

Abstract
The goal of personalized medicine is to match the right drugs to the right patients at the right time. Personalized medicine has been most successful in cases where there is a clear genetic linkage between a disease and a therapy. This is not the case with type 1 diabetes (T1D), a genetically complex immune-mediated disease of β-cell destruction. Researchers over decades have traced the natural history of disease sufficiently to use autoantibodies as predictive biomarkers for disease risk and to conduct successful clinical trials of disease-modifying therapy. Recent studies, however, have highlighted heterogeneity associated with progression, with nonuniform rate of insulin loss and distinct features of the peri-diagnostic period. Likewise, there is heterogeneity in immune profiles and outcomes in response to therapy. Unexpectedly, from these studies demonstrating perplexing complexity in progression and response to therapy, new biomarker-based principles are emerging for how to achieve personalized therapies for T1D. These include therapy timed to periods of disease activity, use of patient stratification biomarkers to align therapeutic target with disease endotype, pharmacodynamic biomarkers to achieve personalized dosing and appropriate combination therapies, and efficacy biomarkers for “treat-to-target” strategies. These principles provide a template for application of personalized medicine to complex diseases.
Introduction
The goal of personalized medicine is to match the right drugs to the right patients at the right time. Without a direct link between genetic etiology and targeted therapy, it is challenging to bring personalized medicine to type 1 diabetes (T1D), a genetically complex immune-mediated disease of β-cell destruction. While heterogeneity of drug responses in some diseases is linked to well-defined genetic or environmental variables, this area remains underdeveloped in T1D. Moreover, as there currently are no approved therapeutic interventions that affect disease course, T1D presents a compelling opportunity to optimize biologic therapies addressing an unmet medical need. It is the very heterogeneity of T1D, both in the natural history and in response to therapy, that can be of use in a data-driven approach to the treatment of disease. Potential benefits of personalized medicine in T1D include matching drugs to the patient population(s) most likely to benefit from treatment, maximizing treatment benefits while minimizing side effects, and minimizing trial-and-error inefficiencies in developing new treatments. The goal of personalized medicine in T1D is to predict the optimal drug, or dose of a drug, for each individual patient.
Researchers over decades have traced the natural history of T1D before and after clinical diagnosis. The data are sufficiently robust for use of autoantibodies as predictive biomarkers in clinical trials to slow disease progression prior to clinical onset (1). After diagnosis, given a baseline value of insulin secretion and age, insulin secretion a year later can be reasonably predicted (2). Like for autoantibodies, such insulin secretion data allow for standardized approaches to clinical trial design. However, within these compelling data is the more complex reality of variability between individuals in natural history and response to therapy and the uneven pattern of functional or actual β-cell loss over time. Similarly, trials of several immune-modifying agents with different mechanism of action generally have yielded similar clinical courses (3). Typically, treatment is associated with a 6- to 12-month period of disease stabilization followed by progression at a rate similar to that in untreated subjects. Unbiased systems approaches using peripheral blood samples from patients in multiple clinical trials have elucidated diverse immunologic mechanisms associated with good versus poor outcome, highlighting previously unknown relationships between disease heterogeneity and response to therapy (4–7). Together, these studies suggest new data-driven approaches to the treatment of T1D and possibly other autoimmune diseases. These strategies will be discussed in the following sections.
Nonuniform β-Cell Loss Over Time: Accelerated Change Is Active Disease
Studies of multiple T1D autoantibody–positive individuals have repeatedly demonstrated impaired insulin secretion many years prior to diagnosis. Longitudinal studies note that for many individuals impaired β-cell function, when measured by C-peptide response to oral glucose tolerance test or first-phase insulin response to i.v. glucose, is persistent, but stable, for many years (8,9). Often, this apparently stable β-cell function begins to fall within 6–12 months prior to clinical diagnosis. Importantly, this increased rate of fall continues as individuals cross the glucose diagnostic threshold (10), until the rate of fall of insulin secretion again appears to level off 6–12 months later, mirroring the pattern prior to clinical diagnosis (11). These data point to a 12- to 18-month interval around the time of clinical diagnosis when a rapid change in insulin secretion capacity is occurring—a period of “active” disease in an individual providing opportunities for personalizing therapy (Fig. 1).
Figure 1.

Nonlinear progression of β-cell loss characterizes stages of T1D. The peri-diagnostic period is highlighted, reflecting a period of accelerated β-cell dysfunction, superimposed on a slow progressive functional decline that begins prior to clinical diagnosis and continues through the early post-diagnostic interval (“Stage 3” disease, or clinical T1D). Observations of immunological acceleration occur within this period, presenting an opportunity for targeted immune intervention based on identification of individualized immune characteristics associated with rapid decline—both prior to diagnosis (blue box, “prevention therapy”) and after diagnosis (green box, “intervention therapy”). Adapted from Greenbaum et al. Strength in numbers: opportunities for enhancing the development of effective treatments for T1D—the TrialNet experience. Diabetes 2018;67:1216–1225.
If a rapid change in insulin secretion defines a period of active disease, there are at least three hypothesis why this could occur (Fig. 2). The window around clinical diagnosis associated with more rapid loss of insulin secretion could be due to an immune “flare.” Several immune therapies administered during this time show partial efficacy (12–17), and in the TrialNet Oral Insulin study, while the overall results were negative, the cohort of individuals with the most rapid progression and thus presumably with active disease responded to therapy (18). A second hypothesis posits that the acceleration in β-cell dysfunction occurs when the system has reached a tipping point, in which a chronic slow loss of function eventually results in “sudden” collapse. Data supporting this concept come from human as well as nonimmune animal models of β-cell destruction, documenting rapid fall in secretion and increase in glucose after 50% loss of β-cell mass (19,20). The third hypothesis blends both these concepts. Rather than this time period initiating from an immune flare of disease, it may instead represent acceleration of β-cell injury from any cause that in turn further promotes immune-mediated damage.
Figure 2.
