

Key Points
Rilzabrutinib oral BTK inhibitor has long residence time and low systemic exposure.
Rilzabrutinib shows multiple mechanisms of adaptive and innate immune responses.
Preclinical data support clinical use of rilzabrutinib for immune-mediated diseases.
Visual Abstract
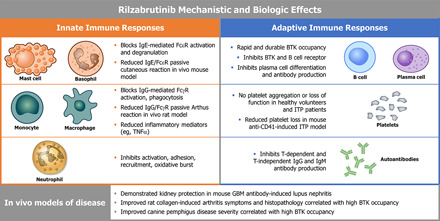
Abstract
Bruton tyrosine kinase (BTK) is expressed in B cells and innate immune cells, acting as an essential signaling element in multiple immune cell pathways. Selective BTK inhibition has the potential to target multiple immune-mediated disease pathways. Rilzabrutinib is an oral, reversible, covalent BTK inhibitor designed for immune-mediated diseases. We examined the pharmacodynamic profile of rilzabrutinib and its preclinical mechanisms of action. In addition to potent and selective BTK enzyme and cellular activity, rilzabrutinib inhibited activation and inflammatory activities of B cells and innate cells such as macrophages, basophils, mast cells, and neutrophils, without cell death (in human and rodent assay systems). Rilzabrutinib demonstrated dose-dependent improvement of clinical scores and joint pathology in a rat model of collagen-induced arthritis and demonstrated reductions in autoantibody-mediated FcγR signaling in vitro and in vivo, with blockade of rat Arthus reaction, kidney protection in mouse Ab-induced nephritis, and reduction in platelet loss in mouse immune thrombocytopenia. Additionally, rilzabrutinib inhibited IgE-mediated, FcεR-dependent immune mechanisms in human basophils and mast cell–dependent mouse models. In canines with naturally occurring pemphigus, rilzabrutinib treatment resulted in rapid clinical improvement demonstrated by anti-inflammatory effects visible within 2 wk and all animals proceeding to complete or substantial disease control. Rilzabrutinib is characterized by reversible covalent BTK binding, long BTK residence time with low systemic exposure, and multiple mechanistic and biological effects on immune cells. Rilzabrutinib’s unique characteristics and promising efficacy and safety profile support clinical development of rilzabrutinib for a broad array of immune-mediated diseases.
Introduction
Bruton tyrosine kinase (BTK), a Tec family tyrosine kinase, is critical in immune pathways as an essential intracellular signaling element (1), participating in both the adaptive and innate arms of the immune response (2, 3). BTK is expressed in many cells of hematopoietic origin, including monocytes, macrophages, neutrophils, mast cells, eosinophils, and platelets but not T cells. BTK is located downstream of the BCR and is required for the maturation, function, and differentiation of B cells and subsequent production of Abs (2).
BTK is also essential for Ab (IgG and IgE) signaling through the FcγR and FcεR signaling pathways (2, 3). Neutrophil recruitment and function during sterile inflammation is also reliant on BTK, facilitating trauma, autoimmunity, ischemic injuries, sterile liver disease mechanisms, and other immunopathology effects caused by excessive neutrophil recruitment (4, 5). Because BTK is expressed in various immune cell types such as B and innate immune cells, it is a key therapeutic target in immune-mediated diseases (2, 3).
There are multiple irreversible, covalent BTK inhibitors (BTKi) such as ibrutinib, acalabrutinib, evobrutinib, which are approved or in development. Ibrutinib was originally approved for mantle cell lymphoma, chronic lymphocytic leukemia, Waldenström macroglobulinemia, marginal zone lymphoma, and has more recently been approved for chronic graft-versus-host disease (6, 7). Acalabrutinib was approved shortly after for mantle cell lymphoma and chronic lymphocytic leukemia/small lymphocytic lymphoma (8–11). Evobrutinib has completed its phase 2 trial in multiple sclerosis patients (12). Despite these advances in BTKi’s, improving selectivity for BTK as well as minimizing systemic exposure are still desirable for reducing adverse off-target side effects. For example, ibrutinib is associated with an increased bleeding risk, with inhibition of collagen-induced platelet aggregation and platelet adhesion via inhibition of intracellular molecules involved in platelet signaling (13). In addition, ibrutinib has been shown to affect PI3K-Akt signaling, which may explain atrial fibrillation events observed in ibrutinib-treated patients (14).
One of the main concerns of irreversible adducts is the hypothesized relationship between covalent drug binding and the potential for idiosyncratic drug toxicity (15, 16). Irreversible modification of off-target cysteines can be particularly problematic in tissues such as the gastrointestinal tract and liver that are exposed to relatively high drug concentrations (often in the micromolar range) after oral administration (17). A highly selective BTKi that manifests minimal adverse side effects could solve many toxicity challenges.
Rilzabrutinib (PRN1008) is an oral, reversible, covalent BTKi with characteristics that may solve many of the selectivity- and reversibility-related concerns accompanying currently available BTKi’s (17, 18). Rilzabrutinib binds in a covalent manner, increasing selectivity by forming a chemical bond to a specific cysteine residue present in BTK (Fig. 1A). This durable covalent engagement allows for maximal efficacy. However, rilzabrutinib’s unique binding mechanism provides the opportunity for a tailored residence time while reducing safety concerns associated with irreversible inhibitors (19). An advantage with rilzabrutinib is its capacity to mimic the long-lasting activity of an irreversible covalent inhibitor, with an in vivo dissociation rate that is governed by BTK target degradation and resynthesis (17). In addition, reversible cysteine engagement may enable fine-tuning of inhibitor residence time, a feature that would facilitate the use of such inhibitors not only in therapeutic applications requiring sustained target engagement, but also in applications where faster target disengagement is preferred (17). Studies to date suggest that rilzabrutinib offers a favorable profile in healthy volunteers (18) and for the treatment of patients with immune-mediated diseases through the inhibition of BCR signaling with low doses of concomitant corticosteroids (CS) (20) (patent WO2014039899).
FIGURE 1.

Biochemical characterization, selectivity, and potency of rilzabrutinib. (A) Rilzabrutinib has both noncovalent and covalent binding regions, as shown at left, which enables binding with high potency and long residence time to BTK and limits binding to off-target kinases, as shown at right. (B) Kinase selectivity was determined at a rilzabrutinib 1 μM concentration using enzymatic inhibition in a panel of 251 kinases. Data were acquired in duplicate and assessed in two independent experiments. The dendrogram illustrates kinase selectivity with >90% inhibition by rilzabrutinib for six kinases (BTK, RLK, TEC, BMX, BLK, ERBB4) shown in red circles. (C) In vitro kinase selectivity for BTK by rilzabrutinib was further supported by inhibition (IC50) of five kinases by rilzabrutinib with potency <10 nM in an ATP competitive enzyme inhibition in vitro assay (left table). Shown at right are the percent target occupancy values of rilzabrutinib toward the same five kinases at both 1 and 24 h, as evaluated using a biochemical residence time assay. Data were acquired in duplicate at each timepoint and assessed in two independent experiments.
Through preclinical in vitro and in vivo studies, the objective of this manuscript is to characterize and demonstrate how the pharmacodynamic (PD) profile of rilzabrutinib translates into biological mechanisms relevant in immune-mediated diseases. We assessed the impact of rilzabrutinib on both adaptive and innate immune mechanisms independent of cell death. The combination of multiple mechanistic and biological effects with favorable PD make rilzabrutinib a promising candidate to treat various immune-mediated diseases, as shown in ongoing clinical studies of pemphigus and immune thrombocytopenia (ITP).
Materials and Methods
All experiments involving animals were conducted in accordance with local laws and regulations concerning animal welfare under protocols approved by local institutional review committees and in compliance with the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals.
Biochemical potency, selectivity, and reversibility
Kinase selectivity was determined at a compound concentration of 1 μM using an enzymatic inhibition panel of 251 kinases (Nanosyn, Santa Clara, CA). The reaction mixture for each kinase assay consisted of enzyme, substrate, and cofactors mixed in a microtiter plate: 100 mM HEPES (pH 7.5), 0.1% BSA, 0.01% Triton X-100, 1 mM DTT, 10 mM MgCl2, 10 μM sodium orthovanadate, 10 μM β-glycerophosphate, a concentration of ATP optimized for each kinase, 1% DMSO (from test compound), 1 μM of a peptide substrate, kinase, and 12 different concentrations of test compound at a 3-fold dilution interval starting with a top concentration of 5 μM. The reaction was incubated at 25°C and then was terminated by the addition of 20 mM EDTA. For experiments that measured the shift in BTK IC50 with different ATP concentrations, IC50 values for the interaction of rilzabrutinib with BTK were determined as described above with variable concentrations of ATP (16, 160, and 800 μM).
Reversibility of binding of rilzabrutinib to BTK
The reversibility assay determined reversible or irreversible compound binding to BTK by assessing if a BTK-bound compound would return unmodified to solution following trypsin digestion. The assay used a buffer of 20 mM HEPES (pH 7.5), 10 mM MgCl2, 150 mM NaCl, 5% glycerol, 0.05% Triton X-100, and 1 mM 2-ME. A solution of 2.2 μM BTK was incubated with 0.6 μM test compound for 60 min. Sixty microliters of the BTK/compound mixture or compound alone was added to 120 μl of 1 mg/ml trypsin and incubated for 60 min. Samples were analyzed via liquid chromatography/mass spectrometry and quantitated via comparison with a standard curve of the test compound.
Binding kinetics of rilzabrutinib to BTK
Kinetics of binding of rilzabrutinib to BTK were determined using a LanthaScreen kinase binding assay (Thermo Fisher Scientific, Waltham, MA). Experiments were performed in an assay buffer of 50 mM HEPES (pH 7.5), 10 mM MgCl2, 0.01% Triton X-100, and 1 mM EGTA. For dissociation studies, 1.5 μM test compound and 0.5 μM BTK were preincubated for 30 min and then diluted 5-fold in assay buffer. Tracer 178 (catalog no. PV5593) and Europium-coupled Anti-6XHis Ab were added to final assay conditions of 50 nM BTK, 150 nM test compound, 15 nM and Europium-coupled Anti-6XHis Ab, and 750 nM Tracer 178. For association studies, a mixture of BTK (2 nM), Tracer 236 (80 nM), and Europium-coupled anti-6XHis Ab (5 nM) was prepared. A concentration series of test compounds was added to this mixture. The final concentration of components was 1 nM BTK, 2.5 nM Europium-coupled Anti-6XHis Ab, 40 nM Tracer 236, and 1–314 nM test compound.
