

Abstract
Melanoma is the deadliest of all skin cancers due to its high metastatic potential. In recent years, advances in targeted therapy and immunotherapy have contributed to a remarkable progress in the treatment of metastatic disease. However, intrinsic or acquired resistance to such therapies remains a major obstacle in melanoma treatment. Melanoma disease progression, beginning from tumor initiation and growth to acquisition of invasive phenotypes and metastatic spread and acquisition of treatment resistance, has been associated with cellular dedifferentiation and the hijacking of gene regulatory networks reminiscent of the neural crest (NC)—the developmental structure which gives rise to melanocytes and hence melanoma. This review summarizes the experimental evidence for the involvement of NC stem cell (NCSC)‐like cell states during melanoma progression and addresses novel approaches to combat the emergence of stemness characteristics that have shown to be linked with aggressive disease outcome and drug resistance.
Keywords: cancer, development, drug resistance, invasion, melanoma, neural crest stem cells (NCSCs), tumor initiation
Significance statement.
This review summarizes the experimental evidence for reemergence of neural crest stem cell (NCSC)‐like cells in melanoma, which hijack embryonic programs to acquire advantages regarding tumor initiation and growth, metastatic spread, and eventually therapy resistance. Furthermore, the authors discuss preclinical efforts to specifically target NCSC‐like melanoma cells to combat drug resistance, which is a major goal in the field and will hopefully soon improve melanoma therapy for patients.
1. INTRODUCTION
The neural crest (NC) is a transient and multipotent stem cell population arising at the dorsal neural tube at early stages of vertebrate development. 1 , 2 , 3 Neural crest stem cells (NCSCs) disseminate into the embryo to differentiate into an array of cell lineages with remarkably different functions. 4 Among the adult NC derivatives are cartilage and bone structures in the head, the outflow tract of the heart, enteric nervous system cells in the gut, neuronal and non‐neuronal cells of the peripheral nervous system, melanocytes and others. 5 , 6
While most of the NC progeny have acquired a fully differentiated fate in the adult, cells with NCSC‐like characteristics have been isolated from several adult mouse tissues 7 as well as human skin 8 , 9 , 10 , 11 , 12 and dental pulp. 13 Even though the isolated cells possessed multipotent capacities in vitro and could give rise to different NC lineages, the question remains whether NCSC‐like cells derived from adult tissues really represent a maintained embryonic stem cell niche or whether these cells reacquire stem cell features due to isolation procedures and cultivation in vitro.
The reemergence of NCSC‐like cell states in vivo has been observed and studied in the context of pathological and physiological conditions such as carcinogenic transformations of NC‐derived adult tissues or wound healing, respectively. Murine tissue injury models revealed that NC‐derived Schwann cells (peripheral glia) dedifferentiate after injury and reacquire a progenitor‐like cell state by downregulating genes crucial for the myelination machinery and upregulating NCSC‐associated factors such as the neurotrophin receptor CD271 (also p75NTR, NGFR). 14 , 18 Injury‐activated glial cells were further shown to crucially assist regeneration of full‐thickness skin wounds or amputated digit tips. 17 , 18 Furthermore, previous reports revealed that Schwann cell precursors (SCPs) during embryonic development harbor NC cell features and are able to generate a variety of neural and non‐neural cell types. 15 , 16 Likewise, carcinogenic lesions of adult NC‐derived tissues such as melanoma or neuroblastoma can both present with cells expressing NCSC‐associated factors that are not normally expressed in the healthy adult tissue. 19 , 20 , 21 This review focuses on melanoma, wherein the aberrant regulation or expression of developmental NCSC genes or pathways is associated with different aspects of malignancy (Figure 1), such as tumor initiation and sustained tumor growth, 22 , 23 , 24 , 25 promotion of metastatic spread, 26 as well as resistance to therapies 27 , 28 , 29 , 30 and immune evasion. 31 , 32
FIGURE 1.
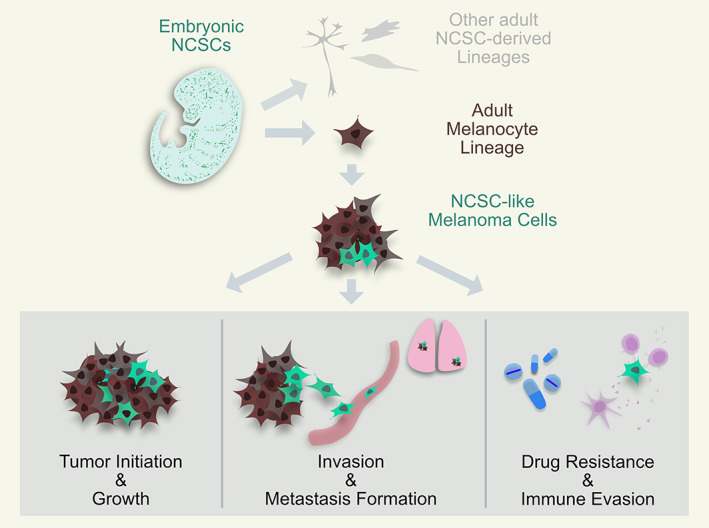
Reacquisition of neural crest stem cell (NCSC)‐like characteristics in melanoma and its implications. Embryonic NCSCs (dark green) arise at the developing dorsal neural tube and migrate into the whole embryo to form different tissues such as craniofacial bone and cartilage, enteric and peripheral nervous system cells (light grey) and, amongst many others, also cells of the melanocyte lineage, specifically melanocyte stem cells (MeSCs), melanoblasts and melanocytes (brown). Due to malignant transformations, those cells can progress into melanoma. Different studies have shown that some melanoma cells (light green) can hijack embryonic NCSC programs, bestowing them with different advantageous characteristics. Melanoma cells with reacquired NCSC features have been associated with the ability to form novel tumors and sustain growth, increased cell invasiveness and metastasis formation, as well as the ability to resist different melanoma therapies and evade immune surveillance
2. DEVELOPMENTAL NCSC GENES REGULATE MELANOMA GROWTH AND HOMEOSTASIS
Cutaneous malignant melanoma, the most aggressive skin cancer, 33 can present with astonishing heterogeneity, ranging from cells expressing typical melanocyte differentiation genes to gene sets typical for other NC‐derived cell lineages like neurons or glia, among others. 34 , 35 , 36 The origin of such heterogeneous tumors has been heavily debated and research was carried out on the expression of factors reminiscent of stem cells, which equips melanoma cells with renewal capacities in vitro and tumor formation potential in vivo. 22 , 37 , 38 For instance, the neurotrophin receptor CD271, which has been used for isolation of rat NCSCs with multipotent capacities in vitro, 14 was found reexpressed in a subset of melanoma cells that were able to initiate tumor formation when grafted into immunocompromised mice, while CD271‐negative cells lacked such potential 22 (Table 1). Furthermore, the tumors grown from single grafted CD271high cells presented with the heterogeneity of the parental tumor from which they were isolated, and not exclusively of CD271‐positive cells as grafted to generate the tumor, supporting the idea that CD271high cells represent stem cell‐like cells. 22 Of importance, CD271 is expressed in few adult cell types 64 , 65 and exerts divergent functions depending on the tissue context, for instance in melanocytes versus transformed malignant melanoma. 66 , 67 While signal transduction networks related to melanocytic differentiation (such as skin development, metabolic hormone processes or cell adhesion) have been found enriched in CD271‐positive human melanocytes, the same pathways were significantly downregulated in CD271‐positive human melanoma cells, 66 supporting the association of CD271high cell states with dedifferentiation and stemness potential in melanoma.
