

Abstract
The majority of crystal structures are determined by the method of molecular replacement (MR). The range of application of MR is limited mainly by the need for an accurate search model. In most cases, pre‐existing experimentally determined structures are used as search models. In favorable cases, ab initio predicted structures have yielded search models adequate for MR. The ORF8 protein of SARS‐CoV‐2 represents a challenging case for MR using an ab initio prediction because ORF8 has an all β‐sheet fold and few orthologs. We previously determined experimentally the structure of ORF8 using the single anomalous dispersion (SAD) phasing method, having been unable to find an MR solution to the crystallographic phase problem. Following a report of an accurate prediction of the ORF8 structure, we assessed whether the predicted model would have succeeded as an MR search model. A phase problem solution was found, and the resulting structure was refined, yielding structural parameters equivalent to the original experimental solution.
Keywords: ab initio, AlphaFold, COVID‐19, deep learning, in silico, ORF8, SARS‐CoV‐2, X‐ray crystallography; molecular replacement
Molecular replacement (MR) is an in silico technique that provides phase information required to solve macromolecular crystal structures. MR relies on the existence of an experimentally determined structure, known as the “search model,” which is similar to the target. Here we show that an ab initio, SARS CoV‐2 ORF8 protein model, generated by Google DeepMind's AlphaFold team, is sufficiently accurate to provide a phase solution by MR, bypassing the need for an experimentally determined search model.
1. INTRODUCTION
Two key pieces of information are required to determine a macromolecular structure from a crystallographic diffraction experiment, namely the amplitudes and phases of the diffracted waves. The amplitudes of the diffracted waves are calculated directly from the measured intensities of the scattered waves, while the phase information is lost. 1 The crystallographic phase problem remains a substantial bottleneck in macromolecular structure determination. In the majority of cases, the phase problem is overcome using an in silico method known as molecular replacement (MR). 2 , 3 , 4 , 5 In MR, a related structure, known as the search model, is used to provide initial phase‐estimates for the target structure. While the advent of MR has rendered phase determination near‐trivial in many cases, it was, until recently, limited to circumstances where structures of homologous proteins already existed.
In principle, advances in in silico protein structure prediction could provide MR search models sufficiently similar to the target structures so as to produce a phase solution. 6 , 7 , 8 Such an advance could, in theory, forever bypass the need for experimentally derived phases. The use of ab initio models as MR search models has seen some success, principally with α helix‐rich proteins. 9 Accurate ab initio prediction of β‐rich folds has long proven to be exceptionally challenging 10 due, in part, to the high proportion of non‐local interactions between β‐strands. To our knowledge only one β‐rich crystal structure has previously been phased using an ab initio generated search model. 11 This approach relied on a highly parallelized trial and error approach where hundreds of input MR ensemble search models were tested. 10
Recent advances in ab initio modeling have come from the long‐accepted principle that evolutionary covariance of residues can aid in inter‐residue contact prediction. This relies on the fact that deleterious point mutations are often paired with compensatory ones during evolutionary development. Multiple sequence alignments (MSA) of many related protein sequences can be used to identify these correlated mutations, with several systems applying neural networks to do so. 12 , 13 , 14 , 15 , 16 Many groups, including Google DeepMind's “AlphaFold,” have taken this principle one step further by predicting specified distances between residue pairs which provide more information about the structure than contact predictions alone. 16 , 17 , 18 , 19 , 20 In the case of AlphaFold, these inter‐residue distances, along with backbone torsion angles, are predicted in a first step using convolutional neural networks. 16 They are then provided as a target in gradient descent algorithm, which aims to bring the 3D structure as close as possible to these predicted distance and torsion angles. These principles were applied to great effect during the 14th biennial Critical Assessment of protein Structure Prediction (CASP) competition, where the AlphaFold2 team contributed a wealth of ab initio predictions, the majority of which were considered highly similar to experimentally determined protein structures with a median Global Distance Test (GDT) score of 92.4 out of 100. 21 , 22
Our group recently determined the crystal structure of ORF8 at 2.04 Å, 23 a SARS‐CoV‐2 protein which has been implicated in immune evasion. The structure revealed that the protein is composed entirely of β‐strands and unstructured regions. While a full native dataset was collected during the early stages of the project, a lack of suitable search model made phase determination by MR unfeasible. Initial phases were obtained by anomalous dispersion.
