

Abstract
Background
Non-coding RNA (ncRNA) and protein interactions play essential roles in various physiological and pathological processes. The experimental methods used for predicting ncRNA–protein interactions are time-consuming and labor-intensive. Therefore, there is an increasing demand for computational methods to accurately and efficiently predict ncRNA–protein interactions.
Results
In this work, we presented an ensemble deep learning-based method, EDLMFC, to predict ncRNA–protein interactions using the combination of multi-scale features, including primary sequence features, secondary structure sequence features, and tertiary structure features. Conjoint k-mer was used to extract protein/ncRNA sequence features, integrating tertiary structure features, then fed into an ensemble deep learning model, which combined convolutional neural network (CNN) to learn dominating biological information with bi-directional long short-term memory network (BLSTM) to capture long-range dependencies among the features identified by the CNN. Compared with other state-of-the-art methods under five-fold cross-validation, EDLMFC shows the best performance with accuracy of 93.8%, 89.7%, and 86.1% on RPI1807, NPInter v2.0, and RPI488 datasets, respectively. The results of the independent test demonstrated that EDLMFC can effectively predict potential ncRNA–protein interactions from different organisms. Furtherly, EDLMFC is also shown to predict hub ncRNAs and proteins presented in ncRNA–protein networks of Mus musculus successfully.
Conclusions
In general, our proposed method EDLMFC improved the accuracy of ncRNA–protein interaction predictions and anticipated providing some helpful guidance on ncRNA functions research. The source code of EDLMFC and the datasets used in this work are available at https://github.com/JingjingWang-87/EDLMFC.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12859-021-04069-9.
Keywords: ncRNA–protein interactions, Multi-scale features combination, Conjoint k-mer, Ensemble deep learning, Independent test, ncRNA–protein networks
Background
Genome sequencing in 2001 showed that only 2% of RNAs encode proteins, and 98% of RNAs do not code for proteins [1, 2], known as non-coding RNAs (ncRNAs). Studies have shown that ncRNAs are closely related to fundamental biological processes by interacting with RNA-binding proteins (RBPs) [3, 4], such as translation [5], splicing [6], chromatin remodeling [7], gene regulation [8], and many other life activities and functions [9–12]. In addition, ncRNAs implicate in cancer and other complex diseases [13–18]. Therefore, accurate prediction of ncRNA–protein interactions (ncRPIs) is crucial for understanding the regulatory function of ncRNAs and the pathogenesis of diseases.
High-throughput experimental techniques (RIP-Chip [19], HITS-CLIP [20], PAR-CLIP [21], etc.) and other experimental techniques of resolving complex structures (X-ray crystal diffraction (X-ray) [22], nuclear magnetic resonance (NMR) [23], electron cryo-microscopy (cryo-EM) [24], etc.) have been developed for revealing ncRPIs. However, experimental methods are time-consuming and labor-intensive [25]. Thus, there is a growing demand for the development of computational methods to predict ncRPIs.
Based on the features they used, computational methods to predict ncRPIs can be divided into two categories: sequence features as inputs and structure features as inputs. For sequence features based methods, lots of studies used machine learning or deep learning methods to learn features for predicting ncRPIs only based on the primary sequence. For instance, Muppirala et al. proposed a model named RPISeq, in which only primary sequence features were used, random forest (RF), or support vector machine (SVM) was used as classifiers to make predictions [26]. Pan et al. proposed a stacked ensemble model called IPMiner [27], learning primary sequence features from 3-mer and 4-mer frequency of protein and ncRNA, respectively. Then, Dai et al. designed a novel method, CFRP [28], put forward to generate complex features generated by non-linear transformations from the traditional k-mer features of ncRNA and protein primary sequences for characterizing ncRNA–protein interaction. RF was selected to reduce the dimensions of complex features and implement ncRNA–protein interaction (ncRPI) prediction tasks. Besides, Wang et al. utilized the deep convolutional neural network (CNN) to learn high-level features from the RNA and protein sequences, further feeding them into an extreme learning machine (ELM) for classification [29]. Furthermore, our group designed DM-RPIs, a classifier that integrated SVM, RF, and CNN to classify ncRPIs by learning the discriminative features from 3-mer and 4-mer frequency of proteins and ncRNAs, respectively [30]. In addition, LightGBM, rpiCOOL, RPIFSE, RPI-SAN, and LPI-CNNCP also made ncRPI predictions based on primary sequence [31–35].
