

Abstract
Perfluorooctanoic acid (PFOA) is a toxic compound that is absorbed and distributed throughout the body by noncovalent binding to serum proteins such as human serum albumin (hSA). Though the interaction between PFOA and hSA has been already assessed using various analytical techniques, a high resolution and detailed analysis of the binding mode is still lacking. We report here the crystal structure of hSA in complex with PFOA and a medium‐chain saturated fatty acid (FA). A total of eight distinct binding sites, four occupied by PFOAs and four by FAs, have been identified. In solution binding studies confirmed the 4:1 PFOA‐hSA stoichiometry and revealed the presence of one high and three low affinity binding sites. Competition experiments with known hSA‐binding drugs allowed locating the high affinity binding site in sub‐domain IIIA. The elucidation of the molecular basis of the interaction between PFOA and hSA might provide not only a better assessment of the absorption and elimination mechanisms of these compounds in vivo but also have implications for the development of novel molecular receptors for diagnostic and biotechnological applications.
Keywords: binding mode, crystal structure, fluoroalkyl substances, human serum albumin, molecular interaction, perfluorooctanoic acid
Short abstract
Abbreviations and symbols
- dhSA
defatted hSA
- Dka
capric acid
- FA
fatty acid
- hSA
human serum albumin
- ITC
isothermal titration calorimetry
- KD
dissociation constant
- Myr
myristic acid
- NaPi
sodium phosphate buffer
- PFAS
perfluoroalkyl substance
- PFOA
Perfluorooctanoic acid
- PFOS
perfluorooctanesulfonic acid
1. INTRODUCTION
Perfluorooctanoic acid (PFOA) is a perfluoroalkyl substance (PFAS) with a carboxyl functional group and seven fluorinated carbon atoms. 1 PFOA is a man‐made organic compound widely found in consumer and industrial products as well as in food items. 2 Its high resistance to degradation, in combination with its ubiquity and global distribution, gives rise to an increasing public concern over its potential risks to human health and its impact on the environment. 2 Health effects associated to PFOA exposure include altered lipid metabolism, modified function of the endocrine system, liver toxicity, affected immune function, tumor formation, prenatal and neonatal toxicity, decreased birth weight and size and even obesity. 2 , 3 Currently, there are no proved medical interventions to facilitate the removal of persistent PFOA from the body. In recent years, a number of toxicokinetic studies in humans and animals reported that PFOA shows low elimination rates and high accumulation levels in the blood and in vital organs such as liver, kidney and lung. 4 , 5 , 6 Indeed, the average half‐life values for serum elimination of PFOA in environmentally exposed human populations are estimated to be in the order of years. 4 , 5 , 7 Notably, PFOA is easily absorbed and distributed throughout the body by noncovalent binding to plasma proteins at concentrations ranging from 5 nM (general population) to 50 μM (highly exposed communities). 8 The primary binding protein for PFASs in the blood is serum albumin (hSA), the most abundant protein in plasma, with an average concentration of ∼40 g L−1 (600 μM). 9 hSA has important physiological roles, including maintenance of colloid osmotic pressure and pH value. Moreover, hSA is capable of binding a large variety of small endogenous and exogenous organic molecules, shielding their hydrophobic character and strongly enhancing their solubility and half‐life in plasma. 9 , 10 In particular, hSA acts as the key lipid delivery vehicle for the tissues, binding up to seven and nine molecules of long‐ and medium‐chain fatty acids (FAs), respectively, for a total of 0.1–2 mol of FAs per mol of protein. 11 Short‐ to medium‐length FAs (6–12 carbons) bind hSA with affinities between 0.5 and 60 μM, while the longest ones (14 to 18 carbons) have ten10fold higher affinities (below 50 nM). 12 , 13 Given the structural similarities of PFOA to endogenous FAs it is not surprising that these fluorinated compounds are capable of binding hSA at physiological concentrations above 90% leading to very high concentrations in blood. 14 , 15 Hence, the investigation of the interaction between PFOA and hSA is crucial for a better understanding of the biological process and toxic mechanism of these compounds in vivo. In recent years the binding of PFOA to hSA has been characterized using multiple analytical techniques 16 and while considerable insights have been accumulated, our understanding of the PFOA‐hSA interactions is far from complete. Though hSA has been reported to bind between 1 to more than 10 PFOA molecules with binding affinities ranging from 10−2 to 10−6 M, 17 , 18 , 19 , 20 , 21 , 22 , 23 a high resolution structural analysis of the interaction between PFOA and hSA is still lacking. In the present study we applied X‐ray crystallography and isothermal titration calorimetry (ITC) to investigate the nature and locations of the PFOA binding sites. A total of four PFOA binding sites with different affinities have been determined. Competition experiments with known hSA‐binding drugs allowed locating the high affinity binding site in sub‐domain IIIA. The elucidation of the molecular basis of the interaction between PFOA and hSA is expected to have implications for the development of superior hSA‐based molecular receptors for diagnostic and biotechnological applications. 24
2. RESULTS
2.1. Overview of the crystal structure of hSA in complex with PFOA and myristic acid
To unveil the binding mode of hSA to PFOA, we applied X‐ray crystallography and determined the structure of the complex. To resemble the physiological conditions, we co‐crystallized hSA in the presence of both PFOA and the representative long FA myristic acid (Myr). The hSA‐PFOA‐Myr complex was prepared by incubating the defatted hSA (dhSA) protein with a two‐fold molar excess of PFOA over Myr. The best crystals diffracted to 2.10 Å maximum resolution and the structure solved by molecular replacement (Table S1, PDB identification code: 7AAE). The polypeptide chain of hSA could be traced unambiguously from His3 to Leu585. The electron density was clearly visible for all the ligands allowing a definite assignment of the positions and orientations of both PFOA (Figure 1a) and Myr (Figure 1b) bound molecules. A total of eight distinct binding sites, four occupied by PFOA and four by Myr, have been identified (Figure 1c). The binding sites occupied by PFOA are located at the Sudlow's drug‐binding site I (subdomain IIA) and II (subdomain IIIA). Here, we refer to these two ligands as PFOA3 and PFOA1. A third molecule, named PFOA2, is positioned at the interface of subdomains IIA and IIB (FA6) whereas a fourth binding site (PFOA4) laid in the cleft at the interface between sub‐domains IB and IIIA. The remaining binding sites FA1, FA2, FA3, and FA5 were all occupied by Myr molecules (Figure 1c). To better compare the binding mode of PFOA with that of Myr, we determined the crystal structure of hSA‐Myr binary complex (Figure 1d, PDB identification code: 7AAI). Superposition of our two crystal structure complexes with those of other hSA‐FA complexes previously described 10 , 11 does not show any striking rearrangements of the main backbone with root mean square deviations of the Cα‐atoms that never exceed 0.85 Å. Importantly, the pattern of binding of all PFOAs is similar to those of medium‐chain FAs. Moreover, no major differences are observed for the side chains of amino acids involved in the binding when compared to other hSA‐FA complexes (Figure S1). Despite several structural analyses reported the presence of different FAs in FA7 site, our binary hSA‐Myr complex shows a 2‐methyl‐2,4‐pentanediol (MPD) molecule bound to it (Figure 1d, S2). 11 This evidence is consistent with earlier studies reporting a poor and truncated electron density lacking the characteristic expansion at one end necessary to locate the carboxylate head‐group of the Myr, thus corroborating the relatively low biding affinity of FAs for this site. 25 Overall, the structure of hSA in complex with PFOA and Myr revealed the presence of four distinct PFOA binding sites and no conformational differences with those of other hSA‐FA complexes.