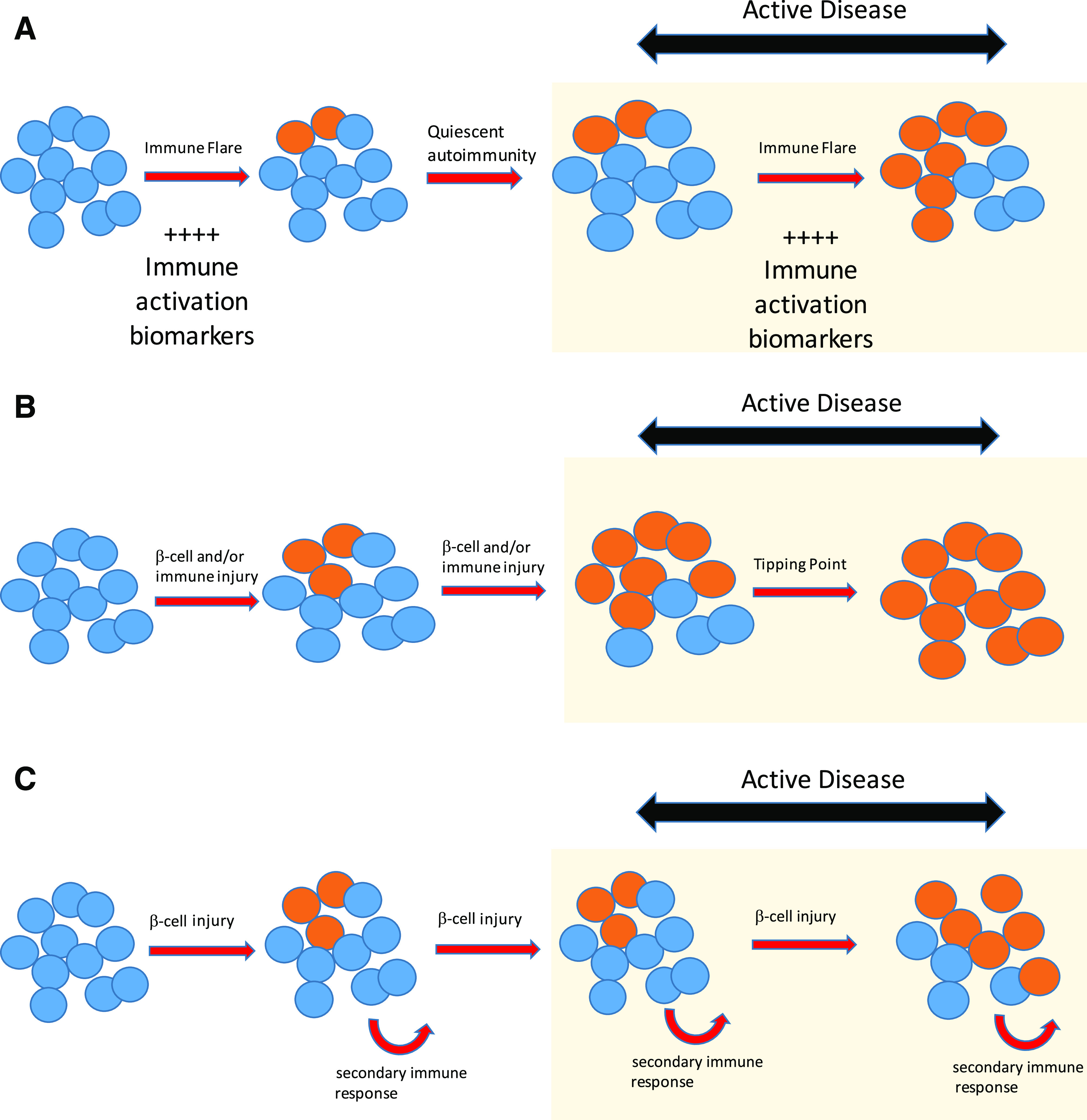
Potential mechanisms accounting for a period of accelerated change in insulin secretion during the development of T1D. Functional loss of β-cells is associated both with intrinsic properties of damaged or stressed islets and with extrinsic properties of activated and aggressive immune responses. The interplay between these two factors manifests in an accelerated phase of disease activity associated with biomarker changes in both intrinsic and extrinsic compartments and manifested by a rapid change in β-cell function. Blue ovals show functional β-cells. Orange ovals show dead or dysfunctional β-cells. A: Hypothesis: Immune activation causes active disease. Prediction: Immune biomarkers will associate only with periods of β-cell change. B: Hypothesis: β-Cell injury and associated autoimmunity continue over time. Active disease occurs due to β-cell destruction at threshold or tipping point. Prediction: No association of immune markers and active disease. Nonlinear changes in markers of β-cell injury. C: Hypothesis: β-Cell injury from nonimmune source is primary cause of dysfunction. Immunity is in response to tissue injury. Active disease occurs due to episodic acute (exogenous/environmental) injury. Prediction: No association of immune markers and active disease. Markers of β-cell injury will be strongly associated with changes of β-cell function.
Measures of Active Disease
β-Cell Health/Islet Function.
To determine whether acceleration of β-cell damage initiates the period of active disease, alternative measures of β-cell health or islet function may be needed to detect subtle injury. This includes physiologic measures of functional β-cell mass through longitudinal assessments of glucose-potentiated arginine or glucagon secretion (21,22), markers of β-cell injury including shifts in proinsulin/insulin (23), and proIAPP-to-amylin ratios (24) that may indicate defects in processing enzymes. Circulating blood measures of cell-free DNA to indicate β-cell death have also been described (25), although recent work with a more sensitive and specific assay highlights that challenges remain in interpreting results from these tests (26). Simultaneous longitudinal measures of exocrine function (27) and size or other characteristics of the whole pancreas detected by imaging studies (28) may provide key data for evaluation of the temporal relationship of β-cell injury to active disease.
Immune Measures.
Evaluating whether there are immune flares at any time during the progression of T1D or in association with the peri-diagnostic period of accelerated dysfunction has proven challenging. Prior to clinical diagnosis, most work has focused on autoantibodies, evaluating patterns of antibody types or titers over time with progression of disease defined as worsening glucose tolerance. However, since insulin secretion measured by oral glucose tolerance test or first-phase insulin response is only moderately associated with glucose tolerance (29), such analytic approaches may limit the ability to find an immune change signaling active disease that manifests as rapid change in β-cell function. Reanalysis of existing immune biomarker data prior to diagnosis in relationship to change in insulin secretion rather than glucose tolerance is needed.
