

Summary
Voltage-gated sodium channels are targets for many analgesic and antiepileptic drugs whose therapeutic mechanisms and binding sites have been well characterized. We describe the identification of a previously unidentified receptor site within the NavMs voltage-gated sodium channel. Tamoxifen, an estrogen receptor modulator, and its primary and secondary metabolic products bind at the intracellular exit of the channel, which is a site that is distinct from other previously characterized sodium channel drug sites. These compounds inhibit NavMs and human sodium channels with similar potencies and prevent sodium conductance by delaying channel recovery from the inactivated state. This study therefore not only describes the structure and pharmacology of a site that could be leveraged for the development of new drugs for the treatment of sodium channelopathies but may also have important implications for off-target health effects of this widely used therapeutic drug.
Keywords: Voltage-gated Sodium Channels, Tamoxifen, Estrogen Receptor, Crystal Structure, Electrophysiology, Drug Binding, Pharmacology, Novel Binding Site, Channelopathies, Off-Target Drug Effects
Graphical abstract
Highlights
-
•
Structure and function of tamoxifen/voltage-gated sodium channel complexes
-
•
Tamoxifen binds to a previously unidentified drug binding site in sodium channels
-
•
Sodium channel tamoxifen binding pocket shares features with the estrogen receptor
X-ray crystallography and electrophysiology reveal the nature of the interactions of the drug tamoxifen (and its metabolic derivatives) with voltage-gated sodium channels.
Introduction
Voltage-gated sodium channels (Navs) form ion-conducting pores responsible for electrical signaling in nerves and cardiac cells. Variants in human voltage-gated sodium channel isoforms (hNav1.1–1.9) are associated with a range of diseases, including epilepsy, chronic pain, and cardiomyopathy. A number of commonly used antiepileptic and analgesic drugs, such as lamotrigine and lidocaine, have been developed to inhibit or modulate their ion-conducting properties. Prokaryotic Navs are homotetramers, with each subunit consisting of a four-helix voltage sensor module (VSM) and a two-helix pore module (PM). Prokaryotic Navs are analogous to human Navs, except that the latter are encoded by a single peptide composed of four domains, each of which contains a VSM and a PM. The structures of a number of prokaryotic and eukaryotic Navs have been elucidated in recent years using X-ray crystallography and cryoelectron microscopy (cryo-EM) methods (Payandeh et al., 2011; Ulmschneider et al., 2013; Sula et al., 2017; Ahuja et al., 2015; Xu et al., 2019; Pan et al., 2019; Shen et al., 2018; Bagnéris et al., 2014). Those studies have demonstrated that prokaryotic and eukaryotic sodium channels share a high degree of similarity in overall fold and assembly, with the exception of their intra- and extracellular loops and termini (Figure S1A). One prokaryotic channel, NavMs from Magnetococcus marinus, has been shown to be a particularly good exemplar for hNav pharmacology in terms of drug potency, mechanism of drug inhibition, and receptor site interactions (Bagnéris et al., 2014).
Tamoxifen was developed as a treatment for breast cancer more than 40 years ago and is currently prescribed to more than 200 million patients worldwide (Errico, 2015; Jordan, 2003; https://clincalc.com/DrugStats/Top300Drugs.aspx). Apart from its therapeutic effects targeting the estrogen receptor (ER) (Shiau et al., 1998), electrophysiology studies have demonstrated that tamoxifen antagonizes sodium currents from metastatic adenocarcinoma and glioma cells (Fraser et al., 2005; Wang and Jiao, 2009). To date, however, its sodium channel receptor site and mechanism of inhibition have not been defined. Its therapeutic activity in cancer cells has been linked to antagonism of the ER, but its off-target effects, such as binding to and inhibiting Navs expressed in excitable cells such as neurons, might be responsible for side effects and limitations in clinical safety (Fraser et al., 2014; Klein et al., 2013; Mürdter et al., 2011; He et al., 2003).
Tamoxifen metabolism occurs mainly through two pathways, involving 4-hydroxylation and N-demethylation, resulting in the primary metabolites 4-hydroxytamoxifen and N-desmethyltamoxifen, and the secondary metabolite endoxifen (Helland et al., 2017; Kisanga et al., 2004; Lien et al., 1991a, 1991b) (Figure S2). For this reason, we have explored the structural and functional effects not only of tamoxifen itself but also those of its metabolic products. X-ray crystallographic studies of the complexes formed with these compounds define two occupancy sites located near the intracellular gate of the channel. Functional studies show that tamoxifen antagonizes the NavMs channel activity through a unique mechanism, in which drug inhibition is dependent on channel entry into the inactivated channel state. Similar potencies for sodium current inhibition for all of these compounds were observed in NavMs and human Nav channels.
Results
An overview of the tamoxifen-bound NavMs structures
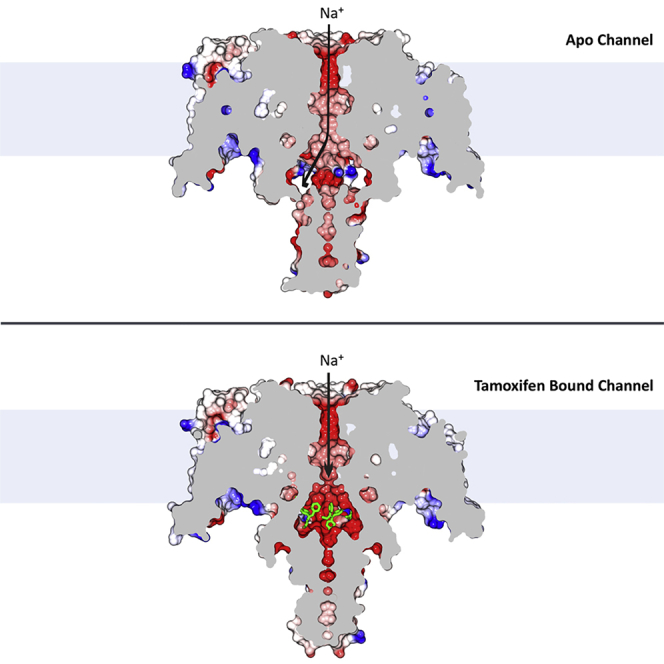
The crystal structures (Figures 1, 2, and 3; Table S1) of the NavMs sodium channel complexes formed with tamoxifen and its metabolites clearly show the positions of two unique drug binding sites in the channel. The drugs are located near the intracellular gate or exit of the transmembrane ion-conducting pore (Figure 1). When compared with the apo-NavMs open state structure, the C-terminal ends (starting at I224) of the S6 helices in the drug-bound NavMs channel structure are displaced toward the center of the ion-conducting pathway (overall root-mean-square deviation [RMSD] of 1.2 Å). This change in the channel conformation creates a binding pocket between pore domains (Figures 2A and 2B) and narrows the pore radius of the intracellular gate from 2.3 Å (in the open channel) to ∼1.4 Å (Figure S3A). As the S4 helices found in the VSMs are in the “up” position and the PMs are in narrow, non-conductive conformations, the new drug-bound structures may represent either the “pre-open” or “inactivated” states. In the channel-drug complexes described in this study, eight tamoxifen (or tamoxifen metabolite) molecules can bind per tetramer (Figure 1A), and each tamoxifen molecule interacts with the S6 helices from two different subunits. Notably, tamoxifen and its analogs do not bind in the relatively wide and hydrophobic cavity located in the middle of the transmembrane pore (Figure 2B), where analgesic and antiepileptic drugs bind, nor do they occupy the transmembrane fenestrations which have been proposed (Hille, 1977) to provide the passageways for hydrophobic/uncharged drugs (Montini et al., 2018; Gamal El-Din et al., 2018) to enter from the membrane lipid phase into the central cavity of the transmembrane pore.