TABLE 1.
Literature overview of NCSC‐associated factors regulating parts of melanomagenesis
| Melanoma implication | Factor (function) | Detailed function in melanoma | References | NCSC function | References |
|---|---|---|---|---|---|
| Tumor initiation/growth | SOX10 (TF) | Broadly expressed in human nevi and melanomas. Depletion leads to abolished tumor formation in a NRAS Q61K Ink4a‐deficient mouse model, associated with reduced numbers of CD271+ cells. | 24 | Specifies murine NCSCs and melanocytic and glial lineages. | 39, 40 |
| YY1 (TF) | Haploinsufficiency is enough to prevent melanoma initiation in a NRAS Q61K Ink4a‐deficient mouse model. Regulates shared metabolic and translational pathways in neural crest and melanoma. | 25 | Essential for early murine NC development and the adult melanocyte lineage. | 25 | |
| CD271/NGFR/p75NTR (receptor tyrosine kinase) | Single CD271+ melanoma patient‐derived cells can form tumors (with the heterogeneity of the parental tumor) upon grafting into immunocompromised mice, while CD271‐ cells cannot. Inhibition of CD271 in human melanoma cells reduces their tumor initiation potential. | 22, 41 | Used to isolate mammalian NCSCs that were multipotent in vitro | 14 | |
| EZH2 (histone methyl transferase) | Upregulated in human malignant melanoma compared to melanocytes. Depletion in a NRAS Q61K Ink4a‐deficient mouse model inhibits melanoma growth. | 42 | Controls differentiation of NC‐derived mesenchymal lineages (bone and cartilage) | 43 | |
| DHODH (pyrimidine metabolism) | Transcriptional elongation of genes crucial for melanomagenesis. | 44 | Transcriptional elongation of neural crest developmental genes. | 44 | |
| DDX21 (RNA helicase) | Controls transcriptional elongation (after nucleotide shortage‐induced stress). | 45 | Controls transcriptional elongation. | 45 | |
| Crestin (unknown) | Marks tumor‐initiating cells in a BRAF V600E p53‐deficient zebrafish melanoma model. | 23 | mRNA widely expressed in zebrafish NCSCs | 46 | |
| Phenotype switch/invasion | MSX1 (TF) | Induces phenotypic switching (E‐cadherinhigh, nonmigratory toward ZEB1high, invasive) in melanoma. Depletion reduces liver metastasis after tail vain injection of human melanoma cells into immunocompromised mice. | 47 | NC induction in xenopus | 48 |
| Twist1/Zeb1 (TF) | NRAS/BRAF activation in melanocytes leads to upregulation of EMT TFs including TWIST1 and ZEB1, dedifferentiation and neoplastic transformation of melanocytes. | 49 |
Twist1: NC specifier; delamination of cranial NC; cell fate decision within cardiac NC Zeb1: upregulated by Zeb2, essential for melanocyte migration and differentiation. |
50, 51, 52 | |
|
FOXD3 PAX3 (TF) |
FOXD3 and PAX3 drive CXCR4 expression in melanoma, which was shown to promote melanoma metastasis formation. | 53, 54, 55 |
FOXD3: NC specifier PAX3: expressed in neural plate border |
56, 57 | |
| CD271 (receptor tyrosine kinase) | Associated with increased metastasis in patients. Transient overexpression induces a reversible phenotype switch in vitro and increased metastatic potential of human melanoma cells grafted onto immunocompromised mice. | 26, 35 | |||
| EZH2 (histone methyl transferase) | Depletion in a NRAS Q61K Ink4a‐deficient mouse model leads to abolished macro‐metastasis formation. | 42 | |||
| Drug Resistance |
FOXD3 (TF) ERBB3 (receptor tyrosine kinase) |
FOXD3 upregulates ERBB3, leading to BRAFi resistance in vitro and in vivo. | 58 | ERBB3: NC differentiation and dev. of sympathetic nervous system | 59 |
| CD271 (receptor tyrosine kinase) | NGFR+ AXL+ melanoma patient cells represent a dormant, MAPKi‐resistant cell population. Long‐term (3 weeks) BRAFi treatment leads to emergence of a drug‐tolerant or drug‐resistant NC‐like cell state in vitro. | 60, 61 | |||
| RXRG (nuclear receptor) | Minimal residual disease in a BRAFi/MEKi‐tolerant PDX model represents as dedifferentiated melanoma (NGFR+ RXRG+ AQP1+ GFRA+). | 28 | Expressed in migrating cranial chick NC cells. | 62 | |
| Immune Evasion | CD271 (receptor tyrosine kinase) |
TNFα induces dedifferentiation of melanoma cells (NGFRhigh) and resistance to adaptive T‐cell therapy in a murine model of adoptive cell transfer therapy. Long‐term exposure of patient‐derived melanoma cells to antigen‐specific cytotoxic T cells leads to enrichment of NGFRhigh cells, which are refractory to T cells as well as to BRAF/MEKi |
31, 32 | ||
| EZH2 (histone methyl transferase) | Intratumoral TNFα and T‐cell accumulation induce Ezh2 in melanoma cells originating from NRAS Q61K Ink4a‐loss or B16 F10 murine models, leading to loss of immunogenicity. | 63 |
Abbreviations: MEK, mitogen‐activated extracellular signal‐regulated kinase; NC, neural crest; NCSC, neural crest stem cell; SOX10, SRY‐related HMG‐box 10; TF, transcription factor; YY1, Ying Yang 1. Transgenic animal models: NRASQ61K Ink4a‐deficient mouse model, Tyr:NrasQ61K Cdkn2a ‐/‐; BRAFV600E p53‐deficient zebrafish, mitfa:BRAFV600E p53‐/‐.