In contrast, studies of immune markers after T1D diagnosis to either predict or associate disease state with rate of loss of secretion are unlikely to reveal an immune flare that initiates or signals the start of active disease. Furthermore, there are few longitudinal multidimensional immune data in individuals before, during, and after the change in slope of insulin secretion defining active disease occurring in the peri-diagnostic period. An additional challenge is that blood testing and measures of insulin secretion and glucose tolerance are customarily done at 6- or 12-month intervals; this infrequent sampling may result in missing signals of active disease.
Implications for Response to Therapy
A large number of immune interventions have now been evaluated in T1D clinical trials, and in each case, the response of subjects receiving an active immunological agent is highly variable. Differences in disease activity, as outlined above, overlap in these trials with differences in baseline and induced immune parameters. Longitudinal multidimensional data with simultaneous assessments of β-cell dysfunction and immune markers are limited, but innovative clinical trial designs can help test the relationship of efficacy to when therapy is administered. Prior to clinical diagnosis, trials could use prerandomization data to stratify those with pretreatment changes in insulin secretion versus those with stable secretion. Or trials could be done where the enrollment criteria require a fall of secretion as entry criteria rather than use of glycemic status to define cohorts—an important change since the hallmark of disease is β-cell failure and there is only a moderate relationship between secretion and glucose tolerance. Aligning our understanding of these subject-specific characteristics with opportunities for use of individualized targeted therapies is an important next step, informed by several recent trials that are discussed below. The more selective the entry criteria, the more challenging trial enrollment may be. However, such practical concerns may be offset by the potential for a greater effect in a more targeted population, such that a smaller number of subjects may be required. Alternatively, if interim markers of drug effect(s) could be identified, shorter trials may be possible.
T-cell autoreactivity is a major component of the immune response in T1D, with activated cells expressing markers of immune memory and self-renewal as hallmarks of disease (30–33). Various drugs that deplete T cells or interfere with T-cell function have been tested in T1D clinical trials, and analysis of T-cell subsets reveals that differences in treatment efficacy are correlated with differential effects on different T-cell subsets. An example is shown in Fig. 3, comparing the effect of anti–T-cell therapy administered to subjects recently diagnosed with T1D. In the Inducing Remission in New-onset Type 1 Diabetes with Alefacept (T1DAL) trial (17), represented in Fig. 3A, depletion of effector T cells (a subset that displays memory markers and effector molecules indicating proinflammatory properties) follows administration of alefacept, a therapeutic agent that targets the CD2 molecule on these T cells. Another T-cell subset, regulatory T cells (functional suppressors of immune responses) is not depleted, due to much lower expression of CD2. As a consequence, the ratio of regulatory to effector cells is increased during each therapeutic administration of alefacept, which corresponds to the time frame in which metabolic stability, measured by preservation of C-peptide, is observed. These data contrast with those shown in Fig. 3B, from the Study of Thymoglobulin to ARrest T1D (START) clinical trial (34), in which similar subjects received administration of antithymocyte globulin (ATG), an anti–T-cell antibody mixture that depleted both effector and regulatory cells. As shown, there was no change in the regulatory-to-effector cell ratio and no preservation of C-peptide from this therapy. The importance of preserving regulatory T cells during anti–T-cell therapy is emphasized by the TrialNet trial that tested a lower dose of ATG (15), which achieved a relative sparing of regulatory cells and also improved clinical outcomes relative to the START trial. T cell–directed therapy in T1D may be viewed as a form of induction regimen, depleting or modulating sufficient effector cells to achieve transient arrest of the autoimmune process while potentially allowing regulatory cells and mechanisms to restore homeostasis. Interestingly, efficacy (measured as preservation of C-peptide) is often, but not always, strongest in younger subjects in the trials, suggesting a correlation with a more active disease state, as discussed in the previous section. This is consistent with an interpretation that a promising way to use T-cell depletion therapy could be to select subjects based on the presence of activated effector T cells, perhaps most prevalent during the disease-acceleration phase described above, while assuring that regulatory T cells are available, and spared, to allow for their preferential survival.
Figure 3.
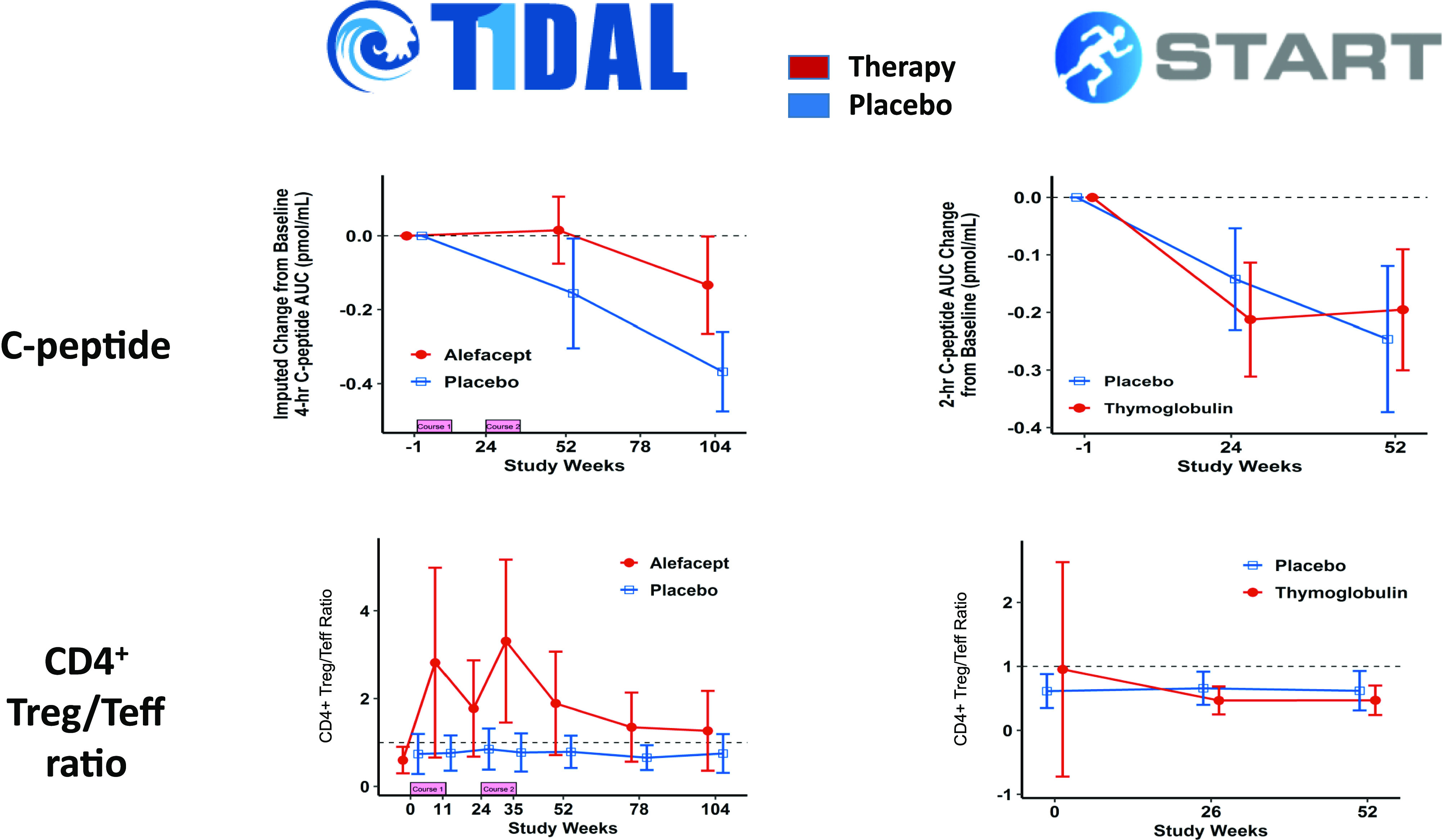
Frequencies of regulatory T cells (Treg) and effector T cells (Teff) following use of anti–T-cell immunomodulatory therapy correlate with persistence or loss of insulin secretory capacity. Shown are stimulated C-peptide area under the curve (AUC) measurements from the T1DAL trial of alefacept (top left panel) and the START trial of ATG (top right panel), documenting transient preservation of β-cell function following alefacept treatment. The bottom panels display a ratio of the frequencies of regulatory and effector T lymphocytes in peripheral blood during these clinical trials, illustrating a marked increase in this ratio during the period of C-peptide preservation in T1DAL but not in START. Data are adapted from Rigby et al. (17) and Gitelman et al. (34).
