

Abstract
Coronavirus disease 2019 (COVID-19) is a new pandemic characterized by quick spreading and illness of the respiratory system. To date, there is no specific therapy for Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2). Flavonoids, especially rutin, have attracted considerable interest as a prospective SARS-CoV-2 main protease (Mpro) inhibitor. In this study, a database containing 2017 flavone analogs was prepared and screened against SARS-CoV-2 Mpro using the molecular docking technique. According to the results, 371 flavone analogs exhibited good potency towards Mpro with docking scores less than −9.0 kcal/mol. Molecular dynamics (MD) simulations, followed by molecular mechanics-generalized Born surface area (MM/GBSA) binding energy calculations, were performed for the top potent analogs in complex with Mpro. Compared to rutin, PubChem-129-716-607 and PubChem-885-071-27 showed better binding affinities against SARS-CoV-2 Mpro over 150 ns MD course with ΔGbinding values of −69.0 and −68.1 kcal/mol, respectively. Structural and energetic analyses demonstrated high stability of the identified analogs inside the SARS-CoV-2 Mpro active site over 150 ns MD simulations. The oral bioavailabilities of probable SARS-CoV-2 Mpro inhibitors were underpinned using drug-likeness parameters. A comparison of the binding affinities demonstrated that the MM/GBSA binding energies of the identified flavone analogs were approximately three and two times less than those of lopinavir and baicalein, respectively. In conclusion, PubChem-129-716-607 and PubChem-885-071-27 are promising anti-COVID-19 drug candidates that warrant further clinical investigations.
Keywords: SARS-CoV-2 main protease, COVID-19, Molecular docking, Drug-likeness, Molecular dynamics
Graphical abstract

1. Introduction
The human severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a universal menace to world health and economy [1,2]. Eventually, the World Health Organization (WHO) officially labeled the COVID-19 outbreak as a pandemic on March 11, 2020 [3]. Based on WHO’s proclamation, more than 118 million people around the world have been infected with COVID-19, including some 2.6 million deaths as of March 12, 2021 [4]. Unfortunately, the numbers of infected cases are on the increase [5]. Until now, no specific drugs are available to eliminate COVID-19 infection [6]. More recently, remdesivir and dexamethasone were declared to have positive influences in clinical trials [[6], [7], [8]]. Ongoing clinical trials are being conducted to assess the efficacy of several drugs to treat COVID-19 disease [9]. Recently, WHO announced 200 vaccine products at various stages of clinical development and issued an Emergency Use Listing (EUL) for Pfizer and AstraZeneca/Oxford COVID-19 vaccines [10]. Although the developed vaccines are currently being distributed and administered, a mass vaccination program will take a long time. Therefore, continued efforts are needed to develop safe and effective anti-COVID-19 drugs.
SARS-CoV-2 Mpro (also called 3CLpro) is one of the most charming targets for preventing viral replication [11]. Very recently, the first crystal structure of SARS-CoV-2 Mpro in complex with a peptidomimetic inhibitor (N3) was resolved to enable the rational design of specific inhibitory compounds towards SARS-CoV-2 Mpro [12].
Utilizing in silico drug discovery techniques, several attempts have been made to repurpose known pharmaceutical drugs as potential therapeutic candidates for the treatment of COVID-19 [[13], [14], [15]]. Virtual screening combined with molecular dynamics (MD) simulations of chemical libraries against SARS-CoV-2 targets was conducted for the sake of hunting the most promising inhibitors that could block the viral replication [16].
Natural products have been the focus of several investigations in the search for discovering anti-COVID-19 drug candidates [17,18]. Among natural products, flavonoids have attracted considerable interest as potential SARS-CoV-2 inhibitors [[19], [20], [21]].
Rutin is a flavonol glycoside molecule extracted from many plants, including buckwheat, tobacco, forsythia, hydrangea, viola, etc. Rutin exemplifies a significant component of many antiviral medicines [22]. Rutin is also known to demonstrate good affinity against avian influenza virus [23], HSV-1 [24], HSV-2 [25], and parainfluenza-3 virus [26]. Several studies have documented that rutin has high efficacy as a potent inhibitor to treat COVID-19 infection [27,28]. The potency of rutin against the SARS-CoV-2 Mpro was underpinned and attributed to the hydroxyl groups within the sugar group involved in its structure, demonstrating several noncovalent interactions with the heteroatoms of amino acids of the Mpro’s active site [21,29,30].
Therefore, the current study was set out to evaluate binding affinities of flavone analogs as SARS-CoV-2 Mpro inhibitors using in silico structure-based drug discovery techniques. A database of 2017 flavone analogs was retrieved, prepared and screened virtually against SARS-CoV-2 Mpro. Based on the predicted docking scores, the most promising hits were then subjected to molecular dynamics (MD) simulations. The stabilities and affinities of the top potent analogs were further investigated over a 150 ns MD course. Prediction of drug-likeness was carried out to disclose the bioavailabilities of the identified drug candidates. The current data highlight the importance of the identified potential anti-SARS-CoV-2 Mpro inhibitors, which can be tested in vitro and in vivo, to fight the global threat of COVID-19.
2. Computational methodology
2.1. Protein preparation
The resolved experimental 3D crystal structure of SARS-CoV-2 main protease (Mpro; PDB code: 6LU7 [12]) in complex with peptidomimetic inhibitor (N3) was chosen for all molecular docking and molecular dynamics simulations. Heteroatoms, water molecules and ions were omitted. H++ server was utilized to assign the protonation state of SARS-CoV-2 Mpro. As well, all missing hydrogen atoms were appropriately added [31]. The pKa for SARS-CoV-2 Mpro residues was investigated under the physical conditions of salinity = 0.15, internal dielectric = 10, pH = 6.5 and external dielectric = 80.
2.2. Database preparation
Prior to molecular docking calculations, a set of 2017 flavone analogs was retrieved in SDF format from the PubChem database (https://pubchem.ncbi.nlm.nih.gov). The investigated flavone analogs were selected based on the chemical skeleton of rutin, as shown in Fig. 1 . The 3D chemical structures of the retrieved analogs, in addition to rutin (PubChem-528-080-5), were generated with the help of Omega2 software [32,33]. For each compound, a conformational search was first carried out and all possible conformers within the energy window value of 10 kcal/mol were generated. The lowest energy conformer was selected and subsequently minimized using an MMFF94S force field with the help of accessible SZYBKI software [34,35]. A schematic representation of the utilized in silico techniques and the filtration process of the database is depicted in Fig. 1.