The idea that stem cell‐like cells exist in melanoma was further strengthened by the finding that apart from CD271, the expression of other genes reminiscent of NCSCs was reported in melanoma and found to be essential for tumor initiation and maintained tumor growth. The transcription factor (TF) SRY‐related HMG‐box 10 (SOX10), for instance, which regulates the melanocytic and glial murine lineages derived from the NC, 39 , 40 was found highly expressed in melanocytic nevi and malignant melanoma. 24 , 68 , 69 Upon depletion of SOX10 in a transgenic NRAS Q61K‐mutant, Ink4a‐deficient murine melanoma model, tumor growth was diminished, and silencing of SOX10 in human melanoma cells drastically reduced the number of cells expressing CD271 and completely abolished tumor growth in vivo 24 (Table 1). Similar to SOX10, the TF Ying Yang 1 (YY1) was found to regulate both murine NC development and melanoma formation in NRAS Q61K‐mutant, Ink4a‐deficient mice 25 (Table 1). The importance of reemerging NCSC‐like cell states during melanoma initiation was highlighted in yet another study, where in BRAF V600E‐mutant, p53‐deficient melanoma‐prone zebrafish, cells capable of melanoma initiation activated a gene regulatory program characterized by the activation of zebrafish embryonic NC regulators such as crestin 23 (Table 1). Similarly, a recent study identified a set of genes expressed in embryonic melanoblasts, which are downregulated in fully differentiated melanocytes but partially reexpressed in human metastasis samples, providing further evidence for melanoma cells reacquiring progenitor‐like cell states. 70 Consequently, some of those genes like KDELR3 indeed proved to be essential for the formation of experimental metastasis in a model of tail vain‐injected murine B16 cells. 70
Nevertheless, the role of some genes involved in NC development remains unclear and heavily debated. SOX2, a member of the SRY TF family like SOX10, for instance, was shown to regulate NCSCs and SCPs together with MITF, while SOX2 downregulation led to precursor differentiation into melanocytes, 71 suggesting that the upregulation of SOX2 in melanoma might orchestrate stem cell‐like traits. However, while some studies report that SOX2 promotes tumor initiation of human cells grafted into immunocompromised mice, 72 , 73 others have shown that it is dispensable for melanomagenesis in genetically engineered mouse models. 74 , 75
While some factors reminiscent of NC development regulate cell cycle regulatory networks to allow tumor initiation and maintenance of adult melanoma, other cellular processes enabling melanoma tumor formation such as transcriptional regulation 44 , 45 or metabolism 25 have been shown to be regulated by NCSC‐associated factors. It was shown, for instance, that dihydroorotate dehydrogenase (DHODH), an enzyme involved in nucleotide biosynthesis, is essential for transcriptional elongation of genes crucial for both NC development and melanoma formation and that hence, inhibition of DHODH suppressed proper NC development as well as melanoma formation, especially when combined with BRAF inhibition 44 (Table 1). Mechanistically, DHODH inhibition‐induced nucleotide stress has recently been linked to the RNA helicase DDX21, which senses nucleotide shortage in NCSCs as well as melanoma cells, upon which it stops transcriptional elongation 45 (Table 1). In addition, DDX21 was found to regulate transcription in melanocyte stem cells (McSCs) in the hair follicle bulge to prevent differentiation into melanocytes. 76
There has been much debate recently about whether the cell at the origin of melanoma indeed represents a stem cell‐like cell. In fact, the above‐mentioned studies somewhat stand in contrast to reports claiming that instead of cells with stem cell features, fully differentiated melanocytes can give rise to melanoma. For instance, it was shown that BRAF V600E‐mutant, Pten‐deficient fully differentiated and melanin‐producing melanocytes could give rise to melanoma. 77 Of importance, Köhler et al 77 state in their study that after a first radial growth phase initiated by fully differentiated melanocytes, a subset of melanoma cells also underwent a transcriptional dedifferentiation process potentially reminiscent of the reemergence of NCSC‐like programs reported by others.
Yet another theory on the origin of melanoma argues that McSCs residing in the bulge of the hair follicle, rather than differentiated melanocytes, could stand at the origin of melanoma primary tumor formation. 78 , 79 Moreover, given that adult Schwann cells have the capacity of dedifferentiation in vivo 18 and that SCPs are able to generate melanocytes, 80 it is conceivable that some melanoma might derive from the peripheral glial lineage.
Several animal models have allowed melanoma induction with specific cues followed by in‐depth analysis and lineage tracing to understand disease initiation and progression. However, such analyses remain challenging in the context of human disease. While single‐cell studies of patient‐derived melanoma allow unprecedented insights into heterogeneous disease evolution, an alternative approach to tackle melanoma origin as such, could be the artificial induction of melanoma in melanocyte lineage cells derived from human embryonic stem cells followed by lineage tracing and single‐cell characterization. Yet, the mystery of how human melanoma arises in vivo remains to be unraveled.
3. NCSC‐LIKE CELLS CAN INDUCE INVASION AND METASTATIC SPREAD OF MELANOMA
Apart from inducing or maintaining melanoma tumor growth, the reacquisition of NCSC‐like characteristics has also been associated with later stages of melanoma progression, such as invasiveness and metastatic spread. 22 , 26 , 35 To become invasive and disseminate from the primary tumor, cancer cells are thought to undergo an epithelial to mesenchymal transition (EMT), rendering them with decreased cell‐to‐cell contacts, increased motility, and an increased potential to remodel extracellular matrix components. 81 , 82
Although, by definition, classical EMT is a process associated with epithelial cancers, which is not the case for melanoma, melanoma cells can undergo an EMT‐like process called “phenotype switching,” where cells transform from a high proliferative/low invasive to a low proliferative/high invasive phenotype. 83 , 84 Even though it has been reported that melanoma invasion and metastasis can progress independently of the ‘classical’ phenotype switching model, 85 EMT‐like phenotype switching is thought to be a crucial driver of melanoma invasiveness and metastasis formation. 86
Migratory NCSCs delaminating from the neural tube to migrate out into the embryo are a paradigm example for EMT during embryonic development. 4 , 5 , 87 Intriguingly, regulatory genes responsible for developmental NC EMT are reexpressed in adult malignant melanoma, 19 , 88 including members of the Snail, Zeb, and Twist families 49 (Table 1). Caramel et al 49 showed that upon NRAS/BRAF activation in melanocytes, the EMT TFs TWIST1 and ZEB1 were upregulated and induced dedifferentiation and neoplastic transformation. They also showed that this EMT‐TF signature, when found in late‐stage melanoma patients, correlated with poor prognosis. Similarly, transforming growth factor beta also acts as a potent inducer of melanoma phenotype switching, 84 , 89 while playing a crucial physiological role in NC development by providing signaling cues for migration and differentiation into several lineages. 90 , 91 , 92
Apart from an invasive and migratory potential, epithelial cancer cells undergoing EMT have been associated with the acquisition of stem cell‐like features. 93 , 94 Similarly, melanoma phenotype switching has been associated with the reemergence of signatures similar to NCSC regulatory networks 47 (Table 1). Expression of the NCSC‐associated factors CD271 and SOX10 39 , 40 in human melanoma correlates with high metastatic potential and worse patient prognosis 35 (Table 1). Furthermore, Msh homeobox 1 (MSX1), which specifies the NC at the neural border of zebrafish and xenopus, 48 when upregulated in melanoma, leads to dedifferentiation of melanoma cells, which upregulate NCSC‐associated factors such as CD271 and induce a phenotype switch toward increased cell migration 47 (Table 1). Vice versa, silencing of MSX1 reduces liver metastasis of tail vein‐injected human melanoma cells in mice. 47 Also the zebrafish and murine NC specifier FOXD3 together with PAX3, which is expressed at the neural plate border, 56 , 57 have been shown to induce human melanoma invasiveness by directly regulating CXCR4, 53 which in turn regulates melanoma metastasis formation 54 , 55 (Table 1). Finally, even though a zebrafish study suggested otherwise, 95 ectopic overexpression of CD271 induces a phenotype switch in human melanoma cells, ultimately leading to an increased metastatic potential of human melanoma cells grafted into immunocompromised mice 26 (Table 1). These findings revealed a functional involvement of single factors reminiscent of NCSCs in melanoma disease progression. Whether, in general, the reemergence of a broader NCSC signature is functionally implicated in melanoma metastasis formation remains to be elucidated.