T-cell immunobiology is complex, and general subset classifications such as “regulatory” and “effector” lymphocytes, as cited above, do not fully reflect important functional properties that may correlate with therapeutic benefit. Examples include a helper T-cell subset known as Tfh (follicular helper T cells), selectively targeted by abatacept, which specialize in communication between T and B lymphocytes supporting antibody production, and an effector T-cell subset known as Tex (exhausted T cells), a state induced by chronic activation that reflects low functional capacity and correlates with response to teplizumab, as discussed below.
The concept of selecting immune subset-specific therapy for particular subjects is not limited to T-cell compartments. A recent systems biology study in T1D using peripheral blood transcriptome analysis identified patterns of B-cell transcripts and of neutrophil transcripts that also fit this paradigm (7). In this study, subjects enrolled in the placebo arms of T1D clinical trials showed increases in peripheral B-cell gene expression profiles correlating with more rapid loss of C-peptide over the course of 2 years after diagnosis; lower neutrophil gene expression profiles were observed in the same samples. This relationship was particularly strong in younger T1D subjects, and opposite profiles were observed in older subjects with slower progression of C-peptide loss. Stratification of subjects with this B-cell profile who were enrolled in the TrialNet clinical trial and receiving B-cell depletion therapy with rituximab (12) showed a significant relationship with the rate of loss of C-peptide, suggesting that rituximab was most effective in those with high B-cell gene expression.
In the examples summarized above, indicators of increased T- and B-cell immunity correlated with age but offer additional value for targeted therapeutic benefit. Using young age as an indicator of suitability for these types of immune modulation therapy is itself a way to increase the likelihood of response but is an imprecise strategy (35). And although fairly simple biomarkers such as effector T-cell and B-cell profiles improve this precision, it is likely that even more specific immune properties can further enhance the value of biomarker stratification (36). For example, one possible refinement indicating T-cell target suitability may be to focus on plasticity or stem cell–like properties of these cells, characteristics that indicate potential for development into autoreactive effector cells (37). Another potential strategy may be to use clonal expansions as indicators of in vivo autoreactive activation, visualized through T-cell receptor sequencing (38,39). Functional properties of autoreactive T cells that make them appropriate targets for directed therapy, such as their resistance to immune regulation (40), are more difficult to measure but likely also represent characteristics that will identify individuals well suited for targeted effector T-cell therapy. In addition, monitoring of the recurrence of these types of effector cells could be very helpful in deciding whether and when to repeat treatment rather than delaying until further metabolic decompensation occurs. Indeed, it is possible that amplification of resistant effector cells accumulates with age and immune experience, providing an additional rationale for focusing induction therapy on younger subjects and early phases of disease.
Selection of subjects likely to respond to induction therapy, directed by T- and/or B-cell characteristics, offers a first step toward improving the likelihood of a clinical response. Even with this advance, however, there remains the equally important challenge of selecting therapies that will maintain a beneficial response. By analogy with the “induction, then consolidation” strategies used in cancer chemotherapy, durable benefit in T1D immune therapy likely will require a consolidation step following successful induction treatment. Indeed, if induction therapy involves lymphocyte depletion, avoiding a rebound phenomenon from reconstituting effector cells is likely to be essential. Maintaining therapeutic benefit may also require biomarker-driven patient stratification, and two distinct types of immune profiles have been associated with successful consolidation in previous clinical trials: CD4 T-cell regulation and CD8 T-cell exhaustion.
As noted in the example in Fig. 3, elevation of the regulatory-to-effector T cell ratio is a correlate of successful preservation of C-peptide in the context of T cell–targeted therapy. Interestingly, Tregs in some T1D subjects have been reported to be deficient in their response to IL2, a cytokine that supports maintenance of regulatory function in these cells (41). This may present an opportunity to use biomarker-driven selection of therapies that target IL2 signaling in individuals who display this type of Treg deficiency, to enhance consolidation of therapeutic benefit. Strategies to enhance Treg capabilities in this context are challenging, with several potential pitfalls such as whether the Tregs need to be targeted to particular islet antigens, whether they will they be long-lived and have stable function, and whether an initial induction therapy has abrogated a hostile tissue environment that could inhibit regulatory activity. Indeed, assessing these types of hazards to Treg utility may provide additional opportunities for personalization of the choice of therapeutic option.