Fig. 1.

Schematic representation of the utilized in silico techniques and the filtration process.
2.3. Molecular docking
AutoDock4.2.6 software was employed to carry out all molecular docking calculations [36]. AutoDock protocol was followed to prepare the pdbqt file of SARS-CoV-2 Mpro [37]. The genetic-algorithm number (GA) and the maximum number of energy evaluations (eval) were adjusted to 250 and 25,000,000, respectively. All other options were kept at their default parameters. The dimensions of the docking grid were defined to enclose the active site of SARS-CoV-2 Mpro (60 Å × 60 Å × 60 Å). As well, the grid spacing value was set to 0.375 Å. The grid center’s coordinates were positioned at −13.069, 9.740, 68.490 (XYZ assignments, respectively). Atomic partial charges of the investigated analogs were assigned using the Gasteiger method [38]. The probable binding modes for each studied analog were processed by built-in clustering analysis (2.0 Å RMSD tolerance) and the conformation with the lowest energy in the largest cluster was picked out as representative.
2.4. Molecular dynamics simulations
All molecular dynamics (MD) simulations were executed for the most promising flavone analogs complexed with SARS-CoV-2 Mpro using AMBER16 software [39]. General AMBER force field (GAFF2) [40] and AMBER force field 14SB [41] were utilized to describe the flavone analogs and Mpro, respectively. In the current study, implicit and explicit MD simulations were conducted. In implicit MD simulations, the AM1-BCC method was employed to calculate atomic partial charges of the analogs [42]. There were no cutoff or periodic boundary conditions applied for nonbonded interactions (more precisely, a cutoff value of 999 Å was utilized). Solvent effect was considered using igb = 1 implicit solvent model [43]. The docked analog-Mpro complexes were initially energy minimized for 500 steps and gradually heated from 0 K to 300 K over 10 ps under NVT condition, utilizing Langevin thermostat. Eventually, 250 ps in addition to 1000 ps production stages were carried out and snapshots were recorded every 1 ps, giving 250 and 1000 snapshots, respectively. The CPU version of pmemd (pmemd.MPI) in AMBER16 was used to perform all implicit MD simulations.
In explicit MD simulations, the restrained electrostatic potential (RESP) approach at the HF/6-31G∗ level was applied to calculate atomic partial charges of the analogs with the assistance of Gaussian09 software [44,45]. The analog-Mpro complexes were solvated in a cubic water box with a minimum distance to the box edge of 15 Å using the TIP3P water model with periodic boundary conditions [46]. Combined steepest descent and conjugate gradient method was employed to perform energy minimization for 5000 steps on the solvated analog-Mpro complexes. The investigated systems were then gently annealed from 0 K to 300 K over 50 ps with a weak restraint of 10 kcal/mol Å−1 on the Mpro. Besides, the systems were adequately equilibrated for 1 ns, and production stages were carried out under the NPT ensemble over simulation times of 5 n, 10 ns, 50 and 150 ns. Snapshots were collected every 10 ps, giving 500, 1000, 5000, and 150000 snapshots, respectively. Long-range electrostatic forces and energies were calculated with Particle Mesh Ewald (PME) method and the Lennard-Jones interactions were estimated with a 12 Å cutoff [47]. Langevin thermostat with a gamma_ln collision frequency set to 1.0 was applied to conserve the temperature at 298 K. A Berendsen barostat was employed for the pressure control with a pressure relaxation time of 2 ps [48]. SHAKE option to constrain all bonds involving hydrogen atoms was used with a time step of 2 fs [49]. Moreover, coordinates and energy values were gathered every 10 ps over the production stage for binding energy calculations and post-dynamics analyses. The GPU version of pmemd (pmemd.cuda) in AMBER16 was employed to execute all explicit MD simulations. All calculations, including molecular docking, molecular dynamics, and quantum mechanics, were performed on the CompChem GPU/CPU cluster (hpc.compchem.net). Molecular graphics were carried out using BIOVIA DS Visualize 2020 [50].
2.5. Relative binding free energy calculation
The binding free energies of the most potent flavone analogs in complex with SARS-CoV-2 Mpro were evaluated with the help of molecular mechanical-generalized Born surface area (MM/GBSA) approach [51]. In the current study, the modified GB model suggested by Onufriev (igb = 2) was utilized to determine the polar solvation energy. Uncorrelated snapshots were taken every 10 ps throughout the MD course, and the MM/GBSA (ΔG binding) energy calculated as follows:
2.6. In silico drug-likeness evaluation
In silico drug-likeness prediction of the most potent flavone analogs was executed using the Molinspiration cheminformatics software (http://www.molinspiration.com) to realize the Lipinski parameters. The following molecular descriptors were inspected for each investigated analog: number of hydrogen bond donors (nOHNH), number of rotatable bonds (nrotb), number of hydrogen bond acceptors (nON), n-octanol/water partition coefficient (P), molecular weight (MWt), topological polar surface area (TPSA), molecular volume (MVol), and percentage of absorption (%ABS). The calculated partition coefficient (P) was determined using the miLogP2.2 software from Molinspiration (https://www.molinspiration.com/services/logp.html). %ABS was estimated as follows [52]: %ABS = 109 − [0.345 × TPSA].
3. Results and discussion
3.1. Virtual screening of flavone analogs
Virtual screening is a comprehensive approach at the initial stage of the drug discovery pipeline that permits discovering promising bioactive inhibitors at a great throughput [53]. Because of its quickness and cost-efficiency, it is a recommended technique to recognize potential drug candidates against the universally spreading SARS-CoV-2 virus, mainly when time is of the essence.
Based on rutin’s chemical structure, the PubChem database was explored, and compounds with similar substructures were identified, giving a total number of 2017 flavone analogs (see Fig. 1). Consequently, all identified analogs were extracted and virtually screened via Autodock4.2.6 software using docking parameters of GA = 250 and eval = 25,000,000 towards SARS-CoV-2 Mpro. The estimated docking scores for all investigated analogs against the SARS-CoV-2 Mpro are summarized in Table S1. It can be seen from the data in Table S1 that 371 analogs displayed docking scores less than −9.0 kcal/mol. Evaluated docking scores, 2D chemical structures, number of conformations in the largest cluster and binding features for nine most potent analogs with SARS-CoV-2 Mpro are summarized in Table 1 . Corresponding data for rutin is also considered for the purpose of comparison. As well, the 2D representations of interactions of those nine potent analogs with the essential amino acid residues of SARS-CoV-2 Mpro are illustrated in Fig. S1. It is worth mentioning that those nine potent analogs were picked out based on the estimated 50 ns MD/MM/GBSA binding energy calculations described in latter sections.