FIGURE 1.
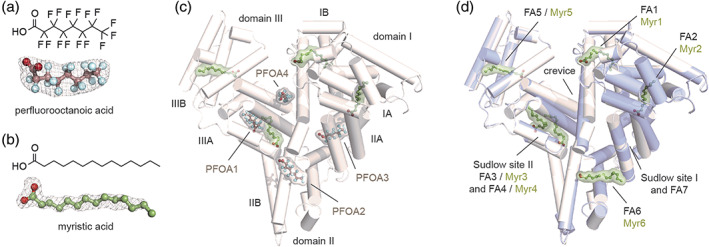
Structure of hSA in complex with PFOA and Myr. Chemical structure (top) and composite omit maps depicting the (F o−F c) electron density (bottom) of PFOA (a) and Myr (b) contoured at 4σ; (c) Crystal structure of hSA‐PFOA‐Myr complex (white) obtained using a twofold molar excess of PFOA over Myr [PDB identification code: 7AAE]; (d) Superimposition of hSA‐PFOA‐Myr ternary complex (white) with aligned hSA‐Myr binary complex (blue white) [PDB identification code: 7AAI]. The structure of hSA is organized in homologues domains (I, II and III), subdomains (A and B), fatty acids (FA) and Sudlow's binding sites. The α‐helices of hSA are represented by cylinders. Bound PFOA and Myr are shown in a ball‐and‐stick representation with a semi‐transparent van der Waals and colored by atom type (PFOA: carbon = dark salmon, oxygen = firebrick, fluorine = palecyan; Myr: carbon = smudge green, oxygen = firebrick). The electron density PFOA and Myr is shown as grey mesh
2.2. Molecular binding mode of PFOA to hSA
The electronic density of all PFOAs and Myr ligands is well defined allowing an unambiguous assignment of the positions of both hydrophilic carboxylate head‐groups and n‐tetradecane (C14) methylene or fluorinated n‐octyl (C8) lipophilic tails. The PFOA1 molecule lies in the long and narrow Sudlow's drug‐binding site II (named FA4), located in sub‐domain IIIA, and is positioned approximately at right angles to the Myr molecule (Myr3), located in the nearby FA3 site (Figure 2a, b). Alike FAs, the carboxylate head‐group of PFOA1 forms a hydrogen bond with the side chain of Ser489 and polar interactions with side chains of adjacent Asn391, Arg410, and Tyr411 (Figure 2b, S3, Table S2). Furthermore, numerous polar interactions are established between fluorine atoms (F3, F11, F12, F14, F17, and F20) and the oxygen and nitrogen atoms of both main and side chains of nearby Tyr411, Phe488, and Ser489 (Figure 2b, S3, Table S2). The rest of the fluorinated n‐octyl tail accommodates in the hydrophobic tunnel and establishes non‐polar contacts with surrounding Leu387, Tyr411, Val415, Leu430, Leu453, Leu457, Leu460, Arg485, Phe488, Ser489, and Leu491 residues (Figure 2b, S3, Table S3). Notably, the positions of the side chains of the amino acids engaged in contacts with PFOA1 are similar to those of other hSA‐FA complexes described before (Figure S1). 11 The PFOA2 occupies FA6 binding site at the interface of subdomains IIA and IIB (Figure 2a, c). The hydrophilic carboxylate head‐group of PFOA2 is involved in polar interactions with adjacent main‐chain nitrogen atoms of amino acids Lys351, Leu481, and Val482, while the fluorinated n‐octyl tail extends linearly within the narrow hydrophobic tunnel making additional stabilizing contacts with the main and side chains of surrounding Phe206, Arg209, Ala210, Ala213, Leu327, Leu347, Ala350, Lys351, Glu354, Ser480, Leu481, and Val482 residues (Figure 2c, S3, Table S2, S3). The remaining space present at the tip of the fluorinated tail appears to be occupied by a small organic molecule. The detected additional electron density can most likely be attributed to a MPD molecule present in the crystallization solution (Figure S2). Again, no difference is observed for the side chains of amino acids involved in the binding to PFOA2 when compared to other hSA‐FA complexes (Figure S1). 11 Interestingly, the extended network of hydrogen bonds and polar contacts, made by the side chains of Arg209, Asp324, and Glu354 residues and an interacting water molecule, forms a molecular “net” that holds and further stabilizes the binding of PFOA2 to FA6 site (Figure 2c, S3, Table S2). The PFOA3 molecule occupies the Sudlow's drug‐binding site I, also named FA7, located in sub‐domain IIA (Figure 2d). The carboxylate head‐group of PFOA3 establishes hydrogen bonds with adjacent Arg218 and Arg222 residues via a bridging water molecule (Figure 2d). One oxygen of the carboxylate head‐group of PFOA3 is further involved in a polar contact with an oxygen of the close tetraethylene glycol (PG4) molecule present in the crystallization solution (Figure 2d, S2). Notably, the same precipitant molecule appears to play an important role in the formation of polar interactions with the side chains of Arg257 and Ser287, forming a “cap” that helps to grip PFOA3 in the pocket (Figure 2d). Again, the fluorinated n‐octyl tail accommodates well within the pocket making additional polar and non‐polar contacts with the main and side chains of surrounding Trp214, Arg218, Leu219, Arg222, Leu238, Arg257, Leu260, Ala261, Ile264, Ser287, Ile290, and Ala291 residues (Figure 2d, S3, Table S2, S3). The majority of the amino acid side chains, involved in the binding to PFOA3 superimposed well with those of other hSA‐FA complexes, except for Arg218 that reorients in between the following Gln221 and Arg222 residues (Figure S1). 11 Finally, PFOA4 molecule lays in a large cleft located at the interface between sub‐domains IB and IIIA (Figure 2e). This site has been reported to bind exclusively short and medium‐chain FAs such as decanoic acid (Dka) and it is therefore not surprising to see a PFOA molecule bound to it instead of a Myr (Figure S1). 11 Interestingly, the PFOA4 molecule runs in opposite direction than Dka. While the carboxylate head‐group of Dka forms a salt bridge with the side chain of Lys436, located at the top of the crevice, the carboxylate head‐group of PFOA4 points at the opposite site, towards the solvent, and engages polar contacts with atoms of nearby Lys190, Arg197, and Val455 residues. Again, a water molecule plays a key role in mediating some of these interactions (Figure 2e, S1). Furthermore, a salt‐bridge between Asp187 (domain IB) and Lys432 (domain IIIA) forms a side chain strap across the top of the crevice that appears to hold PFOA4 in place (Figure 2d). Binding of PFOA4 to this cleft is further stabilized by polar and non‐polar contacts of the fluorinated n‐octyl tail with the main and side chains of surrounding Asp187, Lys190, Ala191, Asn429, Lys432, Lys436, Tyr452, Val455, Val456, and Gln459 residues (Figure 2d, S3, Table S2, S3). Most of the hSA side chains that are engaged in PFOA4 binding displayed similar conformations, except for Arg197 (domain IB) and Lys436 (domain IIIA). In the presence of PFOA4, the Arg197 side chain rotates through about 180° to form polar contacts with the carboxylate head‐group of PFOA4 (Figure S1). Similarly, the presence of the fluorinated tail of PFOA4 appears to push away the side chain of Lys436 which undergoes a 180° rotation toward the solvent (Figure S1). 11 Taken together, our data indicate that the binding mode of PFOAs is similar to those of medium‐chain FAs and involves both the carboxylate head‐group and the fluorinated tail that establish polar and non‐polar contacts with surrounding hSA residues. Data also suggest that PFOA1, located in the FA4 pocket, forms a larger number of inter‐molecular interactions, if compared to other bound PFOA molecules.