A different type of consolidation strategy is suggested by studies in T1D clinical trials of teplizumab, an anti-CD3 monoclonal antibody: In these studies, CD8 T cells acquire an exhaustion-like phenotype after induction therapy with teplizumab, consistent with a mechanism in which partial agonism through the T-cell receptor triggers a cellular program similar to an exhaustion state frequently seen in the immune response to cancers and chronic virus infection. This phenotype is associated with low levels of immune function, and in clinical trials of recent-onset T1D and secondary prevention of T1D (4,16), acquisition of this phenotype is significantly correlated with preservation of C-peptide and delay or prevention of T1D. Furthermore, there is a strong indication that sustaining an exhausted T-cell response is advantageous: In the Autoimmunity-blocking Antibody for Tolerance in Recently Diagnosed Type 1 Diabetes (AbATE) trial, teplizumab was administered in two cycles 1 year apart. Only subjects who responded with induction of an exhaustion profile on both occasions had long-term favorable outcomes, and a retrospective analysis indicated that failure to respond to the second cycle of therapy was correlated with the presence of anti-drug antibodies, providing a likely explanation. This finding suggests the possibility of using T-cell exhaustion markers or other regulatory phenotypes (42,43) to monitor effective consolidation therapies in individuals, potentially guiding choice of drugs and timing of treatment to sustain a response.
There may be an interactive relationship between some of these immune characteristics and the age dependence of T1D progression in children with T1D (11,44). For example, subjects in the TrialNet study of abatacept showed age-related baseline features, characterized by elevated levels of B cells or neutrophils, that accompanied rapid or slow progression, respectively, in both abatacept- and placebo-treated groups (6). Other studies from the same trial reported alterations in T cell subsets associated with response to treatment: A reduction in CD4 central memory T cells (45) and positive association of baseline abundance of circulating Treg cells was associated with slower C-peptide decline (46) in abatacept-treated subjects. In contrast, higher frequencies of follicular helper T cells at baseline were associated with a poor clinical response (47). Together, these data suggest that better therapeutic success may be gained by targeting immunomodulatory therapy in specific T1D populations defined by considering both age and immune characteristics.
Figure 4 summarizes these opportunities to use immune profiling for identification of individualized characteristics that could influence choices of immune therapy in T1D. Whether selecting subjects who could be eligible for personalized therapy, choosing a specific type of targeted intervention, or making a decision about repeating therapy, understanding and monitoring of immunological parameters that differ between individuals offers a way to evaluate the potential for tailoring intervention. In the context of a multitude of immunologically active drug candidates, careful evaluation of characteristics like these should facilitate rational choices, efficient study design, and improved likelihood of good clinical outcomes.
Figure 4.

Opportunities for individualized therapeutic decision-making using T1D immune profiles. Targeted therapeutics for particular T or B lymphocytes, specific cytokines, or other selected immune pathways provide alternatives for disease intervention, tailored to individualized patients. Such personalized approaches will require biomarker stratification that reflects several types of immune characteristics, examples of which are listed here. The choice of biomarker and choice of therapeutic may be different during initial patient assessment compared with later in the disease course, reflecting changes in the immunobiology of T1D during disease progression. Teff, effector T lymphocytes; TCR, T-cell receptor; SCM, stem cell memory; Treg, regulatory T lymphocytes.
Personalized Pharmacodynamics
A surprising finding across multiple studies has been the association of poor outcome with reduced pharmacodynamic activity in some subjects. For example, subjects resistant to treatment with abatacept showed a transient increase in activated B cells, altered costimulatory ligand gene expression, and reduced inhibition of anti-insulin antibodies (6). These findings suggested nonuniform pharmacodynamic activity of abatacept across different subjects. Similarly, studies with rituximab (12) showed that subjects with high levels of T cells showed less suppression of de novo antibodies targeting phiX174, again indicating suboptimal pharmacodynamic activity of rituximab treatment. Finally, in the AbATE study with teplizumab (13), we noted that responders to treatment showed more sustained lymphoreduction after treatment (Fig. 5). Transient lymphoreduction is a hallmark of teplizumab treatment and can be considered a proximal pharmacodynamic biomarker for T-cell targeting. Together, these findings suggest that personalized dosing of a monotherapy based on individualized pharmacodynamic measurements might improve efficacy of multiple biologic agents in T1D.
Figure 5.