Table 1.
Estimated docking scores (in kcal/mol) and binding features for rutin and the top nine potent analogs against SARS-CoV-2 Mpro.
| No. | PubChem code | 2D-Chemical Structure | Docking Score (kcal/mol) | Cluster Populationa | Binding Featuresb |
|---|---|---|---|---|---|
| 1 | Rutin (PubChem-528-080-5) | 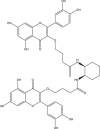 |
−7.2 | 43 | ASP187 (1.90 Å), HIS164 (2.25 Å), GLU166 (2.05 Å), GLN192 (2.86 Å), THR190 (2.19, 1.92 Å) |
| 2 | PubChem-137-399-195 | 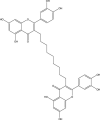 |
−10.9 | 38 | GLN189 (2.22 Å), GLU166 (1.92, 1.90, 2.99 Å), GLY143 (1.95, 2.88 Å), LEU141 (2.57 Å), SER144 (1.83 Å) |
| 3 | PubChem-129-716-607 | 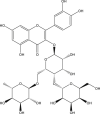 |
−10.7 | 37 | THR26 (2.07 Å), GLU166 (2.06 Å), HIS164 (2.03 Å), TYR54 (2.33 Å), ASP187 (2.32 Å), THR190 (2.20, 2.02 Å) |
| 4 | PubChem-893-333-51 |  |
−9.9 | 47 | THR190 (1.92 Å), ASP187 (1.90 Å), GLU166 (2.02, 1.85, 1.78 Å), HIS164 (2.28 Å), HIS41 (2.40 Å), LEU141 (1.83 Å), SER144 (1.96 Å), GLY143 (2.37 Å), CYS145 (2.59, 2.61 Å) |
| 5 | PubChem-123-170-316 | 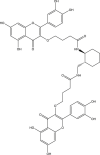 |
−9.9 | 27 | THR190 (1.85 Å), GLU166 (1.94 Å), HIS164 (2.45 Å), ASP187 (1.93 Å) |
| 6 | PubChem-142-513-769 | 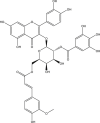 |
−9.8 | 29 | GLY143 (1.62 Å), ASN142 (2.33 Å), PHE140 (2.09 Å), HIS163 (2.08 Å), HIS164 (2.74 Å), GLU166 (2.13, 2.16, 2.96 Å), PRO168 (2.05 Å) |
| 7 | PubChem-101-020-740 | 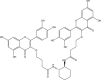 |
−9.7 | 35 | THR190 (1.98, 2.08 Å), GLN189 (1.99 Å), GLU166 (1.93 Å), ASP187 (2.78 Å), HIS164 (2.92 Å), HIS163 (2.09 Å), SER144 (2.12 Å), LEU141 (2.21 Å), LEU141 (1.98, 2.21 Å) |
| 8 | PubChem-142-513-754 | 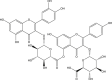 |
−9.7 | 37 | LEU141 (2.79 Å), HIS163 (1.65 Å), GLU166 (2.48, 2.16 Å), ASP187 (2.00 Å), THR190 (2.18, 2.26 Å) |
| 9 | PubChem-885-071-27 | 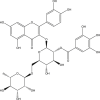 |
−9.5 | 42 | THR26 (2.07 Å), ASN142 (2.06 Å), PHE140 (2.8 Å), HIS163 (1.99 Å), GLU166 (2.07 Å), MET165 (2.4, 2.72 Å), GLN192 (2.73 Å), THR190 (2.21, 1.73 Å), PRO168 (2.20, 1.89 Å) |
| 10 | PubChem-101-133-681 |
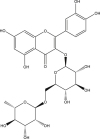 1 1 |
−9.2 | 34 | GLY143 (2.08 Å), GLU166 (1.70, 2.20, 1.86 Å), THR190 (2.22, 2.54 Å) |
Number of conformations in the largest cluster.
Only hydrogen bonds (in Å) were listed.
From docking features in Table 1 and Fig. S1, it is apparent that all identified flavone analogs demonstrated similar binding modes, exhibiting a substantial hydrogen bond with GLU166 and other hydrogen bonds with several amino acid residues inside Mpro’s binding pocket. Other interactions were also spotted, such as van der Waals, hydrophobic and pi-based interactions between the investigated analogs and Mpro, giving a docking score less than −9.0 kcal/mol (Fig. S1).
For instance, PubChem-137-399-195 showed the lowest docking score (i.e., highest binding affinity) against SARS-CoV-2 Mpro with a value of −10.9 kcal/mol, forming multiple hydrogen bonds, hydrophobic as well as van der Waals interactions, and pi-based interactions with the key amino acids within the active site (Fig. 2 ). PubChem-137-399-195 exhibited eight hydrogen bonds with GLN189 (2.22 Å), GLU166 (1.92, 1.90, 2.99 Å), GLY143 (1.95, 2.88 Å), LEU141 (2.57 Å), and SER144 (1.83 Å). Compared to PubChem-137-399-195, rutin displayed a much lower binding affinity with a docking score of −7.2 kcal/mol, forming only six hydrogen bonds with ASP187, HIS164, GLU166, GLN192, and THR190 with bond lengths ranging from 1.90 to 2.86 Å (Fig. 2).
Fig. 2.

3D and 2D representations of predicted binding modes of (a) PubChem-137-399-195 and (b) rutin with SARS-CoV-2 main protease (Mpro).