Here we assessed whether a template‐free, ab initio protein model, generated by the AlphaFold2 group, was of sufficient quality to phase the native ORF8 dataset by MR. No truncation of the model was required nor was there any need to provide an ensemble of search models. Not only is this approach likely to prove useful for future structural determination campaigns where a homologous structure is not available but could aid in the determination of preexisting “unsolvable” datasets.
2. RESULTS
Superposition of the ab initio AlphaFold2 ORF8 model (CASP14 model ID: T1064TS427_1‐D1) with the previously deposited crystal structure protomer (PDBID: 7JTL) showed they are highly similar, exhibiting all atom RMSDs of 1.4 Å and 1.6 Å with chains A and B respectively (Figure 1). This suggests the potential of the AlphaFold2 model as an appropriate MR search model candidate.
FIGURE 1.
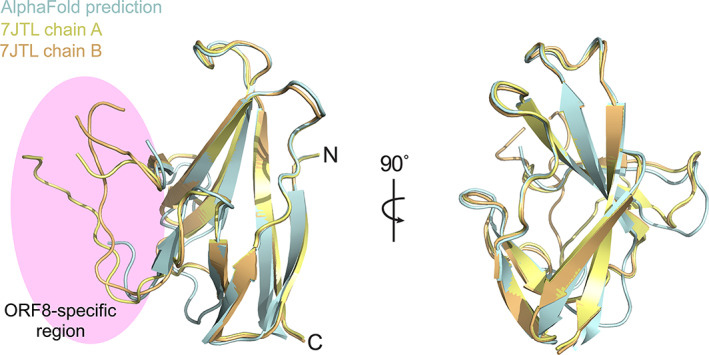
Superposition of experimentally determined CoV‐2 ORF8 structure with AlphaFold2 prediction. Chains A and B of the ORF8 crystal structure (PDBID: 7JTL) are superposed and colored yellow and orange respectively. The AlphaFold2 prediction is colored cyan. N and C termini are labeled accordingly. The ORF8‐specific region is highlighted in pink
To test this, MR was performed using native ORF8 structure factor amplitudes (PDBID: 7JTL) and the unaltered AlphaFold2 ORF8 prediction as a search model. A single MR solution was identified with two copies in the asymmetric unit. The Log Likelihood Gain (LLG) was 167 with values of 120 or greater indicating that a correct solution has almost certainly been found. 24 The MR solution places the search model at the correct position within the crystal lattice (Figure 2).
FIGURE 2.
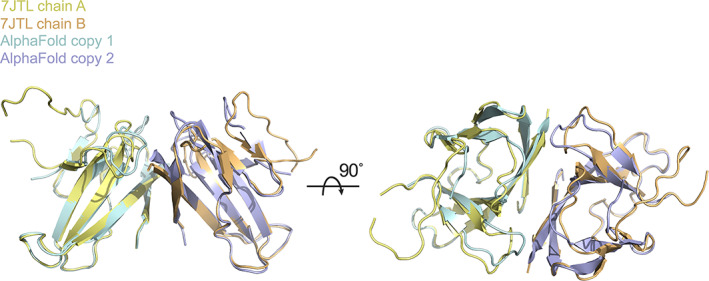
Position of two placed CoV‐2 ORF8 AlphaFold2 search models following MR. MR was performed using native ORF8 diffraction data. The first and second placed copies of the search model are colored cyan and purple respectively. The positions of chains A and B of the experimentally‐phased, previously determined ORF8 crystal structure (PDBID: 7JTL) asymmetric unit are shown for reference and are colored yellow and orange respectively
A 2Fo‐Fc electron density map was generated using phases from the placed but un‐modified AlphaFold2 model and the native structure factor amplitudes (Figure 3). The density was of sufficient quality to allow the majority of sidechains and unmodeled main chain regions to be built unambiguously. The majority of side‐chain positions in the starting model proved to be remarkably close to the final structure (Figure 4a,b). Other map features were present that were in poor agreement with the input model (Figure 3b,c–f). These differences exclude the possibility that the density is dominated by input model‐based phase‐bias. Such differences include incorrect side‐chain positioning/orientation, minor main chain deviations and the presence of unbuilt/unmodeled regions (Figures 3 and 4). Of the side‐chains that were included in the AlphaFold2 model, 18.5% adopted rotamer conformations that were not consistent with the experimentally determined structures (PDBIDs: 7JTL, 7JX6). 23 This value increased to 26.5% when unmodeled regions were considered as having incorrect rotamer assignment. Iterative rounds of manual model building and refinement produced a final structure determination with global quality metrics on par with those of the previously deposited structure (Table 1).