BTK target occupancy and durability in B cells
The effects of rilzabrutinib on BTK target occupancy in a human B cell line were tested in Ramos B cells (CRL-1596; American Type Culture Collection [ATCC]) to model inhibition of BCR signaling. Ramos cells were treated with an eight-point, 3-fold dilution series starting at 1 μM (with final DMSO concentration 0.1% in all wells). Ramos cells were pretreated for 1 h with rilzabrutinib, then treated with a BTK-selective biotinylated probe to measure BTK target occupancy. The probe binds irreversibly to any BTK target molecules unoccupied by rilzabrutinib, and therefore, the signal generated is a function of BTK target occupancy. Ramos cells were treated with biotinylated probe for 1 h in 5% CO2 at 37°C, then lysed (CelLytic M lysis buffer; Sigma-Aldrich) for probe binding detection by the Alphascreen Kit (Perkin Elmer). The Alphascreen signal was generated by the close proximity of a donor and acceptor bead pair. The acceptor bead sandwich was generated by incubating protein A beads from the Alphascreen Kit with mouse anti-human BTK (BD Biosciences) for 30 min. Cell lysate was then added to aliquoted acceptor beads in a 384-well plate and incubated for an additional 30 min to allow binding of BTK with bound probe in the lysate to the Ab. Donor beads coated with streptavidin were then added, and binding to the biotin probe was detected on an Envision Model 2101 Multilabel Reader plate reader (Perkin Elmer) using a 680-nm excitation and 615-nm emission wavelength filter combination. The log rilzabrutinib concentration was plotted as a function of percent inhibition of the biotin-labeled positive control value. IC50 analysis was performed using the Levenberg–Marquardt nonlinear least square–fitting algorithm implemented in the Dotmatics data package.
For assessment of durability, a single concentration of 1 μM rilzabrutinib was tested in Ramos cells plated in a 12-well plate. After treatment for 1 h, cells were washed to remove compound and then incubated in growth medium for either 4 or 18 h. After washout, cells were labeled with a biotin-labeled BTK-selective probe for 1 h, then cell lysates were prepared and binding of rilzabrutinib was determined by Alphascreen, as described above. Percent occupancy was calculated by converting the ratio of probe signal at 1 μM of rilzabrutinib to the signal with no compound present to a percentage and subtracted from 100%.
BTK target occupancy and durability in human PBMCs
BTK target occupancy was determined by first treating multiple whole-blood samples with serial dilutions of rilzabrutinib. Human whole blood (AllCells, Alameda, CA) was treated with 0.0003–10 μM concentrations of rilzabrutinib or DMSO control for 1 h at 37°C, 5% CO2. The PBMCs were then isolated from rilzabrutinib-treated whole-blood samples by Ficoll gradient (Ficoll-Paque, catalog no. 17-1440-03; GE Healthcare). Isolated PBMCs were treated with a bodipy-labeled BTK-selective probe for 1 h at 37°C, 5% CO2, and then lysed with CelLytic M lysis buffer (Sigma-Aldrich). Protein concentration of cell lysates was determined by BCA assay (Thermo Fisher Scientific-Pierce Biotechnology). To measure BTK occupancy and total BTK, bodipy probe–treated cell lysates were resolved by SDS-PAGE. The gel was scanned on a Typhoon 8600 fluorescence scanner (GE Healthcare) using the fluorescein filter to measure BTK probe occupancy. Proteins were then transferred to a nitrocellulose membrane, blocked, and immunoblotted using primary mouse anti-human BTK Ab (BD Biosciences) and secondary goat anti-mouse IgG–Alexa Fluor 647 fluorescent Ab (Cell Signaling Technologies). The blot was scanned on the Typhoon fluorescence scanner using the cyanine filter to measure total BTK. The BTK probe occupancy and total BTK were quantified from the scanned gel and membrane using ImageQuant TL image analysis software (GE Healthcare). The resulting total BTK values were used to normalize the BTK probe occupancy signal in each sample. The percent occupancy was calculated by converting the ratio of probe signal of rilzabrutinib-treatments to the signal with DMSO present to a percentage and subtracting from 100%.
For the assessment of durability, whole blood was treated with a single concentration of 2 μM rilzabrutinib, after which the compound was washed out of the isolated PBMCs and incubated in growth medium for 4 or 18 h to test durability of binding. After washout, cells were labeled with 1 μM PRN933, a bodipy-labeled, BTK-selective probe, for 1 h. After treatment with the probe, cell lysates were prepared, and binding of rilzabrutinib was determined by fluorescent scanning of SDS-PAGE–separated protein blots, as described above. The percent occupancy was calculated by converting the ratio of probe signal at 1 μM of rilzabrutinib to the signal with no compound present to a percentage and subtracting from 100%.
Cellular off-target and cytotoxicity assays
HCT-116 cells (CCL-247; ATCC), a transformed human epithelial cell line (not expressing BTK) derived from a colorectal carcinoma, were used to assess possible BTK-independent effects of rilzabrutinib on cell viability as a measure of cytotoxicity. HCT-116 cells were exposed to nine 3-fold dilutions of rilzabrutinib starting at 30 μM and ending at 0.005 μM with a final DMSO concentration of 0.3% in each dilution. After addition of the compound dilutions, cells were incubated for 48 h at 37°C, 5% CO2. Cell viability was then measured by CellTiter-Glo (Promega) analysis using a Wallac Victor plate reader to measure luminescence or by Presto-blue analysis using a molecular Dynamics HT plate reader to measure fluorescence. Percent inhibition was plotted as a function of log compound concentration. IC50 determination was performed using the Levenberg–Marquardt nonlinear least square–fitting algorithm implemented in the Dotmatics data package.
Ramos B cells stimulated with IL-4 to induce STAT6 were used in the non-BTK–dependent off-target screening. IL-4–induced STAT6 activation was measured, using the Invitrogen Cell-sensor reporter assay system, a β-lactamase–based reporter assay system that uses conversion of a fluorescence resonance energy transfer (FRET)–based substrate to measure transcriptional activity. Cells were treated with ten 3.16-fold dilutions of rilzabrutinib starting at 5 μM and ending at 0.158 μM, with a final DMSO concentration of 0.1%. The cells were pretreated with compound for 30 min, stimulated, and incubated for 5 h at 37°C, 5% CO2. The β-lactamase substrate was then added and incubated for 2 h followed by measurement of substrate conversion by FRET on a fluorescence plate reader. A dose response curve of percent inhibition was fit to a sigmoidal dose–response model in XLfit.
The lack of effect of rilzabrutinib on T cells was assessed in TCR-induced activation (αCD3/αCD28) and calcium flux–induced (thapsigargin) activation assays. Additional cellular selectivity of rilzabrutinib was also assessed in ME-180 cells (a cervical carcinoma cell line) stimulated with EGF and the activation of the transcription factor AP-I was measured in a reporter assay. These assays Invitrogen Cell-sensor, β-lactamase–based reporter assay system that uses conversion of an FRET-based substrate to measure transcriptional activity. Cells were treated with ten 3.16-fold dilutions of rilzabrutinib starting at 5 μM and ending at 0.158 μM with a final DMSO concentration of 0.1%. The cells were pretreated with compound for 30 min, stimulated, and incubated for 5 h at 37°C, 5% CO2. The β-lactamase substrate was then added and incubated for 2 h followed by measurement of substrate conversion by FRET on a fluorescence plate reader. A dose response curve of percent activation was fit to a sigmoidal dose–response model in XLfit.
Ab-dependent cell-mediated cytotoxicity
Assessment of the effect of rilzabrutinib on Ab-dependent cell-mediated cytotoxicity (ADCC) of a rituximab-biosimilar Ab against Jeko-1 (mantle cell lymphoma cells; ATCC) was performed at Eureka Therapeutics (Emeryville, CA). Freshly isolated PBMCs (AllCells), used as effector cells, were first pretreated with eight concentrations of rilzabrutinib at 2-fold the final concentrations of 0.005–10 μM for 1 h at room temperature. Jeko-1 target cells were pretreated with 1 μg/ml rituximab-biosimilar or human IgG1 isotype control for 30 min prior to addition of effector cells. Rilzabrutinib-treated PBMC effector cells were added to the rituximab-treated Jeko-1 target cells at a 25:1 E/T ratio and incubated for 16 h at 37°C, 5% CO2. After the 16 h incubation, the supernatants were assayed for target cell lysis based on lactate dehydrogenase release using the CytoTox 96 Nonradio Lactate Dehydrogenase Assay Kit (Promega).
B cell activation in human whole blood
The ability of rilzabrutinib to block BCR-driven activation of B cells was measured by expression of the surface maker CD69. Human whole blood (AllCells) were pretreated for 1 h with 5% CO2 at 37°C with eleven 3-fold serial dilutions of compound starting at a concentration of 5 μM. Whole blood was then stimulated by addition of 50 μg/ml goat anti-human IgM F(ab’)2 (Southern Biotech, Birmingham, AL) to stimulate the BCR. Stimulated blood was incubated overnight for 18 h in 5% CO2 at 37°C followed by staining for 30 min with labeled anti-CD20 and anti-CD69 (BD Biosciences). The RBCs were then lysed with PharmLyse (BD Biosciences), washed, and analyzed for percent CD69 expression on CD20+ B cells on a Cytek DxP flow cytometer. The log of the rilzabrutinib concentration was plotted as a function of the percent inhibition of the CD69 positive control value. IC50 determination was performed using the Levenberg–Marquardt nonlinear least square–fitting algorithm implemented in the Dotmatics data package. The IC50 values were determined by fitting the inhibition curves using a four-parameter sigmoidal dose–response model.
B cell proliferation
The potency of rilzabrutinib inhibition of anti-IgM–induced human B cell proliferation was assessed at Bioduro (San Diego, CA). B cells were isolated by negative selection from freshly isolated PBMCs using the Dynabeads Untouched Human B cell Kit (Thermo Fisher Scientific). The purified B cells were treated with ten 3-fold dilutions of rilzabrutinib starting at 10 μM and ending at 0.0005 μM with a final DMSO concentration of 0.1% for 30 min at 37°C, 5% CO2. Anti-human IgM (BD Pharmingen) was added at a final concentration of 5 μg/ml to induce B cell proliferation. B cells were incubated for 48 h at 37°C, 5% CO2 and then assayed for proliferation by CellTiter-Glo (Promega, Madison, WI). IC50 value was calculated by plotting the percent inhibition versus compound concentration using a four-parameter fit nonlinear regression model in GraphPad Prism.
B cell Ab production
B cells were enriched from leukocyte concentrate of TrimaAccel LRS chamber recovered after a plateletpheresis procedure from healthy volunteers (Stanford Blood Center, Palo Alto, CA) using the Straight from LRSC CD19 Microbead Kit (Miltenyi Biotech). B cells were treated with 10 concentrations of 3-fold serial dilutions rilzabrutinib in DMSO for 1 h at 37°C, 5% CO2. The rilzabrutinib-treated B cells and DMSO control were then stimulated with TNP-LPS (Santa Cruz Biotechnology), CpG (ODN2006; Invivogen), or anti-CD40 (BD Biosciences) + IL-21 (R&D Systems) for 7 d at 37°C, 5% CO2. Rilzabrutinib inhibition of Ab production was determined by measurement of IgG and IgM in the cell culture lysate using human IgG and IgM AlphaLISA kits (Perkin Elmer). IC50 was calculated in GraphPad Prism with nonlinear regression using “log (inhibitor) versus response − Variable slope (four parameters)” analysis with no constraints.