3.2. Molecular dynamics (MD) simulations
Molecular dynamics (MD) simulations should be applied to puzzle out structural details, conformational flexibilities and stabilities of ligand-enzyme complexes, and to also realize reliable drug-receptor binding affinities [54,55]. Consequently, MD simulations followed by binding energy calculations for the most potent analogs complexed with SARS-CoV-2 Mpro were carried out. To shortlist the top potent analogs, a docking score of less than −9.0 kcal/mol was taken as the threshold value, giving 371 compounds (Table S1). In order to reduce computational costs and time, MD simulations were performed in the implicit solvent over 250 ps. MM/GBSA approach was as well utilized to calculate the corresponding binding energies (see computational methodology section for details). The estimated MM/GBSA binding energies for the top 371 potent analogs are summarized in Table S2.
The data in Table S2 interestingly displayed 227 analogs with binding energies (ΔG binding) less than −40.0 kcal/mol. These identified analogs complexed with SARS-CoV-2 Mpro were further inspected over 1000 ps MD simulations to shortlist prospective potent analogs. MM/GBSA binding energies estimated for the 227 analogs over 1000 ps MD simulations are listed in Table S3. As Table S3 shows, about half of the screened compounds (≈46%) exhibited considerable binding energies (ΔG binding less than −50.0 kcal/mol). Those potent 105 analogs were further subjected to 5 ns MD simulations in explicit water solvent towards more reliable binding affinities of analog-Mpro complexes. The corresponding 5 ns MD/MM/GBSA binding energies calculations were evaluated and presented in Table S4. It can be seen from data in Table S4 that only 33 compounds showed considerable binding energies (ΔG binding less than −55.0 kcal/mol).
Furthermore, 10 ns MD simulations in explicit water solvent, followed by MM/GBSA binding energy calculations, were performed on the top 33 potent analogs complexed with Mpro (Table S5). It is apparent from Table S5 that nine potent analogs showed MM/GBSA binding energies (ΔG binding) less than −55.0 kcal/mol. Finally, those nine potent analogs were picked out and submitted to molecular dynamics (MD) simulations over 50 ns, and the corresponding binding energies were calculated (Table 2 and Fig. 3 ).
Table 2.
Average MM/GBSA binding energies (in kcal/mol) for rutin and the top nine potent analogs against SARS-CoV-2 main protease (Mpro)a.
| No. | PubChem code | MM/GBSA Binding Energy (kcal/mol) |
||||
|---|---|---|---|---|---|---|
| Implicit Solvent |
Explicit Solvent |
|||||
| 250 ps | 1000 ps | 5 ns | 10 ns | 50 ns | ||
| 1 | Rutin | −44.3 | −46.3 | −33.4 | −31.9 | −30.1 |
| 2 | PubChem-129-716-607 | −57.2 | −58.2 | −66.7 | −67.1 | −69.1 |
| 3 | PubChem-885-071-27 | −54.5 | −55.8 | −66.4 | −66.7 | −67.6 |
| 4 | PubChem-893-333-51 | −51.4 | −53.6 | −63.7 | −67.5 | −59.4 |
| 5 | PubChem-137-399-195 | −57.2 | −57.1 | −63.2 | −65.2 | −55.1 |
| 6 | PubChem-101-133-681 | −49.6 | −50.3 | −64.7 | −62.7 | −53.1 |
| 7 | PubChem-123-170-316 | −44.3 | −45.6 | −57.4 | −56.0 | −53.1 |
| 8 | PubChem-142-513-754 | −52.1 | −51.0 | −55.5 | −56.8 | −51.6 |
| 9 | PubChem-101-020-740 | −51.7 | −51.2 | −54.7 | −55.7 | −50.1 |
| 10 | PubChem-142-513-769 | −62.0 | −58.3 | −55.4 | −57.8 | −48.4 |
Data sorted according to MM/GBSA binding energy over 50 ns MD simulations.
Fig. 3.

Average MM/GBSA binding energies for rutin and the top nine potent flavone analogs in complex with SARS-CoV-2 main protease (Mpro) over 5 ns, 10 ns, 50 ns and 150 ns MD simulations in explicit solvent.
As shown in Table 2 and Fig. 3, only two analogs, namely PubChem-129-716-607 and PubChem-885-071-27, demonstrated stable binding energies over the 5, 10, and 50 ns MD courses; while variations were observed in the estimated MM/GBSA binding energies for all other analogs. For instance, MM/GBSA binding energies for PubChem-142-513-769 with Mpro were −55.4, −57.8, and −48.4 kcal/mol over 5, 10, and 50 ns MD simulations, respectively. This revealed the necessity of long MD simulations to predict reliable analog-Mpro binding affinities. Therefore, MD simulations for PubChem-129-716-607 and PubChem-885-071-27 in complex with Mpro were extended to 150 ns and the corresponding MM/GBSA binding energies were estimated (Fig. 3).
Interestingly, Fig. 3 showed that there was no considerable difference between estimated MM/GBSA binding energies for PubChem-129-716-607 and PubChem-885-071-27 with Mpro over both the 50 ns and 150 ns MD simulations. Compared to rutin, PubChem-129-716-607 and PubChem-885-071-27 displayed binding affinities over 150 ns MD against SARS-CoV-2 Mpro with ΔG binding of −69.0 and −68.1 kcal/mol, respectively. The outperformance potentialities of PubChem-129-716-607 and PubChem-885-071-27 as SARS-CoV-2 Mpro resulted from their capabilities of exhibiting numerous hydrogen bonds and van der Waals interactions, in addition to hydrophobic and pi-based interactions, with the proximal amino acid residues inside the SARS-CoV-2 Mpro active site. More precisely, PubChem-129-716-607 and PubChem-885-071-27 form ten and nine hydrogen bonds, respectively, with the key amino acid residues of Mpro (Fig. 4 ). The reference compound (i.e., rutin) exhibited high binding energy with an average MM/GBSA binding energy of −28.4 kcal/mol over the 150 ns MD simulation, forming only four hydrogen bonds with the key amino acid residues of Mpro (Fig. 4). Most striking is that the calculated binding affinities of PubChem-129-716-607and PubChem-885-071-27 are two-fold higher than rutin.
Fig. 4.

3D and 2D representations of binding modes of (a) PubChem-129-716-607, (b) PubChem-885-071-27, and (c) rutin with SARS-CoV-2 main protease (Mpro) based on the last snapshot of the 150 ns MD simulations.