FIGURE 3.
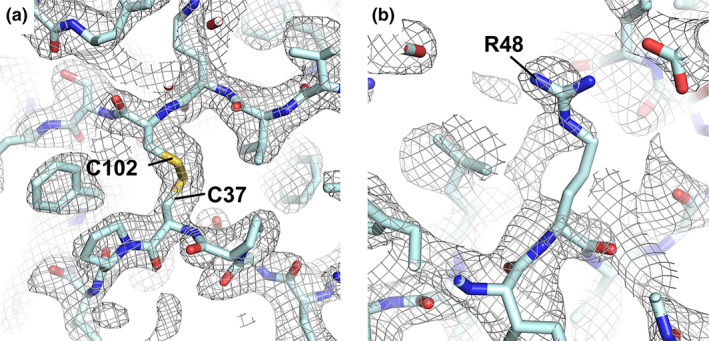
Representative regions of 2Fo‐Fc electron density following MR with AlphaFold2 CoV‐2 ORF8 search model. Panel (a) shows a region of density that is in good agreement with the unmodified AlphaFold2 search model. Panel (b) provides an example where there is an obvious discrepancy between the experimental data and the search model, suggesting that the position and orientation of the Arg48 side‐chain should be modified. Map is contoured at 1.2 σ and represented as a grey mesh
FIGURE 4.
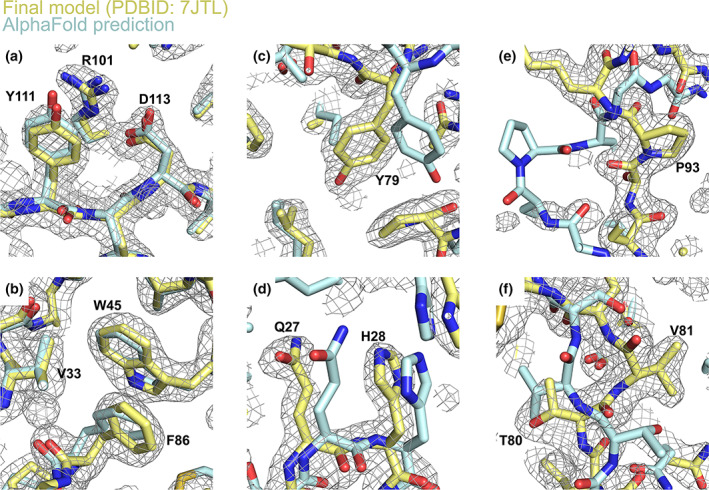
Detailed structural comparison of the fully refined, ORF8 model with the unmodified AlphaFold prediction. Panels (a) and (b) show example regions where the AlphaFold prediction is excellent agreement with the experimentally determined model. Panels (c) and (d) show less‐accurate regions displaying incorrect side chain conformations (Tyr79, panel c) and minor main chain displacements (Gln27 and His28, panel d). Panels (e) and (f) show regions where the AlphaFold prediction is in poor agreement with the final model, both exhibiting major main‐chain deviations. The fully refined, final model (PDBID: 7JTL) and AlphaFold prediction are colored yellow and cyan respectively. 2Fo‐Fc electron density was calculated using the previously deposited ORF8 coordinates and structure factor amplitudes (PDBID: 7JTL), contoured at 1.2 σ and represented as a grey mesh. Notable residues are labeled accordingly
TABLE 1.