Figure 1.

Overview of crystal structures of the NavMs sodium channel complexes with tamoxifen or its metabolic products
The top of each panel corresponds to the cell exterior, and the bottom corresponds to the cell interior. The approximate positions of the cell membrane surfaces are denoted by the horizontal black lines. The three sodium ions present in the selectivity filter are shown as pink spheres. In all panels, the protein is depicted in gold ribbon motif, and the bound compounds are in stick representations (green, carbon; red, oxygen; blue, nitrogen). A total of eight copies of either tamoxifen or a metabolite can bind into a single channel in two sets (designated “inner” and “outer”) of four symmetry-related positions. The structures of the proteins in all of these complexes are essentially identical, having overall RMSDs ranging from 0.21 to 0.5 Å.
(A) NavMs complex with 4-hydroxytamoxifen, indicating the locations of all eight copies (four “inner” and four “outer” sites) of the drug in the structure. This illustrates the close packing of the compounds in the protein cavity that lies just above the intracellular exit of the channel.
(B) As in (A), except that this depicts (left) the wild-type NavMs complex with 4-hydroxytamoxifen and (right) the F208L mutant (NavMsL) complex with 4-hydroxytamoxifen. Both the wild-type and mutant proteins were examined for comparison, as this binding site residue is an F in two domains of the corresponding human Nav1.2 protein and L in the two other domains of the human protein (Figure S6). For clarity, in these structures, only one copy of the drug is shown in each of the two distinct “inner” and “outer” sites.
(C) NavMsL showing (for clarity) only one copy of tamoxifen bound in the “outer” site.
(D) As in (C), except that this depicts the NavMsL complexes with the other metabolites: (left) N-desmethyltamoxifen in the “inner” site and (right) NavMsL with endoxifen in the “inner” site.
See also Figures S1 and S6 and Table S1.
Figure 2.

Comparisons of NavMs structures in the absence and presence of tamoxifen and its metabolites
(A) Overlaid ribbon representations of the polypeptide backbones of single monomers of apo-NavMsL (gray), tamoxifen-NavMsL (green), 4-hydroxytamoxifen-NavMsL (red), N-desmethyltamoxifen-NavMsL (coral), and endoxifen-NavMsL (blue), showing the main differences in the polypeptide chains that occur upon binding any of the compounds. Although the overall conformations of the apo and bound structures are essentially identical throughout their transmembrane domains (with an overall C-alpha RMSD of 1.35 Å), in all of the complexed structures, the C-terminal tails differ considerable from the apo structure, starting at the C-terminal end of the S6 helix (bottom of figure) (residue I224) (denoted by the black circle) reaching an RMSD of 5.9 Å at residue P240, the last residue before the intracellular coil-coiled region (denoted by the dashed orange box), which is clearly displaced from the location of this region in the apo structure. All of the rest of the polypeptide chain structures are essentially identical for all of the complexes (maximum deviations of 0.2–0.5 Å).
(B) Electrostatic surface representations of (left) the apo-NavMs crystal structure, and (right) the 4-hydroxytamoxifen-NavMs complex. The drug (depicted in green stick representation) is just visible in the opening above the C-terminal intracellular domain denoted within the orange box. The lower right panel shows a vertical slice through the middle of the 4-hydroxytamoxifen complex, where the compound (in green stick representation) is clearly visible (in the purple box) at the bottom of the transmembrane channel pore. This is a region distinct from, and below, the central hydrophobic cavity (denoted by the black box) which is considered to be the binding sites for many analgesic and anesthetic compounds.
See also Figure S3.
Figure 3.

Detailed views of the “inner” and “outer” binding sites in the 4-hydroxytamoxifen NavMs complex
There are no steric clashes between any of the sites, so all eight positions can be occupied simultaneously. For clarity, in this figure only one of the four equivalent positions of each type of site is shown.
(A) The “inner” binding site. Left: the crystal structure of NavMs (in ribbon depiction for the four polypeptide chains, which are colored differently), with the 4-hydroxytamoxifen molecule shown in stick depiction overlaying (in blue mesh) the (2Fo – Fc, contoured at 1.5 sigma) electron density map. Right: detailed view of the binding site and its location between two different polypeptide chains (colored in blue and coral).
(B) More detailed view of the “inner” binding site, showing the (2Fo – Fc, contoured at 1.5 sigma) electron density map (blue mesh) of the 4-hydroxytamoxifen overlaying the protein structure, with the adjacent protein side chains in stick depiction (colored by chain, with residue numbers labeled). The right and left panels depict two views of the same site, rotated to show a clear view of the molecule.
(C and D) As in (A) and (B), except depicting the “outer” sites with their (2Fo – Fc, contoured at 1 sigma) electron density maps.
(E) 4-Hydroxytamoxifen dose-response curves generated for the NavMs, D220E and E227D mutant channels, which disrupt the “inner” binding. n = 4 cells tested at each concentration, and error bars are equal to SD.
(F) Change in free energy of 4-hydroxytamoxifen binding to D220E and E227D mutants relative to WT NavMs channels.
See also Figure S4.
Tamoxifen analogs occupy inner and outer sites near the channel gate
The structures of the NavMs channels in the complexes formed in the presence of the three tamoxifen metabolic products (4-hydroxytamoxifen, N-desmethyltamoxifen, and endoxifen) (Figures 1B–1D) are very similar to one another (protein Cα RMSDs of ∼0.35 Å) and to the complex with the parent tamoxifen molecules (RMSDs of ∼0.5 Å). All of these complexes exhibit two sets of four symmetry-related binding sites, making a total of eight sites per channel (Figures 1A and S4). The “outer” set of sites for 4-hydroxytamoxifen is closer to the intracellular C-terminal portion of the channel and does not involve any hydrogen bond interactions between the drugs and the polypeptides (Figures 3C and 3D). The positions of the drug molecules vary slightly among the different tamoxifen metabolite structures (Figure S4), which along with their higher crystallographic temperature (B) factors (Table S1) suggests that drug coordination in this site is somewhat flexible. By comparison, the 4-hydroxytamoxifen “inner” sites (Figures 1, 3A, 3B, and S4A–S4C) are better defined (lower average temperature factors for the drug molecules; Table S1) and are closer to the ion-conducting pathway. Binding to the “outer” site does not significantly change the pore radius (Figure S3), indicating that drug binding would not interfere with sodium ion egress from the channel. In contrast, the “inner” binding sites for 4-hydroxytamoxifen (Figure 3B) and the other metabolites specifically involve residues M222 (the intracellular gate residue), K226, E227, and I241 of one monomer and residues D220, L245, and L246 of the other monomer. Drug binding at these sites narrows the pore diameter at the cell interior end of the channel (Figures S3A and S3B); comparisons of tamoxifen docked into the corresponding site in hNav1.2 (Figures S3C and S3D) show a similar narrowing. The drug hydroxyl groups form hydrogen bonds with the E227 side chain of one polypeptide, and the drug amide groups form hydrogen bonds with D220 side chain of a different polypeptide chain (Figures 3 and S4).