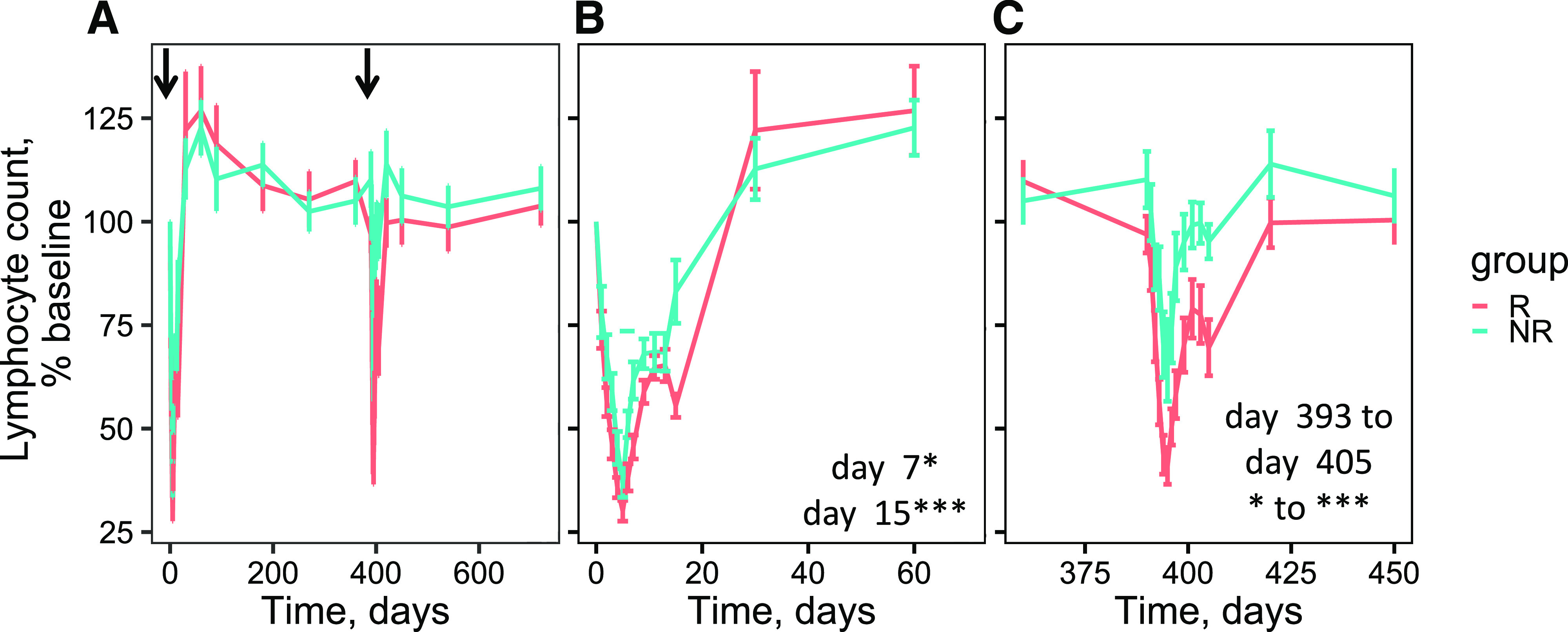
The extent of lymphocyte reduction is linked to therapeutic outcome after teplizumab treatment outcome in the AbATE study. Shown are mean lymphocyte counts ± SE (error bars) as determined by complete blood count at various intervals after treatment with teplizumab. Patient groups corresponding to responders (R) and nonresponders (NR) to teplizumab treatment were designated as previously described (13). A: Complete study. Arrows represent initiation of the two treatment cycles. B: First treatment cycle. C: Second treatment cycle. P values for individual time points: ***P value >1e-4 and <1e-3; *P value >1e-2 and <0.05. Unless otherwise indicated, P values were > 0.05 and considered not significant.
Individual variation in drug response is perhaps inevitable, and even though clinical trials generally focus on relatively homogenous patient populations, there may still be interindividual variation in optimal dosing. The concept of “precision dosing” is difficult to achieve in practice, with difficulty perhaps exacerbated when drugs developed on older individuals are used for younger patients, as is prevalent in T1D. One possible strategy is the implementation of “treat-to-target” (TTT) methodologies for the use of biologics in T1D. TTT is a therapeutic concept that uses specific physiologic targets to control disease pathophysiology. With type 2 diabetes, TTT has been used to compare investigational insulins with a standard insulin by titration of insulin dosages for achievement of a prespecified treatment goal (48). While, to our knowledge, TTT has not been applied for biologics in T1D, it is conceivable that biologic agents could be titrated to a desired clinical end point, or even that biomarkers or pharmacodynamic measures could be used as proxies for clinical measurements. Additionally, as we move forward in personalizing therapy, it may be possible to develop markers that separate generalized immune suppression from therapeutic efficacy.
Combination Therapies
Another conceptual implication evolving from such in-depth immunological characterization of individual outcomes in the trials is a focus on data-driven, pathway-targeted approaches for combination therapy in individual patients. In the case of biologic therapies for T1D, the case could be made for testing combinations of agents that as monotherapies have complementary mechanisms. We will here consider two pathways that can be targeted for beneficial effects in T1D: B cell–T cell interactions and CD8 T-cell exhaustion (Fig. 6).
Figure 6.

Combination therapy options to overcome mechanisms of resistance to monotherapies. Shown are therapeutic options for T1D based on targeting B cell–T cell interactions (A) and T-cell exhaustion (B). Black font, currently tested monotherapies; gray font, mechanism of resistance to monotherapy; red font, proposed combination (Combo) mechanism (B cell–T cell interactions [49] and T-cell exhaustion [4,50]); blue font, specific combination options. IR+, inhibitory receptor-positive.
Collaborative interactions between B cells and T cells are essential for successful adaptive immune responses. Therapeutically, these interactions can be modulated by altering levels of B cells or T cells (by rituximab and teplizumab, respectively) or by blocking cognate interactions (by abatacept). One observation from mechanism of action and resistance studies of biologic agents in T1D is that resistance to agents that modify B cell–T cell interactions may be accompanied by overproduction of a cognate cell partner. For example, suboptimal therapy with B cell–targeting rituximab was accompanied by elevated peripheral blood T-cell levels (5). Likewise, suboptimal therapy with the T-cell costimulation blocker, abatacept, was linked to elevated peripheral blood B cells and altered expression of costimulatory molecules (6). In both cases, suboptimal clinical treatment was associated with incomplete blocking of de novo antibody production, which can be regarded as a downstream pharmacodynamic assay. Considering these observations in terms of a model for B cell–T cell interactions (Fig. 6A) suggests that suboptimal treatment with either single agent could be augmented using the other agent, with a complementary mechanism of resistance. This is the rationale underlying the planned TrialNet study testing sequential administration of rituximab and abatacept (available from ClinicalTrials.gov, clinical trial reg. no. NCT03929601). Furthermore, these studies recall early studies of biologic agents in organ transplantation (49), where best graft acceptance was obtained using a combination of agents blocking T-cell costimulation (abatacept) and B-cell activation (anti-CD40LG). Although such an agent has not yet been tested in combination with abatacept, organ transplantation studies suggest that this combination may be beneficial.
Another pathway that may be targeted for beneficial effects in T1D is induction of CD8 T-cell exhaustion using the anti–CD3-targeting monoclonal antibody teplizumab (Fig. 6B). Studies to date have not shown untoward tolerability issues with teplizumab, suggesting that induction of T-cell exhaustion does not trigger dangerous levels of immunosuppression. Current thinking based on studies in cancer and chronic viral infection invokes a three-signal model for development of T-cell exhaustion, with persistent antigen load, negative costimulation, and chronic inflammation comprising signals 1–3, respectively (50). In this model, teplizumab likely displays agonist effects on T-cell receptor signaling. This suggests that suboptimal effects of teplizumab may be complemented by enhancing one of the other signals triggering T-cell exhaustion (negative costimulation or chronic inflammation). For example, negative costimulation could be enhanced using either abatacept to block positive costimulation or an inhibitory receptor agonist, such as a PD-1 agonist, for negative signaling.