Data collection and refinement statistics
| SARS‐CoV‐2 ORF8 | ||
|---|---|---|
| Experimentally phased | AlphaFold2 MR | |
| Data collection | ||
| Space group | P41212 | |
| Cell dimensions | ||
| a, b, c (Å) | 44.3 44.3264.1 | |
| α, β, γ (°) | 90, 90, 90 | |
| Resolution (Å) | 43.65–2.04 (2.113–2.04) | |
| R pim | 0.032 (0.555) | |
| I/σI | 14.92 (1.20) | |
| Completeness (%) | 97.8 (90.2) | |
| Redundancy | 10.0 (7.6) | |
| Refinement | ||
| Resolution (Å) | 43.65–2.04 | 43.65–2.04 |
| No. reflections | 17,005 (1399) | 17,005 (1399) |
| R work/R free (%) | 22.0 (35.2)/26.4 (38.4) | 22.3 (35.4)/27.5 (42.2) |
| No. atoms | ||
| Protein | 1,609 | 1,625 |
| Water | 201 | 96 |
| B‐factors | ||
| Protein | 45.4 | 44.3 |
| Water | 44.3 | 45.7 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.008 | 0.010 |
| Bond angles (°) | 1.00 | 1.30 |
Note: Values in parentheses are for highest‐resolution shell.
The region corresponding to residues 62–74 was absent from the AlphaFold2 prediction. This missing region is located within a ~35 amino acid insertion known as the “ORF8‐specific region” which has been proposed to be involved in the formation of higher‐order assemblies (Figure 1). 23 This majority of this region is well ordered in both our structure (PDBID: 7JTL) and a subsequently determined crystal structure produced by another group (PDBID: 7JX6). Despite being ordered, this region is largely devoid of secondary structure with each individual chain adopting a unique conformation in each of the two molecules within the asymmetric unit, suggesting that it is dynamic and context‐dependent in nature.
MR was systematically attempted with the nine next‐best ranked ORF8 predictions that were submitted to CASP14 (Table 2). Other than the AlphaFold2 prediction, none of the top 10 ranking models produced a convincing, single, MR solution. Top Phaser LLG scores ranged from 43.0 to 45.7 and visual comparison with the experimentally phased model (PDBID: 7JTL) confirmed that they were incorrectly placed, with the resulting electron densities dominated by input model phase bias.
TABLE 2.
Top 10 CASP14 ORF8 predictions ranked by GDT_TS score
| CASP14 group | ORF8 prediction rank | GDT_TS score | Top solution Phaser LLG | Number of potential MR solution(s) |
|---|---|---|---|---|
| AlphaFold2 | 1 | 86.96 | 167.0 | 1 a |
| Xianmingpan | 2 | 42.94 | 43.0 | 220 |
| PerezLab_Gators | 3 | 33.15 | 45.2 | 341 |
| HMSCasper‐MSA | 4 | 29.62 | 44.0 | 243 |
| DeepPotential | 5 | 29.35 | 44.6 | 350 |
| Zhang‐TBM | 6 | 27.17 | 43.3 | 86 |
| Bilbul2020 | 7 | 26.63 | 43.2 | 145 |
| Zhang‐CEthreader | 8 | 26.63 | 45.7 | 375 |
| Zhang | 9 | 26.63 | 43.7 | 143 |
| Zhang‐Server | 10 | 26.63 | 44.2 | 242 |
Top phaser solution correctly placed.
Immunoglobulin domains from the Drosophila neural receptor Dscam1 (PDBID: 4X83) and Murine Natural Killer Cell Receptor 2B4 (PDBID: 2PTU) 25 , 26 were retrospectively identified as the most‐similar existing PDB entries to chain A (RMSD: 3.4 Å, sequence identity: 11%) and chain B (RMSD: 3.7 Å, sequence identity 9%) of SARS CoV‐2 ORF8 (PDBID: 7JTL) respectively (Table 3). These models also failed to produce convincing MR phase‐solutions, with top LLG scores of 43.4 and 44.1 for Dscam1 and Natural Killer Cell Receptor 2B4 respectively. Visual comparison of these top‐ranking solutions with the completed ORF8 structure (PDBID: 7JTL) confirmed that a correct solution had not been found.
TABLE 3.
Existing PDB entries with the highest similarity to SARS CoV‐2 ORF8
| Search model | RMSD chain A/B (Å) | Sequence identity (%) | DALI Z‐score chain A/B | Top solution Phaser LLG | Number of potential MR solution(s) |
|---|---|---|---|---|---|
| Dscam1 (PDBID: 4X83) | 3.4/3.6 | 11 | 6.2/6.3 | 43.4 | 291 |
| Natural killer cell receptor 2B4 (PDBID: 2PTU) | 3.6/3.7 | 9 | 6.1/6.4 | 44.1 | 229 |
3. DISCUSSION
The success of the AlphaFold2 predictions at CASP14 has led to considerable excitement and discussion of the relative roles of ab initio prediction and experimental structure determination in the future. 22 Our own assessment found that the AlphaFold2 predicted structure of SARS‐CoV‐2 ORF8 was of sufficient quality to yield a correct MR solution. This represents both a stringent test of accuracy and bodes well for the practical utility of AlphaFold2 predictions in MR. It will clearly be important for the AlphaFold2 method, and the computing power needed to support it, to become available to the structural biology community.