FIGURE 2.
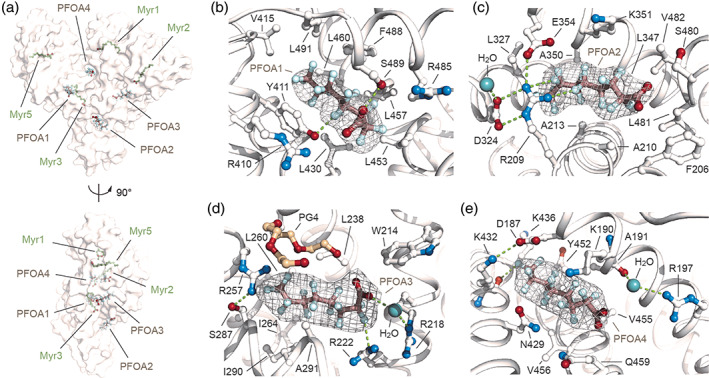
Details on the binding mode of PFOA to hSA. (a) Molecular surface representation of the overall hSA‐PFOA‐Myr complex shown in two orientations (90° rotation); (b) PFOA1 bound to FA4 in sub‐domain IIIA; (c) PFOA2 bound to FA6 at the interface of subdomains IIA and IIB; (d) PFOA3 bound to FA7 in sub‐domain IIA; (e) PFOA4 bound to the crevice between sub‐domains IB and IIIA. The α‐helices of hSA are shown in white and the selected amino acid side chains are represented as ball‐and‐stick and colored by atom type (carbon = white, oxygen = firebrick, nitrogen = skyblue). Bound PFOA molecules are depicted as ball‐and‐stick models (PFOA: carbon = dark salmon, oxygen = firebrick, fluorine = palecyan) and the composite omit maps, representing the (<i > F</i > <sub>o</sub> − <i > F</i > <sub>c</sub>) electron density contoured at 4σ, are shown as grey mesh. Bound water molecule is shown in lightteal. Bound tetraethylene glycol (PG4) molecule is shown as ball‐and‐stick model and colored by atom type (carbon = wheat, oxygen = firebrick). Hydrogen bonds, salt bridges and polar interactions are shown as splitpea dashed lines. For visualization, only inter‐molecular polar interactions below 3.0 Å are shown
2.3. Differences between PFOA and PFOS interaction with hSA
We next compared our structure with that of hSA in complex with a similar fluoroalkyl substance, the perfluorooctane sulfonate (PFOS). 26 Comparison of the hSA‐PFOA‐Myr and hSA‐PFOS (PDB identification code: 4E99) structures revealed differences in the respective binding modes. Primarily, the overall conformations of the two protein complexes diverge from each other. These major differences can be ascribed to either presence or absence of Myr in the complex. Our ternary complex is wider and superimposes well with that of other hSA‐FA structures. Contrariwise, the hSA‐PFOS complex resembles better that of the dhSA with a large downward shift of subdomain IA and IIIB, resulting in a more compact structure (Figure 3a, S4). Moreover, our ternary complex reveals four PFOA binding sites, while the binary hSA‐PFOS complex identified solely two PFOS binding sites, one located at Sudlow's drug‐binding site II (FA4) and the other at the FA6 binding site (Figure 3a–c). Interestingly, despite being very similar molecules and occupying the same sites, the binding mode of PFOA and PFOS ligands into FA4 and FA6 pockets varies significantly. While the PFOS3 molecule stretches across both FA3 and FA4 binding sites, the PFOA1 ligand occupies solely the FA4 site, with the FA3 pocket being filled by a Myr molecule (Myr3, Figure 3b). Though the two fluorinated n‐octyl tails run in opposite directions, the hydrophilic head‐groups converges toward the Ser489 residue that establishes a polar interaction with both PFOA and PFOS ligands (Figure 3b, S5). The sulfonate head‐group of PFOS3 forms a salt bridge with Lys414 and a hydrogen bond with Tyr411, whereas in the case of PFOA1 the Tyr411 is shifted upward and the rotated Lys414 is situated too far to form contacts with the carboxylate head‐group (Figure 3c). Importantly, PFOS3 forms a larger number of inter‐molecular interactions with the FA4 pocket residues, if compared to PFOA1. Alike PFOA2 molecule, PFOS6 occupies FA6 binding site at the interface of subdomains IIA and IIB (Figure 3d). The hydrophilic head‐groups of both PFOA2 and PFOS6 point toward the same direction of the carboxylate head‐group of Myr6. While the sulfonate head‐group of PFOS3 halts at the center of the tunnel forming hydrogen bonds with nearby Arg209 and Glu354, the carboxylate head‐group of PFOA1 slides toward subdomains IIIA by roughly 7 Å (head‐to‐head spacing) letting the fluorine atoms of the tail to establish polar interactions with Arg209 and Glu354 (Figure 3e). Overall, we conclude that while being similar molecules, PFOA and PFOS occupy FA4 and FA6 sites by exploiting different directions and hSA residues. Though hSA appears to bind more PFOA molecules than PFOSs, the latter establish a larger number of inter‐molecular contacts and cover larger surfaces of interaction (Figure S5, Table S4).
FIGURE 3.