Concluding Remarks
Individual variation presents a challenge to “one-size-fits-all” approaches to immune-based therapies for T1D. Recent biomarker studies indicate new opportunities for patient stratification based on individualized parameters, including age and particular immune phenotypes, which merge timing of active disease with targeted treatment options (Table 1). Additional studies identifying biomarkers for pharmacodynamic activity and resistance mechanisms will enable better dosing and therapeutic combinations. These studies presage an era of personalized therapy for complex autoimmune diseases, with near-term opportunities to improve study designs that will accelerate more targeted and effective therapy for clinical use.
Table 1.
Key concepts
| 1. T1D is a progressive disease, with acceleration in the peri-diagnostic period. This phase provides a targeted—and individualized opportunity for therapeutic intervention. |
| 2. Integration of immune biomarker data with β-cell secretion measurements offers a personalized alternative to current classification of disease status. |
| 3. Immune characteristics that define an individual’s autoimmune response have distinct properties that invite targeted immune therapy. |
| 4. Use of longitudinal insulin secretion and immune acceleration biomarkers offers opportunities for efficient clinical trial enrollment and randomization. |
| 5. Alignment between biomarkers and specific therapy options targeting T- or B-cell compartments suggests strategies for improved induction therapy of T1D. |
| 6. Rational combinations of therapeutic agents, informed by biomarker assessments, may be essential for avoiding disease recurrence after initial immune therapy. |
| 7. Individualized “treat-to-target” methodologies, using immune biomarkers, can enable precision dosing that is likely to improve efficacy of multiple biologic agents in T1D. |
Article Information
Acknowledgments. The authors thank the numerous colleagues and collaborators who have shared ideas and participated in the work cited and thank the patients and families who have volunteered for clinical studies.
Funding. The authors’ work was supported in part by National Institutes of Health grants UM1AI09565 and DP3 DK104465-0.
Duality of Interest. P.S.L. has consulting agreements with Bristol-Myers Squibb and Viela Bio. C.J.G. consulted with Viela Bio this past year and received in-kind support for ongoing research studies from Bristol-Myers Squibb and Janssen. No other potential conflicts of interest relevant to this article were reported.
References
- 1.Bingley PJ, Wherrett DK, Shultz A, Rafkin LE, Atkinson MA, Greenbaum CJ. Type 1 Diabetes TrialNet: a multifaceted approach to bringing disease-modifying therapy to clinical use in type 1 diabetes. Diabetes Care 2018;41:653–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bundy BN; Type 1 Diabetes TrialNet Study Group . A quantitative measure of treatment response in recent-onset type 1 diabetes. Endocrinol Diabetes Metab 2020;3:e00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenbaum CJ, Schatz DA, Haller MJ, Sanda S. Through the fog: recent clinical trials to preserve β-cell function in type 1 diabetes. Diabetes 2012;61:1323–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long SA, Thorpe J, DeBerg HA, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol 2016;1:eaai7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linsley PS, Greenbaum CJ, Rosasco M, Presnell S, Herold KC, Dufort MJ. Elevated T cell levels in peripheral blood predict poor clinical response following rituximab treatment in new-onset type 1 diabetes. Genes Immun 2019;20:293–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linsley PS, Greenbaum CJ, Speake C, Long SA, Dufort MJ. B lymphocyte alterations accompany abatacept resistance in new-onset type 1 diabetes. JCI Insight 2019;4:e126136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dufort MJ, Greenbaum CJ, Speake C, Linsley PS. Cell type-specific immune phenotypes predict loss of insulin secretion in new-onset type 1 diabetes. JCI Insight 2019;4:e125556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans-Molina C, Sims EK, DiMeglio LA, et al.; Type 1 Diabetes TrialNet Study Group . β cell dysfunction exists more than 5 years before type 1 diabetes diagnosis. JCI Insight 2018;3:e120877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sosenko JM, Skyler JS, Beam CA, et al.; Type 1 Diabetes TrialNet and Diabetes Prevention Trial–Type 1 Study Groups . Acceleration of the loss of the first-phase insulin response during the progression to type 1 diabetes in Diabetes Prevention Trial–Type 1 participants. Diabetes 2013;62:4179–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogun MM, Bundy BN, Goland RS, Greenbaum CJ. C-peptide levels in subjects followed longitudinally before and after type 1 diabetes diagnosis in TrialNet. Diabetes Care 2020;43:1836–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenbaum CJ, Beam CA, Boulware D, et al.; Type 1 Diabetes TrialNet Study Group . Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes 2012;61:2066–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al.; Type 1 Diabetes TrialNet Anti-CD20 Study Group . Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 2009;361:2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herold KC, Gitelman SE, Ehlers MR, et al.; AbATE Study Team . Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013;62:3766–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orban T, Bundy B, Becker DJ, et al.; Type 1 Diabetes TrialNet Abatacept Study Group . Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 2011;378:412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haller MJ, Long SA, Blanchfield JL, et al.; Type 1 Diabetes TrialNet ATG-GCSF Study Group . Low-dose anti-thymocyte globulin preserves C-peptide, reduces HbA1c, and increases regulatory to conventional T-cell ratios in new-onset type 1 diabetes: two-year clinical trial data. Diabetes 2019;68:1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herold KC, Bundy BN, Long SA, et al.; Type 1 Diabetes TrialNet Study Group . An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med 2019;381:603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigby MR, Harris KM, Pinckney A, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest 2015;125:3285–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krischer JP, Schatz DA, Bundy B, Skyler JS; Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study Group . Effect of oral insulin on prevention of diabetes in relatives of patients with type 1 diabetes: a randomized clinical trial. JAMA 2017;318:1891–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC. Relationship between β-cell mass and fasting blood glucose concentration in humans. Diabetes Care 2006;29:717–718 [DOI] [PubMed] [Google Scholar]