Despite the impressive accuracy of the fold assignment and backbone structure in the AlphaFold2 prediction, critical details were missing and could only be resolved by crystallographic structure solution. Side‐chain conformations were often inaccurate in the predicted structure. Moreover, dimerization of ORF8 is thought to be important for function, yet the AlphaFold2 prediction only provided the structure of the monomer. Even before AlphaFold2, high quality ab initio structure predictions were accurate enough to reduce, if not eliminate, the need for experimental determinations of the overall fold of single domain proteins. 27 In our view, the real utility of the “accuracy revolution” in structure prediction will be to increase synergy with experimentation. Not only will this be invaluable for crystallographic MR, as assessed here, but in the interpretation of density in cryo‐EM as well.
4. METHODS
4.1. Molecular replacement
The AlphaFold2 ORF8 prediction was obtained from the CASP14 website (www.predictioncenter.org; model ID: T1064TS427_1‐D1) and prepared for Molecular Replacement using the Phenix software suite. 28 Molecular replacement was performed using the program Phaser with default search parameters. The prepared AlphaFold2 ORF8 prediction was provided as a search model and native ORF8 structure factors as experimental data. The search was limited to the known space group P41212. A single solution was obtained with two copies in the asymmetric unit, an LLG value of 167 and TFZ score of 14.7 indicating that the correct solution had been found. Identical search parameters were used for all other search models.
4.2. Model building and refinement
Iterative sounds of manual model building and refinement were performed using Coot 29 and Phenix Refine 28 respectively (for statistics see Table 1). Figures were produced using the program PyMOL (https://pymol.org/2/).
4.3. Rotamer analysis
Rotamer analysis was performed using the Structure Comparison Tool within the Phenix Suite which assigns rotamer IDs according to the Ultimate Rotamer Library designation. 30 An AlphaFold2 rotamer was considered incorrect when its assigned Ultimate Rotamer Library ID failed to match those of any of the equivalent residues within the experimentally determined structures (all chains from PDBIDs: 7JTL, 7JX6).
AUTHOR CONTRIBUTIONS
Thomas G. Flower: Conceptualization; formal analysis; investigation; writing‐original draft. James H. Hurley: Conceptualization; project administration; supervision; writing‐original draft; writing‐review & editing.
ACKNOWLEDGMENTS
We thank Marc Allaire, Cosmo Buffalo, and Richard Hooy for helpful discussions and comments on the manuscript. This work was supported by UCOP emergency seed grant R00RG2347 (JHH), and National Institutes of Health grant R37 AI112442 (JHH).