Monocyte IgG/FcγR activation assay
TNF-α was measured in IgG-activated human monocytes enriched from PBMCs purified from buffy coats obtained from healthy volunteers at Stanford Blood Center (Palo Alto, CA) over a Ficoll-histopaque gradient. The purified PBMCs were incubated in complete RPMI 1640 at 37°C, 5% CO2 in tissue culture flasks for 2 h to allow monocytes to adhere. After the 2 h incubation, nonadherent cells were gently washed away, and adherent monocytes scraped into fresh media. Monocytes were treated with ten 3-fold dilutions of rilzabrutinib starting at 10 μM and ending at 0.0005 μM with a final DMSO concentration of 0.1%. Monocytes were pretreated with rilzabrutinib for 1 h at 37°C, 5% CO2, then transferred to 96-well plates coated with 25 μg/ml goat anti-human F(ab′)2 IgG (Jackson ImmunoResearch). After a 4-h stimulation at 37°C, 5% CO2, the supernatant was removed and assayed for TNF-α production using the Human TNF α AlphaLISA Kit (Perkin Elmer). IC50 values were calculated in GraphPad Prism with nonlinear regression using “log(inhibitor) versus response − Variable slope (four parameters)” analysis with no constraints.
Basophil IgE/FcεR activation assay
Human whole blood from healthy donors was purchased from AllCells and assayed for rilzabrutinib inhibition of IgE basophil activation. Whole blood was treated with ten 3-fold dilutions of rilzabrutinib starting at 10 μM and ending at 0.0005 μM with a final DMSO concentration of 0.1%. Whole blood was pretreated with compound for 1 h at 37°C, 5% CO2, basophils were activated with anti-IgE Ab (Beckman Coulter) for 15 min in a 37°C water bath, and then stopped by the addition of EDTA. Basophil activation was analyzed by flow cytometry using CD63-FITC/CD123-PE/Anti–HLA-DR–PerCP Triple-Ab Mixture (BD Biosciences). Quantitative determination of activated basophils was measured as the percentage of CD63+ expression on CD123+, low-side scatter, HLA-DR– cells.
Platelet aggregation assays
Five healthy subjects and five patients diagnosed with primary ITP were recruited for the study. Citrated whole blood was drawn from donors and centrifuged to isolate platelet-rich plasma (PRP). PRP platelet counts were adjusted to 200,000–300,000 in healthy subjects and >125,000 in ITP patient samples. PRP was pretreated with 0.3 or 1 μM of rilzabrutinib, or controls ibrutinib, 1% DMSO, and 10 μM roxifiban for 15 min. Samples were then transferred to platelet aggregation test wells and a panel of platelet aggregation agonists were added: 10 μM ADP, 5 μM TRAP, 5 μM U46619, saline, 1.5 mg/ml ristocetin, 5 μg/ml collagen, and 2.5 μg collagen. Platelet aggregation was measured in a lumiaggregometer for 6 min for all agonists except saline, where aggregation was measured for 15 min. Agonist-induced platelet aggregation was indicated by maximum aggregation caused by agonist. The effect of rilzabrutinib was analyzed by conducting a two-sample (equal variance), two-tailed t test between the DMSO samples and each inhibitor-treated group.
Lupus nephritis model
The effects of rilzabrutinib were evaluated in a model of sheep anti-rat glomerular basement membrane (GBM) Ab-induced nephritis in 129X1/SvJ mice. On study day −5, mice were immunized by i.p. injection with 10 mg/kg sheep IgG (catalog no. 642851; MP Biomedicals) previously emulsified in an equal volume of CFA (catalog no. I5131; Sigma-Aldrich). On day 0, disease was induced; group 1 (nondisease; vehicle) was injected i.v. with 100 μl of normal sheep serum (catalog no. PTX-000S; Probotex), and groups 2–8 (disease; vehicle and treated) were injected i.v. with 40 μl of sheep anti-rat GBM serum (catalog no. PTX-001S; Probotex) mixed with 60 μl of normal sheep serum. Mice were dosed orally once daily on days −1 through 10 with vehicle or rilzabrutinib (10, 20, or 40 mg/kg), or orally twice a day with vehicle or rilzabrutinib (20 mg/kg). Positive controls were dosed orally once daily on days −1 to 9 with dexamethasone (1 mg/kg). Efficacy evaluations compared with disease controls were based on animal body weights, urine protein analysis, organ weights (kidneys and spleens), serum blood urea nitrogen (BUN) levels, and blinded histopathologic evaluation of the kidneys.
Anti-CD41–induced ITP model
BALB/c mice were dosed with rilzabrutinib (10, 20, or 40 mg/kg, orally once daily) or IVIG (Gamunex-C; Grifols Therapeutics) at 24 h prior to and 1 h prior to ITP model induction with anti-CD41; 1.3 μg of anti-CD41 (rat anti-mouse CD41 IgG1, catalog no. sc-19963; Santa Cruz Biotechnology) was administered i.p. Blood was collected at 6 h post–Ab challenge into EDTA-treated tubes, and platelet counts were measured and compared with vehicle control for all treatment groups. The unmanipulated/naive mice that did not receive anti-CD41 treatment served as a baseline measure of normal mouse platelet counts.
Passive Arthus reaction model
Sprague Dawley rats were shaved and injected i.v. with Evans blue dye (EBD; catalog no. E2129; Sigma-Aldrich) and OVA (catalog no. A5378; Sigma-Aldrich). Based on methods previously described by Chang et al. (21), the Arthus reaction was induced 10 min later with intradermal injection in three sites on the back with 50 μg/site rabbit anti-OVA IgG (catalog no. R1101; Acris Antibodies) and 50 μg/site at three sites on the contralateral side of the back with rabbit IgG (I8140; Sigma-Aldrich). Rats were dosed with rilzabrutinib (10, 20, or 40 mg/kg, orally once daily) or prednisolone (10 mg/kg) for 3 d with the final dose 1 h prior to Arthus induction. The IgG-mediated Arthus reaction was quantified by the level of EBD extravasation at 4 h after Ab administration. The mice were euthanized, the skin was removed from the back and reversed, and the diameter of EBD extravasation was recorded with a digital caliper by a blinded researcher. An 8-mm skin punch centered on the intradermal injection sites was prepared; skin punches were incubated with 1 ml of formamide (catalog no. 15515-026; Invitrogen) in an 80°C water bath overnight, and extravasation was recorded as OD measurement at 610-nm wavelength.
Passive cutaneous anaphylaxis model
Based on methods previously described by Chang et al. (21), the passive cutaneous anaphylaxis (PCA) model evaluated BALB/c mice that were shaved on their dorsal surface and flanks prior to being sensitized intradermally in the back with IgE anti-DNP (catalog no. D8406; Sigma-Aldrich) at three sites (10 ng IgE anti-DNP per site) and in three different sites with saline. After 24 h, mice were injected i.v. with 0.2 ml of EBD containing 500 ng of DNP-BSA (catalog no. E2129; Sigma-Aldrich; catalog no. D-5050-100; LCG Biosearch Technologies). Mice were dosed once daily with vehicle or rilzabrutinib (40 or 80 mg/kg) or twice a day with vehicle or rilzabrutinib (20 or 40 mg/kg) for 3 d, with the final dose given 1 h prior to DNP/EBD administration. Positive controls were cyproheptadine 25 mg/kg once daily i.p. (catalog no. C6022; Sigma-Aldrich) and prednisolone 10 mg/kg once daily orally (catalog no. P4153; Sigma-Aldrich). IgE-mediated reaction was assessed by dye extravasation area (square millimeters) and intensity of EBD extravasation in the skin at 30 min after challenge. The mice were euthanized, the skin was removed from the back and reversed, and the extravasation area was calculated from the diameter of EBD extravasation recorded with a digital caliper by a blinded researcher. An 8-mm skin punch centered on the intradermal injection sites was prepared; skin punches were incubated with 1 ml of formamide (catalog no. 15515-026; Invitrogen) in an 80°C water bath overnight, and extravasation was recorded as OD measurement at 610-nm wavelength.
In vivo pharmacokinetics/PD
Assessment of PD, measured as BTK occupancy, was evaluated in female Lewis rats who were dosed orally with vehicle or rilzabrutinib 10, 20, or 40 mg/kg (n = 3 groups per timepoint). At 1, 14, or 24 h postdose, blood was collected to assess plasma drug levels, and spleens were harvested for PD analysis. Spleens were processed to a single-cell suspension and incubated with an irreversible fluorescent bodipy-labeled BTK occupancy probe according to methods described previously. The percent BTK occupancy was calculated for each patient by dividing the normalized rilzabrutinib-treated samples from the vehicle-treated sample, then multiplying by 100 and subtracting from 100. The resulting percent occupancy values were then used to measure the amount of rilzabrutinib bound to target BTK in the treated samples.
Collagen-induced arthritis model
Female Lewis rats with established collagen-induced arthritis (CIA) (n = 12 per group) were treated on study days 10–20 (arthritis days 1–11) with vehicle (citric acid in water), rilzabrutinib 10, 20, or 40 mg/kg once daily or 20 mg/kg oral twice a day, or the reference compound dexamethasone 0.075 mg/kg oral twice a day. Animals were terminated on day 11 of arthritis (study day 20). Efficacy evaluation was based on animal body weights, daily ankle caliper measurements, ankle diameter expressed as area under the curve, clinical arthritis scores of ankles, arthritis scores area under the curve, terminal hind paw weights, and blinded histopathologic evaluation of ankles and knees. Select paws from vehicle controls and from the rilzabrutinib 20 mg/kg twice-a-day group were processed for microcomputed tomography assessment. Drug pharmacokinetics (PK) levels were assessed, and spleen percentage of BTK occupancy was measured by binding of an irreversible fluorescent probe to unbound BTK in prepared splenic cell samples collected from animals at 1 or 24 h (12 h for twice-a-day groups) after final dose. In a second study, rats (n = 8 per group) were dosed orally twice a day on study days 13–20 (arthritis days 4–11) with vehicle or rilzabrutinib (20 mg/kg) to determine effects on reversal of disease.
Rilzabrutinib crossover for canines with pemphigus foliaceus
The design of this crossover study was open label for cases of newly presenting canine pemphigus foliaceus (PF). Four dogs were treated with rilzabrutinib at ∼15 mg/kg orally once daily as monotherapy, with intrasubject dose adjustment based on clinical response and BTK occupancy. Dosing was adjusted to the maximum allowable dosage of 30 mg/kg/d as a twice-a-day–divided dosage if minimal clinical improvement was seen after 2–8 wk treatment at the 15 mg/kg dose. After 12 wk, the dose was reduced to ∼50%, if clinically acceptable, as Monday, Wednesday, and Friday dosing of the same prior once daily. If a dog subsequently relapsed, owners had the choice to commence conventional treatment or recommence full-dose rilzabrutinib therapy for a 3–6 mo period. Clinical improvement was measured through canine pemphigus disease activity index (cPDAI), which was modified from the validated human pemphigus disease activity index scoring system (22) that is considered a reliable, quick, and easy-to-use method to capture the extent of skin and mucosal lesions in mild-to-moderate pemphigus cases (23). cPDAI measured the number and size of lesions and the surface area affected at baseline, day 15, and 4, 8, 12, 16, and 20 wk. The scoring system ranged from 0 to 10, with 0 representing absence of lesions, and 10 standing for lesions covering entire body area. The study was conducted in collaboration with University of California, Davis.