In order to unveil the main driving force in the binding of the identified flavone analogs with SARS-CoV2 Mpro, decomposition of the MM/GBSA binding energies was executed (Table 3 ). As can be seen from Table 3, E vdw was the major favorable contributor in PubChem-129-716-607-, PubChem-885-071-27-, and rutin-SARS-CoV-2 Mpro binding energies with an average value of −74.3, −86.1, and −41.7 kcal/mol, respectively. Besides, E ele was also favorable with an average value of −64.9, −71.2, and −32.3 for PubChem-129-716-607-, PubChem-885-071-27- and rutin-SARS-CoV-2 Mpro binding energies, respectively. Intriguingly, E vdw, and E ele of the two analogs were nearly two times less than those of rutin.
Table 3.
Components of the MM/GBSA binding energies for PubChem-129-716-607, PubChem-885-071-27 and rutin with SARS-CoV-2 main protease (Mpro) through the MD course of 150 ns.
| PubChem code | Estimated MM/GBSA binding energy (kcal/mol) |
||||||
|---|---|---|---|---|---|---|---|
| ΔEVDW | ΔEele | ΔEGB | ΔESUR | ΔGgas | ΔGSolv | ΔGbinding | |
| Rutin | −41.7 | −32.3 | 51.1 | −5.4 | −74.1 | 45.7 | −28.4 |
| PubChem-129-716-607 | −74.3 | −64.9 | 77.8 | −7.6 | −132.0 | 70.2 | −69.0 |
| PubChem-885-071-27 | −86.1 | −71.2 | 98.0 | −8.9 | −147.8 | 89.0 | −68.1 |
3.3. Post-dynamics analyses
Structural and energetic analyses were executed over 150 ns MD simulations to demonstrate the analog’s stability inside the SARS-CoV-2 Mpro active site. The structural and energetic analyses involved binding energy per frame, hydrogen bond length, root-mean-square deviation (RMSD), and center-of-mass (CoM) distance.
3.3.1. Binding energy per frame
The correlation between the binding energy per frame and time for PubChem-129-716-607- and -PubChem-885-071-27-Mpro complexes were inspected and compared to that of rutin over 150 ns MD simulations to inspect the stability of the inhibitor inside the active site of the SARS-CoV-2 Mpro (Fig. 5 ). From data in Fig. 5, apparently overall stabilities were observed for PubChem-129-716-607, PubChem-885-071-27 and rutin inside the Mpro active site over 150 ns MD simulations with average binding energies (ΔG binding) of −69.0, −68.1 and −28.4 kcal/mol, respectively. The current results pointed out favorable stabilities of analog-SARS-CoV-2 Mpro complexes.
Fig. 5.

Evaluated MM/GBSA binding energy per frame for PubChem-129-716-607 (in red), PubChem-885-071-27 (in blue) and rutin (in black) towards SARS-CoV-2 main protease (Mpro) over 150 ns MD simulations. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3.2. Hydrogen bond length
Hydrogen bond analysis was performed on the assembled trajectories over the 150 ns MD course. The results are listed in Table 4 and the figures strikingly indicate the great stability of PubChem-129-716-607 and PubChem-885-071-27 compared to rutin against SARS-CoV-2 Mpro. Indeed, PubChem-129-716-607 and PubChem-885-071-27 exhibited two stable hydrogen bonds with GLU166 and VAL186 with an average bond length of 2.6 and 2.7, and 2.7 and 2.8 Å, respectively. In comparison, rutin formed an essential hydrogen bond with GLU166 with an average of 2.7 Å and showed a persistent 75.6% of the MD trajectory snapshots. Interestingly, PubChem-129-716-607 and PubChem-885-071-27 displayed permanent hydrogen bonds in 97.5 and 93.3, and 92.9, and 70.5% with GLU166 and VAL186, respectively. Overall, these post-dynamics results gave evidence for the stability of PubChem-129-716-607 and PubChem-885-071-27 in complex with SARS-CoV-2 Mpro.
Table 4.
Hydrogen bonds formed between the key residues and the identified flavone analogs towards SARS-CoV-2 main protease (Mpro).
| PubChem code | Acceptor | Donor | Distance (Å)a | Angle (degree)a | Occupied (%)b |
|---|---|---|---|---|---|
| Rutin | GLU166@O | Rutin@O12–H28 | 2.7 | 158 | 75.6 |
| PubChem-129-716-607 | GLU166@O | PubChem-129-716-607@O5–H29 | 2.6 | 165 | 97.5 |
| VAL186@O | PubChem-129-716-607@O16–H29 | 2.7 | 156 | 93.3 | |
| PubChem-885-071-27 | GLU166@O | PubChem-885-071-27@O16–H29 | 2.7 | 164 | 92.9 |
| VAL186@O | PubChem-885-071-27@O12– H25 | 2.8 | 148 | 70.5 |
The hydrogen bonds are investigated by the acceptor-donor atom distance of less than 3.5 Å and acceptor-H-donor angle of higher than 120°.
Only hydrogen bonds with occupancy higher than 50% were listed.
3.3.3. Center-of-mass distance
In order to obtain a more in-depth insight into the stability of inhibitor-Mpro throughout the MD simulations, center-of-mass (CoM) distances were investigated between PubChem-129-716-607, PubChem-885-071-27 and rutin and GLU166 residue (Fig. 6 ). The graph most excitingly illustrated that CoM distances were more narrow-fluctuated for PubChem-129-716-607- and PubChem-885-071-27-Mpro complexes than rutin-Mpro complex, with average values of 6.2, 6.5 and 8.2 Å, respectively. The current results revealed that PubChem-129-716-607 and PubChem-885-071-27 bound more tightly with the Mpro complex compared to rutin.
Fig. 6.

Center-of-mass (CoM) distances (in Å) between PubChem-129-716-607 (in red), PubChem-885-071-27 (in blue) and rutin (in black) and GLU166 of SARS-CoV-2 main protease (Mpro) over 150 ns MD simulations. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3.4. Root-mean-square deviation
To estimate the stability of the identified analogs over the MD simulations, root-mean-square deviations (RMSDs) for the complex backbone atoms relative to the initial structures during the MD simulations were inspected (Fig. 7 ). Fig. 7 shows the overall stability for PubChem-129-716-607-, PubChem-885-071-27- and rutin-Mpro complexes with average RMSD values of 0.27, 0.28, and 0.19 nm, respectively. These findings substantiated that the identified analogs are tightly bonded and do not affect the overall topology of SARS-CoV-2 Mpro.
Fig. 7.