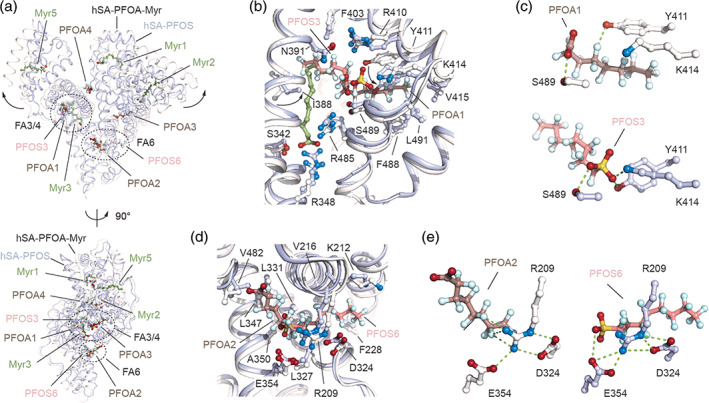
Structural comparison of the ligand binding modes of PFOA and PFOS to hSA. (a) The superimposed hSA‐PFOA‐Myr (white/dark salmon/smudge green) and hSA‐PFOS (blue white/salmon; PDB identification code 4E99) complexes are shown in two orientations (90° rotation); (b) Detailed view of the superimposed PFOA1 and PFOS3 molecules bound to FA4 in sub‐domain IIIA of hSA; (c) Comparison of polar interaction network formed by PFOA1 (top) and PFOS3 (bottom) molecules bound to FA4; (d) Detailed view of the superimposed PFOA2 and PFOS6 molecules bound to FA6 in domain II hSA; (e) Comparison of polar interaction network formed by PFOA2 (left) and PFOS6 (right) molecules bound to FA6. The α‐helices of hSA in complex with PFOA and PFOS are represented by ribbon diagram and shown in white and light blue, respectively. The selected amino acid side chains are represented as ball‐and‐stick and colored by atom type (carbon = white for hSA/PFOA/Myr complex and blue white for hSA/PFOS complex, oxygen = firebrick, nitrogen = skyblue). Bound PFOA, PFOS and Myr are shown in a ball‐and‐stick representation and colored by atom type (PFOA: carbon = dark salmon, oxygen = firebrick, fluorine = pale cyan; PFOS: carbon = salmon, oxygen = firebrick, fluorine = pale cyan, sulfur = yellow orange; Myr: carbon = smudge green, oxygen = firebrick). For visualization, only inter‐molecular polar interactions below 3.0 Å are shown
2.4. Binding stoichiometry of PFOA to hSA in solution
The molecular interaction between PFOA and hSA was further characterized using ITC. In solution studies confirmed a 4:1 binding stoichiometry observed in the X‐ray structure of the complex. The four PFOA binding sites are grouped into two pairs of different strength: a high‐affinity site (K D = 0.357 μM) and three low‐affinity ones (K D = 27.1 μM) (Figure 4a, Table S5). For all the four sites, the energetic of PFOA binding followed an exothermic reaction. The high affinity site shows a favorable energetic contribution of both enthalpy (∆H) and entropy (∆S), suggesting that the interaction is achieved through electrostatic and hydrophobic interactions. Inversely, binding of the PFOAs to the three low‐affinity sites is mainly characterized by a positive entropic contribution, which can be ascribed to hydrophobic interactions (Figure S6, Table S5, S6). Next, we assessed the effect of the temperature on the binding of PFOA to hSA. Interestingly, no significant variations were observed in the stoichiometry nor in the binding affinities when ITC studies were performed at temperatures ranging from 12 to 30°C (Figure S7, Table S5). On the contrary, a change in the stoichiometry of the low‐affinity binding sites was instead observed at physiological temperature (37°C) with one or two low‐affinity sites that have diminished their binding affinities from micromolar to millimolar values. Though, these sites appear to recur when longer titrations are performed (Figure S7, Table S5). Further native electrospray ionization mass spectrometry (ESI‐MS) analysis, using increasing molar ratio of PFOA to dhSA, revealed the presence of up to eight PFOA binding sites (Figure S6). This is in agreement with previous studies 21 and confirmed the ability of PFOA to occupy additional hSA pockets at higher concentrations. A consistent trend in the binding stoichiometry was observed when the analysis was performed using dhSA or untreated hSA (Figure S6). To assign the high affinity binding site, we performed competitive ITC studies using a range of commercially available drugs with known binding affinity and site‐selectivity. 27 These include ibuprofen (FA4 and FA6), 3‐carboxy‐4‐methyl‐5‐propyl‐2‐furanpropionic acid (CMPF; FA4 and FA7), warfarin (FA7) and iodipamide (FA7 and cleft; Figure 4b). Titration profiles of the four compounds to dhSA confirmed the binding to the protein and allowed us to assess the optimal saturating conditions for each drug (Figure S8, Table S6). The experimental binding parameters were in good agreement with those reported in the literature. 28 , 29 , 30 The presence of small amounts of DMSO, needed to solubilize some drugs, did not alter the binding parameters of PFOA to dhSA (Figure S8, Table S6). The overlay of the individual titration profiles of PFOA to dhSA saturated with single drug enabled the identification of the highest binding affinity site for PFOA. While the titration profiles obtained in the presence of warfarin‐ and iodipamide‐saturated dhSA are comparable to that of dhSA, those obtained in the presence of ibuprofen‐ and CMPF‐saturated dhSA revealed a nearly saturated flat curve, suggesting a direct competition and preservation of the only lower affinity sites (Figure 4c, S8, Table S7). In addition, the thermodynamics parameters of binding of PFOA to dhSA in the presence of ibuprofen or CMPF show little or null enthalpic contribution, which is instead present when the protein is saturated with iodipamide or warfarin and comparable to that of dhSA (Figure S9). Given that ibuprofen and CMPF share the same FA4 binding site and that among all the compounds tested ibuprofen is the only one that does not bind FA7, we can conclude that FA4 is a high affinity site for PFOA, whereas FA6, FA7 and the cleft are the low affinity ones. The results are consistent with the comparative analysis of the interactions of each single molecule of PFOA to the residues of hSA which shows that PFOA1 molecule bound to FA4 site establishes a higher number of inter‐molecular contacts if compared to other sites (Figure S3, Table S2, S3).
FIGURE 4.