- 20.Saisho Y, Butler AE, Manesso E, et al. Relationship between fractional pancreatic beta cell area and fasting plasma glucose concentration in monkeys. Diabetologia 2010;53:111–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao W, Woodwyk A, Beam C, Bahnson HT, Palmer JP, Greenbaum CJ. Assessment of β cell mass and function by AIRmax and intravenous glucose in high-risk subjects for type 1 diabetes. J Clin Endocrinol Metab 2017;102:4428–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandemeulebroucke E, Keymeulen B, Decochez K, et al.; Belgian Diabetes Registry . Hyperglycaemic clamp test for diabetes risk assessment in IA-2-antibody-positive relatives of type 1 diabetic patients. Diabetologia 2010;53:36–44 [DOI] [PubMed] [Google Scholar]
- 23.Sims EK, Chaudhry Z, Watkins R, et al. Elevations in the fasting serum proinsulin–to–C-peptide ratio precede the onset of type 1 diabetes. Diabetes Care 2016;39:1519–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Courtade JA, Klimek-Abercrombie AM, Chen YC, et al. Measurement of pro-islet amyloid polypeptide (1-48) in diabetes and islet transplants. J Clin Endocrinol Metab 2017;102:2595–2603 [DOI] [PubMed] [Google Scholar]
- 25.Lehmann-Werman R, Neiman D, Zemmour H, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A 2016;113:E1826–E1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neiman D, Gillis D, Piyanzin S, et al. Multiplexing DNA methylation markers to detect circulating cell-free DNA derived from human pancreatic β cells. JCI Insight 2020;5:e136579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell-Thompson M, Rodriguez-Calvo T, Battaglia M. Abnormalities of the exocrine pancreas in type 1 diabetes. Curr Diab Rep 2015;15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Virostko J, Williams J, Hilmes M, et al. Pancreas volume declines during the first year after diagnosis of type 1 diabetes and exhibits altered diffusion at disease onset. Diabetes Care 2019;42:248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greenbaum CJ, Cuthbertson D; Disease Prevention Trial of Type I Diabetes Study Group . Type I diabetes manifested solely by 2-h oral glucose tolerance test criteria. Diabetes 2001;50:470–476 [DOI] [PubMed] [Google Scholar]
- 30.Habib T, Long SA, Samuels PL, et al.; Type 1 Diabetes TrialNet Study Group . Dynamic immune phenotypes of B and T helper cells mark distinct stages of T1D progression. Diabetes 2019;68:1240–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barcenilla H, Åkerman L, Pihl M, Ludvigsson J, Casas R. Mass cytometry identifies distinct subsets of regulatory T cells and natural killer cells associated with high risk for type 1 diabetes. Front Immunol 2019;10:982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viisanen T, Ihantola EL, Näntö-Salonen K, et al. Circulating CXCR5+PD-1+ICOS+ follicular T helper cells are increased close to the diagnosis of type 1 diabetes in children with multiple autoantibodies. Diabetes 2017;66:437–447 [DOI] [PubMed] [Google Scholar]
- 33.Yeo L, Woodwyk A, Sood S, et al. Autoreactive T effector memory differentiation mirrors β cell function in type 1 diabetes. J Clin Invest 2018;128:3460–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gitelman SE, Gottlieb PA, Felner EI, et al.; ITN START Study Team . Antithymocyte globulin therapy for patients with recent-onset type 1 diabetes: 2 year results of a randomised trial. Diabetologia 2016;59:1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Battaglia M, Ahmed S, Anderson MS, et al. Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care 2020;43:5–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed S, Cerosaletti K, James E, et al. Standardizing T-cell biomarkers in type 1 diabetes: challenges and recent advances. Diabetes 2019;68:1366–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdelsamed HA, Zebley CC, Nguyen H, et al. Beta cell-specific CD8+ T cells maintain stem cell memory-associated epigenetic programs during type 1 diabetes. Nat Immunol 2020;21:578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerosaletti K, Barahmand-Pour-Whitman F, Yang J, et al. Single-Cell RNA sequencing reveals expanded clones of islet antigen-reactive CD4+ T cells in peripheral blood of subjects with type 1 diabetes. J Immunol 2017;199:323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobsen LM, Posgai A, Seay HR, Haller MJ, Brusko TM. T cell receptor profiling in type 1 diabetes. Curr Diab Rep 2017;17:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol 2008;181:7350–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Long SA, Cerosaletti K, Bollyky PL, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4+CD25+ regulatory T-cells of type 1 diabetic subjects. Diabetes 2010;59:407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Zhong T, Tang R, et al. PD-1 and PD-L1 expression in peripheral CD4/CD8+ T cells is restored in the partial remission phase in type 1 diabetes. J Clin Endocrinol Metab 2020;105:dgaa130. [DOI] [PubMed] [Google Scholar]
- 43.Terrazzano G, Bruzzaniti S, Rubino V, et al. T1D progression is associated with loss of CD3+CD56+ regulatory T cells that control CD8+ T cell effector functions. Nat Metab 2020;2:142–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wherrett DK, Chiang JL, Delamater AM, et al.; Type 1 Diabetes TrialNet Study Group . Defining pathways for development of disease-modifying therapies in children with type 1 diabetes: a consensus report. Diabetes Care 2015;38:1975–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orban T, Beam CA, Xu P, et al.; Type 1 Diabetes TrialNet Abatacept Study Group . Reduction in CD4 central memory T-cell subset in costimulation modulator abatacept-treated patients with recent-onset type 1 diabetes is associated with slower C-peptide decline. Diabetes 2014;63:3449–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cabrera SM, Engle S, Kaldunski M, et al.; Type 1 Diabetes TrialNet CTLA4-Ig (Abatacept) Study Group . Innate immune activity as a predictor of persistent insulin secretion and association with responsiveness to CTLA4-Ig treatment in recent-onset type 1 diabetes. Diabetologia 2018;61:2356–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edner NM, Heuts F, Thomas N, et al. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat Immunol 2020;21:1244–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garber AJ. Treat-to-target trials: uses, interpretation and review of concepts. Diabetes Obes Metab 2014;16:193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larsen CP, Elwood ET, Alexander DZ, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature 1996;381:434–438 [DOI] [PubMed] [Google Scholar]
- 50.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol 2019;37:457–495 [DOI] [PubMed] [Google Scholar]