Statistical analyses
Evaluations for statistically significant differences in quantitative variables comparing rilzabrutinib (or positive controls) versus vehicle or disease controls were analyzed using a one-way ANOVA with a Dunnett post hoc analysis, Student two-tailed t test for measured (parametric) data, Kruskal–Wallis test with a Dunn post hoc analysis, or Mann–Whitney U test for scored (nonparametric) data. Statistical analysis was performed using GraphPad Prism version 6.0 software. A p value <0.05 was considered statistically significant; a p value <0.001 was also included to convey an additional level of statistical significance.
Results
Biochemical potency, selectivity, and durability of rilzabrutinib
Rilzabrutinib binds via two components that fit the noncovalent and covalent elements of the target, allowing for high selectivity and reduced off-target activity. The covalent bond between rilzabrutinib and the target molecule was fully reversible without risk of permanent modification. This reversibility also allows for a tailored residence time (Fig. 1A). Rilzabrutinib was assessed for potency and selectivity in a broad kinase enzyme inhibition panel. At a concentration of 1 μM, rilzabrutinib demonstrated >90% inhibition of only six of 251 kinases screened (Fig. 1B). Enzyme IC50 values were determined for all kinases showing >50% inhibition at 1 μM (Supplemental Table I). Rilzabrutinib potently inhibited BTK (IC50 = 1.3 ± 0.5 nM) and also displayed potent inhibitory activity against receptor-like kinase (RLK), tyrosine protein kinase TEC, bone marrow tyrosine kinase on chromosome X (BMX), and B cell lymphocyte kinase (BLK) (IC50 = 1.2 ± 0.3, 0.8 ± 0.1, 1.0 ± 0.1, and 6.3 ± 0.7 nM, respectively). Each of these kinases share a common cysteine at the same active site location as BTK. Rilzabrutinib showed minimal inhibition of other kinases such as EGFR, MKK7, ITK, and JAK3.
Rilzabrutinib engaged BTK in a more durable manner than the other TEC family members, with selectivity further improving with time. Rilzabrutinib demonstrated a rapid on-rate and slow off-rate for BTK by achieving over 80% occupancy within the first hour and maintaining higher occupancy levels over 24 h relative to the decreases observed by non-BTKs (Fig. 1C). Because rilzabrutinib is an ATP competitive inhibitor of BTK, IC50 values for BTK inhibition were anticipated to shift to weaker potency with increasing ATP concentration. Indeed, IC50 values were 1.3 ± 0.5, 3.1 ± 0.7, and 9.8 ± 0.7 nM at ATP concentrations of 16, 160, and 800 μM, respectively. Rilzabrutinib demonstrated complete reversibility (>100%), as measured by percent recovery of compound after digestion with trypsin. In comparison, ibrutinib, an irreversible BTKi, showed minimal reversibility (2%). Biochemically, because of the cysteine reversible covalent bonding, rilzabrutinib exhibited a fast on-rate of 5.1 ± 2.1 × 104 per molar per second and a slow off-rate of 1.2 ± 0.1 × 10−6 per second, with BTK occupancy diminishing only modestly from 1 h (95% ± 1%), to 6 h (91% ± 1%), and 24 h (85% ± 2%).
Cellular BTK occupancy and durability of rilzabrutinib
To confirm the durability of BTK target binding in cells, a series of cellular washout studies were performed. Results confirmed slow off-rate from BTK, as demonstrated by retention of drug bound to BTK for prolonged periods. Rilzabrutinib potently bound to BTK in Ramos B cell line, achieving an IC50 for BTK occupancy of 8 ± 2 nM. In washout experiments, rilzabrutinib maintained 72% occupancy at 18 h following the removal of compound (Fig. 2A). Similarly, in PBMCs, rilzabrutinib maintained 79% ± 2% BTK occupancy at 18 h after washout of compounds from the assay system (Fig. 2B). In PBMCs, the irreversible BTKi ibrutinib also sustained BTK occupancy, showing 98% ± 5% BTK occupancy at 18 h after washout. The durable binding of rilzabrutinib was further confirmed in vivo in female Lewis rats who were dosed orally with 10–40 mg/kg rilzabrutinib (discussed below).
FIGURE 2.

Reversible, covalent binding and durable BTK inhibition with rilzabrutinib in cells. (A) BTK occupancy, potency, and durability was evaluated in human Ramos B cells treated with rilzabrutinib or DMSO, then with a competitive BTK-selective biotinylated probe to measure BTK occupancy by competitive binding. Rilzabrutinib treatment in an eight-point concentration curve (starting at 1 μM; final DMSO concentration 0.1%) resulted in potent BTK occupancy of 8 ± 2 nM (n = 4). In a washout experiment, rilzabrutinib 1 μM treatment was given for 1 h and washed out, and cells were further incubated to measure durability of biotin-labeled BTK-selective probe for 4 or 18 h. BTK occupancy was durable at 72% at 18 h postwashout (n = 2). (B) BTK occupancy in human whole-blood PBMCs treated with rilzabrutinib 2 μM for 1 h; PBMCs were isolated, washed three times, and further incubated for 4 and 18 h postwashout. Cells were labeled with a bodipy-labeled, BTK-selective probe for 1 h, processed, and measured through fluorescent scanning of SDS-PAGE–separated protein blots. BTK occupancy showed strong durability in PBMCs, with 79% BTK occupancy at 18 h postwashout (n = 3).
Cellular selectivity and lack of off-target effects
To confirm the high level of selectivity in cells toward BTK and to assess any undesired effects of rilzabrutinib, cytotoxicity, activation of T cells, and other non-BTK–dependent cellular pathways were assessed. Rilzabrutinib demonstrated no evidence of cytotoxicity in non-BTK–expressing cell lines, and weak activation of T cells and other non-BTK–dependent cellular pathways (Table I). The addition of rilzabrutinib tested at a concentration range of 0.5–1000 μM did not inhibit ADCC (mean target cell lysis ranged from 53 to 61%) relative to the effects of rituximab-biosimilar Ab alone (59%; data not shown).
Table I. Cellular characterization of rilzabrutinib confirms selectivity and limited off-target effects.
| Target | Cell Type and Assay | IC50 ± SD, nM |
|---|---|---|
| On-target activity | ||
| BCR | B cell: human proliferation | 5 ± 2 |
| BTK | B cell: Ramos occupancy | 8 ± 2 |
| FcγR | Monocyte: IgG activation | 56 ± 45 |
| BCR | B cell: human whole blood activation | 123 ± 38 |
| FcεR | Basophil: IgE activation in human whole blood | 490 ± 130 |
| Off-target activity | ||
| TCR | T cell: Jurkat receptor signaling | >5,000 |
| IL-4/STAT6 | B cell: Ramos IL-4–induced STAT6 | >5,000 |
| EGFR | Cellular reporter assay | >5,000 |
| Cytotoxicity | HCT116 cells | >16,000 |
Cells were selected based on the presence or absence of BTK, BCR, FcR, and non-BTK–related functions to determine rilzabrutinib selectivity. Following treatment with rilzabrutinib, cell-based assays examined the on-target and off-target activity (measured by IC50 levels) of rilzabrutinib on B cell activation, Fc-receptor signaling, T cells, and other non-BTK–dependent cellular pathways.
Rilzabrutinib also did not block IL-4 stimulation of B cells or exhibit cytotoxicity in an epithelial cell line HCT-116 (Table I). Rilzabrutinib did not inhibit either TCR-induced activation or calcium flux–induced activation of T cells (>5000 nM), and rilzabrutinib was inactive in the non-BTK–dependent off-target screening assay. Furthermore, rilzabrutinib had no activity on EGFR signaling.
Rilzabrutinib did not interfere with normal platelet aggregation
Platelets express BTK; however, alternative signaling pathways exist that bypass BTK signaling to retain normal platelet functions and thrombus formation (10, 24). The effect of clinically relevant concentrations of rilzabrutinib (0.3 and 1 μM) on normal platelet aggregation was assessed against a panel of platelet aggregation agonists on both healthy volunteer blood samples, and samples from patients diagnosed with primary ITP. Unlike ibrutinib, rilzabrutinib did not interfere with normal platelet aggregation responses to any platelet agonists in healthy subjects (Fig. 3A). This was further confirmed in platelet samples from patients with ITP, demonstrating no impact on normal platelet aggregation function in platelets treated with rilzabrutinib (Fig. 3B, Supplemental Table II). In contrast, ibrutinib treatment significantly impacted collagen platelet aggregation responses (Fig. 3C) in confirmation with published reports (25). In these studies, a positive control platelet inhibitor roxifiban inhibited platelet aggregation >80% as expected.
FIGURE 3.
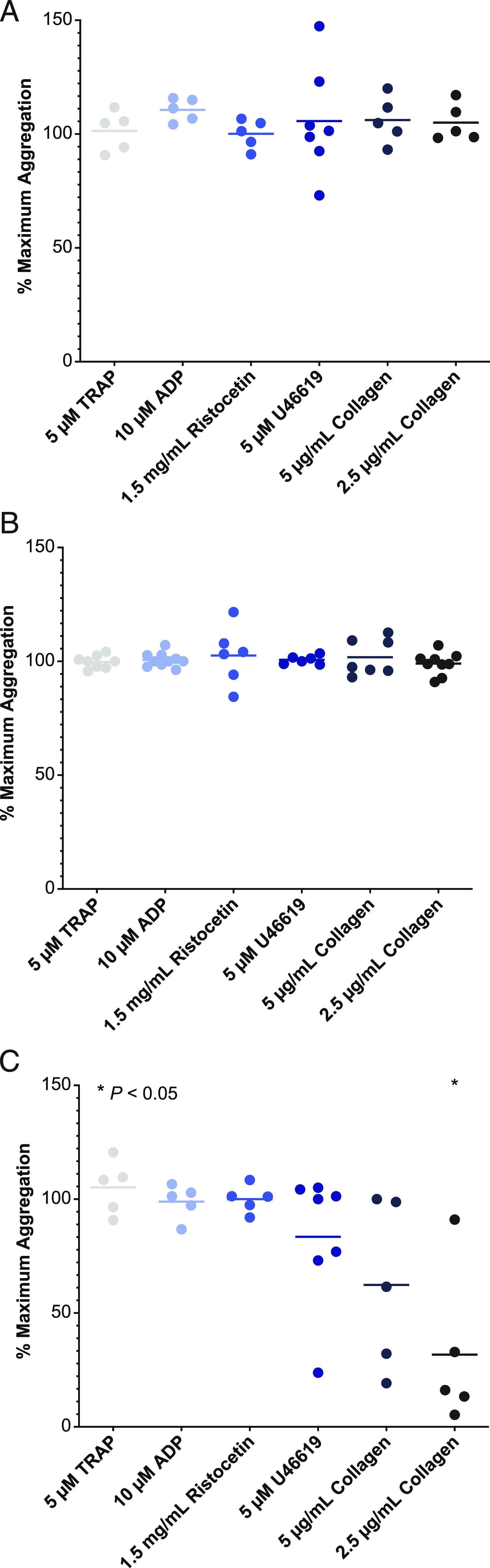
Platelet aggregation and function in healthy volunteers and ITP patients treated with rilzabrutinib. Plasma from human healthy volunteers [HVs; n = 5 (A)] or ITP patients treated with rilzabrutinib 1 μM [n = 7 (B)] or with ibrutinib 1 μM in HVs [n = 5 (C)] were studied to evaluate their impact on platelet aggregation. Plotted is the percent of maximum platelet aggregation of compound-treated samples normalized to that of untreated samples for each of the indicated platelet agonists and compared using a two-tailed t test versus DMSO control. Only the ibrutinib-treated 2.5 μg/ml collagen group (in C) was statistically significant at *p < 0.05.