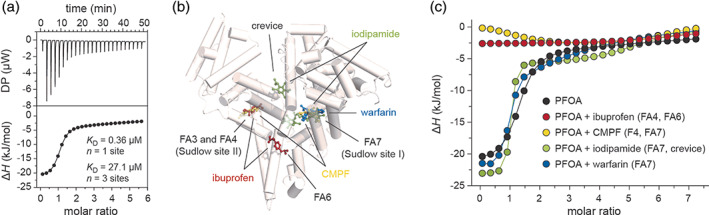
Isothermal titration calorimetry analysis of PFOA binding to hSA. (a) Representative raw trace (top) of the calorimetric titration of PFOA into dhSA and integrated binding isotherm (bottom); (b) Superimposition of hSA structure (white) with aligned hSA‐ibuprofen [PDB identification code: 2BXG], hSA‐CMPF [PDB identification code: 2BXA], hSA‐iodipamide [PDB identification code: 2BXN] and hSA‐warfarin [PDB identification code: 2BXD] complexes. The α‐helices of hSA are represented by cylinders. Bound molecules are shown in a ball‐and‐stick representation and colored as follow: ibuprofen = red, CMPF = yellow, iodipamide = green and warfarin = blue; (c) Overlay of the individual titration profiles of PFOA to hSA saturated with ibuprofen (red), CMPF (yellow), iodipamide (green) and warfarin (blue). Titration profile of PFOA to hSA alone is shown in black
3. DISCUSSION AND CONCLUSIONS
PFOA is a man‐made toxic compound that is easily absorbed and circulate for years throughout the body by noncovalent binding to serum proteins, such as hSA. Though the interaction of PFOA to hSA has been already assessed using various analytical techniques, a high resolution and detailed characterization of their binding mode is still lacking. Here we report the biochemical analysis of hSA in complex with PFOA and the medium‐chain saturated FA Myr. The crystal structure revealed four PFOA binding sites, namely FA4 (Sudlow binding site I), FA6, FA7 (Sudlow binding site II) and crevice. The protein complex superimposed well with that of other hSA‐FAs. The binding mode of the PFOAs is similar to those of medium‐chain FAs and involves direct or water‐mediated interactions of the carboxylate head‐groups to nearby basic or polar residues. The lipophilic tails accommodate within the hydrophobic cavities. Though shorter than Myr, PFOA molecules appear to establish a larger number of both polar and non‐polar contacts with hSA residues. Such property could be attributed to the ability of the carbon–fluorine (C—F) bond to participate in multiple non‐covalent intermolecular interactions (e.g., C—H···F—C, C—F···F—C, C—F···π, C—F···X where X = N, O, S, halogen). 31 Further in solution binding studies confirmed the 4:1 PFOA‐hSA stoichiometry and revealed the presence of one high and three low affinity binding sites. Similarly to FAs, the binding of PFOA to hSA appears to be characterized by a favorable exothermic process and by a gain in entropy most probably due to desolvatation of the fluoroalkyl tail. 32 While the exothermic process is mainly driven by van der Waals interactions, hydrogen bonding and electrostatic interactions, the gain in entropy is the result of hydrophobic interactions that are stronger than ones with their hydrocarbon analogs, which reflects the size of the structured water cage required to solvate the fluorocarbon groups and the entropy gained from desolvating upon protein binding. 33 Though numerous studies indicate the presence of additional PFOA binding sites, our structural and in solution characterization revealed that these are unlikely to be physiologically relevant and were probably observed to be occupied only because of the lack of FAs or the relatively high concentrations of PFOA used in the studies. This is in agreement with the fact that most hSA sites have greater affinity for long‐chain FAs than for PFOA and that the former are normally most prevalent in the circulation. Competition experiments with known hSA‐binding drugs identified the FA4 pocket in sub‐domain IIIA as the high affinity binding site for PFOA. Consistent with these evidences, structural analysis of the different PFOA‐binding sites revealed that PFOA1 located in FA4 pocket forms a larger number of inter‐molecular interactions, if compared to other sites. Interestingly, the carboxylate head‐group of PFOA1 is the only one establishing a hydrogen bond with adjacent hSA residues. The ability of PFOA1 to outcompete Myr for the FA4 binding, a primary site for medium and long‐chain FAs, is remarkable and further supports the key role of hSA in enhancing the half‐life of PFOA in plasma. Contrariwise, the low binding of affinities of PFOAs to FA6, FA7 and crevice site is anticipated as all these pockets lack positive and polar amino acid side chains necessary for the anchoring of the carboxylic group of PFOA. Our observations are consistent with previous studies, showing that these three binding sites may be the primary site for shorter‐chain FAs and are occupied only at high molar ratio of FAs to have. 11 Finally, comparison of the crystal structures of hSA in complex with PFOA and PFOS enables to appreciate analogies and differences in the binding mode of two, yet similar, fluoroalkyl substances. While the structure of hSA in complex with PFOA is wider and displays four bound molecules, the structure of hSA in complex with PFOS is more compact and includes only two bound ligands. Similarly to PFOAs, PFOSs occupy FA4 and FA6 pockets and their binding to hSA appear to be exothermic and driven by both polar and non‐polar interactions. 34 , 35 Yet, the orientations and positions exploited by PFOA and PFOS in FA4 and FA6 pockets differs significantly. Finally, detailed structural analysis reveals that the greater binding affinity of PFOS toward hSA reported in literature 20 , 21 could be explained by the presence of an additional oxygen of sulfonate head‐group of PFOS and its larger van der Waals volume, which ultimately facilitates the formation of a greater number of inter‐molecular interactions. In conclusion, we report here a detailed analysis of the molecular interaction of PFOA with hSA, the main protein carrier of these widely found toxic compounds in vivo. The crystallography data well agree with the in‐solution biochemical characterization and provide a reliable map of the locations and the binding modes of PFOA to hSA. Although many challenges still remain, the elucidation of the interaction between PFOA and hSA might provide a better assessment of the absorption and elimination mechanisms of these compounds in vivo. Moreover, in light of the urgent need to implement better strategies to detect and reduce exposure to PFOA, our study is also expected to have implications for the development of novel molecular receptors for biosensor, bioremediation and biomedical applications.
4. MATERIALS AND METHODS
4.1. Proteins and chemicals
Recombinant human serum albumin (hSA, Albagen XL; UniProt ID: P02768) was purchased from Albumin Bioscience (Alabama). The charcoal was purchased by Caesar & Loretz GmbH. PFOA, sodium myristate (Myr) and warfarin were purchased form Sigma Aldrich, ibuprofen and 3‐carboxy‐4‐methyl‐5‐propyl‐2‐furanpropionic acid (CMPF) from Cayman Chemical, and iodipamide from MedChemExpress (MCE). All the reagents were of analytical grade and solutions were prepared using double distilled deionized water.
4.2. Protein preparation and purification
The defatted recombinant human serum albumin (dhSA) was obtained by adsorption onto activated charcoal as previously described. 36 Briefly, the water‐washed charcoal (0.4 mg per mg of hSA) was initially dissolved in PBS pH 7.4 and the pH further lowered to 3 using a 1 M HCl solution. The resulting suspension was incubated for at least 3 hr under gentle shaking at 4°C. The pH of the suspension was then adjusted to 7.4 by using a 2 M NaOH solution and filtered using a 0.22 μm membrane filter. The protein aggregates and the disulfide‐bridged dimers formed during this treatment were removed by size exclusion chromatography (SEC) using a HiLoad 16/600 Superdex 200 prep grade column (GE Healthcare) connected to an ÄKTA pure 25 M system (GE Healthcare) equilibrated with 50 mM sodium phosphate buffer (NaPi), 100 mM NaCl, pH 7.4. The fractions containing monomeric dhSA protein were pooled and further concentrated by using 10,000 NMWL Amicon Ultra‐15 ultrafiltration devices (Merck Life Science) at 4000 g and 4°C on a Heraeus Multifuge X1R centrifuge (Thermo Fisher Scientific) to a final protein concentration of 25 mg ml−1 (375 μM). Protein concentration was determined using a mySPEC spectrophotometer (VWR). Purified dhSA protein was flash frozen in liquid nitrogen and stored at −80°C. The monodisperse state of concentrated dhSA protein was confirmed by SEC using a Superdex 200 10/300 GL column (GE Healthcare) connected to an ÄKTA pure 25 M system and equilibrated with 50 mM NaPi, 100 mM NaCl, pH 7.4. Purified dhSA proteins were eluted as a single peak at elution volumes that corresponds to apparent molecular mass of about 66 kDa (monomer).