4. DEDIFFERENTIATED MELANOMA CELLS DISPLAY RESISTANCE TO DIFFERENT THERAPIES
While traditionally the most common therapy for melanoma has been surgical removal of primary tumors plus radiation and chemotherapy, 96 the advent of immune and targeted therapies significantly improved the survival rate, especially of patients with metastatic melanoma. 33 Targeted therapies for melanoma are mostly directed against the serine/threonine kinase BRAF or the mitogen‐activated extracellular signal‐regulated kinase (MEK), leading to inhibition of the mitogen‐activated protein kinase (MAPK) pathway, an oncogenic pathway mutated and constitutively active in most melanomas. 97 , 98 Immunotherapies on the other hand aim at boosting the antitumoral activity of cytotoxic T lymphocytes (CTLs) to combat melanoma. 29 However, one of the major remaining challenges is the acquisition or preexistence of melanoma cells resistant to such therapies, 99 , 100 , 101 , 102 , 103 which ultimately lead to relapse.
Resistance to different melanoma therapies has been associated with cells undergoing phenotype switching and lacking pigmentation‐related differentiation genes while expressing genes reminiscent of NC development. 28 , 29 , 30 , 32 , 104 , 105 Specifically, MAPK pathway inhibition was shown to promote the de novo generation or expansion of subpopulations of melanoma cells expressing NCSC‐associated factors like CD271 28 , 106 or the NC specifier gene FOXD3. 58 In addition, targeted therapy led to increased expression of genes linked to invasiveness like the receptor tyrosine kinase AXL 106 , 107 (Table 1). NCSC‐like melanoma cell subpopulations were further reported to contribute to minimal residual disease and, ultimately, to disease relapse. 28 , 108 Conversely, another study showed that melanoma cells expressing the melanocyte differentiation gene Dopachrome tautomerase (DCT, also TYRP2) were intrinsically resistant to BRAF inhibition. 109 However, increasing evidence supports the hypothesis that it is particularly the dedifferentiated melanoma cell population, expressing genes reminiscent of NCSCs, that can resist different treatments, including immunotherapies.
Along that line, a recent study showed that long‐term exposure of patient‐derived melanoma cells to antigen‐specific (MART1) T cells led to an enrichment of CD271high melanoma cells, which showed increased resistance to cytotoxic T cells (which recognized differentiation and non‐differentiation antigens), as well as to BRAF and MEK inhibitors 31 (Table 1). In line with these findings, another study revealed that in a mouse adoptive T cell therapy model, tumor necrosis factor alpha (TNFα)‐induced inflammation led to dedifferentiation of patient‐derived transplanted melanoma by upregulation of CD271 and downregulation of melanocyte‐specific antigens, which resulted in reduced tumor recognition by infiltrating T cells 32 (Table 1). Furthermore, while Boshuizen et al 31 interfered with CD271 upregulation to combat T cell resistance and relapse in their preclinical model via an unspecific heat shock protein inhibitor, another study developed a CD271‐specific monoclonal humanized antibody to counteract CD271 function. 110 The authors showed that treatment of CD271‐positive human melanoma grafts in NOD/scid mice with this CD271 antibody in combination with natural killer or peripheral blood mononuclear effector cells achieved a significant antitumor effect. 110 Whether such a CD271‐specific antibody could combat therapy resistance of melanoma tumors toward targeted or immune therapy remains to be answered, but previous findings appear to support such an approach. 28 , 31 , 32
Immunotherapy‐induced T‐cell accumulation and TNFα have also been shown to induce other factors reminiscent of NC development in melanoma, such as the histone methyltransferase Ezh2, which regulates mesenchymal fates during murine NC development. 43 Ezh2 upregulation in melanoma led to reduced immunogenicity of B16 F10 or Nras Q61K‐mutant Ink4a‐deficient murine tumors autologously grafted onto C57BL/6 mice, while pharmacological inhibition of Ezh2 attenuated this effect and synergized with anti‐CTLA‐4 and IL‐2 immunotherapies in mice 63 (Table 1).
5. TARGETING THE REEMERGENCE OF NCSC‐LIKE MELANOMA STATES
In line with NCSC‐like cell states observed in melanoma during disease progression and treatment, increasing evidence supports approaches targeting melanoma cells that have hijacked developmental programs. Indeed, several preclinical studies have succeeded in targeting or inhibiting the emergence of dedifferentiated melanoma cells to combat therapy resistance (Figure 2).
FIGURE 2.
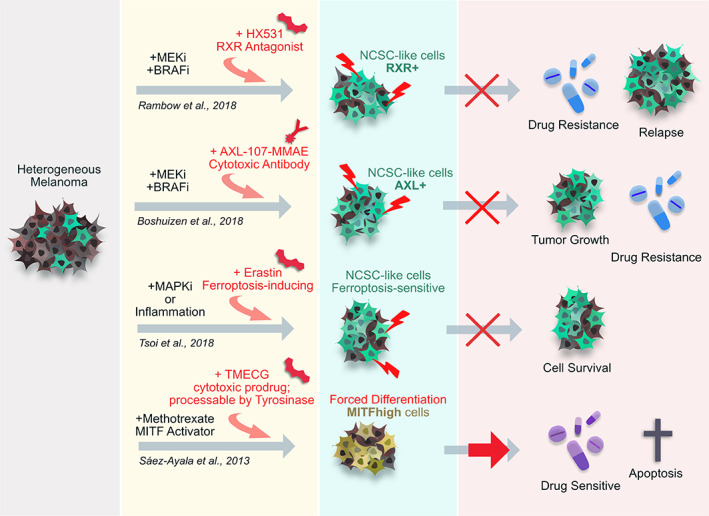
Interfering with dedifferentiated, neural crest stem cell (NCSC)‐like melanoma cells to combat drug resistance. Different preclinical studies successfully managed to circumvent therapy resistance of melanoma cells or patient‐derived grafts in mice by targeting the reemergence of NCSC‐like melanoma states. Rambow et al 28 were able to attenuate the accumulation of NCSC‐like cells after BRAF/MEK inhibition by treating patient‐derived melanoma with an antagonist toward RXRG, a NCSC‐associated gene strongly upregulated within their drug resistant melanoma subpopulation. Similarly, Boshuizen et al 107 successfully targeted tumor growth by combining MAPK inhibition with a cytotoxic antibody against AXLhigh cells emerging as resistant cells after BRAF/MEK inhibition. AXL is associated with invasiveness and drug resistance in melanoma 60 but has also been associated with reemergence of NCSC states in melanoma. 106 Also, Tsoi et al 106 could show that MAPK inhibition as well as pro‐inflammatory signaling from immunotherapies promoted drug resistant, dedifferentiated melanoma cells, which were characterized by increased sensitivity to ferroptosis. The authors subsequently co‐treated melanoma cells with targeted therapy and the ferroptosis‐inducing drug Erastin, which led to decreased melanoma cell survival. Sáez‐Ayala et al 111 achieved to circumvent drug resistance by forced differentiation of melanoma cells due to treatment with methotrexate (MTX), which induced the expression of the melanocyte differentiation marker MITF and inhibited invasiveness. This drug was further combined with the cytotoxic prodrug TMECG, activated by tyrosinase (a target of MITF), which is expressed in differentiated melanocytes
For instance, inhibiting CD271 in melanoma cells restored their susceptibility to BRAF inhibitors. 27 Likewise, suppressing the emergence of dedifferentiated, NCSC‐like melanoma cells upon MEK and BRAF inhibition (MEKi and BRAFi) interfered with resistance formation in vivo. 28 Specifically, this elegant study showed that a set of NCSCs genes, such as the Retinoid X receptor gamma (RXRG), which is expressed in chicken NCSCs, 62 was induced by MEKi and BRAFi treatment, and that pharmacological inhibition of RXRG prevented disease relapse of patient‐derived melanoma in immunocompromised mice 28 (Figure 2). Yet another study used a cytotoxic antibody approach to target AXLhigh melanoma cells resistant to MEKi and BRAFi, which led to reduced tumor growth and restored drug susceptibility 107 (Figure 2). Of note, AXL has not been identified as a regulator of the embryonic NC as such, however its expression has been associated with NCSC‐reminiscent factors such as CD271. 60 Specifically the receptor tyrosine kinases AXL and CD271 (together with EGFR and others) have been identified as marker genes for BRAF inhibitor resistance of human melanoma cells in vitro, 30 suggesting that targeting AXLhigh drug resistant melanoma cells putatively affects cells with NCSC‐reminiscent features.