For structure features based methods, besides sequence features, the often-used structure-derived features include secondary structure sequences, physicochemical properties, and others. Bellucci et al. proposed catRAPID [36, 37], which was based on the physiochemical properties of proteins and long non-coding RNAs (lncRNAs), including secondary structure, hydrogen bonding, and van der Waals propensities. Lu et al. proposed lncPro [38], using the same input features as Bellucci’s and fisher linear discriminant approach to implementing lncRNAs and proteins interaction predictions. Then, Suresh et al. proposed RPI-Pred [39], which combined the primary sequence and tertiary structure information of ncRNAs and proteins to predict ncRPIs. Lately, Peng et al. designed a hierarchical deep learning framework, RPITER [40], added more primary sequence information and sequence structure information by the improved conjoint triad feature coding method, which improved the classification performance of ncRPIs. Besides, Fan et al. considered pseudo nucleotide/amino acid composition and designed a novel computational method LPI-BLS by integrating logistic regression with five broad learning system classifiers [41], which performed a better classification performance than other state-of-the-art methods.
In the studies above, there are still few ones involving high-order 3D structural features. Our group found that the structural features play important roles in RNA-binding sites prediction, these structural characteristics reflect the properties around the binding sites, the clustering properties of the conserved interfacial residues, and the binding tendency [42]. We think structural features can also be used to predict ncRPIs. Furthermore, overwhelming majority of these relied on shallow machine learning techniques to implement classification tasks, such as fisher linear discriminant, RF, SVM, and logistic regression: lncPro employed fisher linear discriminant; RPI-seq employed RF and SVM; and LPI-BLS employed logistic regression. However, deep learning provides an approach to more effectively learn features from inputs and form high-level representations for more accurate prediction. One reason is that the increasing number of training samples can be derived from high-throughput sequencing techniques, which is highly beneficial for training deep learning models. The other is deep learning-based methods (especially CNN) that are powerful for analyzing spatial structure buried in data. And bi-directional long short-term memory network (BLSTM) is a widely used recurrent neural networks (RNN) with the memory cells, which can learn long dependency on the sequential data. Currently, CNN and BLSTM has been widely applied on computational biology and achieved superior performance in various biological sequence analysis problem [43], such as DNA function [44], RNA–protein binding sites [45] and protein–RNA binding preferences predictions [46].
Therefore, we proposed EDLMFC, a multi-scale features combination-based approach to predict ncRPIs through an ensemble deep learning model, which utilizes not only the primary sequence features of ncRNAs and proteins but also the structural features. These features are learned by layered networks, including CNN and BLSTM layers. Compared with the other three state-of-the-art methods, the comprehensive results demonstrate that EDLMFC has the best classification performance for ncRPI predictions.
Results
Performance comparison on EDLMFC with existing state-of-the-art methods
To evaluate the performance of EDLMFC, we compared our method with the other three state-of-the-art methods. Since the work link of RPI-Pred was not available, and lncPro only provided the source code for the predictive model that has been trained on their dataset. Therefore, we chose RPITER, IPMiner, and CFRP to localize for comparation on RPI1807, NPInter v2.0, and RPI488 datasets under five-fold cross-validation (5CV), respectively. Seven performance metrics: accuracy (ACC), true positive rate (TPR), true negative rate (TNR), positive predictive value (PPV), F1-score (F1), Matthews correlation coefficient (MCC), and area under the curve (AUC) of the receiver operation characteristic (ROC), were employed to evaluate the above four methods comprehensively. The experimental results on RPI1807, NPInter v2.0, and RPI488 datasets are shown in Fig. 1a–c, respectively. And the detailed results are all listed in Table 1.
Fig. 1.