4.3. Crystallization
Crystallization trials of dhSA in complex with PFOA and/or sodium myristate (Myr) were carried out at 285 K in a MRC maxi 48‐well crystallization plate (Hampton Research) using the sitting‐drop vapor‐diffusion method and the Morpheus MD1‐46 protein crystallization screen kit (Molecular Dimensions Ltd.). Droplets of 1.6 μl volume (0.8 μl of protein complex and 0.8 μl of reservoir solution) were set up using an Oryx 8 crystallization robot (Douglas Instruments Ltd.) and equilibrated against 120 μl reservoir solution. In all the cases, the largest crystals were obtained by streak‐ or micro‐seeding into drops that had been allowed to equilibrate for 5–7 days. Best crystals of dhSA (1 mM) incubated with a 10‐fold molar excess of PFOA (10 mM) and a fivefold molar excess of Myr (5 mM) were obtained using the following precipitant agent: 50 mM HEPES, 50 mM MOPS, 30 mM diethylene glycol, 30 mM triethylene glycol (PGE), 30 mM tetraethylene glycol (PG4), 30 mM pentaethylene glycol, 12.5% vol/vol MPD, 12.5% wt/vol PEG 1000, 12.5% wt/vol PEG 3350 pH 7.5. Best crystals of dhSA (1 mM) incubated with a 10‐fold molar excess of Myr (10 mM) were obtained using the following precipitant agent: 50 mM HEPES, 50 mM MOPS, 30 mM sodium fluoride, 30 mM sodium bromide, 30 mM sodium iodide, 12.5% vol/vol MPD, 12.5% wt/vol PEG 1000, 12.5% w/v PEG 3350 pH 7.5. For X‐ray data collection, crystals were mounted on LithoLoops (Molecular Dimensions Ltd.), soaked in cryoprotectant solution (crystallization buffer added with 20% vol/vol ethylene glycol) and flash frozen in liquid nitrogen.
4.4. X‐ray diffraction data collection and processing
X‐ray diffraction data of the complexes were collected at the i03 and i04 beamline of Diamond Light Source Ltd (DLS, Oxfordshire, UK). The best crystals of the ternary complex hSA‐PFOA‐Myr (1:10:5 ratio), obtained using an excess of PFOA over Myr, diffracted to 2.10 Å maximum resolution. Crystals belong to the C2 space group, with unit cell parameters: a = 184.79 Å, b = 38.51 Å, c = 95.59 Å, α = 90°, β = 104.95° and γ = 90°. The asymmetric unit contains 1 molecule, corresponding to a Matthews coefficient of 2.45 Å3/Da and a solvent content of 49.84% of the crystal volume. The best crystals of the binary complex hSA‐Myr (1:10 ratio) diffracted to 1.80 Å maximum resolution. Crystals belong to the I2 space group, with unit cell parameters: a = 95.31 Å, b = 38.54 Å, c = 184.28 Å, α = 90°, β = 104.45° and γ = 90°. The asymmetric unit contains 1 molecule, corresponding to a Matthews coefficient of 2.45 Å3/Da and a solvent content of 49.71% of the crystal volume. Frames were indexed and integrated with software XIA2, merged and scaled with AIMLESS (CCP4i2 crystallographic package). 37
4.5. Structure determination and model refinement
The structures were solved by molecular replacement with software PHASER 38 using as a template the model 1BJ5. 25 Refinement was carried on using REFMAC 39 and PHENIX. 40 Rebuilding and fitting of the PFOA, Myr and precipitant/buffer molecules (ethylene glycol, EDO; 2‐methyl‐2,4‐pentanediol, MPD; 3‐(N‐morpholino)propanesulfonic acid, MOPS; tetraethylene glycol, PG4; triethylene glycol, PGE) was performed manually with graphic software COOT. 41 Since the first cycles of refinement, the electron density corresponding to the bound PFOA and/or Myr molecules was clearly visible in the electron density map. The final model of the ternary complex hSA‐PFOA‐Myr contains 4,650 protein atoms, 100 PFOA ligand atoms, 64 Myr ligand atoms, 88 water molecules and 88 atoms of other molecules. The final crystallographic R factor is 0.21 (Rfree 0.28). The final model of the binary complex hSA‐Myr contains 4,642 protein atoms, 96 Myr ligand atoms, 23 water molecules and 48 atoms of other molecules. The final crystallographic R factor is 0.22 (Rfree 0.29). Geometrical parameters of the two models are as expected or better for this resolution. The solvent excluded volumes and the corresponding buried surfaces were calculated using PISA software 42 and a spherical probe of 1.5 Å radius. Intra‐molecular and inter‐molecular hydrogen bond interactions were analyzed by PROFUNC, 43 LIGPLOT+ 44 and PYMOL 45 software. The interactions established by PFOA atoms were calculated using CLICK server. 46 Structural alignments were performed using GESAMT. 47 Protein Data Bank (PDB) identification codes for hSA‐PFOA‐Myr ternary complex and for hSA‐Myr binary complex are 7AAE and 7AAI, respectively.
4.6. Isothermal titration calorimetry
ITC experiments were performed using a Microcal PEAQ‐ITC instrument (Malvern Panalytical). Recombinant dhSA (120 μM), PFOA (4 mM), ibuprofen (4 mM) and 3‐carboxy‐4‐methyl‐5‐propyl‐2‐furanpropionic acid (CMPF, 4 mM) were dissolved in 50 mM NaPi, 100 mM NaCl, pH 7.4, while warfarin (8 mM) and iodipamide (4 mM) were dissolved in the same buffer including 2.5% vol/vol DMSO to improve solubility. All working solutions were properly degassed. Titrations were carried out at different temperature values ranging from 12 to 37°C and 750 rpm stirring rate to ensure rapid mixing. A volume of 280 μl of dhSA in the cell was titrated with 40 μl of PFOA, ibuprofen, CMPF, warfarin or iodipamide. The injection volume was 1.5 μl and a 120 s interval between injections was applied to guarantee the equilibrium at each titration point. Each titration involved a total of 25 independent stepwise additions. Initial injection volume (0.4 μl) was excluded from the analysis. The heats of ligands dilutions were subtracted in all experiments. Competition experiments were conducted by adding in the cell saturating concentrations of each single molecule (ibuprofen = 250 μM; CMPF = 250 μM; warfarin = 200 μM; iodipamide = 600 μM) and titrating PFOA (4 mM). Data were analyzed using the MicroCal PEAQ‐ITC Evaluation software (Malvern). Integrated heat signals were fitted to a “one set of sites” or a “two set of sites” binding models. Values for the affinity constant (K A = K D ‐1) and enthalpy change (ΔH) together with the stoichiometry of the PFOA‐dhSA reaction were obtained from the curve fitting. Free energy and the entropy change (ΔS) were calculated from the Gibbs free energy (ΔG) relationships: –RTlnK A = ΔG = ΔH–TΔS.
AUTHOR CONTRIBUTIONS
Lorenzo Maso: Conceptualization; data curation; formal analysis; investigation; methodology; writing‐original draft; writing‐review and editing. Matteo Trande: Conceptualization; data curation; formal analysis; investigation; methodology; writing‐original draft; writing‐review and editing. Stefano Liberi: Data curation; formal analysis; investigation; methodology; writing‐review and editing. Giulia Moro: Data curation; formal analysis; investigation; methodology; writing‐review and editing. Elise Daems: Data curation; formal analysis; investigation; methodology; writing‐original draft. Sara Linciano: Data curation; formal analysis; investigation; methodology; writing‐original draft. Frank Sobott: Data curation; formal analysis; investigation; methodology; writing‐original draft. Sonia Covaceuszach: Data curation; formal analysis; investigation; methodology; writing‐original draft. Alberto Cassetta: Data curation; formal analysis; investigation; methodology; writing‐original draft. Silvano Fasolato: Investigation; methodology; resources; writing‐original draft. Ligia Moretto: Project administration; resources; supervision; writing‐original draft. Karolien De Wael: Project administration; resources; writing‐original draft; writing‐review and editing. Laura Cendron: Conceptualization; project administration; resources; supervision; writing‐original draft; writing‐review and editing. Alessandro Angelini: Conceptualization; project administration; resources; supervision; writing‐original draft; writing‐review and editing.
