1). N.K. acknowledges funding from BMBF (ANI-CoV, 01KI2074A). H.S. acknowledges funding from the Ministry for Science, Research and the Arts of Baden-Württemberg, Germany and the European Commission (HORIZON2020 Project SUPPORT-E, no. 101015756). A.S.H. acknowledges funding from the German Research Foundation (HA 6013/6 - 1). H.-M.J. acknowledges funding from the BMBF (COVIM and 01KI2043A), the German Research Foundation (TRR130), the Bayerische Forschungsstiftung (CORAdc), and the Bavarian Ministry of Science and Art. We thank Lu Lu for advice with EK1 and EK1C4 synthesis.
Author contributions
Conceptualization, M.H., J.M., and S.P.; funding acquisition, S.P. and J.M.; investigation, M.H., P.A., R.G., A.S., B.F.H., A.S.H., N.K., L.G., H.H.-W., and A.K.; essential resources, M.S.W., S.S., H.-M.J., B.J., H.S., M.M., and A.K.; writing, M.H. and S.P., review and editing, all authors.
Declaration of interests
The authors declare no competing interests.
Published: March 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.03.036.
Supplemental information
References
- Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S.A., Rouphael N., Creech C.B., et al. COVE Study Group Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021;384:403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosch B., Dubuisson J., Cosset F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Ajithdoss D., Copin R., Zhou A., Lanza K., Negron N., Ni M., Wei Y., Mohammadi K., Musser B., et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science. 2020;370:1110–1115. doi: 10.1126/science.abe2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014–1018. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Rentsch M., Zimmer G. A vesicular stomatitis virus replicon-based bioassay for the rapid and sensitive determination of multi-species type I interferon. PLoS ONE. 2011;6:e25858. doi: 10.1371/journal.pone.0025858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher-Friebertshäuser E., Stein D.A., Klenk H.D., Garten W. Inhibition of influenza virus infection in human airway cell cultures by an antisense peptide-conjugated morpholino oligomer targeting the hemagglutinin-activating protease TMPRSS2. J. Virol. 2011;85:1554–1562. doi: 10.1128/JVI.01294-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass A.L., Huang I.C., Benita Y., John S.P., Krishnan M.N., Feeley E.M., Ryan B.J., Weyer J.L., van der Weyden L., Fikrig E., et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann C., Hoffmann M., Lübke A., Nehlmeier I., Krämer-Kühl A., Winkler M., Pöhlmann S. The glycoprotein of vesicular stomatitis virus promotes release of virus-like particles from tetherin-positive cells. PLoS ONE. 2017;12:e0189073. doi: 10.1371/journal.pone.0189073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchrieser J., Dufloo J., Hubert M., Monel B., Planas D., Rajah M.M., Planchais C., Porrot F., Guivel-Benhassine F., Van der Werf S., et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2021;40:e107405. doi: 10.15252/embj.2020107405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M., Jr., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369:1586–1592. doi: 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E., Mallapaty S. Novavax offers first evidence that COVID vaccines protect people against variants. Nature. 2021;590:17. doi: 10.1038/d41586-021-00268-9. [DOI] [PubMed] [Google Scholar]
- CDC . 2021. Emerging SARS-CoV-2 Variants.https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html [Google Scholar]
- Chen P., Nirula A., Heller B., Gottlieb R.L., Boscia J., Morris J., Huhn G., Cardona J., Mocherla B., Stosor V., et al. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N. Engl. J. Med. 2020;384:229–237. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claro I.M., da Silva Sales F.C., Ramundo M.S., Candido D.S., Silva C.A.M., de Jesus J.G., Manuli E.R., de Oliveira C.M., Scarpelli L., Campana G., et al. Local Transmission of SARS-CoV-2 Lineage B.1.1.7, Brazil, December 2020. Emerg. Infect. Dis. 2021;27:970–972. doi: 10.3201/eid2703.210038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco R.D.S., Jr., Benites L.F., Lamarca A.P., de Almeida L.G.P., Hansen A.W., Gularte J.S., Demoliner M., Gerber A.L., de C Guimarães A.P., Antunes A.K.E., et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 2021;296:198345. doi: 10.1016/j.virusres.2021.198345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway S.E., Paul P., MacCannell D.R., Johansson M.A., Brooks J.T., MacNeil A., Slayton R.B., Tong S., Silk B.J., Armstrong G.L., et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage - United States, December 29, 2020-January 12, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021;70:95–99. doi: 10.15585/mmwr.mm7003e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb R.L., Nirula A., Chen P., Boscia J., Heller B., Morris J., Huhn G., Cardona J., Mocherla B., Stosor V., et al. Effect of Bamlanivimab as Monotherapy or in Combination With Etesevimab on Viral Load in Patients With Mild to Moderate COVID-19: A Randomized Clinical Trial. JAMA. 2021;325:632–644. doi: 10.1001/jama.2021.