Root-mean-square deviation (RMSD) of the backbone atoms from the initial structure for PubChem-129-716-607 (in red), PubChem-885-071-27 (in blue) and rutin (in black) towards the SARS-CoV-2 main protease (Mpro) over 150 ns MD simulations. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. In silico drug-likeness
In order to assess the activity and bioavailability of the discovered flavone analogs, Lipinski’s rules were utilized using Molinspiration cheminformatics online software (http://www.molinspiration.com). The in silico drug-likeness prediction of PubChem-129-716-607 and PubChem-885-071-27 was carried out and compared to rutin. Lipinski’s parameters, topological polar surface area (TPSA), and percentage of absorption (%ABS) were predicted and listed in Table 5 .
Table 5.
Predicted physiochemical parameters of rutin and identified flavone analogs as promising SARS-CoV-2 main protease (Mpro) inhibitors and their different structural descriptors.
| PubChem code | miLogP | TPSA | nON | nOHNH | Nrotb | MVol | MWt | %ABS |
|---|---|---|---|---|---|---|---|---|
| Rutin | 1.1 | 269.4 | 16 | 10 | 6 | 496.1 | 610.5 | 16.0% |
| PubChem-129–716–607 | 4.4 | 222.2 | 12 | 8 | 12 | 602.7 | 696.7 | 32.3% |
| PubChem-885–071–27 | 1.5 | 203.7 | 24 | 13 | 10 | 721.1 | 922.7 | 38.7% |
A striking feature of the figures in Table 5 is the milogP being less than five for the three investigated compounds (calc. 1.1–4.4), suggesting that these compounds have good permeability through the cell membrane. Molecular weights of the analogs were more than 500 (calc. 610.5–922.7). The number of hydrogen bond donors (nON) and the number of hydrogen bond acceptors (nOHNH) were in the range of 12–24 and 8 to 13, respectively. It has been reported that the high molecular weight and numbers of hydrogen bond donors and hydrogen bond acceptors do not have dramatic influences on the compound’s transportation and diffusion as several FDA-approved drugs exceed the standard Lipinski’s values of molecular weight, hydrogen bond donors, and hydrogen bond acceptors [56]. Besides, the calculated %ABS values were in the range of 16.0%–38.7%, and the TPSA values were noticed in the range of 203.7–269.4 Å, demonstrating the high bioavailability of these compounds.
3.5. Identified flavones vs. clinical trial and experimental inhibitors
Lopinavir (PubChem-927-27) is a human immunodeficiency virus (HIV) protease inhibitor and has been lately submitted to clinical trial as an anti-COVID-19 drug [57]. Baicalein is a flavonoid-based noncovalent SARS-CoV-2 Mpro inhibitor and its complex structure with Mpro has been recently resolved (PDB code: 6M2N [58]).
To assess the potentiality of the identified flavone analogs, the binding modes and affinities of PubChem-129-716-607 and PubChem-885-071-27 were compared to those of lopinavir and baicalein against SARS-CoV-2 Mpro. Therefore, molecular docking calculations and MD simulations followed by MM/GBSA binding energy calculations were executed for lopinavir and baicalein in complex with Mpro (Fig. S2 and Table 6 ). A comparison of the re-docked structure of baicalein inside the active site of Mpro with the experimental structure revealed that Autodock4.2.6 meticulously predicted the correct binding mode of baicalein with an RMSD value of 0.22 Å (Fig. S2a), forming six fundamental hydrogen bonds with LEU141 (3.06 Å), ASN142 (2.70 Å), GLY143 (2.09, 2.26 Å), SER144 (2.99 Å) and GLU166 (2.14 Å).
Table 6.
Calculated molecular docking scores (in kcal/mol), binding features and MM/GBSA binding energies over 150 ns MD simulation for the identified potent flavones, lopinavir and baicalein in complex with SARS-CoV-2 main protease (Mpro).
| PubChem code | Docking Score (kcal/mol) | Binding Features (Hydrogen bond length in Å) | Calculated MM/GBSA binding energy (kcal/mol) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔEVDW | ΔEele | ΔEGB | ΔESUR | ΔGgas | ΔGSolv | ΔGbinding | |||
| PubChem-129-716-607 | −10.7 | THR26 (2.07 Å), GLU166 (2.06 Å), HIS164 (2.03 Å), TYR54 (2.33 Å), ASP187 (2.32 Å), THR190 (2.20, 2.02 Å) |
−74.3 | −64.9 | 77.8 | −7.6 | −132.0 | 70.2 | −69.0 |
| PubChem-885-071-27 | −9.5 | THR26 (2.07 Å), ASN142 (2.06 Å), PHE140 (2.8 Å), HIS163 (1.99 Å), GLU166 (2.07 Å), MET165 (2.4, 2.72 Å), GLN192 (2.73 Å), THR190 (2.21, 1.73 Å), PRO168 (2.20, 1.89 Å) |
−86.1 | −71.2 | 98.0 | −8.9 | −147.8 | 89.0 | −68.1 |
| Lopinavir (PubChem-927-27) | −9.8 | HIS164 (2.62 Å), GLY143 (2.01 Å), LEU141 (1.96 Å), SER144 (3.09 Å) |
−45.6 | −22.1 | 39.9 | −5.6 | −67.8 | 34.2 | −33.6 |
| Baicalein | −7.3 | LEU141 (3.06 Å), ASN142 (2.70 Å), GLY143 (2.09, 2.26 Å), SER144 (2.99 Å), GLU166 (2.14 Å). |
−27.2 | −6.3 | 20.2 | −3.4 | −33.6 | 16.8 | −16.8 |
As can be observed from data given in Table 6 and Fig. S2, PubChem-129-716-607, PubChem-885-071-27, and lopinavir share the same binding modes with Mpro, exhibiting hydrogen bonds with the key amino acid residues inside the active site of Mpro. More exactly, lopinavir demonstrated four hydrogen bonds with GLY143, LEU141, HIS164 and SER144 with bond lengths of 2.01, 1.96, 2.62 and 3.09 Å, respectively. Interestingly, PubChem-129-716-607, PubChem-885-071-27 and lopinavir manifested similar binding affinities towards Mpro with docking scores of −10.7, −9.5 and −9.8 kcal/mol, respectively. Compared to PubChem-885-071-27, and PubChem-885-071-27, baicalein exhibited low binding affinity with Mpro with a docking score of −7.3 kcal/mol.