CONFLICT OF INTEREST
The authors declare no conflicting interests.
Supporting information
Table S1 Statistics on X‐ray diffraction data collection and refinement
Table S2. Inter‐molecular polar interactions between atoms in the complexes between hSA and PFOA, Myr and Dka
Table S3. Inter‐molecular non‐polar interactions in the complexes between hSA and PFOA, Myr and Dka
Table S4. Volumes and surface areas of PFOA, Myr, Dka and PFOS ligands
Table S5. Thermodynamic and stoichiometric data for the binding of hSA to PFOA at different temperatures
Table S6. Thermodynamic and stoichiometric data for the binding of hSA to PFOA and four known hSA‐binding drugs
Table S7. Thermodynamic and stoichiometric data for the competitive binding of hSA to PFOA in the presence of hSA‐binding drugs
Figure S1. Superimposition of the ligand‐bind sites of hSA in complex with PFOA and two saturated fatty acids
Figure S2. Comparison of FA6 and FA7 ligand occupancy in hSA‐PFOA‐Myr and hSA‐Myr complexes
Figure S3. Comparative analysis of the interactions of hSA residues with PFOA, Dka and Myr ligands
Figure S4. Superimposition of hSA‐PFOA‐Myr and hSA‐PFOS complexes
Figure S5. Comparative analysis of the interactions of hSA residues with PFOA and PFOS ligands
Figure S6. Characterization of the binding of PFOA to hSA
Figure S7. Isothermal titration calorimetry analysis of hSA binding to PFOA at different temperatures
Figure S8. Isothermal titration calorimetry analysis of hSA binding to four commercially available drugs in the absence and in the presence of PFOA
Figure S9. Summary of thermodynamic parameters for the binding of PFOA to hSA in the presence of four commercially available drugs.
ACKNOWLEDGMENTS
We thank Prof. G. Pasqual and Prof. G. Zanotti for critical reading of this manuscript. Furthermore, we are grateful to Filippo Vascon all of the group members for helpful discussions and technical assistance. The authors would like to thank the staff of beamline I03 and I04 of Diamond Light Source Ltd (DLS, Oxfordshire, UK; proposal MX21741) for assistance with crystal testing and data collection. The financial contribution from the Fund for Scientific Research (FWO) Flanders to E.D. and K.D.W is gratefully acknowledged.
Maso L, Trande M, Liberi S, et al. Unveiling the binding mode of perfluorooctanoic acid to human serum albumin. Protein Science. 2021;30:830–841. 10.1002/pro.4036
Contributor Information
Laura Cendron, Email: laura.cendron@unipd.it.
Alessandro Angelini, Email: alessandro.angelini@unive.it.
DATA AVAILABILITY STATEMENT
Atomic coordinates and structure factors were deposited into the Protein Data Bank under the accession codes 7AAE and 7AAI.
REFERENCES
- 1. Buck RC, Franklin J, Berger U, et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr Environ Assess Manag. 2011;7:513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sznajder‐Katarzyńska K, Surma M, Cieślik I. A review of perfluoroalkyl acids (PFAAs) in terms of sources, applications, human exposure, dietary intake, toxicity, legal regulation, and methods of determination. J Chem. 2019;2019:2717528. [Google Scholar]
- 3. Li K, Gao P, Xiang P, Zhang X, Cui X, Ma LQ. Molecular mechanisms of PFOA‐induced toxicity in animals and humans: Implications for health risks. Environ Int. 2017;99:43–54. [DOI] [PubMed] [Google Scholar]
- 4. US Environmental Protection Agency . (2016) Health Effects Support Document for Perfluorooctanoic Acid (PFOA).
- 5. ATSDR . (2018) Toxicological profile for Perfluoroalkyls. (Draft for Public Comment).
- 6. Pérez F, Nadal M, Navarro‐Ortega A, et al. Accumulation of perfluoroalkyl substances in human tissues. Environ Int. 2013;59:354–362. [DOI] [PubMed] [Google Scholar]
- 7. Pizzurro DM, Seeley M, Kerper LE, Beck BD. Interspecies differences in perfluoroalkyl substances (PFAS)toxicokinetics and application to health‐based criteria. Regul Toxicol Pharmacol. 2019;106:239–250. [DOI] [PubMed] [Google Scholar]
- 8. Steenland K, Fletcher T, Savitz DA. Epidemiologic evidence on the health effects of perfluorooctanoic acid (PFOA). Environ Health Perspect. 2010;118:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peters T. All about albumin: Biochemistry, genetics, and medical applications. Diego, CA: Acad. Press, 1996. [Google Scholar]
- 10. Curry S. Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug Metab Pharmacokinet. 2009;24:342–357. [DOI] [PubMed] [Google Scholar]
- 11. Bhattacharya AA, Grüne T, Curry S. Crystallographic analysis reveals common modes of binding of medium and long‐chain fatty acids to human serum albumin. J Mol Biol. 2000;303:721–732. [DOI] [PubMed] [Google Scholar]
- 12. Spector AA. Fatty acid binding to plasma albumin. J Lipid Res. 1975;16:165–179. [PubMed] [Google Scholar]
- 13. Kragh‐Hansen U, Watanabe H, Nakajou K, Iwao Y, Otagiri M. Chain length‐dependent binding of fatty acid anions to human serum albumin studied by site‐directed mutagenesis. J Mol Biol. 2006;363:702–712. [DOI] [PubMed] [Google Scholar]
- 14. Jones PD, Hu W, De Coen W, Newsted JL, Giesy JP. Binding of perfluorinated fatty acids to serum proteins. Environ Toxicol Chem. 2003;22:2639–2649. [DOI] [PubMed] [Google Scholar]
- 15. Forsthuber M, Kaiser AM, Granitzer S, et al. Albumin is the major carrier protein for PFOS, PFOA, PFHxS, PFNA and PFDA in human plasma. Environ Int. 2020;137:105324. [DOI] [PubMed] [Google Scholar]
- 16. Liu X, Fang M, Xu F, Chen D. Characterization of the binding of per‐ and poly‐fluorinated substances to proteins: A methodological review. Trends Anal Chem. 2019;116:177–185. [Google Scholar]
- 17. Wu LL, Gao HW, Gao NY, Chen FF, Chen L. Interaction of perfluorooctanoic acid with human serum albumin. BMC Struct Biol. 2009;9:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. MacManus‐Spencer LA, Tse ML, Hebert PC, Bischel HN, Luthy RG. Binding of perfluorocarboxylates to serum albumin: A comparison of analytical methods. Anal Chem. 2010;82:974–981. [DOI] [PubMed] [Google Scholar]
- 19. Bischel HN, MacManus‐Spencer LA, Luthy RG. Noncovalent interactions of long‐chain perfluoroalkyl acids with serum albumin. Environ Sci Technol. 2010;44:5263–5269. [DOI] [PubMed] [Google Scholar]
- 20. Beesoon S, Martin JW. Isomer‐specific binding affinity of perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) to serum proteins. Environ Sci Technol. 2015;49:5722–5731. [DOI] [PubMed] [Google Scholar]
- 21. Chi Q, Li Z, Huang J, Ma J, Wang X. Interactions of perfluorooctanoic acid and perfluorooctanesulfonic acid with serum albumins by native mass spectrometry, fluorescence and molecular docking. Chemosphere. 2018;198:442–449. [DOI] [PubMed] [Google Scholar]