Potency of Nav inhibition and validation of the inner tamoxifen receptor site
The whole-cell patch-clamp method was used to determine the efficacy and potency of sodium current inhibition by tamoxifen and its metabolic products in cells expressing either NavMs or hNav1.2 channels (Figures 4A and 4B; Table 1). Among the metabolites tested, 4-hydroxytamoxifen was the most potent for both NavMs and hNav1.2. Both Z and E isomers of 4-hydroxytamoxifen inhibited the currents, but the Z form was the more potent (half maximal inhibitory concentrations [IC50] of 239 nM for NavMs and 1.2 μM for Nav1.2), indicating a possible stereoselectivity of the receptor in Nav1.2 and NavMs channels (Table 1). As the drug interactions with the channel were best resolved within the inner binding site, where they appeared to involve H-bond formation with two of the protein side chains (D220 and E227), these residues were individually mutated to alanines, but no voltage-dependent Na+ currents (INa) could be measured from such channels. However, when the residues at these sites were replaced with the alternative acidic residue (D swapped for E, and vice versa), the channels retained functionality, but the potency of 4-hydroxytamoxifen INa inhibition (IC50) was ∼7 times less than observed for the wild-type channel (Figure 3E; Table 1). The changes in apparent affinity of 4-hydroxytamoxifen for these D220E and E227D channels correspond to losses in the free energies of binding (ΔG°) of 1.5 and 1.4 kcal/mol, respectively (Figure 3F). The structural and functional findings therefore suggest that these substitutions disrupt the optimal hydrogen bond coordination distance made between the side chain carboxylates and the amine and hydroxyl moieties of 4-hydroxytamoxifen (Figure 3B). It is notable that NavMs and hNav1.2 share similar folds (Figure S1A) and that these NavMs tamoxifen receptor residues are conserved in domains III and IV of hNav1.2 (Figure S1E).
Figure 4.

Functional studies showing sodium current inhibition by tamoxifen analogs
(A and B) HEK293 cells transfected with either (A) NavMs or (B) hNav1.2 were voltage-clamped in the whole-cell configuration. Exemplar voltage-gated sodium currents (INa) recorded in control conditions and in the presence of increasing concentrations of extracellular tamoxifen derivatives. Currents were activated by a 0.5 Hz train of depolarizations to the indicated potentials (gray), and INa inhibition was evaluated after 2 min in each condition. Right panels: the resulting drug concentration-percentage sodium current inhibition (% INa inhibition) relationships were fit to the Hill equation to estimate the half maximal inhibitory concentration (IC50). Error bars are equal to 1 SD, and four to seven cells were tested for each concentration of the drug.
Table 1.
Potency of sodium current inhibition (IC50) by tamoxifen and related compounds
| Drug | NavMs, HEK cells | D220E, HEK cells | E227D, HEK cells | NaV1.2, HEK cells | MDA-MD-231 cells |
|---|---|---|---|---|---|
| Tamoxifen | 830 ± 85 nM (1.08 ± 0.1) | NT | NT | 2.4 ± 0.11 μM (1.05 ± 0.1) | NT |
| Endoxifen | 328 ± 89 nM (1.09 ± 0.1) | NT | NT | 3.1 ± 0.13 μM (0.97 ± 0.1) | NT |
| 4OH-Tam | 297 ± 75 nM (1.14 ± 0.1) | 3.6 ± 0.14 μM (1.17 ± 0.1) | 2.8 ± 0.16 μM (1.13 ± 0.1) | 2.1 ± 0.14 μM (0.96 ± 0.1) | 889 ± 98 nM (0.98 ± 0.1) |
| NM-Tam | 1.1 ± 0.16 μM (1.11 ± 0.1) | NT | NT | 3.2 ± 0.12 μM (0.96 ± 0.1) | NT |
| E-4OH-Tam | 2.3 ± 0.18 μM (1.15 ± 0.1) | NT | NT | 4.2 ± 0.15 μM (0.94 ± 0.1) | NT |
| Z-4OH-Tam | 239 ± 87 nM (1.05 ± 0.1) | NT | NT | 1.2 ± 0.12 μM (0.95 ± 0.1) | NT |
| TTX | NT | NT | NT | NT | 2.6 ± 0.17 μM (1.01 ± 0.1) |
Listed are the average IC50 values derived from fitting the dose-response data to the Hill equation. The average slopes of the fits are indicated in parentheses. The drugs tested in the study were tamoxifen, endoxifen, N-desmethyltamoxifen (NM-Tam), 4-hydroxytamoxifen (4OH-Tam), and the E and Z isomers of 4OH-Tam. NT indicates conditions not tested. Error is equal to SD.
Tamoxifen and its primary metabolic products bind in similar sites
Tamoxifen metabolism occurs mainly through two pathways, 4-hydroxylation and N-demethylation, resulting in the very potent secondary metabolite, endoxifen (Figure S2) (Helland et al., 2017; Kisanga et al., 2004; Lien et al., 1991a, 1991b). The primary metabolite, 4-hydroxytamoxifen, was originally shown to be ∼30- to 100-fold more potent in vivo as an anti-estrogen than tamoxifen itself. However, the pathway that produces it only contributes minimally to tamoxifen metabolism. The conversion of tamoxifen by N-demethylation to N-desmethyltamoxifen is catalyzed primarily by CYP3A4 and CYP3A5 liver enzymes and contributes to ∼92% of tamoxifen metabolism. However, N-desmethyltamoxifen binds to ER with an affinity ∼100 times lower than that of tamoxifen itself. The secondary metabolite endoxifen, which contains both the 4-hydroxylation and N-desmethylation modifications, is the primary binder to ERs in vivo (Helland et al., 2017; Kisanga et al., 2004; Lien et al., 1991a, 1991b). For these reasons the binding sites of not only tamoxifen itself but that of its primary metabolic products, 4-hydroxytamoxifen and N-desmethyltamoxifen, and its secondary metabolite, endoxifen, were studied. All of these compounds formed crystal complexes with NavMs (Figure 1; Table S1), and the “inner” and “outer” drug binding sites were located in the same regions of the molecule, near the bottom of the transmembrane region (Figure 2). These pockets are also present in the structure of hNav1.2 (Figures S1C and S1D), but instead of being formed from the four polypeptide chains in NavMs, they are formed from different regions of the four human channel domains.