Furthermore, targeted as well as immunotherapy have been shown to induce drug resistance, along with acquisition of NCSC‐like gene expression programs, that lead to an increased sensitivity to ferroptosis, 106 a type of programmed cell death. 112 The study authors subsequently managed to inhibit melanoma dedifferentiation and therapy resistance by addition of ferroptosis‐inducing drugs 106 (Figure 2).
The opposite approach of pushing melanoma into a fully differentiated cell fate to circumvent resistance formation, has been achieved in vitro through application of methotrexate (MTX), which activates the expresison of microphthalmia‐induced TF (MITF), a key regulator of differentiated melanocytes 113 . MTX treatment further increased melanoma cell susceptibility to a cytotoxic prodrug (TMECG), activated by tyrosinase, a target of MITF and expressed by fully differentiated melanocytes 111 (Figure 2).
All in all, these studies have demonstrated that interference with melanoma dedifferentiation or, vice versa, the promotion of a fully differentiated melanoma state can yield increased drug susceptibility and prevention of disease relapse in preclinical models, which make this a highly promising approach for patient therapy.
6. CONCLUDING REMARKS
Several approaches to target the reemergence of NCSC‐like cell states in treatment‐resistant melanomas (Figure 2) have shown great promise in preclinical settings. However, further in‐depth studies and proper characterization of such NCSC‐like melanoma cell states are needed to unravel the exact nature of gene regulatory networks that lead to the most therapy‐resilient, and hence aggressive, tumors. Unfortunately, inter‐patient and intratumoral heterogeneity has always posed a substantial challenge in melanoma treatment and also complicates the identification of exact NCSC‐like programs emerging within melanoma patients.
Currently, most studies performed on NCSC‐like cells in melanoma and cited within this review are preclinical studies 114 performed on human material in vitro 30 , 106 or in murine models, where NC‐reminiscent factors were shown to be crucial for tumor formation and disease progression in genetically engineered melanoma models 24 , 25 , 63 as well as in patient‐derived xenograft models. 22 , 26 , 28 , 47 However, extensive analyses of patient materials, which are important to support the clinical relevance of the above discussed findings, are often missing due to limited access to samples reflecting specific stages of melanoma progression. Also, some of the first in‐depth single‐cell analyses of patient tissues have been single case reports 28 rather than studies involving big patient cohorts. Therefore, the possibility remains that the reemergence of aggressive, therapy‐resistant NCSC‐like cell states observed in preclinical models does not or not always occur during disease progression in human melanoma patients and that metastatic disease and therapy resistance could still be established by alternative pathways.
Another remaining question concerns whether the mutational landscape predisposes melanoma subtypes toward the potential for dynamic remodeling into NCSC‐like cell states. For now, it is unclear whether some of the most frequent melanoma mutations, namely, BRAF V600E and NRAS Q61K, preferably favor the reemergence of NCSC‐like cell states. Work by Zon and colleagues has shown that in zebrafish, BRAF V600E‐mutated melanomas activate a NCSC progenitor program essential for melanoma initiation, 23 while in NRAS Q61K‐mutated melanomas, such a NCSC signature did not emerge at early stages of disease but only after transformation into malignant melanoma. 115 Whether this discrepancy is reflective of the human disease physiology remains unclear, since tumor initiation and onset of invasion and metastatic spread cannot be properly monitored or modeled in humans. Furthermore, most preclinical studies that have associated NCSC‐like melanoma subpopulations with resistance to therapy have addressed BRAF inhibitor resistance and accordingly used mostly BRAF‐mutated melanoma material, 28 , 31 leading to a bias toward BRAF‐ vs NRAS‐mutated material under investigation.
In conclusion, future studies including human material from bigger, more representative patient cohorts and collected at different time points of disease progression, specifically also during response to therapies, are needed to address the relevance of NCSC‐like melanoma cell states in humans. Hopefully, further single‐cell studies will allow us to answer whether NCSC‐reminiscent melanoma subpopulations indeed emerge in patients and whether they substantially interfere with melanoma treatment. This could open promising new avenues for designing novel therapies.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
J.D., L.S.: wrote this manuscript.
ACKNOWLEDGMENT
We thank Rishika Pandya for critical reading of this manuscript.