- 22. Chen H, Wang Q, Cai Y, Yuan R, Wang F, Zhou B. Investigation of the interaction mechanism of perfluoroalkyl carboxylic acids with human serum albumin by spectroscopic methods. Int J Environ Res Public Health. 2020;17:1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen YM, Guo LH. Fluorescence study on site‐specific binding of perfluoroalkyl acids to human serum albumin. Arch Toxicol. 2009;83:255–261. [DOI] [PubMed] [Google Scholar]
- 24. Moro G, Bottari F, Liberi S, et al. Covalent immobilization of delipidated human serum albumin on poly(pyrrole‐2‐carboxylic) acid film for the impedimetric detection of perfluorooctanoic acid. Bioelectrochemistry. 2020;134:107540. [DOI] [PubMed] [Google Scholar]
- 25. Curry S, Mandelkow H, Brick P, Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat Struct Biol. 1998;5:827–835. [DOI] [PubMed] [Google Scholar]
- 26. Luo Z, Shi X, Hu Q, Zhao B, Huang M. Structural evidence of perfluorooctane sulfonate transport by human serum albumin. Chem Res Toxicol. 2012;25:990–992. [DOI] [PubMed] [Google Scholar]
- 27. Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug‐binding specificity of human serum albumin. J Mol Biol. 2005;353:38–52. [DOI] [PubMed] [Google Scholar]
- 28. Ràfols C, Zarza S, Bosch E. Molecular interactions between some non‐steroidal anti‐inflammatory drugs (NSAID's) and bovine (BSA) or human (HSA) serum albumin estimated by means of isothermal titration calorimetry (ITC) and frontal analysis capillary electrophoresis (FA/CE). Talanta. 2014;130:241–250. [DOI] [PubMed] [Google Scholar]
- 29. Zaidi N. Khan RH. A biophysical insight into structural and functional state of human serum albumin in uremia mimic milieu. Int J Biol Macromol. 2019;31:697–705. [DOI] [PubMed] [Google Scholar]
- 30. Ràfols C, Amézqueta S, Fuguet E, Bosch E. Molecular interactions between warfarin and human (HSA) or bovine (BSA) serum albumin evaluated by isothermal titration calorimetry (ITC), fluorescence spectrometry (FS) and frontal analysis capillary electrophoresis (FA/CE). J Pharm Biomed Anal. 2018;150:452–459. [DOI] [PubMed] [Google Scholar]
- 31. Panini P, Chopra D. Understanding of noncovalent interactions involving organic fluorine, in hydrogen bonded supramolecular structures. Lecture Notes Chem. 2015;87:37–67. [Google Scholar]
- 32. Bojesen E, Bojesen IN. Albumin binding of long‐chain fatty acids: Thermodynamics and kinetics. J Phys Chem. 1996;100:17981–17985. [Google Scholar]
- 33. Dalvi VH, Rossky PJ. Molecular origins of fluorocarbon hydrophobicity. Proc Natl Acad Sci U S A. 2010;107:13603–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X, Chen L, Fei XC, Ma YS, Gao HW. Binding of PFOS to serum albumin and DNA: Insight into the molecular toxicity of perfluorochemicals. BMC Mol Biol. 2009;10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Cao Z, Zong W, Liu R. Interaction rule and mechanism of perfluoroalkyl sulfonates containing different carbon chains with human serum albumin. RSC Adv. 2017;7:24781–24788. [Google Scholar]
- 36. Chen RF. Removal of fatty acids from serum albumin by charcoal treatment. J Biol Chem. 1967;242:173–181. [PubMed] [Google Scholar]
- 37. Potterton L, Agirre J, Ballard C, Cowtan K, Dodson E, Evans PR, Jenkins HT, Keegan R, Krissinel E, Stevenson K, Lebedev A, McNicholas SJ, Nicholls RA, Noble M, Pannu NS, Roth C, Sheldrick G, Skubak P, Turkenburg J, Uski V, Von Delft F, Waterman D, Wilson K, Winn M, Wojdyr M. CCP4i2: The new graphical user interface to the CCP4 program suite. Acta Cryst D 2018;74:68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vagin AA, Steiner RA, Lebedev AA, et al. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Cryst D. 2004;60:2184–2195. [DOI] [PubMed] [Google Scholar]
- 40. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: A comprehensive Python‐based system for macromolecular structure solution. Acta Cryst D. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krissinel E, Henrick K. (2007) inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- 43. Laskowski RA, Watson JD, Thornton JM. ProFunc: A server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laskowski RA, Swindells MB. LigPlot+: Multiple ligand‐protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. [DOI] [PubMed] [Google Scholar]
- 45. The PyMOL Molecular Graphics System, Version 2.0. Schrodinger LLC.
- 46. Nguyen MN, Tan KP, Madhusudhan MS. CLICK–topology‐independent comparison of biomolecular 3D structures. Nucleic Acids Res. 2011;39:W24–W28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krissinel E. Enhanced fold recognition using efficient short fragment clustering. J Mol Biochem. 2012;1:76–85. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Statistics on X‐ray diffraction data collection and refinement
Table S2. Inter‐molecular polar interactions between atoms in the complexes between hSA and PFOA, Myr and Dka
Table S3. Inter‐molecular non‐polar interactions in the complexes between hSA and PFOA, Myr and Dka
Table S4. Volumes and surface areas of PFOA, Myr, Dka and PFOS ligands
Table S5. Thermodynamic and stoichiometric data for the binding of hSA to PFOA at different temperatures
Table S6. Thermodynamic and stoichiometric data for the binding of hSA to PFOA and four known hSA‐binding drugs
Table S7. Thermodynamic and stoichiometric data for the competitive binding of hSA to PFOA in the presence of hSA‐binding drugs
Figure S1. Superimposition of the ligand‐bind sites of hSA in complex with PFOA and two saturated fatty acids
Figure S2. Comparison of FA6 and FA7 ligand occupancy in hSA‐PFOA‐Myr and hSA‐Myr complexes
Figure S3. Comparative analysis of the interactions of hSA residues with PFOA, Dka and Myr ligands
Figure S4. Superimposition of hSA‐PFOA‐Myr and hSA‐PFOS complexes
Figure S5. Comparative analysis of the interactions of hSA residues with PFOA and PFOS ligands
Figure S6. Characterization of the binding of PFOA to hSA
Figure S7. Isothermal titration calorimetry analysis of hSA binding to PFOA at different temperatures
Figure S8. Isothermal titration calorimetry analysis of hSA binding to four commercially available drugs in the absence and in the presence of PFOA
Figure S9. Summary of thermodynamic parameters for the binding of PFOA to hSA in the presence of four commercially available drugs.
Data Availability Statement
Atomic coordinates and structure factors were deposited into the Protein Data Bank under the accession codes 7AAE and 7AAI.