In an attempt to obtain more reliable results and shed light on the identified flavone analogs, lopinavir and baicalein were further inspected over 150 ns molecular dynamics simulations. As well, their corresponding MM/GBSA binding energies were consequently calculated over 150 ns MD simulations (Table 6). Decomposition of average MM/GBSA binding energy over 150 ns MD simulation was carried out to unveil the nature of predominant interactions in the lopinavir- and baicalein-Mpro complexes (Table 6). It is apparent from Table 6 that the average MM/GBSA binding energies (ΔG binding) for lopinavir and baicalein with Mpro were only −33.6 and −16.8 kcal/mol, respectively, compared to −69.0 and −68.1 kcal/mol for PubChem-129-716-607 and PubChem-885-071-27, respectively. Similar to PubChem-129-716-607 and PubChem-885-071-27, binding energies for lopinavir and baicalein with Mpro were dominated by E vdw interactions with an average value of −45.6 and −27.2 kcal/mol, respectively. The single most striking observation to emerge from the data comparison was that total MM/GBSA binding energies of the identified flavone analogs were approximately three and two times less than those of lopinavir and baicalein, respectively.
4. Conclusion
The absence of specific drugs against COVID-19 infection guides the world to discover novel and potent drugs. SARS-CoV-2 main protease (Mpro) is considered one of the fundamental targets because of its vital role in viral replication. In order to identify prospective SARS-CoV-2 Mpro inhibitors, a total of 2017 flavones were screened towards SARS-CoV-2 Mpro. Combined molecular docking and molecular dynamics (MD) simulations, followed by molecular mechanics-generalized Born surface area (MM/GBSA) binding energy calculations, were applied to filtrate the scrutinized flavones. According to the MM/GBSA binding energy calculations, PubChem-129-716-607 and PubChem-885-071-27 showed promising binding affinities with ΔG binding less than −68.0 kcal/mol. The identified flavones’ stability in complex with Mpro was approved using energetic and structural analyses over the 150 ns MD simulations. In silico prediction of drug-like features demonstrated the potentiality of the prospective flavones towards Mpro as drug candidates. The current results provide a high prospect that two flavones could be promising for further in vivo and in vitro research.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Dr. Mahmoud F. Moustafa extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding this work under Grant No. R.G.P. 1/143/42. The computational work was completed with resources supported by the Science and Technology Development Fund, STDF, Egypt, Grants No. 5480 & 7972 (Granted to Dr. Mahmoud A. A. Ibrahim). Dr. Mahmoud A. A. Ibrahim extends his appreciation to the Academy of Scientific Research and Technology (ASRT), Egypt, for funding Graduation Projects conducted at CompChem Lab, Egypt.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmgm.2021.107904.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Hui D.S., I Azhar E., Madani T.A., Ntoumi F., Kock R., Dar O., et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health — the latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020;91:264–266. doi: 10.1016/j.ijid.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO director-general’s opening remarks at the media briefing on COVID-19–11th March 2020. https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19 accessed on 15 March 2020.
- 4.WHO coronavirus disease (COVID-19) dashboard. https://covid19.who.int/ accessed on 20 July 2020.
- 5.Yang W., Cao Q., Qin L., Wang X., Cheng Z., Pan A., et al. Clinical characteristics and imaging manifestations of the 2019 novel coronavirus disease (COVID-19):A multi-center study in Wenzhou city, Zhejiang, China. J. Infect. 2020;80:388–393. doi: 10.1016/j.jinf.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrison C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020;38:379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- 7.Spinner C.D., Gottlieb R.L., Criner G.J., Lopez J.R.A., Cattelan A.M., Viladomiu A.S., et al. Effect of remdesivir vs standard care on clinical status at 11 Days in patients with moderate COVID-19 A randomized clinical trial. J. Am. Med. Assoc. 2020;324:1048–1057. doi: 10.1001/jama.2020.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Group R.C., Horby P., Lim W.S., Emberson J.R., Mafham M., Bell J.L., et al. Dexamethasone in hospitalized patients with covid-19 - preliminary report. N. Engl. J. Med. 2020 doi: 10.1056/NEJMoa2021436. [DOI] [Google Scholar]
- 9.Thorlund K., Dron L., Park J., Hsu G., Forrest J.I., Mills E.J. A real-time dashboard of clinical trials for COVID-19. Lancet Digit. Health. 2020 doi: 10.1016/S2589-7500(20)30086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.World Health Organization . 2020. DRAFT Landscape of COVID-19 Candidate Vaccines. [Google Scholar]
- 11.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 13.Ibrahim M.A.A., Abdelrahman A.H.M., Hegazy M.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1791958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sencanski M., Perovic V., Pajovic S.B., Adzic M., Paessler S., Glisic S. Drug repurposing for candidate SARS-CoV-2 main protease inhibitors by a novel in silico method. Molecules. 2020;25:3830–3842. doi: 10.3390/molecules25173830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ibrahim M.A.A., Abdelrahman A.H.M., Allemailem K.S., Almatroudi A., Moustafa M.F., Hegazy M.F. Silico Evaluation of Prospective Anti-COVID-19 Drug Candidates as Potential SARS-CoV-2 Main Protease Inhibitors, Protein J. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gentile D., Patamia V., Scala A., Sciortino M.T., Piperno A., Rescifina A. Putative inhibitors of SARS-CoV-2 main protease from A library of marine natural products: a virtual screening and molecular modeling study. Mar. Drugs. 2020;18:225–233. doi: 10.3390/md18040225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ibrahim M.A.A., Abdeljawaad K.A.A., Abdelrahman A.H.M., Hegazy M.F. Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: in-silico drug discovery. J. Biomol. Struct. Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1790037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibrahim M.A.A., Abdelrahman A.H.M., Hussien T.A., Badr E.A.A., Mohamed T.A., El-Seedi H.R., et al. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors, Comput. Biol. Med. 2020;126:104046–104055. doi: 10.1016/j.compbiomed.2020.104046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zakaryan H., Arabyan E., Oo A., Zandi K. Flavonoids: promising natural compounds against viral infections. Arch. Virol. 