Comparing tamoxifen sites within NavMs and the ER
The widely accepted molecular target of tamoxifen and its derivatives is the intracellular ER (Shiau et al., 1998), where the ER-tamoxifen complex binds to DNA and regulates gene transcription. Most interestingly, the three-dimensional binding sites of the 4-hydroxytamoxifen metabolite in the on-target (ER) and off-target (sodium channel) proteins show similar features (Figure 5A). Although there is effectively no sequence or overall structural or even fold similarity between NavMs and the ER (Shiau et al., 1998), comparisons of the crystal structures of their binding sites reveal that specific geometric details of the residues involved in the binding sites in both are similar, including hydrogen bonds with an aspartic acid (D351 in ER and D220 in NavMs) at one end and a glutamate (E353 in ER and E227 in NavMs) at the other, which correspond with the critical residues identified structurally and functionally, as described above. Both the NavMs and ER pockets are of sufficient size to accommodate the large and hydrophobic 4-hydroxytamoxifen molecules. This strong local similarity would thus potentially enable tamoxifen and its derivatives to bind to and inhibit both the target ER and the unrelated Navs and could be the origin of its adventitious off-target effects in Navs previously reported in adenocarcinoma cells. To test this, voltage clamping of MDA-MB-231 cells (a triple-negative breast cancer cell line that functionally expresses Nav1.7 and the neonatal splice form of Nav1.5 [nNav1.5] but does not express the ER [Gradek et al., 2019; Brackenbury et al., 2007]) produced endogenous sodium currents which exhibit low sensitivity to tetrodotoxin (TTX) (IC50 = 2.6 μM), suggesting that the preponderance of the current is conducted by TTX-insensitive Nav subtypes (Figures 5B and 5C). 4-Hydroxytamoxifen produces dose-dependent inhibition of the INa with an IC50 of 890 nM (Figure 5C; Table 1), which demonstrates that the efficacy and potency of this drug against endogenous nNav1.5 in these cancer cells is independent of ER modulation. This study thus provides a structural basis for the observations of its effects on sodium transport in these adenocarcinoma cells.
Figure 5.

Tamoxifen binding in the estrogen receptor and the NavMs channel and its inhibition of sodium currents recorded from adenocarcinoma cells
(A) Stereo image of overlays of the 4-hydroxytamoxifen binding sites in NavMs (PDB: 6SXC) (dark green) and in the estrogen receptor (dark pink) (PDB: 3ERT). This shows the similarities of the binding site geometries and residues that form hydrogen bonds (Asp at one end and Glu at other end) involved in the interactions of the compound (shown in corresponding light green and light pink stick depictions) with both target proteins. The side chains of the NavMs and estrogen receptor protein molecules are depicted in dark green and dark pink, respectively.
(B) Endogenous sodium currents recorded from the adenocarcinoma cell line MDA-MB-231 cells. Left: exemplar voltage-gated sodium currents activated by successively increasing the depolarizing potential by 5 mV from a −100 mV holding potential before and after TTX treatment. Right: resulting current densities plotted as a function of voltage.
(C) Left: exemplar INa recorded from MDA-MB-231 cells and their inhibition by 4-hydroxytamoxifen using the same protocol described in Figure 4. Right, drug concentration-percentage sodium current inhibition relationships for TTX and 4-hydroxytamoxifen as described in Figure 4. Error bars denote SD, and number of cells tested is indicated in parentheses.
See also Figure S2.
The mechanism of sodium current inhibition by 4-hydroxytamoxifen
Therapeutic drugs inhibit sodium channels by either blocking the ion-conducting pathway or stabilizing the non-conducting (closed or inactivated) conformations of the channel (Sampson and Kass, 2011; McNamara, 2011; Catterall and Mackie, 2011). The kinetics of 4-hydroxytamoxifen INa inhibition (τon) and recovery (τoff) were 16 and 765 times slower, respectively, than those observed for the pore-blocking metal ion cadmium (Figures 6A and 6B). The delay in INa recovery (τoff = 6.6 min) is consistent with high-affinity drug occupancy of the channel. To examine the mechanism of INa inhibition, the voltage dependence of activation and inactivation were compared before and after 3 μM 4-hydroxytamoxifen treatment, but no significant shifts in either parameter (Figure S5A) were observed. However, the rate of recovery from inactivation (τrec) did show a ∼10-fold delay following drug treatment (Figure S5B). This suggests that the primary mechanism of INa inhibition by 4-hydroxytamoxifen is a stabilization of the non-conducting inactivated channel state. To further explore the mechanism of drug action, we tested INa inhibition from cells subjected to long (150 ms) depolarization trains, where ∼95% of total current is inactivated by the end of the voltage step (Figure 6C). NavMs channels recover from inactivation in the control condition, as the magnitude of INa stayed the same for each depolarization. Then, the depolarization train was terminated, and the channels were held in the closed state (−180 mV holding potential) while the cell was exposed to 1 μM 4-hydroxytamoxifen. Cells were then returned to the control condition and the depolarization train was re-initiated. INa was unchanged in the first depolarization, but surprisingly, clear inhibition was observed in subsequent depolarizations even though the cell was no longer in the presence of 4-hydroxytamoxifen. Repeating these experiments, but limiting the percentage of channels entering the inactivated state (∼5%) using a shorter depolarization time (7 ms) resulted in 3.4 times less INa inhibition, which clearly demonstrates that the drug effect is dependent on channel entry into the inactivated state (Figures 6D–6F). These results provide clear evidence that tamoxifen binds to sodium channels with high affinity and inhibits the sodium current using a unique mechanism and establish a functional link with the structural studies.
Figure 6.

The mechanism of NavMs sodium current inhibition by 4-hydroxytamoxifen
Voltage protocols are shown above each panel.
(A) The onset (τon) and recovery (τoff) from 4-hydroxytamoxifen inhibition was recorded from voltage-clamped HEK cells expressing NavMs under the indicated conditions. Top: exemplar NavMs INa (black) activated by 150 ms depolarizations from −180 mV before, during, and after drug application.
(B) The time constants were estimated from a single exponential fit upon addition and removal of either 100 μM Cd2+ or 3 μM 4-hydroxytamoxifen.
(C and D) 4-Hydroxytamoxifen inhibition of NavMs sodium currents is dependent on entry into the inactivated state. Graphs indicate the time course of peak sodium current inhibition from exemplar cells subjected to (C) 150 ms and (D) 7 ms 0 mV depolarization trains at 0.5 Hz, where 97% and 6% of INa was inactivated by the end the pulse (Iinact), respectively. Extracellular 1 μM 4-hydroxytamoxifen was applied for 1 min (blue region) while the cell was held at −180 mV, where NavMs channels remain in the closed state. Cells were returned to the control perfusate and depolarized using the same voltage protocol.
(E) Comparing percentage sodium current inhibition by 4-hydroxytamoxifen from cells depolarized with 7 or 150 ms depolarization voltage protocols. Error bars denote SD, and the number of cells tested for each condition (n) is indicated in parentheses.