Blockade of B cell activation, IgM, and IgG Ab production with rilzabrutinib treatment
Rilzabrutinib demonstrated inhibition of B cell activation, BCR-dependent B cell proliferation, and de novo Ab production. To determine the functional activity of rilzabrutinib, the ability of the compound to inhibit B cell activation as assessed by anti-IgM–induced CD69 expression in CD20+ B cells in human whole blood was determined (Fig. 4A). The potency of rilzabrutinib in the B cell activation assay was well correlated with BTK target occupancy. The IC50 determinations were 126 ± 32 nM and 233 ± 75 nM for inhibition of CD69 B cell expression and BTK target occupancy, respectively. As an additional measure of the effects of rilzabrutinib on B cell function, the ability of rilzabrutinib to inhibit BCR-induced human B cell proliferation was determined. The IC50 of rilzabrutinib for inhibition of human B cell proliferation was 5 ± 2.4 nM. Rilzabrutinib did not block ADCC in combination with anti-CD20 Abs, indicating potential for combination therapies (26).
FIGURE 4.

Cellular characterization of rilzabrutinib by evaluating inhibition of B cell activation and in vitro IgG and IgM Ab production. (A) Inhibition of B cell activation as measured by anti-IgM–induced CD69 expression on CD20+ B cells from human whole blood treated with increasing concentrations of rilzabrutinib, as measured by flow cytometry (n = 4). (B) Inhibition of IgG and IgM production. Rilzabrutinib-treated enriched human B cells were stimulated with CpG (n = 4), TNP-LPS (n = 4–6) or anti-CD40 + IL-21 (n = 2–7) for 7 d. Measurement of IgG and IgM showed that both T-dependent and T-independent Ab production were inhibited by rilzabrutinib. Curves are representative experiments. Table provides IC50 profiles of IgG and IgM production in rilzabrutinib-treated B cells.
Rilzabrutinib significantly inhibited IgM and IgG Ab production when stimulated in vitro. Rilzabrutinib inhibited IgG Ab production with an IC50 of 20 ± 20 nM and IgM Ab production with an IC50 of 800 ± 1000 nM when stimulated by a T cell–dependent pathway with anti-CD40 in combination with IL-21 (Fig. 4B). Rilzabrutinib also inhibited IgG and IgM Ab production when stimulated by T-independent pathways with the TLR-9 agonist CpG, with an IgG IC50 of 50 ± 90 nM, and IgM IC50 of 1 ± 1 nM, as well as with TNP-LPS–stimulated Ab production with IgG IC50 of 300 ± 600 nM and IgM IC50 of 200 ± 600 nM (Fig. 4B).
Rapid neutralization of pathogenic IgG autoantibodies through inhibition of FcγR pathways
To confirm the essential role of BTK on IgG-mediated FcγR pathways, human monocytes enriched from PBMCs were activated with IgG Abs and the resultant TNF-α production was analyzed. The results show that rilzabrutinib had an IC50 for inhibition of TNF-α production of 56 ± 45 nM (n = 6), confirming a significant reduction in pathogenic IgG Ab signaling through inhibition of FcγR pathways, thereby preventing inflammatory effects that would normally occur (Fig. 5A, Table I).
FIGURE 5.

Ab-mediated Fc-receptor pathway effects in innate immune cells (A), passive Arthus reaction measured by reduction in diameter (B), and OD of intradermal dye extravasation (C) following Ab challenge. (A) Inhibition of IgG/FcγR–stimulated human monocytes as measured by TNF-α production following treatment with increasing doses of rilzabrutinib (n = 6). Rilzabrutinib treatment resulted in potent inhibition of FcγR-mediated monocyte stimulation with an IC50 of 56 ± 45 nM. (B and C) Sprague Dawley rats were treated for 3 d with oral vehicle, prednisolone (10 mg/kg once daily, positive control), or rilzabrutinib (10, 20, or 40 mg/kg once daily) followed by IgG-mediated Arthus induction (n = 5–8 per group). The reaction was measured 4 h after Ab administration by the diameter of EBD extravasation (B) and dye density was recorded at an OD (C) measurement at 610-nm wavelength.
To demonstrate the role of BTK in autoantibody destructive processes, we assessed rilzabrutinib in the IgG immune complex driven Arthus reaction model (27), which is dependent on IgG Abs and FcγR on macrophages and neutrophils. Rilzabrutinib demonstrated significant dose-dependent inhibition of passive Arthus reaction in a rat model, as measured by reduction in diameter (Fig. 5B) and OD (Fig. 5C) of intradermal dye extravasation following an Ab challenge. Significant inhibitory effects were observed at all doses. At the highest dose of rilzabrutinib 40 mg/kg, a reduction in dye extravasation magnitude was achieved similar to high-dose CS treatment.
Rilzabrutinib was assessed in a model of lupus nephritis, a passive IgG Ab-induced form of nephritis with Abs specifically targeting the GBM, was used to assess downstream Ab effector responses inducing inflammation and kidney damage. Rilzabrutinib dose-dependently inhibited anti-GBM nephritis and significantly protected mice from kidney inflammation and pathology (Fig. 6). Key identifiers of kidney inflammation and pathology included glomerulus diameter, percent glomeruli with crescents, interstitial inflammation, protein casts scores, and vasculitis. Treatment with 40 mg/kg rilzabrutinib once daily significantly reduced glomerulus scores (58% reduction), percent glomeruli with crescents (92%), and interstitial inflammation (92%) compared with once daily vehicle controls (Fig. 6A). These results were similar to those seen in the dexamethasone control group: 64% reduction in glomerulus scores, 60% reduction glomerulus diameters, 64% reduction in glomerular subjective scores of inflammation and necrosis. Additional improvements in kidney function as measured by mean serum BUN levels (Fig. 6B) and lower incidence of severe proteinuria (Fig. 6C) were observed with increasing doses of rilzabrutinib treatment.
FIGURE 6.
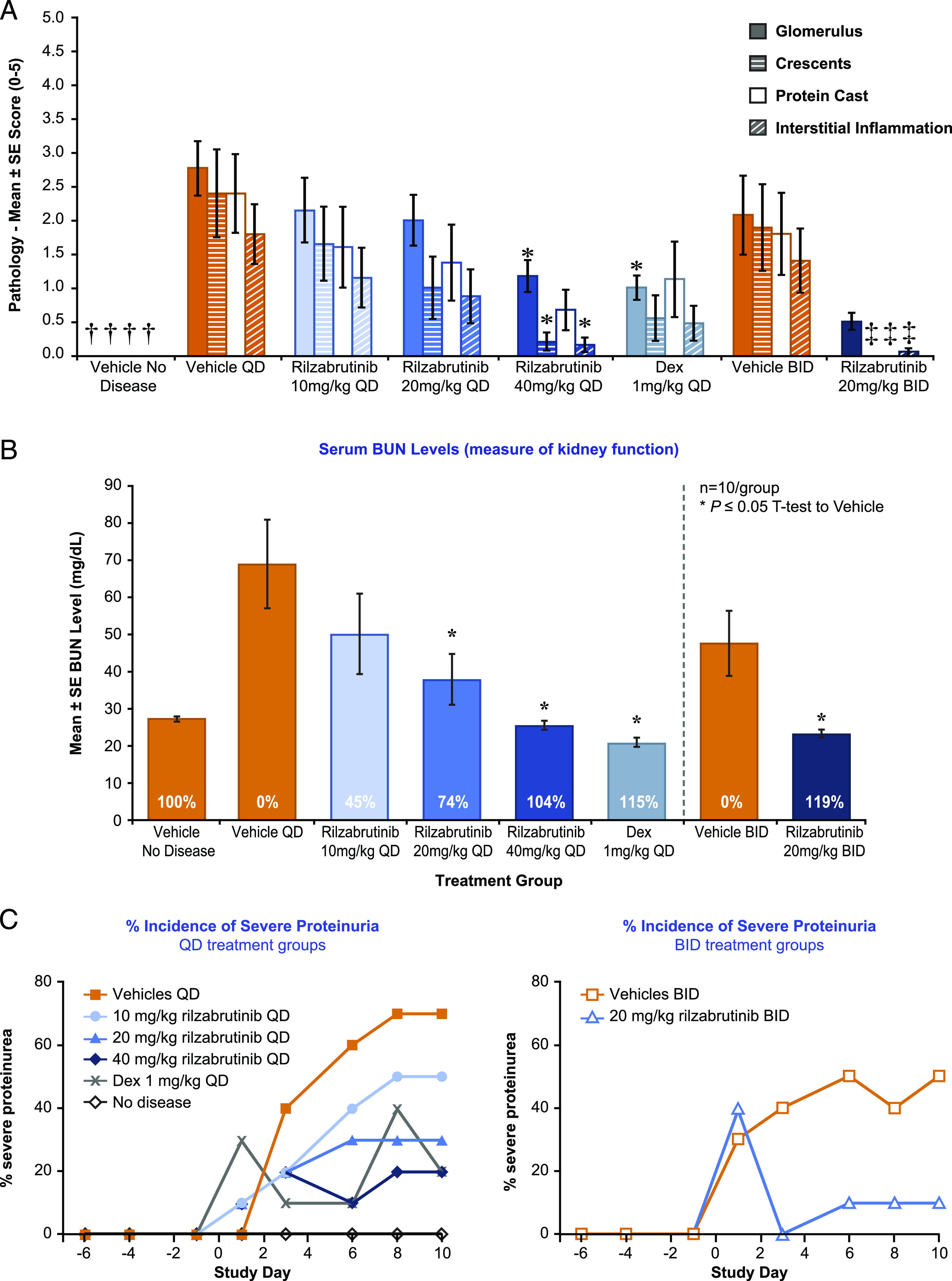
Effect of rilzabrutinib in a sheep anti-rat GBM Ab-induced lupus nephritis model of female 129X1/SvJ mice. A mouse model of GBM Ab-induced lupus nephritis was treated with vehicle, dexamethasone (1 mg/kg; positive control), or rilzabrutinib (once daily or twice-a-day dosages) on days −1 through 10 (through day 9 for positive control) (n = 10 per group). Representative photomicrograph images for each of the groups are shown in Supplemental Fig. 1. (A) Efficacy evaluations were based on glomerulus, crescents, protein casts scores, and interstitial inflammation compared with vehicle controls (n = 10 per group; n = 9 rilzabrutinib 20 mg/kg once daily). *p < 0.05 ANOVA (Dunnett post hoc) versus vehicle once daily; †p < 0.05 Student t test versus vehicle once daily; ‡p < 0.05 Student t test versus vehicle twice a day. (B) Kidney function was measured by serum BUN levels (n = 7–10 per group). *p < 0.05 by ANOVA (Dunnett post hoc) versus vehicle once daily or Student t test versus vehicle twice a day. (C) Incidence of severe proteinuria (>300 mg/dl) was assessed across all treatment groups (n = 10 per group). Urine protein levels to evaluate severe proteinuria were measured over time, with vehicle control mice showing generally expected increased proteinuria scores over the course of the study. At study termination, 7 out of 10 once daily vehicle mice and 5 out of 10 twice-a-day vehicle mice had >300 mg/dl urine protein. Mice treated with rilzabrutinib (once daily or twice a day) or dexamethasone demonstrated a lower incidence of severe proteinuria, but did not differ significantly in absolute urine protein score from their respective vehicle controls over time.