2017;162:2539–2551. doi: 10.1007/s00705-017-3417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherrak S.A., Merzouk H., Mokhtari-Soulimane N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: a molecular docking and simulation studies. PloS One. 2020;15:e0240653. doi: 10.1371/journal.pone.0240653. 0240666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jo S., Kim S., Kim D.Y., Kim M.-S., Shin D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020;35:1539–1544. doi: 10.1080/14756366.2020.1801672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganeshpurkar A., Saluja A.K. The pharmacological potential of rutin. Saudi Pharmaceut. J. 2017;25:149–164. doi: 10.1016/j.jsps.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naicker P., Achilonu I., Fanucchi S., Fernandes M., Ibrahim M.A., Dirr H.W., et al. Structural insights into the South African HIV-1 subtype C protease: impact of hinge region dynamics and flap flexibility in drug resistance. J. Biomol. Struct. Dyn. 2013;31:1370–1380. doi: 10.1080/07391102.2012.736774. [DOI] [PubMed] [Google Scholar]
- 24.Tao J., Hu Q., Yang J., Li R., Li X., Lu C., et al. In vitro anti-HIV and -HSV activity and safety of sodium rutin sulfate as a microbicide candidate. Antivir. Res. 2007;75:227–233. doi: 10.1016/j.antiviral.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Lyu S.Y., Rhim J.Y., Park W.B. Antiherpetic activities of flavonoids against herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) in vitro. Arch Pharm. Res. (Seoul) 2005;28:1293–1301. doi: 10.1007/BF02978215. [DOI] [PubMed] [Google Scholar]
- 26.Orhan D.D., Ozcelik B., Ozgen S., Ergun F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol. Res. 2010;165:496–504. doi: 10.1016/j.micres.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Das P., Majumder R., Mandal M., Basak P. In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (M(pro)) from flavonoid based phytochemical constituents of Calendula officinalis. J. Biomol. Struct. Dyn. 2020:1–16. doi: 10.1080/07391102.2020.1796799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chikhale R.V., Sinha S.K., Patil R.B., Prasad S.K., Shakya A., Gurav N., et al. In-silico investigation of phytochemicals from Asparagus racemosus as plausible antiviral agent in COVID-19. J. Biomol. Struct. Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1784289. [DOI] [PubMed] [Google Scholar]
- 29.Hu X.P., Cai X., Song X., Li C.Y., Zhao J., Luo W.L., et al. Possible SARS-coronavirus 2 inhibitor revealed by simulated molecular docking to viral main protease and host toll-like receptor. Future Virol. 2020;15:359–368. doi: 10.2217/fvl-2020-0099. [DOI] [Google Scholar]
- 30.Huynh T., Wang H., Luan B. Structure-based lead optimization of herbal medicine rutin for inhibiting SARS-CoV-2’s main protease. Phys. Chem. Chem. Phys. 2020;22:25335–25343. doi: 10.1039/d0cp03867a. [DOI] [PubMed] [Google Scholar]
- 31.Gordon J.C., Myers J.B., Folta T., Shoja V., Heath L.S., Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:W368–W371. doi: 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hawkins P.C., Skillman A.G., Warren G.L., Ellingson B.A., Stahl M.T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010;50:572–584. doi: 10.1021/ci100031x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.OMEGA 2.5.1.4. OpenEye Scientific Software; Santa Fe, NM, USA: 2013. [Google Scholar]
- 34.SZYBKI 1.9.0.3. OpenEye Scientific Software; Santa Fe, NM, USA: 2016. [Google Scholar]
- 35.Halgren T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999;20:720–729. doi: 10.1002/(Sici)1096-987x(199905)20:7<720::Aid-Jcc7>3.0.Co;2-X. [DOI] [PubMed] [Google Scholar]
- 36.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gasteiger J., Marsili M. Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. doi: 10.1016/0040-4020(80)80168-2. [DOI] [Google Scholar]
- 39.Case D.A., Betz R.M., Cerutti D.S., Cheatham T.E., Darden T.A., Duke R.E., et al. University of California; San Francisco, USA: 2016. AMBER. [Google Scholar]
- 40.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 41.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 43.Roux B., Simonson T. Implicit solvent models. Biophys. Chem. 1999;78:1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 44.Bayly C.I., Cieplak P., Cornell W.D., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges - the RESP model. J. Phys. Chem. 1993;97:10269–10280. doi: 10.1021/j100142a004. [DOI] [Google Scholar]
- 45.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., et al. Gaussian Inc.; Wallingford CT, USA.: 2009. Gaussian 09. [Google Scholar]
- 46.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 47.Darden T., York D., Pedersen L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 48.Berendsen H.J.C., Postma J.P.M., Vangunsteren W.F., Dinola A., Haak J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 49.Miyamoto S., Kollman P.A. Settle - an analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. doi: 10.1002/jcc.540130805. [DOI] [Google Scholar]
- 50.Dassault B.D.S.V. Dassault Systèmes BIOVIA; San Diego, CA, USA: 2019. Systèmes BIOVIA, Version 2019. [Google Scholar]
- 51.Massova I., Kollman P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000;18:113–135. doi: 10.1023/A:1008763014207. [DOI] [Google Scholar]
- 52.Zhao Y.H., Abraham M.H., Le J., Hersey A., Luscombe C.N., Beck G., et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. (N. Y.) 2002;19:1446–1457. doi: 10.1023/a:1020444330011. [DOI] [PubMed] [Google Scholar]
- 53.Bajorath F. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002;1:882–894. doi: 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- 54.De Vivo M., Masetti M., Bottegoni G., Cavalli A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016;59:4035–4061. doi: 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- 55.Kerrigan J.E. In: Silico Models for Drug Discovery. Kortagere S., editor. Humana Press; Totowa, NJ: 2013. Molecular dynamics simulations in drug design; pp. 95–113. [Google Scholar]
- 56.Mullard A. Re-assessing the rule of 5, two decades on. Nat. Rev. Drug Discov. 2018;17:777. doi: 10.1038/nrd.2018.197. 777. [DOI] [PubMed] [Google Scholar]
- 57.De Meyer S., Bojkova D., Cinatl J., Van Damme E., Buyck C., Van Loock M., et al. Lack of antiviral activity of darunavir against SARS-CoV-2. Int. J. Infect. Dis. 2020;97:7–10. doi: 10.1016/j.ijid.2020.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su H.-x., Yao S., Zhao W.-f., Li M.-j., Liu J., Shang W.-j., et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.