(F) Proposed model of sodium channel inhibition by tamoxifen and its metabolites. Tamoxifen binds to sodium channel but does not block the ion-conducting pore. Drug occupancy of the sodium channel impairs recovery from the inactivated state, resulting in fewer channels available for reopening by subsequent depolarizations. The sodium channel models used in this figure are based on structures of open apoNavMs (PDB: 5HVX) and closed apo NavAb (PDB: 5VB2).
See also Figure S5.
Discussion
In summary, this study has demonstrated that tamoxifen and its metabolic products bind to the prokaryotic sodium channel NavMs through a binding pocket that is structurally conserved in eukaryotic Nav homologs. The tamoxifen-NavMs receptor site is entirely different from both the hydrophobic transmembrane cavity used by analgesic and antiepileptic drugs as binding sites and the voltage sensor receptor sites used by toxins and other hydrophilic inhibitors. Furthermore, the electrophysiology studies have shown that the NavMs-tamoxifen interaction is high affinity and demarked by slow dissociation, which delays the channel recovery from inactivation. These pharmacological features are shared by neuronal sodium channels and endogenous sodium channel currents in metastatic breast cancer cells. The measured binding affinities are consistent with the structural observation that the residues that form the binding pockets for tamoxifen in sodium channels are geometrically similar to those in the target ERs. These results define a new sodium channel drug receptor site, which could be targeted in future drug development studies, as well as highlighting a new mechanism of channel inhibition involving inactivation, which could also be exploited for the future development of novel on-target drugs to treat sodium channelopathies. The observed effects on sodium channel structure and function also have potentially important ramifications for the therapeutic use and consequent side effects of tamoxifen in cancer treatments.
Limitations
This study has demonstrated potential interactions of Navs to bind and be modulated by tamoxifen and its principal metabolic products in vitro at concentrations that approximate ranges commensurate with dosages used for in vivo prophylaxis and chemotherapeutic treatments. However, as with all such studies, the translation of molecular characterizations to in vivo applications is potentially less straightforward. Nevertheless, the newly characterized interactions described in this study may open up a future avenue for development of other pharmaceuticals for Nav targets.
STAR★Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial strains | ||
| Escherichia coli NEB 5-alpha | New England Biolabs | Cat# C2987H |
| Escherichia coli OverExpress C41 (DE3) | Sigma-Aldrich | Cat# CMC0017 |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Tamoxifen Citrate | Sigma-Aldrich | Cat# T9262 |
| 4-Hydroxytamoxifen hydrochloride | Sigma-Aldrich | Cat# H6278 |
| Endoxifen hydrochloride | Sigma-Aldrich | Cat# E8284 |
| N-Desmethyltamoxifen hydrochloride | Sigma-Aldrich | Cat# D9069 |
| Z-4-Hydroxytamoxifen hydrochloride | Abcam | Cat# ab141943 |
| E-4-Hydroxytamoxifen hydrochloride | Abcam | Cat# ab143637 |
| Z-Endoxifen hydrochloride | Generon | Cat# HY-18719A |
| E-Endoxifen hydrochloride | Generon | Cat# HY-18719C |
| Polyethylene Glycol 400 | Sigma-Aldrich | Cat# 202398 |
| Ethylene Glycol | Molecular Dimensions | Cat# MD2-100-158 |
| Dimethyl Sulphoxide (DMSO) | Sigma-Aldrich | Cat# D4540 |
| n-Dodecyl-B-D-maltoside (DDM) | Anatrace | Cat# D310S |
| Decanoyl-N-Hydroxyethylglucamide (HEGA-10) | Anatrace | Cat# H110 |
| Cesium Fluoride | Sigma-Aldrich | Cat# 198323 |
| Cesium Hydroxide | Fisher Chemical | Cat# C97 |
| Calcium Chloride dihydrate | Sigma-Aldrich | Cat# C3306 |
| Ethylene glycol-bis(2-amino ethylether)- N,N,N’,N’-tetraacetic acid (EGTA) | Sigma-Aldrich | Cat# E4378 |
| HEPES | Fisher Chemical | Cat# BP310 |
| D-Mannitol | Acros Organics | Cat# 69658 |
| Deposited data | ||
| Crystal structure of NavMs | Sula et al., 2017 | PDB: 5HVX |
| CryoEM Structure hNav1.2 | Pan et al., 2019 | PDB: 6J8E |
| NavMS F208L (NavMsL) | This Study | PDB: 6SX5 |
| NavMs/4-Hydroxytamoxifen | This Study | PDB: 6SXG |
| NavMsL/Tamoxifen | This Study | PDB: 6SXF |
| NavMsL/4-Hydroxytamoxifen | This Study | PDB: 6SXC |
| NavMsL/Endoxifen | This Study | PDB: 6SXE |
| NavMsL/N-Desmethytamoxifen | This Study | PDB: 6Z8C |
| NavMsL/DMSO | This Study | PDB: 6SX7 |
| Experimental models: cell lines | ||
| HEK293 | ATCC | CRL-3216 |
| MDA-MD-231 | ATCC | HTB-26 |
| Oligonucleotides | ||
| F208L_F CTCACCACCCTGACCGT GCTCAACCTGTTTATTGG |
Eurofins Genomics | N/A |
| F208L_R GAGCACGGTCAGGGTG GTGAGCATGATGAACGGGATG |
Eurofins Genomics | N/A |
| D220E_F ATTGGCATTATTGTAGA AGCAATGGCAATCAC |
IDT | N/A |
| D220E_R GTGATTGCCATTGC TTCTACAAT AATGCCAAT |
IDT | N/A |
| E227D_F ATGGCAATCACCAA GGATCAGGAGGAAGAGGCC |
IDT | N/A |
| E227D_R GGCCTCTTCCTCCT GATCCTTGGTGATTGCCAT |
IDT | N/A |
| Recombinant DNA | ||
| pET15b-NavMS | Sula et al., 2017 | N/A |
| pET15b-NavMS F208L | This Study | N/A |
| pTracer-CMV2-NavMS | Sula et al., 2017 | Addgene #100004 |
| pTracer-CMV2-NavMS D220E E227D | This Study | N/A |
| Human Nav1.2/Pcdna8 | This Study | N/A |
| Software and algorithms | ||
| XDS | Kabsch., 2010 | http://xds.mpimf-heidelberg.mpg.de/html_doc/XDS.html |
| Collaborative Computational Project No.4 | Winn et al., 2011 | https://www.ccp4.ac.uk/ |
| Aimless | Evans and Murshudov, 2013 | https://www.ccp4.ac.uk/dist/html/aimless.html |
| Phaser | McCoy et al., 2007 | https://www.ccp4.ac.uk/html/phaser.html |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Refmac | Murshudov et al., 2011 | https://www.ccp4.ac.uk/html/refmac5.html |
| Buster version 1.10.0 | Bricogne et al., 2011 | https://www.globalphasing.com/buster/ |
| MolProbity | Chen et al., 2010 | molprobity.biochem.duke.edu |
| CCP4mg | McNicholas et al., 2011 | https://www.ccp4.ac.uk/MG/ |
| Clustal Omega | Sievers et al., 2011 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
| HOLE | Smart et al., 1996 | http://www.holeprogram.org/ |
| VMD | Humphrey et al., 1996 | https://www.ks.uiuc.edu/Research/vmd/ |
| pClamp 10 | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite |
| Igor Pro 7.00 | Wavemetrics Inc | https://www.wavemetrics.com/products/igorpro |