Diener J, Sommer L. Reemergence of neural crest stem cell‐like states in melanoma during disease progression and treatment. STEM CELLS Transl Med. 2021;10:522–533. 10.1002/sctm.20-0351
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Baggiolini A, Varum S, Mateos JM, et al. Premigratory and migratory neural crest cells are multipotent in vivo. Cell Stem Cell. 2015;16(3):314‐322. 10.1016/j.stem.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 2. Bronner‐Fraser M, Fraser SE. Cell lineage analysis reveals multipotency of some avian neural crest cells. Nature. 1988;335:161–164. 10.1038/335161a0. [DOI] [PubMed] [Google Scholar]
- 3. Le Douarin, N. , & Kalcheim, C. (1999). The Neural Crest. Cambridge, England: Cambridge University Press. 10.1017/CBO9780511897948 [DOI] [Google Scholar]
- 4. Szabó A, Mayor R. Mechanisms of neural crest migration. Annu Rev Genet. 2018;52(1):43‐63. 10.1146/annurev-genet-120417-031559. [DOI] [PubMed] [Google Scholar]
- 5. Etchevers HC, Dupin E, Le Douarin NM. The diverse neural crest: from embryology to human pathology. Development (Cambridge). 2019;146:dev169821. 10.1242/dev.169821. [DOI] [PubMed] [Google Scholar]
- 6. Simões‐Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142(1):242‐257. 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dupin E, Sommer L. Neural crest progenitors and stem cells: from early development to adulthood. Dev Biol. 2012;366(1):83‐95. 10.1016/j.ydbio.2012.02.035. [DOI] [PubMed] [Google Scholar]
- 8. Fernandes KJL, McKenzie IA, Mill P, et al. A dermal niche for multipotent adult skin‐derived precursor cells. Nat Cell Biol. 2004;6:1082‐1093. 10.1038/ncb1181. [DOI] [PubMed] [Google Scholar]
- 9. Sieber‐Blum M, Grim M, Hu YF, Szeder V. Pluripotent neural crest stem cells in the adult hair follicle. Dev Dyn. 2004;72(2):162‐172. 10.1002/dvdy.20129. [DOI] [PubMed] [Google Scholar]
- 10. Toma JG, Akhavan M, Fernandes KJL, et al. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3(9):778‐784. 10.1038/ncb0901-778. [DOI] [PubMed] [Google Scholar]
- 11. Toma JG, McKenzie IA, Bagli D, Miller FD. Isolation and characterization of multipotent skin‐derived precursors from human skin. Stem Cells. 2005;23(6):727‐737. 10.1634/stemcells.2004-0134. [DOI] [PubMed] [Google Scholar]
- 12. Wong CE, Paratore C, Dours‐Zimmermann MT, et al. Neural crest‐derived cells with stem cell features can be traced back to multiple lineages in the adult skin. J Cell Biol. 2006;175(6):1005‐1015. 10.1083/jcb.200606062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al‐Zer H, Apel C, Heiland M, et al. Enrichment and Schwann cell differentiation of neural crest‐derived dental pulp stem cells. In Vivo. 2015;29(3):319‐326. [PubMed] [Google Scholar]
- 14. Stemple DL, Anderson DJ. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992;71:973‐985. [DOI] [PubMed] [Google Scholar]
- 15. Furlan A, Adameyko I. Schwann cell precursor: a neural crest cell in disguise? Dev Biol. 2018;444:S25‐S35. 10.1016/j.ydbio.2018.02.008. [DOI] [PubMed] [Google Scholar]
- 16. Kameneva P, Kastriti ME, Adameyko I. Neuronal lineages derived from the nerve‐associated Schwann cell precursors. Cell Mol Life Sci. 2020. 10.1007/s00018-020-03609-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnston APW, Yuzwa SA, Carr MJ, et al. Dedifferentiated Schwann cell precursors secreting paracrine factors are required for regeneration of the mammalian digit tip. Cell Stem Cell. 2016;19(4):433‐448. 10.1016/j.stem.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 18. Parfejevs V, Debbache J, Shakhova O, et al. Injury‐activated glial cells promote wound healing of the adult skin in mice. Nat Commun. 2018;9(1):1‐16. 10.1038/s41467-017-01488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bailey CM, Morrison JA, Kulesa PM. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment Cell Melanoma Res. 2012;25(5):573‐583. 10.1111/j.1755-148X.2012.01025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnsen JI, Dyberg C, Wickström M. Neuroblastoma—a neural crest derived embryonal malignancy. Front Mol Neurosci. 2019;12(January):1‐11. 10.3389/fnmol.2019.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vega FM, Colmenero‐Repiso A, Gómez‐Muñoz MA, et al. CD44‐high neural crest stem‐like cells are associated with tumour aggressiveness and poor survival in neuroblastoma tumours. EBioMedicine. 2019;49:82‐95. 10.1016/j.ebiom.2019.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boiko AD, Razorenova OV, Van De Rijn M, et al. Human melanoma‐initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133‐137. 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaufman CK, Mosimann C, Fan ZP, et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science. 2016;351(6272):aad2197. 10.1126/science.aad2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shakhova O, Zingg D, Schaefer SM, et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat Cell Biol. 2012;14(8):882‐889. 10.1038/ncb2535. [DOI] [PubMed] [Google Scholar]
- 25. Varum S, Baggiolini A, Zurkirchen L, et al. Yin Yang 1 orchestrates a metabolic program required for both neural crest development and melanoma formation. Cell Stem Cell. 2019;24(4):637‐653.e9. 10.1016/j.stem.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 26. Restivo G, Diener J, Cheng PF, et al. The low affinity neurotrophin receptor CD271 regulates phenotype switching in Melanoma. Nat Commun. 2017;8(1):1‐16. 10.1038/s41467-017-01573-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lehraiki A, Cerezo M, Rouaud F, et al. Increased CD271 expression by the NF‐kB pathway promotes melanoma cell survival and drives acquired resistance to BRAF inhibitor vemurafenib. Cell Discov. 2015;1:15030. 10.1038/celldisc.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rambow F, Rogiers A, Marin‐Bejar O, et al. Toward minimal residual disease‐directed therapy in melanoma. Cell. 2018;174(4):843‐855.e19. 10.1016/j.cell.2018.06.025. [DOI] [PubMed] [Google Scholar]
- 29. Schatton T, Scolyer RA, Thompson JF, Mihm MC. Tumor‐infiltrating lymphocytes and their significance in melanoma prognosis. Methods Mol Biol. 2014;1102:287‐324. 10.1007/978-1-62703-727-3_16. [DOI] [PubMed] [Google Scholar]
- 30. Shaffer SM, Dunagin MC, Torborg SR, et al. Rare cell variability and drug‐induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546(7658):431‐435. 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boshuizen J, Vredevoogd DW, Krijgsman O, et al. Reversal of pre‐existing NGFR‐driven tumor and immune therapy resistance. Nat Commun. 2020;11(1):1‐13. 10.1038/s41467-020-17739-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Landsberg J, Kohlmeyer J, Renn M, et al. Melanomas resist T‐cell therapy through inflammation‐induced reversible dedifferentiation. Nature. 2012;490(7420):412‐416. 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- 33. Lo JA, Fisher DE. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science. 2014;346(6212):945‐949. 10.1126/science.1253735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Banerjee SS, Eyden B. Divergent differentiation in malignant melanomas: a review. Histopathology. 2008;52(2):119‐129. 10.1111/j.1365-2559.2007.02823.x. [DOI] [PubMed] [Google Scholar]
- 35. Civenni G, Walter A, Kobert N, et al. Human CD271‐positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long‐term growth. Cancer Res. 2011;71(8):3098‐3109. 10.1158/0008-5472.CAN-10-3997. [DOI] [PubMed] [Google Scholar]
- 36. Ennen M, Keime C, Kobi D, et al. Single‐cell gene expression signatures reveal melanoma cell heterogeneity. Oncogene. 2015;34:3251‐3263. 10.1038/onc.2014.262. [DOI] [PubMed] [Google Scholar]