In addition, the effect of rilzabrutinib was assessed in a mouse ITP model passively induced by IgG Abs that bind glycoprotein IIb/IIIa (CD41) on the surface of platelets, resulting in rapid destruction of platelets through splenic macrophages and potentially liver Kupffer cells. Pretreatment with rilzabrutinib was sufficient to rapidly and dose-dependently reduce platelet loss (when measured at 6 h post–anti-CD41 administration) compared with vehicle control–treated animals (Fig. 7). These results demonstrate that rilzabrutinib can rapidly inhibit Ab-mediated innate immune mechanisms in addition to effects on Ab production, supporting the rationale for the use of rilzabrutinib in the treatment of ITP and other autoantibody-mediated diseases.
FIGURE 7.

Evaluation of platelet loss in mouse anti-CD41–induced ITP model. BALB/c mice (n = 8 per group) were treated with vehicle, rilzabrutinib (10, 20, or 40 mg/d once daily), or IVIG 1000 mg/kg i.v. prior to ITP model induction with anti-CD41 (given i.p.). Blood was collected 6 h post–Ab challenge, and platelets were evaluated versus vehicle control. Unmanipulated naive mice were included as a reference control. *p < 0.05, **p < 0.001 compared with vehicle control.
Rapid neutralization of IgE autoantibody responses through inhibition of FcεR pathways
The activation of basophils through cross-linking of FcεR is a BTK-dependent function (28, 29). Rilzabrutinib was able to inhibit IgE-mediated basophil activation in whole blood as measured by degranulation and changes in surface CD63 expression (Fig. 8A), achieving an IC50 of 490 ± 130 nM. Similar inhibition of mast cells was also observed (data not shown). Inhibition of basophil activation was subsequently confirmed clinically in phase 1 healthy volunteer subjects following multiday dosing (18, 30).
FIGURE 8.

IgE/FcεR–stimulated basophils and a mouse PCA-reaction model. (A) Inhibition of IgE/FcεR–stimulated basophils. Rilzabrutinib-treated human whole blood was stimulated with IgE, then assessed for CD63 activation on CD123+/HLA-DR basophils (n = 9). Rilzabrutinib inhibited FcεR activation in basophils with an IC50 of 490 ± 130 nM. (B) PCA-reaction model was induced in BALB/c mice who were treated for 3 d with oral vehicle, prednisolone (10 mg/kg once daily, positive control), cyproheptadine (25 mg/kg once daily i.p., positive control), or rilzabrutinib (20 or 40 mg/kg once daily) followed by IgE-mediated Arthus induction (n = 5 per group). The reaction was measured 4 h after Ab administration by the diameter of EBD extravasation (B) and dye density was recorded at an OD measurement at 610-nm wavelength (C).
Similar to the IgE-dependent PCA model, rilzabrutinib showed significant, rapid, and dose-dependent neutralization of pathogenic IgE Abs by inhibiting allergic IgE/FcεR pathways. Once-daily oral administration with 40 and 80 mg/kg rilzabrutinib resulted in 26 and 34% of reduction in the extravasation area, respectively, and 26 and 55% reduction in the extravasation density, respectively, with no adverse effects (Fig. 8B, 8C). Twice-daily oral administration of rilzabrutinib at 40 mg/kg yielded the greatest response: a 64% reduction in the extravasation area and a 65% reduction in extravasation intensity compared with 96 and 69% in the steroid control group (daily oral administration of 10 mg/kg prednisolone), respectively.
BTK target occupancy confirmed, demonstrating durable binding of rilzabrutinib
To examine in vivo occupancy, the relationship between PD (measured as occupancy of BTK) and PK (measured as plasma concentration) of rilzabrutinib was evaluated in female Sprague Dawley and Lewis rats. In general, BTK occupancy increased with increasing oral doses, and occupancies observed at 14 h after dosing in the two rat strains were similar (Fig. 9A). The study demonstrated that BTK occupancy was maintained after the plasma levels of rilzabrutinib dropped to low levels <3 ng/ml (at 14 h), consistent with durable binding of rilzabrutinib to BTK in vivo.
FIGURE 9.
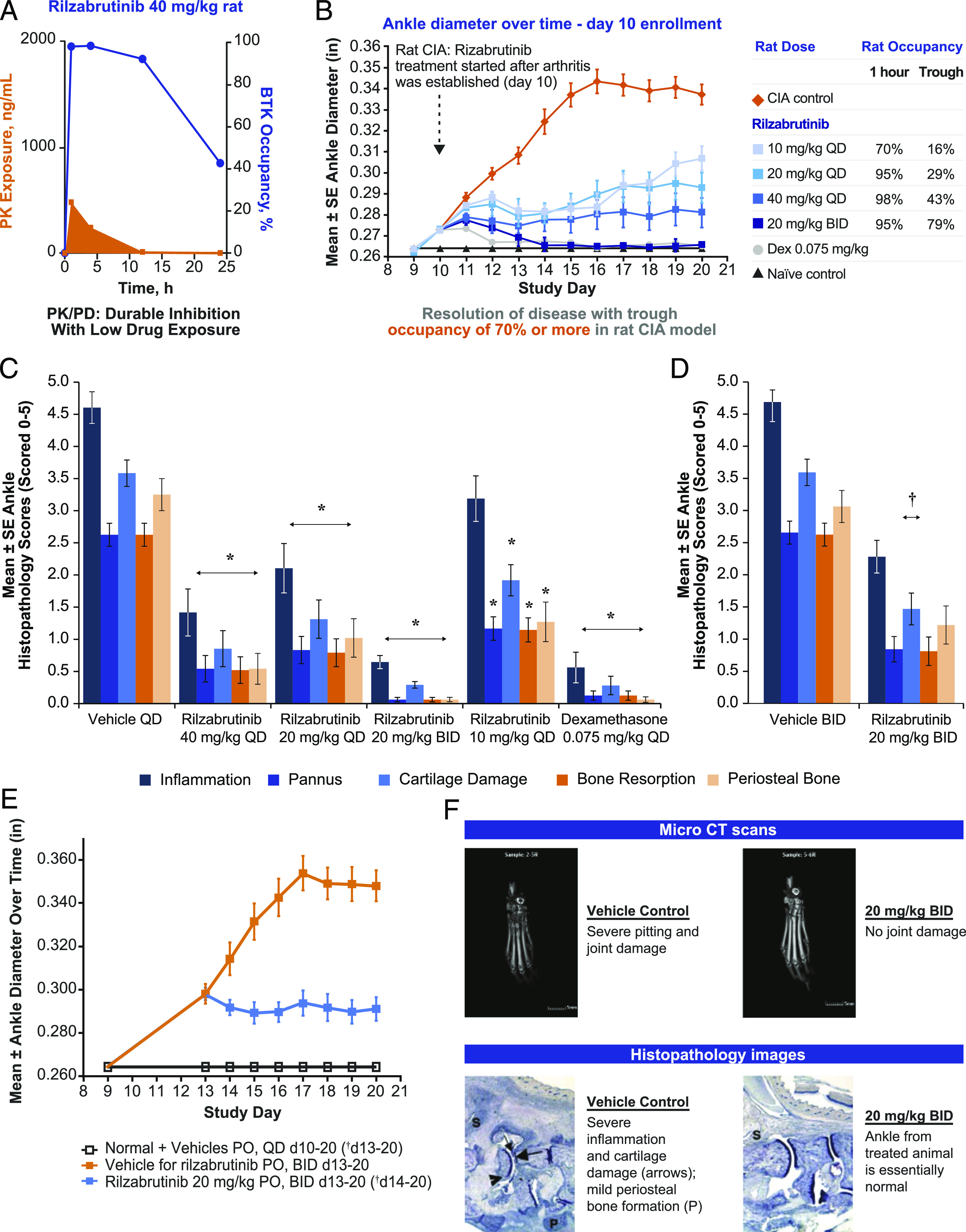
Effects of rilzabrutinib on BTK occupancy, PK, and inflammation in CIA in rats. (A) BTK occupancy in female Lewis rat splenocytes was measured following oral administration of 40 mg/kg rilzabrutinib (mean, n = 3 per timepoint per group) and normalized to vehicle control. Rilzabrutinib PK levels were assessed, and spleen percent BTK occupancy was measured by binding of an irreversible fluorescent probe to unbound BTK in splenic cell samples. (B) Female Lewis rats with established CIA were treated orally with vehicle, dexamethasone (positive control), or rilzabrutinib (10, 20, or 40 mg/kg once daily or 20 mg/kg twice a day) on study days 10–20 (arthritis days 1–11; n = 12 per group). On study day 11, efficacy evaluations were performed on ankle diameter. Sustained BTK occupancies ≥79% achieved with twice-a-day dosing resulted in complete reversal of experimental arthritis scores as shown by ankle diameter. All treatment groups were significantly reduced p < 0.05 compared with vehicle control (orange diamond). (C) Mean individual ankle histopathology scores for CIA rats treated orally with vehicle daily (n = 4), dexamethasone 0.075 mg/kg daily (n = 8), or rilzabrutinib 10, 20, or 40 mg/kg once daily or 20 mg/kg twice a day (n = 12 per group) on study days 10–20. *p < 0.05 compared with vehicle daily control. (D) Mean individual ankle histopathology scores for CIA rats treated orally with vehicle twice a day (n = 4) or rilzabrutinib 20 mg/kg twice a day (n = 8) on study days 13–20. †p < 0.05 compared with vehicle twice-a-day control. (E) Reversal of experimental arthritis disease was achieved with delayed dosing initiated at study day 13 in established rat CIA model. Error bars represent SD; n = 4 normal controls; and n = 8 per treatment group. †p < 0.05 compared with vehicle for rilzabrutinib twice-a-day control. (F) Microcomputed tomography and histopathology imaging showed that joint damage was prevented by treatment with rilzabrutinib.