| Other | ||
| His Trap FF | GE Healthcare | Cat# 17-5255-01 |
| Superdex 10/300 200 GL Increase | GE Healthcare | Cat# 28990944 |
| Amicon Ultra-15 Centrifuge 100K | Merck | Cat# UFC910024 |
| Amicon Ultra-6 Centrifuge 100K | Merck | Cat# UFC810024 |
| MemMeso Crystallization Screen | Molecular Dimensions | Cat# MD1-86 |
| MRC 2 Well Crystallization Plate | Molecular Dimensions | Cat# MD11-00-10 |
| Borosilicate Glass Capillaries | World Precision Instruments, Inc. | Cat# IB150F-4 |
| Patch-Clamp Amplifier | Axon Instruments | Axopatch 200B |
| Data Acquisition Digitizer | Axon Instruments | Axon Digidata 1550B |
| Perfusion Fast-step | Warner Instrument Corporation | SF-77B |
| Inverted Microscope | Olympus | IX73 |
| Thrombin | Sigma-Aldrich | Cat# 69671 |
| Trypsin | Thermo Fisher Scientific | Cat# R001100 |
| DMEM/F12 + GlutaMAX | Thermo Fisher Scientific | Cat# 10565018 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140122 |
| USA Origin Fetal Bovine Serum (FBS) | Sigma-Aldrich | Cat# F2442 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668027 |
| Complete EDTA free Protease inhibitor tables | Sigma-Aldrich | Cat# 5056489001 |
| KAPA HiFi Hotstart ReadyMix (2x) | Roche | Cat# KK2601 |
| Collagenase | Thermo Fisher Scientific | Cat# 17018029 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, B.A. Wallace (b.wallace@cryst.bbk.ac.uk).
Materials availability
Reagents generated in this study will be made available on request, but may require a completed Materials Transfer Agreement and/or payment, if there is potential for commercial application. The pTracer-CMV2, IRES GFP plasmid which contains the gene encoding for N-terminal his-tagged NavMs is available through addgene (http://www.addgene.org/100004), whereas the pcdna8 plasmid containing the human gene encoding for the Nav1.2 is also available upon request.
Data and code availability
The accession numbers for the crystal structure coordinates and structure factors deposited in the Protein Data Bank are: PDB: 6SX5, 6SXF, 6SXG, 6SXC, 6SXE, 6Z8C, and 6SX7.
Experimental model and subject details
Microbe strains
E. coli DH5 alpha was cultured at 37°C in LB medium supplemented with 100 mg/ml of ampicillin for plasmid DNA extraction. E. coli C41 (DE3) was cultured at 37°C in LB medium supplemented with 100 mg/ml of ampicillin for protein overexpression.
Cell lines
HEK293 (Human Embryonic Kidney) and MDA-MD-231 cells were propagated and maintained in Dulbecco’s Modified Eagle Medium (DMEM)/F12 + GlutaMAX supplemented with 10% fetal bovine serum and 1% penicillin streptomycin solution.
Method details
Protein expression and purification
NavMs proteins were expressed and purified as previously described (Sula et al., 2017). The F208L (NavMsL) mutation (Figure S6) was introduced using the SLIM site-directed mutagenesis protocol (Chiu et al., 2004). The sequence was verified by Source Bioscience, UK.
Crystallization
Proteins were concentrated to 10 mg/ml for crystallizations in 20 mM Tris-HCl, 300 mM NaCl, 14 mM HEGA-10 detergent, pH 7.5, and stored at −80°C. Crystals were grown at 4°C using the sitting drop vapor diffusion method by mixing 2:1 ratios of protein and reservoir solution, which typically contained 30% v/v PEG400, 0.1 M NaCl, 0.1 M MES, pH 6.5. Crystal conditions were refined for each crystal type using the four-corner screen method. Details of the precise conditions for individual crystals are documented in the PDB files for their structures. For co-crystallizations, prior to setting up the drops, 50 mM stocks of tamoxifen, 4-hydroxytamoxifen, N-desmethyltamoxifen, or endoxifen (Sigma-Aldrich) were prepared in 100% DMSO and added to the protein solutions to produce final concentrations of 1.25 mM drug and 2.5% v/v DMSO. DMSO-only controls were also set up in the same way and yielded crystals identical to the apo-NavMs and apo-NavMsL crystals grown in the absence of any additives. Crystals were flash-frozen prior to data collection. All crystallization conditions contained low molecular weight PEG which acted as the cryoprotectant. Both wild-type and NavMsL proteins were examined because while the native residue at the site of the mutation is phenylalanine in NavMs and the equivalent residues in two of the domains of all hNavs are also phenylalanines, in the other two domains of hNavs the residue type is a leucine (Figures S1 and S6). Comparisons of the apo-NavMs and apo-NavMsL proteins (PDB: 6SX5, 5HVX) and 4-hydroxytamoxifen-bound structures (PDB 6SXC, 6SXG) (Figure S6; Table S1) demonstrated no effect on the apo-protein structure (Figure S6) nor on the drug interactions (Figure S6). However, the NavMsL crystals were generally of slightly higher quality (better resolution), and for that reason the other structures of crystal-drug complexes reported in this study are for NavMsL.
Crystallography
X-ray diffraction data were collected at either the EMBL Hamburg synchrotron (Germany) P13 beamline or at the I04, I03, or I24 beamlines at the Diamond (UK) synchrotron. More than 100 datasets were collected from the various crystal types. The highest resolution crystals of the native and NavMsL proteins and of the protein/drug complexes were 2.2 Å. Because there was a substantial variation in the unit cell dimensions and resolution between different crystals produced under the same conditions, as we have seen previously (Naylor et al., 2016) we were unable to merge datasets between crystals. The diffraction images were integrated and scaled using the XDS software package (Kabsch, 2010) and merged using Aimless (Evans and Murshudov, 2013) in the CCP4 program suite (Winn et al., 2011). The structures were determined by molecular replacement using the full-length NavMs structure (PDB: 5HVX) as the search model. Phases were obtained with Phaser (McCoy et al., 2007) and model building was performed in Coot (Emsley et al., 2010). The structures were initially refined in Refmac (Murshudov et al., 2011) and then the refinement was continued in Buster (Bricogne et al., 2011). The data collection, processing and refinement statistics are listed in Table S1. The refinement quality was checked using MolProbity (Chen et al., 2010) which indicated that 100% of residues were in allowed conformations. Figures were created in CCP4mg (McNicholas et al., 2011) unless otherwise noted.