- 37. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13(10):727‐738. 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 38. Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res. 2008;21(1):39‐55. 10.1111/j.1755-148X.2007.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38:17‐31. 10.1016/S0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 40. Paratore C, Goerich DE, Suter U, Wegner M, Sommer L. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development. 2001;128(20):3949‐3961. [DOI] [PubMed] [Google Scholar]
- 41. Redmer T, Welte Y, Behrens D, et al. The nerve growth factor receptor CD271 is crucial to maintain tumorigenicity and stem‐like properties of melanoma cells. PLoS One. 2014;9(5):e92596. 10.1371/journal.pone.0092596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zingg D, Debbache J, Schaefer SM, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6(May 2014):1‐17. 10.1038/ncomms7051. [DOI] [PubMed] [Google Scholar]
- 43. Schwarz D, Varum S, Zemke M, et al. Ezh2 is required for neural crest‐derived cartilage and bone formation. Development (Cambridge). 2014;141(4):867‐877. 10.1242/dev.094342. [DOI] [PubMed] [Google Scholar]
- 44. White RM, Cech J, Ratanasirintrawoot S, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518‐522. 10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Santoriello C, Sporrij A, Yang S, et al. RNA helicase DDX21 mediates nucleotide stress responses in neural crest and melanoma cells. Nat Cell Biol. 2020;22(4):372‐379. 10.1038/s41556-020-0493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo R, An M, Arduini BL, Henion PD. Specific pan‐neural crest expression of zebrafish crestin throughout embryonic development. Dev Dyn. 2001;220(2):169‐174. . [DOI] [PubMed] [Google Scholar]
- 47. Heppt MV, Wang JX, Hristova DM, et al. MSX1‐induced neural crest‐like reprogramming promotes melanoma progression. J Investig Dermatol. 2018;138(1):141‐149. 10.1016/j.jid.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tríbulo C, Aybar MJ, Nguyen VH, Mullins MC, Mayor R. Regulation of Msx genes by a Bmp gradient is essential for neural crest specification. Development. 2003;130(26):6441‐6452. 10.1242/dev.00878. [DOI] [PubMed] [Google Scholar]
- 49. Caramel J, Papadogeorgakis E, Hill L, et al. A switch in the expression of embryonic EMT‐inducers drives the development of malignant melanoma. Cancer Cell. 2013;24(4):466‐480. 10.1016/j.ccr.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 50. Bildsoe H, Loebel DAF, Jones VJ, Chen YT, Behringer RR, Tam PPL. Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev Biol. 2009;331(2):176‐188. 10.1016/j.ydbio.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 51. Vincentz JW, Firulli BA, Lin A, Spicer DB, Howard MJ, Firulli AB. Twist1 controls a cell‐specification switch governing cell fate decisions within the cardiac neural crest. PLoS Genet. 2013;9(3):e1003405. 10.1371/journal.pgen.1003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Denecker G, Vandamme N, Akay Ö, et al. Identification of a ZEB2‐MITF‐ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014;21(8):1250‐1261. 10.1038/cdd.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kubic JD, Lui JW, Little EC, et al. PAX3 and FOXD3 promote CXCR4 expression in melanoma. J Biol Chem. 2015;290(36):21901‐14. 10.1074/jbc.M115.670976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bartolomé RA, Galvez BG, Longo N, et al. Stromal cell‐derived factor‐1 a promotes melanoma cell invasion across basement membranes involving stimulation of membrane‐type 1 matrix metalloproteinase and Rho GTPase activities. Cancer Res. 2004;64(7):2534‐2543. [DOI] [PubMed] [Google Scholar]
- 55. Murakami T, Maki W, Cardones AR, et al. Expression of CXC chemokine receptor‐4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Cell. 2002;1:7328‐7334. [PubMed] [Google Scholar]
- 56. Betancur P, Bronner‐Fraser M, Sauka‐Spengler T. Assembling neural crest reegulatory circuits into a gene regulatory network. Annu Rev Cell Dev Biol. 2010;26:581‐603. 10.1146/annurev.cellbio.042308.113245.ASSEMBLING. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meulemans D, Bronner‐Fraser M. Gene‐regulatory interactions in neural crest evolution and development. Develop Cell. 2004;7(3):291‐299. 10.1016/j.devcel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 58. Abel EV, Basile KJ, Kugel CH, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3‐mediated upregulation of ERBB3. J Clin Investig. 2013;123(5):2155‐2168. 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Britsch S, Li L, Kirchhoff S, et al. The ErbB2 and ErbB3 receptors and their ligand, neuregulin‐1, are essential for development of the sympathetic nervous system. Genes Dev. 1998;12(12):1825‐1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science. 2016;352(6282):189‐196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Su Y, Wei W, Robert L, et al. Single‐cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug‐induced resistance. Proc Natl Acad Sci USA. 2017;114(52):13679‐13684. 10.1073/pnas.1712064115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rowe A, Brickell PM. Expression of the chicken retinoid X receptor‐γ gene in migrating cranial neural crest cells. Anat Embryol. 1995;192(1):1‐8. 10.1007/BF00186986. [DOI] [PubMed] [Google Scholar]
- 63. Zingg D, Arenas‐Ramirez N, Sahin D, et al. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 2017;20(4):854‐867. 10.1016/j.celrep.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 64. Barilani M, Banfi F, Sironi S, et al. Low‐affinity nerve growth factor receptor (CD271) heterogeneous expression in adult and fetal mesenchymal stromal cells. Sci Rep. 2018;8(1):9321. 10.1038/s41598-018-27587-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jiang Y, Powers J, Muschler GF, Maciejewski JP. Expression of CD271 in normal and malignant hematopoietic progenitor and stem cells. Blood. 2006;108(11):4271. 10.1182/blood.v108.11.4271.4271. [DOI] [Google Scholar]
- 66. Filipp FV, Li C, Boiko AD. CD271 is a molecular switch with divergent roles in melanoma and melanocyte development. Sci Rep. 2019;9(1):1‐11. 10.1038/s41598-019-42773-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kasemeier‐Kulesa JC, Kulesa PM. The convergent roles of CD271/p75 in neural crest‐derived melanoma plasticity. Dev Biol. 2018;444:S352‐S355. 10.1016/j.ydbio.2018.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Agnarsdóttir M, Sooman L, Bolander Å, et al. SOX10 expression in superficial spreading and nodular malignant melanomas. Melanoma Res. 2010;20(6):468‐478. 10.1097/CMR.0b013e3283403ccd. [DOI] [PubMed] [Google Scholar]
- 69. Mohamed A, Gonzalez RS, Lawson D, Wang J, Cohen C. SOX10 expression in malignant melanoma, carcinoma,and normal tissues. Appl Immunohistochem Mol Morphol. 2013;21(6):506‐510. 10.1097/PAI.0b013e318279bc0a. [DOI] [PubMed] [Google Scholar]
- 70. Marie KL, Sassano A, Yang HH, et al. Melanoblast transcriptome analysis reveals pathways promoting melanoma metastasis. Nat Commun. 2020;11(1):333. 10.1038/s41467-019-14085-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Adameyko I, Lallemend F, Furlan A, et al. Sox2 and Mitf cross‐regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development. 2012;139(2):397‐410. 10.1242/dev.065581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Menon DR, Luo Y, Arcaroli JJ, et al. CDK1 interacts with SOX2 and promotes tumor initiation in human melanoma. Cancer Res. 2018;78(23):6561‐6574. 10.1158/0008-5472.CAN-18-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Santini R, Pietrobono S, Pandolfi S, et al. SOX2 regulates self‐renewal and tumorigenicity of human melanoma‐initiating cells. Oncogene. 2014;33(38):4697‐4708. 10.1038/onc.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cesarini V, Guida E, Todaro F, et al. Sox2 is not required for melanomagenesis, melanoma growth and melanoma metastasis in vivo. Oncogene. 2017;36(31):4508‐4515. 10.1038/onc.2017.53. [DOI] [PubMed] [Google Scholar]
- 75. Schaefer SM, Segalada C, Cheng PF, et al. Sox2 is dispensable for primary melanoma and metastasis formation. Oncogene. 2017;36(31):4516‐4524. 10.1038/onc.2017.55. [DOI] [PubMed] [Google Scholar]