Flower TG, Hurley JH. Crystallographic molecular replacement using an in silico‐generated search model of SARS‐CoV‐2 ORF8. Protein Science. 2021;30:728–734. 10.1002/pro.4050
Funding information National Institute of Allergy and Infectious Diseases, Grant/Award Number: R37 AI112442; National Institutes of Health; UCOP, Grant/Award Number: R00RG2347
REFERENCES
- 1. Taylor G. The phase problem. Acta Crystallogr D. 2003;59:1881–1890. [DOI] [PubMed] [Google Scholar]
- 2. Rossmann MG, Blow DM. The detection of sub‐units within the crystallographic asymmetric unit. Acta Crystallogr. 1962;15:24–31. [Google Scholar]
- 3. Rossmann MG. Molecular replacement—Historical background. Acta Crystallogr D. 2001;57:1360–1366. [DOI] [PubMed] [Google Scholar]
- 4. Scapin G. Molecular replacement then and now. Acta Crystallogr D. 2013;69:2266–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abergel C. Molecular replacement: Tricks and treats. Acta Crystallogr D. 2013;69:2167–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giorgetti A, Raimondo D, Miele AE, Tramontano A. Evaluating the usefulness of protein structure models for molecular replacement. Bioinformatics. 2005;21(Suppl 2):ii72–ii76. [DOI] [PubMed] [Google Scholar]
- 7. DiMaio F. Advances in rosetta structure prediction for difficult molecular‐replacement problems. Acta Crystallogr D. 2013;69:2202–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DiMaio F. Rosetta structure prediction as a tool for solving difficult molecular replacement problems. Methods Mol Biol. 1607;2017:455–466. [DOI] [PubMed] [Google Scholar]
- 9. Simpkin AJ, Thomas JMH, Simkovic F, Keegan RM, Rigden DJ. Molecular replacement using structure predictions from databases. Acta Crystallogr D. 2019;75:1051–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bibby J, Keegan RM, Mayans O, Winn MD, Rigden DJ. Ample: A cluster‐and‐truncate approach to solve the crystal structures of small proteins using rapidly computed ab initio models. Acta Crystallogr D. 2012;68:1622–1631. [DOI] [PubMed] [Google Scholar]
- 11. Rämisch S, Pramhed A, Tillgren V, Aspberg A, Logan DT. Crystal structure of human chondroadherin: Solving a difficult molecular‐replacement problem using de novo models. Acta Crystallogr D. 2017;73:53–63. [DOI] [PubMed] [Google Scholar]
- 12. Jones DT, Kandathil SM. High precision in protein contact prediction using fully convolutional neural networks and minimal sequence features. Bioinformatics. 2018;34:3308–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones DT, Singh T, Kosciolek T, Tetchner S. Metapsicov: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics. 2015;31:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Skwark MJ, Raimondi D, Michel M, Elofsson A. Improved contact predictions using the recognition of protein like contact patterns. PLoS Comput Biol. 2014;10:e1003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang S, Sun S, Li Z, Zhang R, Xu J. Accurate de novo prediction of protein contact map by ultra‐deep learning model. PLoS Comput Biol. 2017;13:e1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Senior AW, Evans R, Jumper J, et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. [DOI] [PubMed] [Google Scholar]
- 17. Xu J, Wang S. Analysis of distance‐based protein structure prediction by deep learning in casp13. Proteins. 2019;87:1069–1081. [DOI] [PubMed] [Google Scholar]
- 18. Kandathil SM, Greener JG, Jones DT. Prediction of interresidue contacts with deepmetapsicov in casp13. Proteins. 2019;87:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao F, Xu J. A position‐specific distance‐dependent statistical potential for protein structure and functional study. Structure. 2012;20:1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aszódi A, Gradwell MJ, Taylor WR. Global fold determination from a small number of distance restraints. J Mol Biol. 1995;251:308–326. [DOI] [PubMed] [Google Scholar]
- 21. Senior AW, Evans R, Jumper J, et al. Protein structure prediction using multiple deep neural networks in the 13th critical assessment of protein structure prediction (casp13). Proteins. 2019;87:1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Callaway E. ‘It will change everything’: Deepmind's ai makes gigantic leap in solving protein structures. Nature. 2020;588:203–204. [DOI] [PubMed] [Google Scholar]
- 23. Flower TG, Buffalo CZ, Hooy RM, Allaire M, Ren X, Hurley JH. Structure of sars‐cov‐2 orf8, a rapidly evolving immune evasion protein. Proc Natl Acad Sci U S A. 2021;118:e2021785118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li SA, Cheng L, Yu Y, Wang JH, Chen Q. Structural basis of dscam1 homodimerization: Insights into context constraint for protein recognition. Sci Adv. 2016;2:e1501118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Velikovsky CA, Deng L, Chlewicki LK, Fernández MM, Kumar V, Mariuzza RA. Structure of natural killer receptor 2b4 bound to cd48 reveals basis for heterophilic recognition in signaling lymphocyte activation molecule family. Immunity. 2007;27:572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song Y, DiMaio F, Wang RY, et al. High‐resolution comparative modeling with rosettacm. Structure. 2013;21:1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Afonine PV, Poon BK, Read RJ, et al. Real‐space refinement in phenix for cryo‐em and crystallography. Acta Crystallogr D. 2018;74:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hintze BJ, Lewis SM, Richardson JS, Richardson DC. Molprobity's ultimate rotamer‐library distributions for model validation. Proteins. 2016;84:1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]