Bioinformatics
Sequence alignments (Figure S1) were undertaken using the Clustal Omega multiple sequence alignment tool (Sievers et al., 2011), with Uniprot codes A0L5S6 (NavMs) and Q99250 (hNav 1.2). The inner pore dimensions were calculated using the HOLE suite of programmes (Smart et al., 1996) and visualized with the Visual Molecular Dynamics (VMD) program (Humphrey et al., 1996), Molecular overlays/comparisons of the NavMs/4-hydroxytamoxifen crystal structure (PDB: 6SXG) and hNav1.2 cryo-EM structure (PDB: 6J8E) were accomplished using CCP4mg software (McNicholas et al., 2011). Structural overlays of 4-hydoxytamoxifen complexes of NavMsL (PDB: 6SXC) and the human estrogen receptor (PDB: 3ERT) were done using CCP4mg (McNicholas et al., 2011) software to maximize the overlay of the tamoxifen molecules in each structure.
Electrophysiology
The MDA-MB-231 and HEK cell lines were obtained from the American Type Culture Collection. The previously described pTracer-CMV2, IRES GFP plasmid which contains the gene encoding for N-terminal his-tagged NavMs used for our whole-cell electrophysiology studies. Standard quick-change site directed mutagenesis protocols were used to create receptor site mutations in the NavMs gene. Transient transfection of exogenous sodium channels (NavMs or hNav1.2) was performed using lipofectamine 24-48 hours prior to electrophysiology recordings. Plasma membrane currents were recorded using borosilicate glass electrodes polished to resistances of 2–4.5 MΩ filled with an internal solution containing 110 mM CsF, 30 mM NaCl, 10 mM HEPES, and 5 mM EGTA (ethylene glycol-bisβ-aminoethyl ether-N,N,N′,N′-tetraacetic acid); the pH was adjusted to 7.3 using CsOH. The extracellular recording solution contained 150 mM NaCl, 10 mM HEPES, and 1.8 mM CaCl2, with the pH adjusted to 7.4 using NaOH. All solutions were osmotically balanced to 300 (±10) mOsm with mannitol. Data were collected using an Axopatch 200B patch-clamp amplifier, Digidata 1550B, and pClamp 10 software. Currents were digitized at 25 kHz and low-pass filtered at 5 kHz. Tamoxifen, 4-hydroxytamoxifen, endoxifen and N-desmethyltamoxifen were purchased from Sigma Aldrich. The 4-hydroxytamoxifen stocks contained at least 70% of the Z- isomer according to manufacturer documentation. The purified E- and Z- isomers of 4-hydroxytamoxifen (Abcam) were also tested. All drug reagents were formulated in DMSO at 10 mM and stored at −20°C. Rapid extracellular drug application was achieved using a Warner Perfusion Fast-Step system in which the patched cells were held in the perfusate stream.
Quantification and statistical analysis
Analyses of electrophysiological data
Data were analyzed by Igor Pro 7.00 (Wavemetrics, Lake Oswego, OR). Conductance (G)-voltage relationships were derived from the peak sodium current measured during the prepulse of Figure S5 using the following equation: G = I/(V-Vr), where Vr is equal to the reversal potential. Half-activation and inactivation voltages (V1/2) were derived by fitting the datasets by using a Boltzmann function: f(x) = 1/[1+exp(V−V1/2)/k]. Percent inhibition was determined by taking the ratio of the current at steady state drug block (Idrug) and the control current (Icontrol), and expressed as: %INa inhibition = 100 - (Idrug/Icontrol)∗100. Drug concentration-response curves were fit to the Hill equation: f(x) = base+(max-base)/{1+(IC50/[Drug]} to estimate the concentration of drug (IC50) required to half-maximally inhibit the sodium current. Gibbs law of free energy was used to calculate the free energy of drug binding: ΔG = -R⋅T⋅Ln(Kd), where R = 0.008314 kJ/mol, temperature T = 297 K°, and Kd is the apparent association constant estimated by the IC50. After an inactivating prepulse, the recovery from inactivation (τrec) was estimated by fitting the normalized current activated by the test pulse to an exponential equation: f(x) = base+amplitude⋅exp[(1/τ)x]. This equation was also used to estimate the rate of channel inhibition (τon) and recovery from inhibition (τoff) by cadmium and 4-hydroxytamoxifen.
Estimations
Protein concentrations were estimated from Nanodrop absorbances measured at 280 nm, using an extinction coefficient of 29450 M-1 cm-1.
The resolution criteria for the crystal structures presented herein were based on having an I/sigma greater than 1 and a CC(1/2) greater than 0.7.
Acknowledgments
We thank the staff at the P13 (European Molecular Biology Laboratory [EMBL], Hamburg, Germany) and I04, I03, I24 (Diamond Light Source) beamlines for assistance during data collection and Dina Molner (Birkbeck) for help in making the F208L mutant. This work was supported by grants BB/L006790, BB/N012763, and BB/R001294 from the U.K. Biotechnology and Biological Science Research Council (BBSRC) to B.A.W.; a grant from the Rosetrees Trust to B.A.W.; a grant from the National Institute of Academic Anaesthesia to B.A.W., H. Hemmings, and K.F. Herold; crystallography beamtime grants to the Birkbeck/UCL BAG consortium from the EMBL, Hamburg, and the Diamond Light Source; and an iNEXT grant for travel to the P13 beamline. P.G.D. was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (1R56DK119709-01, 4R00DK106655, 1R01 DK123463-01, and P30 DK090728), NU GoKidney George M. O’Brien Kidney Research Core Center (P30 DK11485), and the American Society of Nephrology (Carl W. Gottschalk Scholar Award).
Author contributions
B.A.W. and P.G.D. designed and supervised the research. A.S., D.H., L.C.T.N., and P.G.D. performed the research. A.S., L.C.T.N., D.H., M.L., P.G.D., and B.A.W. analyzed the data. B.A.W. wrote the initial version of the paper. All authors contributed to and approved of the final version.
Declaration of interests
The authors declare no competing interests.
Published: January 26, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2020.12.048.
Contributor Information
Paul G. DeCaen, Email: paul.decaen@northwestern.edu.
B.A. Wallace, Email: b.wallace@mail.cryst.bbk.ac.uk.