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J., Baum A., Pascal K.E., Russo V., Giordano S., Wloga E., Fulton B.O., Yan Y., Koon K., Patel K., et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369:1010–1014. doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurich A., Hofmann-Winkler H., Gierer S., Liepold T., Jahn O., Pöhlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014;88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Müller M.A., Drexler J.F., Glende J., Erdt M., Gützkow T., Losemann C., Binger T., Deng H., Schwegmann-Weßels C., et al. Differential sensitivity of bat cells to infection by enveloped RNA viruses: coronaviruses, paramyxoviruses, filoviruses, and influenza viruses. PLoS ONE. 2013;8:e72942. doi: 10.1371/journal.pone.0072942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Pöhlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell. 2020;78:779–784.e5. doi: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Schroeder S., Kleine-Weber H., Müller M.A., Drosten C., Pöhlmann S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020;64 doi: 10.1128/AAC.00754-20. e00754-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörnich B.F., Großkopf A.K., Schlagowski S., Tenbusch M., Kleine-Weber H., Neipel F., Stahl-Hennig C., Hahn A.S. SARS-CoV-2 and SARS-CoV spike-mediated cell-cell fusion differ in the requirements for receptor expression and proteolytic activation. J. Virol. 2021 doi: 10.1128/JVI.00002-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B.E., Brown-Augsburger P.L., Corbett K.S., Westendorf K., Davies J., Cujec T.P., Wiethoff C.M., Blackbourne J.L., Heinz B.A., Foster D., et al. LY-CoV555, a rapidly isolated potent neutralizing antibody, provides protection in a non-human primate model of SARS-CoV-2 infection. bioRxiv. 2020 doi: 10.1101/2020.09.30.318972. [DOI] [Google Scholar]
- Kidd M., Richter A., Best A., Cumley N., Mirza J., Percival B., Mayhew M., Megram O., Ashford F., White T., et al. S-variant SARS-CoV-2 lineage B1.1.7 is associated with significantly higher viral loads in samples tested by ThermoFisher TaqPath RT-qPCR. J. Infect. Dis. 2021 doi: 10.1093/infdis/jiab082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine-Weber H., Elzayat M.T., Hoffmann M., Pöhlmann S. Functional analysis of potential cleavage sites in the MERS-coronavirus spike protein. Sci. Rep. 2018;8:16597. doi: 10.1038/s41598-018-34859-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine-Weber H., Elzayat M.T., Wang L., Graham B.S., Müller M.A., Drosten C., Pöhlmann S., Hoffmann M. Mutations in the Spike Protein of Middle East Respiratory Syndrome Coronavirus Transmitted in Korea Increase Resistance to Antibody-Mediated Neutralization. J. Virol. 2019;93:e01381-18. doi: 10.1128/JVI.01381-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K., Shum M.H., Leung G.M., Lam T.T., Wu J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill. 2021;26:2002106. doi: 10.2807/1560-7917.ES.2020.26.1.2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Liu J., Xia H., Zhang X., Fontes-Garfias C.R., Swanson K.A., Cai H., Sarkar R., Chen W., Cutler M., et al. Neutralizing Activity of BNT162b2-Elicited Serum - Preliminary Report. N. Engl. J. Med. 2021 doi: 10.1056/NEJMc2102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteil V., Kwon H., Prado P., Hagelkruys A., Wimmer R.A., Stahl M., Leopoldi A., Garreta E., Hurtado Del Pozo C., Prosper F., et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell. 2020;181:905–913.e7. doi: 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik A., Wallisch A.K., Sänger B., Swanson K.A., Mühl J., Chen W., Cai H., Maurus D., Sarkar R., Türeci Ö., et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. Science. 