The CIA study was conducted to translate occupancy and efficacy and to assess rilzabrutinib’s potential ability to reverse established disease. When dosing was initiated therapeutically on study day 10 (at first signs of joint inflammation), rilzabrutinib treatment resulted in reduced clinical arthritis scores in a dose-dependent manner. Fig. 9B showed reduction of disease activity when BTK occupancy was between 16 and 80% at trough plasma concentrations, with maximal disease reversal achieved at a dosage of 20 mg of rilzabrutinib administered every 12 h. This dose achieved 100% inhibition following oral administration, falling to no <79% at trough, which equated to an average daily BTK occupancy level of ∼90%. The overall level of efficacy correlated closely with the trough occupancy levels achieved in each dosing level (Supplemental Table III). All ankle histopathology parameters were significantly reduced toward normal for rats given rilzabrutinib 20 mg/kg once daily (64% reduction of summed scores), rilzabrutinib 40 mg/kg once daily (77%), rilzabrutinib 20 mg/kg twice a day (93%), or dexamethasone (93%) compared with day 10–20 vehicle control (Fig. 9C), and for rats given rilzabrutinib 20 mg/kg twice a day (60%) compared with day 13–20 vehicle control (Fig. 9D). Significantly reduced individual ankle scores for the rilzabrutinib 10 mg/kg once daily dosage were observed in ankle pannus (56%), cartilage damage (47%), bone resorption (56%), and periosteal bone formation (61%; Fig. 9C compared with day 10–20 vehicle). When dosing was initiated on day 13, when disease levels were more severe and established (day 4 of arthritis), rilzabrutinib at 20 mg/kg twice a day resulted in disease reversal (Fig. 9E); treatment with rilzabrutinib achieved equivalent efficacy to dexamethasone when dosed at 40 mg/kg once daily or 20 mg/kg twice a day, as confirmed by evaluation of histopathology. All rats given rilzabrutinib had significant reduction in ankle histopathology scores and survived to study termination. As shown in the microcomputed tomography scans and histopathology images, rilzabrutinib prevented joint damage as compared with vehicle control (Fig. 9F) and indicated an ability to inhibit inflammation progression and reverse damage of CIA in rats.
Proof-of-concept achieved with rilzabrutinib in naturally occurring canine pemphigus, achieving rapid clinical responses when given as monotherapy
In four companion dogs with naturally occurring pemphigus, treatment with rilzabrutinib as monotherapy resulted in immediate and rapid clinical improvement, which was significant for three dogs after dosage adjustments to twice a day. Visible improvement in outward features in one representative dog (no. 1) is shown in Fig. 10A. The clinical cPDAI scores showed 77–100% improvement within 2 wk of rilzabrutinib treatment (Fig. 10B). A BTK occupancy range of 50–93% (Fig. 10C), although within the target range, did not correlate with partial versus full clinical response. Rilzabrutinib induced rapid anti-inflammatory effects within 2 wk, as all four animals had an immediate clinical improvement, and subsequently went on to either complete or substantial disease control with the monotherapy, demonstrating that treatment with rilzabrutinib can potentially spare the use of systemic CS (31).
FIGURE 10.

Rilzabrutinib monotherapy (without CSs) was effective for treatment of canines with naturally occurring PF. As an open-label crossover study, four dogs were treated with rilzabrutinib 15 mg/kg oral once daily (or 30 mg/kg divided twice a day per clinical response and BTK occupancy). (A) A representative 13-y-old, yellow Labrador retriever with an initial cPDAI score of 69.5 began with a 500 mg/d dosage of rilzabrutinib and was escalated to 500 mg every 12 h at week 4. Her cPDAI score was 14.5 at week 8 (80% improvement) and 12 at week 20. Significant decreases in lesions and regrowth of nearly all her coat was achieved by week 20. (B) cPDAI scores over time showed 77–100% improvement with rilzabrutinib dosing as monotherapy (n = 4 dogs). (C) BTK occupancy was measured similar to the above in vivo studies; measurements ranged from 50 to 93% at the 4-h (peak) and 24-h (trough) timepoints. Although within the target range, occupancy did not correlate with partial versus full clinical response.
Discussion
The goal of immune-mediated treatments is rapid and durable efficacy with safety profiles suitable for long-term administration. Current therapies for immune-mediated diseases include CS monotherapy or CS combined with a steroid-sparing immunosuppressant drug, such as rituximab (anti-CD20 mAb). However, these therapies present with some pharmacological challenges that compromise efficacy and elicit safety concerns (e.g., serious, and often long-term, morbidities). CS is routinely administered with rituximab to avoid reactions and compensate for the months-long delay in its onset of action (32). The critical role of BTK in multiple immune signaling pathways make it a suitable target for immune therapies. However, marketed covalent BTKi irreversibly bind to off-target kinases, such as EGFR, MKK7, ITK, and JAK3 (17) and potentially to many off-target proteins beyond kinases (33). For instance, ibrutinib concomitantly blocks the PI3K-Akt signaling pathway. Such off-target affinity may lead to clinically observed, bleeding-related adverse events (34) and atrial fibrillation (14), hindering its application and development in the immunology disease space.
Rilzabrutinib was identified as a potent, selective, reversible covalent and orally available inhibitor of BTK and was the first to be tested in the immune-mediated disease setting. These characteristics may address the goal of improved efficacy via high selectivity and limited off-targeted adverse events without the risk of permanent modification. Rilzabrutinib was evaluated in various experimental systems to characterize the pharmacological profile, including selectivity, reversibility, BTK occupancy, biological mechanism of action, and understanding these effects translate into efficacy benefits. Through these studies, we gain detailed insights about rilzabrutinib’s pharmacological profile, mechanism of action, and how rilzabrutinib differentiates from other irreversible covalent BTKi and standard of care treatments such as CS and anti-CD20 B cell–depleting Abs.
Rilzabrutinib proved potent and highly selective for BTK when tested against a broad panel of kinases and cellular assays, while leaving EGFR signaling, T cell activation, and ADCC unaffected. Rilzabrutinib demonstrates more durable binding to BTK than other closely related TEC kinases with additional selectivity enhancement with time, further reducing any potential off-target–binding events. Rilzabrutinib demonstrated that covalent cysteine binding can be completely reversed after denaturation of the target. A reversible covalent inhibitor enables modification of dissociation rates as required for target engagement and fine-tuning of inhibitor residence time, making it suitable for therapeutic applications that require sustained target engagement and/or faster target disengagement. Existing covalent BTKi are characterized by irreversible binding. Drugs that rely intrinsically on irreversible chemistry (e.g., acrylamides) may form permanent covalent adducts with off-target proteins, such as closely related kinases with a homologous cysteine or unrelated targets with hyperreactive cysteines, potentially being less tolerable for patients with chronic immune-mediated diseases. Rilzabrutinib’s reversible covalent inhibition could allow for long-term, systemic treatment of immune-mediated diseases while avoiding the permanent modification of proteins and peptides (17, 19).
Rilzabrutinib demonstrated substantial cellular occupancy that was confirmed in vivo, where high and prolonged BTK occupancy was retained in rodents even after rilzabrutinib plasma concentrations reached the lower limit of detection. This effect was obtained through a combination of high specificity with a long residence time, which can be attributed to the chemical design (18). The advantage of such favorable characteristics is that they allow for lower doses and/or less frequent administration, while achieving optimal results to improve the patient’s condition. Although bound rilzabrutinib remained attached and active for prolonged periods of time, its free, unbound form had a short plasma half-life (18). Relative to noncovalent drugs, rilzabrutinib demonstrates that only short exposure is required to realize clinical benefit. This creates an environment of very low systemic exposure, which reduces the chance of adverse side effects and enhances tolerability, while remaining effective over extended periods of time. This provides the advantage of achieving both efficacy and safety without trade-off.
The BTK inhibitory activities of rilzabrutinib act on a variety of immune cells and pathways to prohibit dysfunctional and potentially harmful inflammatory responses. The results and conclusions of multiple in vitro and in vivo studies reported in this study provide a solid foundation for translation to several ongoing human clinical trials. Rilzabrutinib inhibits B cell activation through inhibition of the BCR; however, it does not lead to B cell depletion or cellular cytotoxicity. This is a key differentiator from current therapies such as rituximab, as maintaining B cells may allow fast reversal of inhibition and alleviate concerns for prolonged immune suppression. Beyond the B cell, BTK is also expressed in many innate immune cells, which are primed for rapid immune responses. Rilzabrutinib rapidly and effectively inhibits Ab-mediated immune cell activation via Fc-receptor signaling. The activation of monocytes and macrophages through cross-linking of IgG and FcγR is a BTK-dependent function; the ability of rilzabrutinib to inhibit IgG-mediated FcγR activation was demonstrated in rodent models of the Arthus reaction, lupus nephritis, and ITP. Rilzabrutinib can also neutralize the IgE response although inhibition of FcεR signaling to halt basophil or mast cell activation, with no adverse effects. Its effect on the IgE pathway, supported by preclinical and clinical evidence of BTK inhibition in basophils (30), highlights a potential use for rilzabrutinib for treatment of allergy and anaphylaxis, in which this is a significant unmet need for effective therapeutics (29). The combination of these two critical aspects of efficient autoantibody suppression, both the rapid effects on halting self-reactive Ab signaling, and the longer-term impact on preventing the formation of new autoantibodies, gives rilzabrutinib an unmatched dual Ab mechanistic approach over the current standard treatments for immune-mediated diseases.
Rilzabrutinib’s inhibition of BTK also results in the disruption of a signaling pathway for neutrophil recruitment. As overproduction of reactive oxygen species by neutrophils has been linked to tissue damage, mitigation of neutrophil recruitment may be beneficial for patients with immune-mediated disorders (35). However, rilzabrutinib is not active on B cells stimulated via a non-BTK–mediated pathway or T cells, sparing other receptors and signaling pathways that are essential for maintaining lymphocyte counts, antiviral responses, and immunity in immune-compromised patients. Additionally, rilzabrutinib did not impact normal platelet aggregation and does not seem to share the increased risk of bleeding associated with an irreversible BTKi, ibrutinib, which is likely because of impaired platelet function and dysfunctional thrombus formation (36).
Rilzabrutinib specifically inhibits a range of inflammatory cellular activities in mast cells, basophils, neutrophils, and other cells activated in immune-mediated diseases, without killing those cells. This response is fast, as evidenced by rilzabrutinib’s rapid efficacy in a mouse model of ITP and in rats with CIA. Significant and dose-dependent inhibition of arthritis scores were achieved with therapeutic treatment after joint inflammation was established, and reversal of disease was apparent when initiation of treatment was further delayed until animals had more severe established disease. A strong relationship of BTK occupancy and efficacy was confirmed, with data supporting that high and sustained levels of BTK occupancy (maximal 100% occupancy at peak, falling to no lower than 79%, equating to a daily average ∼90%) was required to achieve maximal efficacy. A level of target inhibition that cannot easily be reached and maintained by noncovalent approaches. Rilzabrutinib, through reversible covalent binding, was designed to achieve these high and sustained occupancy levels, while minimizing off-target activities and potential dose-limited toxicology through reversibility and enhanced selectivity. Importantly, this concept of occupancy and efficacy relationship translated to the human clinical setting, where high and sustained BTK occupancy levels were achieved with rilzabrutinib treatment in the clinic in phase 1 (18) and informed dose selection for further clinical studies. This occupancy and efficacy relationship was confirmed in patients with pemphigus, demonstrating that high and sustained inhibition of BTK, consistent with the preclinical and early clinical data, was required for maximal therapeutic effect (20).
Another example of rapid disease modulation with rilzabrutinib treatment was achieved clinically in canine pemphigus, a disease in dogs that is analogous to the autoimmune blistering disease in humans (31). The therapeutic potency of rapid anti-inflammatory effects, neutralization of pathogenic autoantibodies, and blockade of Ab production was further supported by the results of rilzabrutinib administration to newly diagnosed dogs afflicted with naturally occurring PF. Conventional treatment comprises a relatively high dosage of 1 mg/kg prednisolone twice daily, combined with steroid-sparing immunosuppressive agents. Response is typically seen within 4 wk, and some level of maintenance therapy is usually required. With rilzabrutinib treatment, a high response rate was achieved within just 2 wk without the need of CS, suggesting that rilzabrutinib may provide a safe alternative therapy to the current standard of care for human patients. Rilzabrutinib’s rapid action may reduce or potentially completely abolish the need for concomitant administration of CS, thus minimizing possible CS adverse side effects.