Supplemental information
References
- Ahuja S., Mukund S., Deng L., Khakh K., Chang E., Ho H., Shriver S., Young C., Lin S., Johnson J.P., Jr. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science. 2015;350:aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- Bagnéris C., DeCaen P.G., Naylor C.E., Pryde D.C., Nobeli I., Clapham D.E., Wallace B.A. Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc. Natl. Acad. Sci. U S A. 2014;111:8428–8433. doi: 10.1073/pnas.1406855111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury W.J., Chioni A.M., Diss J.K., Djamgoz M.B. The neonatal splice variant of Nav1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2007;101:149–160. doi: 10.1007/s10549-006-9281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G. Global Phasing; 2011. BUSTER version 1.10.0. [Google Scholar]
- Catterall W.A., Mackie K. Local anesthetics. In: Brunton L., Chabner B.A., Knollmann B.C., editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; 2011. pp. 565–582. [Google Scholar]
- Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu J., March P.E., Lee R., Tillett D. Site-directed, ligase-independent mutagenesis (SLIM): a single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 2004;32:e174. doi: 10.1093/nar/gnh172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A. Breast cancer: tamoxifen—offering a long-term prevention option. Nat. Rev. Clin. Oncol. 2015;12:66. doi: 10.1038/nrclinonc.2014.236. [DOI] [PubMed] [Google Scholar]
- Evans P.R., Murshudov G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser S.P., Diss J.K., Chioni A.M., Mycielska M.E., Pan H., Yamaci R.F., Pani F., Siwy Z., Krasowska M., Grzywna Z. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 2005;11:5381–5389. doi: 10.1158/1078-0432.CCR-05-0327. [DOI] [PubMed] [Google Scholar]
- Fraser S.P., Ozerlat-Gunduz I., Brackenbury W.J., Fitzgerald E.M., Campbell T.M., Coombes R.C., Djamgoz M.B. Regulation of voltage-gated sodium channel expression in cancer: hormones, growth factors and auto-regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014;369:20130105. doi: 10.1098/rstb.2013.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamal El-Din T.M., Lenaeus M.J., Zheng N., Catterall W.A. Fenestrations control resting-state block of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. U S A. 2018;115:13111–13116. doi: 10.1073/pnas.1814928115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradek F., Lopez-Charcas O., Chadet S., Poisson L., Ouldamer L., Goupille C., Jourdan M.L., Chevalier S., Moussata D., Besson P., Roger S. Sodium channel Nav1.5 controls epithelial-to-mesenchymal transition and invasiveness in breast cancer cells through its regulation by the salt-inducible kinase-1. Sci. Rep. 2019;9:18652. doi: 10.1038/s41598-019-55197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Kargacin M.E., Kargacin G.J., Ward C.A. Tamoxifen inhibits Na+ and K+ currents in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2003;285:H661–H668. doi: 10.1152/ajpheart.00686.2002. [DOI] [PubMed] [Google Scholar]
- Helland T., Henne N., Bifulco E., Naume B., Borgen E., Kristensen V.N., Kvaløy J.T., Lash T.L., Alnæs G.I.G., van Schaik R.H. Serum concentrations of active tamoxifen metabolites predict long-term survival in adjuvantly treated breast cancer patients. Breast Cancer Res. 2017;19:125. doi: 10.1186/s13058-017-0916-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jordan V.C. Tamoxifen: a most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanga E.R., Gjerde J., Guerrieri-Gonzaga A., Pigatto F., Pesci-Feltri A., Robertson C., Serrano D., Pelosi G., Decensi A., Lien E.A. Tamoxifen and metabolite concentrations in serum and breast cancer tissue during three dose regimens in a randomized preoperative trial. Clin. Cancer Res. 2004;10:2336–2343. doi: 10.1158/1078-0432.ccr-03-0538. [DOI] [PubMed] [Google Scholar]
- Klein D.J., Thorn C.F., Desta Z., Flockhart D.A., Altman R.B., Klein T.E. PharmGKB summary: tamoxifen pathway, pharmacokinetics. Pharmacogenet. Genomics. 2013;23:643–647. doi: 10.1097/FPC.0b013e3283656bc1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E.A., Solheim E., Ueland P.M. Distribution of tamoxifen and its metabolites in rat and human tissues during steady-state treatment. Cancer Res. 1991;51:4837–4844. [PubMed] [Google Scholar]
- Lien E.A., Wester K., Lønning P.E., Solheim E., Ueland P.M. Distribution of tamoxifen and metabolites into brain tissue and brain metastases in breast cancer patients. Br. J. Cancer. 1991;63:641–645. doi: 10.1038/bjc.1991.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara J. Pharmacotherapy of the epilepsies. In: Brunton L., Chabner B.A., Knollmann B.C., editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; 2011. pp. 583–608. [Google Scholar]
- McNicholas S., Potterton E., Wilson K.S., Noble M.E.M. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini G., Booker J., Sula A., Wallace B.A. Comparisons of voltage-gated sodium channel structures with open and closed gates and implications for state-dependent drug design. Biochem. Soc. Trans. 2018;46:1567–1575. doi: 10.1042/BST20180295. [DOI] [PubMed] [Google Scholar]
- Mürdter T.E., Schroth W., Bacchus-Gerybadze L., Winter S., Heinkele G., Simon W., Fasching P.A., Fehm T., Eichelbaum M., Schwab M., Brauch H. Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin. Pharmacol. Ther. 2011;89:708–717. doi: 10.1038/clpt.2011.27. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Skubák P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor C.E., Bagnéris C., DeCaen P.G., Sula A., Scaglione A., Clapham D.E., Wallace B.A. Molecular basis of ion permeability in a voltage-gated sodium channel. EMBO J. 2016;35:820–830. doi: 10.15252/embj.201593285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Li Z., Huang X., Huang G., Gao S., Shen H., Liu L., Lei J., Yan N. Molecular basis for pore blockade of human Na+ channel Nav1.2 by the μ-conotoxin KIIIA. Science. 2019;363:1309–1313. doi: 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- Payandeh J., Scheuer T., Zheng N., Catterall W.A. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–358. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson K.J., Kass R.S. Anti-arrhythmic drugs. In: Brunton L., Chabner B.A., Knollmann B.C., editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; 2011. pp. 815–848. [Google Scholar]
- Shen H., Li Z., Jiang Y., Pan X., Wu J., Cristofori-Armstrong B., Smith J.J., Chin Y.K.Y., Lei J., Zhou Q. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science. 2018;362:eaau2596. doi: 10.1126/science.aau2596. [DOI] [PubMed] [Google Scholar]
- Shiau A.K., Barstad D., Loria P.M., Cheng L., Kushner P.J., Agard D.A., Greene G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O.S., Neduvelil J.G., Wang X., Wallace B.A., Sansom M.S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996;14:354–360, 376. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Sula A., Booker J., Ng L.C.T., Naylor C.E., DeCaen P.G., Wallace B.A. The complete structure of an activated open sodium channel. Nat. Commun. 2017;8:14205. doi: 10.1038/ncomms14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider M.B., Bagnéris C., McCusker E.C., DeCaen P.G., Delling M., Clapham D.E., Ulmschneider J.P., Wallace B.A. Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc. Natl. Acad. Sci. U S A. 2013;110:6364–6369. doi: 10.1073/pnas.1214667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Jiao B.-H. The inhibitory effects of tamoxifen on glioma cells SHG-44. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2009;25:207–210. [PubMed] [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Li T., Rohou A., Arthur C.P., Tzakoniati F., Wong E., Estevez A., Kugel C., Franke Y., Chen J. Structural basis of Nav1.7 inhibition by a gating-modifier spider toxin. Cell. 2019;176:702–715.e14. doi: 10.1016/j.cell.2018.12.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the crystal structure coordinates and structure factors deposited in the Protein Data Bank are: PDB: 6SX5, 6SXF, 6SXG, 6SXC, 6SXE, 6Z8C, and 6SX7.