2021;371:1152–1153. doi: 10.1126/science.abg6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwenda M., Saasa N., Sinyange N., Busby G., Chipimo P.J., Hendry J., Kapona O., Yingst S., Hines J.Z., Minchella P., et al. Detection of B.1.351 SARS-CoV-2 Variant Strain - Zambia, December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021;70:280–282. doi: 10.15585/mmwr.mm7008e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Mentzer A.J., Liu G., Yao X., Yin Z., Dong D., Dejnirattisai W., Rostron T., Supasa P., Liu C., et al. Oxford Immunology Network Covid-19 Response T cell Consortium. ISARIC4C Investigators Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020;21:1336–1345. doi: 10.1038/s41590-020-0782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante J.A., Liu Y., Liu J., Xia H., Johnson B.A., Lokugamage K.G., Zhang X., Muruato A.E., Zou J., Fontes-Garfias C.R., et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2020 doi: 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Perez Marc G., Moreira E.D., Zerbini C., et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riepler L., Rössler A., Falch A., Volland A., Borena W., von Laer D., Kimpel J. Comparison of Four SARS-CoV-2 Neutralization Assays. Vaccines (Basel) 2020;9:13. doi: 10.3390/vaccines9010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda L.B., Netland J., Shehata L., Pruner K.B., Morawski P.A., Thouvenel C.D., Takehara K.K., Eggenberger J., Hemann E.A., Waterman H.R., et al. Functional SARS-CoV-2-Specific Immune Memory Persists after Mild COVID-19. Cell. 2020;184:169–183.e17. doi: 10.1016/j.cell.2020.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer A.K., Liang C.H., Stech J., Peeters B., Quéré P., Schwegmann-Wessels C., Wu C.Y., Wong C.H., Herrler G. Characterization of the sialic acid binding activity of influenza A viruses using soluble variants of the H7 and H9 hemagglutinins. PLoS ONE. 2014;9:e89529. doi: 10.1371/journal.pone.0089529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt F., Weisblum Y., Muecksch F., Hoffmann H.H., Michailidis E., Lorenzi J.C.C., Mendoza P., Rutkowska M., Bednarski E., Gaebler C., et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020;217:e20201181. doi: 10.1084/jem.20201181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surleac M., Casangiu C., Banica L., Milu P., Florea D., Sandulescu O., Streinu-Cercel A., Vlaicu O., Tudor A., Hohan R., et al. Evidence of novel SARS-CoV-2 variants circulation in Romania. AIDS Res. Hum. Retroviruses. 2021 doi: 10.1089/AID.2021.0009. [DOI] [PubMed] [Google Scholar]
- Umair M., Ikram A., Salman M., Alam M.M., Badar N., Rehman Z., Tamim S., Khurshid A., Ahad A., Ahmad H., Ullah S. Importation of SARS-CoV-2 Variant B.1.1.7 in Pakistan. J. Med. Virol. 2021 doi: 10.1002/jmv.26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz E., Hill V., McCrone J.T., Price A., Jorgensen D., O’Toole A., Southgate J., Johnson R., Jackson B., Nascimento F.F., et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell. 2021;184:64–75.e11. doi: 10.1016/j.cell.2020.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajnberg A., Amanat F., Firpo A., Altman D.R., Bailey M.J., Mansour M., McMahon M., Meade P., Mendu D.R., Muellers K., et al. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science. 2020;370:1227–1230. doi: 10.1126/science.abd7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Schmidt F., Weisblum Y., Muecksch F., Barnes C.O., Finkin S., Schaefer-Babajew D., Cipolla M., Gaebler C., Lieberman J.A., et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021 doi: 10.1038/s41586-021-03324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman D., Alameh M.G., de Silva T., Collini P., Hornsby H., Brown R., LaBranche C.C., Edwards R.J., Sutherland L., Santra S., et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe. 2021;29:23–31.e4. doi: 10.1016/j.chom.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . 2020. Weekly epidemiological update - 9 February 2021.https://www.who.int/publications/m/item/weekly-epidemiological-update---9-february-2021 [Google Scholar]
- Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Liu Y., Liu J., Zhang X., Zou J., Fontes-Garfias C.R., Xia H., Swanson K.A., Cutler M., Cooper D., et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021 doi: 10.1038/s41591-021-01270-4. [DOI] [PubMed] [Google Scholar]
- Yadav P.D., Nyayanit D.A., Sahay R.R., Sarkale P., Pethani J., Patil S., Baradkar S., Potdar V., Patil D.Y. Isolation and characterization of the new SARS-CoV-2 variant in travellers from the United Kingdom to India: VUI-202012/01 of the B.1.1.7 lineage. J. Travel Med. 2021;28:taab009. doi: 10.1093/jtm/taab009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Lou J., Cao L., Zheng H., Chong M.K.C., Chen Z., Chan R.W.Y., Zee B.C.Y., Chan P.K.S., Wang M.H. Quantifying the transmission advantage associated with N501Y substitution of SARS-CoV-2 in the UK: an early data-driven analysis. J. Travel Med. 2021;28:taab011. doi: 10.1093/jtm/taab011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study did not generate unique datasets or code.