Because the BTK protein is highly conserved across species, the efficacy of BTKis in canine PF may translate well to the analogous autoimmune blistering disease in humans. Although these animal studies are relatively small, they provide sufficient translation to human studies for the evaluation of clinical efficacy and safety with high confidence in a successful outcome. Combined, these results suggest that rilzabrutinib may be a promising first-line treatment for pemphigus to quickly and effectively alleviate symptoms in human patients without the downsides of existing therapies.
In conclusion, our results show that, pharmacologically, rilzabrutinib has selectivity, complete reversibility, and high and prolonged BTK occupancy with low systemic exposure, thus providing the advantage of high sustained inhibition, minimal off-target activity, and low drug levels. Additionally, data show that rilzabrutinib has multiple immune mechanisms of action across both adaptive and innate immune cells, including rapid anti-inflammatory effects, neutralization of pathogenic autoantibody signaling, and prevention of new autoantibody production. These results support rilzabrutinib’s potential development in immunology as a promising oral, reversible covalent BTKi for the treatment of immune-mediated disease.
Supplementary Material
Acknowledgments
These studies were supported by Principia Biopharma Inc.
The online version of this article contains supplemental material.
- ADCC
- Ab-dependent cell-mediated cytotoxicity
- ATCC
- American Type Culture Collection
- BTK
- Bruton tyrosine kinase
- BTKi
- BTK inhibitor
- BUN
- blood urea nitrogen
- CIA
- collagen-induced arthritis
- cPDAI
- canine pemphigus disease activity index
- CS
- corticosteroid
- EBD
- Evans blue dye
- FRET
- fluorescence resonance energy transfer
- GBM
- glomerular basement membrane
- ITP
- immune thrombocytopenia
- PCA
- passive cutaneous anaphylaxis
- PD
- pharmacodynamics
- PF
- pemphigus foliaceus
- PK
- pharmacokinetics
- PRP
- platelet-rich plasma.
Disclosures
C.L.L., J.M.B., T.D.O., M.R.F., Y.X., J.S., J.L., K.A.B., D.M.G., P.A.N. are employees of and receive stock ownership from Principia Biopharma Inc. A.B. is an employee of and receives stock ownership from Precision for Medicine. R.J.H. is an employee of and receives stock ownership from AbbVie. The other authors have no financial conflicts of interest.
References
- 1.Mohamed, A. J., Yu L., Bäckesjö C. M., Vargas L., Faryal R., Aints A., Christensson B., Berglöf A., Vihinen M., Nore B. F., Smith C. I.. 2009. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 228: 58–73. [DOI] [PubMed] [Google Scholar]
- 2.Crofford, L. J., Nyhoff L. E., Sheehan J. H., Kendall P. L.. 2016. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 12: 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rip, J., Van Der Ploeg E. K., Hendriks R. W., Corneth O. B. J.. 2018. The role of Bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit. Rev. Immunol. 38: 17–62. [DOI] [PubMed] [Google Scholar]
- 4.Herter, J. M., Margraf A., Volmering S., Correia B. E., Bradshaw J. M., Bisconte A., Hill R. J., Langrish C. L., Lowell C. A., Zarbock A.. 2018. PRN473, an inhibitor of Bruton’s tyrosine kinase, inhibits neutrophil recruitment via inhibition of macrophage antigen-1 signalling. Br. J. Pharmacol. 175: 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volmering, S., Block H., Boras M., Lowell C. A., Zarbock A.. 2016. The neutrophil Btk signalosome regulates integrin activation during sterile inflammation. Immunity 44: 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pharmacyclics LLC . 2020. Imbruvica (ibrutinib) Prescribing Information. Pharmacyclics LLC, Sunnyvale, CA. [Google Scholar]
- 7.Miklos, D., Cutler C. S., Arora M., Waller E. K., Jagasia M., Pusic I., Flowers M. E., Logan A. C., Nakamura R., Blazar B. R., et al. 2017. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood 130: 2243–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.AstraZeneca Pharmaceuticals LP . 2019. Calquence (acalabrutinib) Prescribing Information. AstraZeneca Pharmaceuticals LP, Wilmington, DE. [Google Scholar]
- 9.Awan, F. T., Schuh A., Brown J. R., Furman R. R., Pagel J. M., Hillmen P., Stephens D. M., Woyach J., Bibikova E., Charuworn P., et al. 2019. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 3: 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byrd, J. C., Harrington B., O’Brien S., Jones J. A., Schuh A., Devereux S., Chaves J., Wierda W. G., Awan F. T., Brown J. R., et al. 2016. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 374: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jurczak, W., Długosz-Danecka M., Wang M.. 2019. Acalabrutinib for adults with mantle cell lymphoma. Expert Rev. Clin. Pharmacol. 12: 179–187. [DOI] [PubMed] [Google Scholar]
- 12.Montalban, X., Gold R., Thompson A. J., Otero-Romero S., Amato M. P., Chandraratna D., Clanet M., Comi G., Derfuss T., Fazekas F., et al. 2018. ECTRIMS/EAN Guideline on the pharmacological treatment of people with multiple sclerosis. [Published erratum appears in 2020 Mult. Scler. 26: 517.] Mult. Scler. 24: 96–120. [DOI] [PubMed] [Google Scholar]
- 13.Aguilar, C. 2018. Ibrutinib-related bleeding: pathogenesis, clinical implications and management. Blood Coagul. Fibrinolysis 29: 481–487. [DOI] [PubMed] [Google Scholar]
- 14.McMullen, J. R., Boey E. J., Ooi J. Y., Seymour J. F., Keating M. J., Tam C. S.. 2014. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood 124: 3829–3830. [DOI] [PubMed] [Google Scholar]
- 15.Johnson, D. S., Weerapana E., Cravatt B. F.. 2010. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2: 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou, S., Chan E., Duan W., Huang M., Chen Y. Z.. 2005. Drug bioactivation, covalent binding to target proteins and toxicity relevance. Drug Metab. Rev. 37: 41–213. [DOI] [PubMed] [Google Scholar]
- 17.Bradshaw, J. M., McFarland J. M., Paavilainen V. O., Bisconte A., Tam D., Phan V. T., Romanov S., Finkle D., Shu J., Patel V., et al. 2015. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 11: 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith, P. F., Krishnarajah J., Nunn P. A., Hill R. J., Karr D., Tam D., Masjedizadeh M., Funk J. O., Gourlay S. G.. 2017. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 83: 2367–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serafimova, I. M., Pufall M. A., Krishnan S., Duda K., Cohen M. S., Maglathlin R. L., McFarland J. M., Miller R. M., Frödin M., Taunton J.. 2012. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 8: 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murrell, D. F., Patsatsi A., Stavropoulos P., Baum S., Zeeli T., Kern J. S., Sinclair R., Neale A., Werth V. P., Caux F., Joly P.. 2020. BELIEVE-PV phase II part B study: extended treatment with PRN1008 improves outcomes for patients with pemphigus. American Academy of Dermatology Virtual Meeting, June 2020. Available at: https://aad.wistia.com/medias/fwkv0fhv5r. Accessed: February 23, 2021. [Google Scholar]
- 21.Chang, B. Y., Huang M. M., Francesco M., Chen J., Sokolove J., Magadala P., Robinson W. H., Buggy J. J.. 2011. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther.13: R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenbach, M., Murrell D. F., Bystryn J. C., Dulay S., Dick S., Fakharzadeh S., Hall R., Korman N. J., Lin J., Okawa J., et al. 2009. Reliability and convergent validity of two outcome instruments for pemphigus. J. Invest. Dermatol. 129: 2404–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grover, S. 2011. Scoring systems in pemphigus. Indian J. Dermatol. 56: 145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Futatani, T., Watanabe C., Baba Y., Tsukada S., Ochs H. D.. 2001. Bruton’s tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br. J. Haematol. 114: 141–149. [DOI] [PubMed] [Google Scholar]
- 25.Levade, M., David E., Garcia C., Laurent P. A., Cadot S., Michallet A. S., Bordet J. C., Tam C., Sié P., Ysebaert L., Payrastre B.. 2014. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood 124: 3991–3995. [DOI] [PubMed] [Google Scholar]
- 26.Pirunsarn, A., Kijrattanakul P., Chamnanchanunt S., Polprasert C., Rojnuckarin P.. 2018. A randomized multicenter trial comparing low-dose prednisolone versus observation for prevention of recurrences in adult immune thrombocytopenia. Clin. Appl. Thromb. Hemost. 24: 867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ravetch, J. V. 2002. A full complement of receptors in immune complex diseases. J. Clin. Invest. 110: 1759–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Advani, R. H., Buggy J. J., Sharman J. P., Smith S. M., Boyd T. E., Grant B., Kolibaba K. S., Furman R. R., Rodriguez S., Chang B. Y., et al. 2013. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 31: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dispenza, M. C., Krier-Burris R. A., Chhiba K. D., Undem B. J., Robida P. A., Bochner B. S.. 2020. Bruton’s tyrosine kinase inhibition effectively protects against human IgE-mediated anaphylaxis. J. Clin. Invest. 130: 4759–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith, P. F., Krishnarajah J., Nunn P. A., Hill R. J., Karr D., Tam D., Masjedizadeh M., Gourlay S. G.. 2015. A phase 1 clinical trial of PRN1008, an oral, reversible, covalent BTK inhibitor demonstrates clinical safety and therapeutic levels of BTK occupancy without sustained systemic exposure. EULAR, Rome. [Google Scholar]
- 31.Goodale, E. C., White S. D., Bizikova P., Borjesson D., Murrell D. F., Bisconte A., Francesco M., Hill R. J., Masjedizadeh M., Nunn P., et al. 2020. Open trial of Bruton’s tyrosine kinase inhibitor (PRN1008) in the treatment of canine pemphigus foliaceus. Vet. Dermatol. 31: 410-e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ling, Y., Qian X., Cao X.. 2013. Combination therapy of rituximab and corticosteroids for patients with refractory chronic immune thrombocytopenic purpura: report of two cases. Contemp. Oncol. (Pozn.) 17: 222–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanning, B. R., Whitby L. R., Dix M. M., Douhan J., Gilbert A. M., Hett E. C., Johnson T. O., Joslyn C., Kath J. C., Niessen S., et al. 2014. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 10: 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shatzel, J. J., Olson S. R., Tao D. L., McCarty O. J. T., Danilov A. V., DeLoughery T. G.. 2017. Ibrutinib-associated bleeding: pathogenesis, management and risk reduction strategies. J. Thromb. Haemost. 15: 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mittal, M., Siddiqui M. R., Tran K., Reddy S. P., Malik A. B.. 2014. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 20: 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bye, A. P., Unsworth A. J., Desborough M. J., Hildyard C. A. T., Appleby N., Bruce D., Kriek N., Nock S. H., Sage T., Hughes C. E., Gibbins J. M.. 2017. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. [Published erratum appears in 2018 Blood Adv. 2: 3515.] Blood Adv. 1: 2610–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.