- 76. Johansson JA, Marie KL, Lu Y, et al. PRL3‐DDX21 transcriptional control of endolysosomal genes restricts melanocyte stem cell differentiation. Dev Cell. 2020;54(3):317‐332.e9. 10.1016/j.devcel.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Köhler C, Nittner D, Rambow F, et al. Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell. 2017;21(5):679‐693.e6. 10.1016/j.stem.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 78. Moon H, Donahue LR, Choi E, et al. Melanocyte stem cell activation and translocation initiate cutaneous melanoma in response to UV exposure. Cell Stem Cell. 2017;21(5):665‐678.e6. 10.1016/j.stem.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sun Q, Lee W, Mohri Y, et al. A novel mouse model demonstrates that oncogenic melanocyte stem cells engender melanoma resembling human disease. Nat Commun. 2019;10(1):1‐16. 10.1038/s41467-019-12733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Adameyko I, Lallemend F, Aquino JB, et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell. 2009;139(2):366‐379. 10.1016/j.cell.2009.07.049. [DOI] [PubMed] [Google Scholar]
- 81. Nieto MA, Huang RYYJ, Jackson RAA, Thiery JPP. EMT: 2016. Cell. 2016;166(1):21‐45. 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 82. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial‐mesenchymal transitions in development and disease. Cell. 2009;139(5):871‐890. 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 83. Hoek KS, Eichhoff OM, Schlegel NC, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68(3):650‐656. 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 84. Schlegel NC, von Planta A, Widmer DS, Dummer R, Christofori G. PI3K signalling is required for a TGFβ‐induced epithelial‐mesenchymal‐like transition (EMT‐like) in human melanoma cells. Exp Dermatol. 2015;24:22‐28. 10.1111/exd.12580. [DOI] [PubMed] [Google Scholar]
- 85. Tuncer E, Calçada RR, Zingg D, et al. SMAD signaling promotes melanoma metastasis independently of phenotype switching. J Clin Investig. 2019;129(7):2702‐2716. 10.1172/JCI94295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li FZ, Dhillon AS, Anderson RL, McArthur G, Ferrao PT. Phenotype switching in melanoma: implications for progression and therapy. Front Oncol. 2015;5(FEB):1‐7. 10.3389/fonc.2015.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bronner ME, Simões‐costa M. The neural crest migrating into the 21st century Marianne. Curr Top Dev Biol. 2016;116:115‐134. 10.1016/bs.ctdb.2015.12.003.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vandamme N, Berx G. Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front Oncol. 2014;4:352. 10.3389/fonc.2014.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fang R, Zhang G, Guo Q, et al. Nodal promotes aggressive phenotype via Snail‐mediated epithelial‐mesenchymal transition in murine melanoma. Cancer Lett. 2013;333(1):66‐75. 10.1016/j.canlet.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 90. Conway SJ, Kaartinen V. TGFβ superfamily signaling in the neural crest lineage. Cell Adhes Migr. 2011;5(3):232‐236. 10.4161/cam.5.3.15498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. John N, Cinelli P, Wegner M, Sommer L. Transforming growth factor β‐mediated sox10 suppression controls mesenchymal progenitor generation in neural crest stem cells. Stem Cells. 2011;29(1):689‐699. 10.1002/stem.607. [DOI] [PubMed] [Google Scholar]
- 92. Wurdak H, Ittner LM, Lang KS, et al. Inactivation of TGFβ signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005;19(5):530‐535. 10.1101/gad.317405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69‐84. 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 94. Mani SA, Guo W, Liao MJ, et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704‐715. 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Saltari A, Truzzi F, Quadri M, et al. CD271 down‐regulation promotes melanoma progression and invasion in three‐dimensional models and in zebrafish. J Investig Dermatol. 2016;136(10):2049‐2058. 10.1016/j.jid.2016.05.116. [DOI] [PubMed] [Google Scholar]
- 96. Eggermont AMM, Kirkwood JM. Re‐evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? Eur J Cancer. 2004;40(12):1825‐1836. 10.1016/j.ejca.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 97. Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251‐263. 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32(19):2373‐2379. 10.1038/onc.2012.345. [DOI] [PubMed] [Google Scholar]
- 99. Garrido F, Cabrera T, Concha A, Glew S, Ruiz‐Cabello F, Stern PL. Natural history of HLA expression during tumour development. Immunol Today. 1993;14(10):491‐499. 10.1016/0167-5699(93)90264-L. [DOI] [PubMed] [Google Scholar]
- 100. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714‐726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 101. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999‐1005. 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401‐1409. 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 103. Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118‐122. 10.1038/nature13121. [DOI] [PubMed] [Google Scholar]
- 104. Reinhardt J, Landsberg J, Schmid‐Burgk JL, et al. MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res. 2017;77(17):4697‐4709. 10.1158/0008-5472.CAN-17-0395. [DOI] [PubMed] [Google Scholar]
- 105. Roesch A, Fukunaga‐Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow‐cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583‐594. 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tsoi J, Robert L, Paraiso K, et al. Multi‐stage differentiation defines melanoma subtypes with differential vulnerability to drug‐induced iron‐dependent oxidative stress. Cancer Cell. 2018;33(5):890‐904.e5. 10.1016/j.ccell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Boshuizen J, Koopman LA, Krijgsman O, et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody‐drug conjugate and BRAF/MEK inhibitors. Nat Med. 2018;24(2):203‐212. 10.1038/nm.4472. [DOI] [PubMed] [Google Scholar]
- 108. Travnickova J, Wojciechowska S, Khamseh A, et al. Zebrafish MITF‐low melanoma subtype models reveal transcriptional subclusters and MITF‐independent residual disease. Cancer Res. 2019;79(22):5769‐5784. 10.1158/0008-5472.CAN-19-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ho YJ, Anaparthy N, Molik D, et al. Single‐cell RNA‐seq analysis identifies markers of resistance to targeted BRAF inhibitors in melanoma cell populations. Genome Res. 2018;28(9):1353‐1363. 10.1101/gr.234062.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Morita S, Mochizuki M, Wada K, et al. Humanized anti‐CD271 monoclonal antibody exerts an anti‐tumor effect by depleting cancer stem cells. Cancer Lett. 2019;461:144‐152. 10.1016/j.canlet.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 111. Sáez‐Ayala M, Montenegro MF, Sánchez‐del‐Campo L, et al. Directed phenotype switching as an effective antimelanoma strategy. Cancer Cell. 2013;24(1):105‐19. 10.1016/j.ccr.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 112. Li J, Cao F, Yin HL, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88. 10.1038/s41419-020-2298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Mort RL, Jackson IJ, Elizabeth Patton E. The melanocyte lineage in development and disease. Development (Cambridge). 2015;142(4):620‐632. 10.1242/dev.106567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rebecca VW, Somasundaram R, Herlyn M. Pre‐clinical modeling of cutaneous melanoma. Nat Commun. 2020;11(1):2858. 10.1038/s41467-020-15546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. McConnell AM, Mito JK, Ablain J, et al. Neural crest state activation in NRAS driven melanoma, but not in NRAS‐driven melanocyte expansion. Dev Biol. 2019;449(2):107‐114. 10.1016/j.ydbio.2018.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.