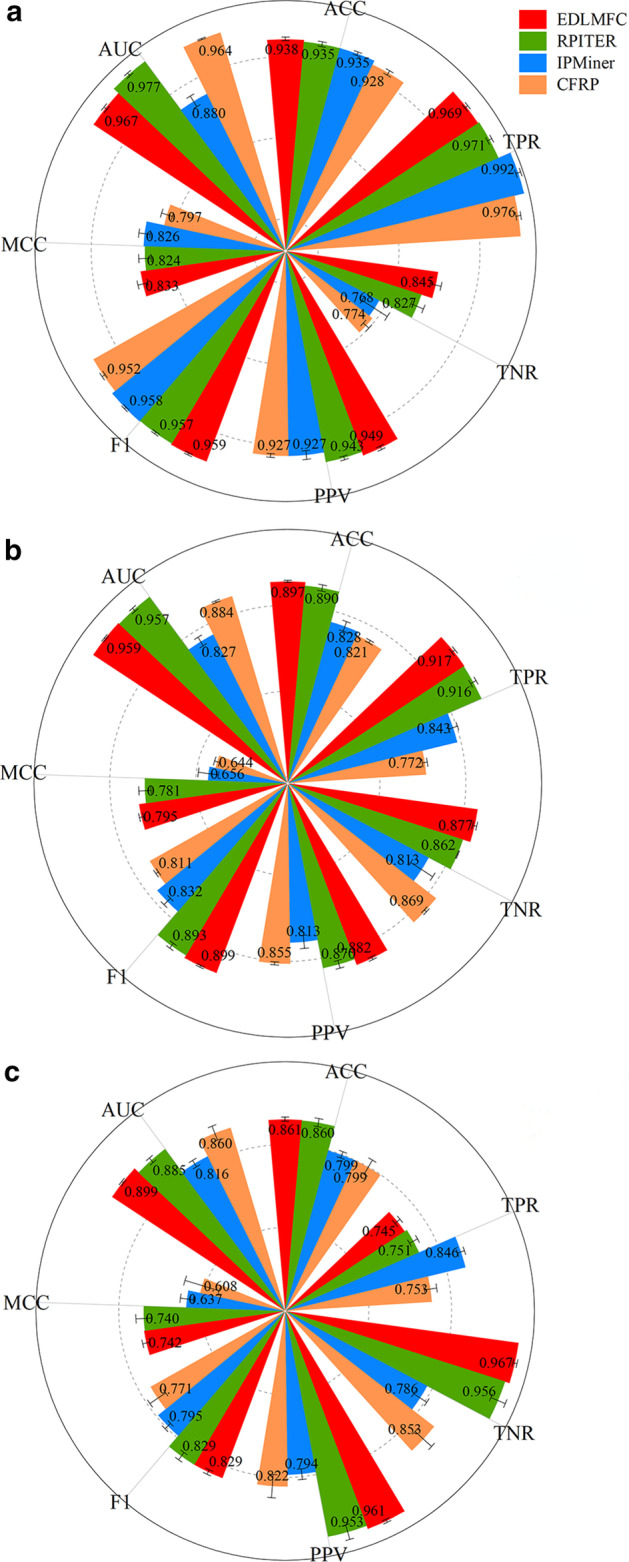
Performance comparison among different ncRPI prediction methods. a Performance comparison on RPI1807 dataset. b Performance comparison on NPInter v2.0 dataset. c Performance comparison on RPI488 dataset
Table 1.
Performance comparison between EDLMFC and other ncRPI prediction methods on RPI1807, NPInter v2.0, and RPI488
| Dataset | Method | ACC (%) | TPR (%) | TNR (%) | PPV (%) | F1 (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|---|---|
| RPI1807 | EDLMC | 93.8 ± 0.3 | 96.9 ± 0.3 | 84.5 ± 0.9 | 94.9 ± 0.3 | 95.9 ± 0.2 | 83.3 ± 0.8 | 96.7 ± 0.3 |
| RPITER | 93.5 ± 0.4 | 97.1 ± 0.4 | 82.7 ± 1.1 | 94.3 ± 0.3 | 95.7 ± 0.2 | 82.4 ± 1.0 | 97.7 ± 0.3 | |
| IPMinter | 93.5 ± 0.3 | 99.2 ± 0.4 | 76.8 ± 2.4 | 92.7 ± 0.7 | 95.8 ± 0.2 | 82.6 ± 0.9 | 88.0 ± 1.0 | |
| CFRP | 92.8 ± 0.4 | 97.6 ± 0.4 | 77.4 ± 0.6 | 92.7 ± 0.3 | 95.2 ± 0.3 | 79.7 ± 0.9 | 96.4 ± 0.1 | |
| NPInter v2.0 | EDLMFC | 89.7 ± 0.2 | 91.7 ± 0.4 | 87.7 ± 0.4 | 88.2 ± 0.3 | 89.9 ± 0.2 | 79.5 ± 0.4 | 95.9 ± 0.2 |
| RPITER | 89.0 ± 0.6 | 91.6 ± 0.6 | 86.2 ± 0.1 | 87.0 ± 0.8 | 89.3 ± 0.6 | 78.1 ± 1.2 | 95.7 ± 0.4 | |
| IPMinter | 82.8 ± 1.0 | 84.3 ± 0.9 | 81.3 ± 2.6 | 81.3 ± 1.3 | 83.2 ± 0.9 | 65.6 ± 2.0 | 82.7 ± 1.0 | |
| CFRP | 82.1 ± 0.3 | 77.2 ± 0.5 | 86.9 ± 0.3 | 85.5 ± 0.3 | 81.1 ± 0.3 | 64.4 ± 0.5 | 88.4 ± 0.2 | |
| RPI488 | EDLMC | 86.1 ± 0.5 | 74.5 ± 0.8 | 96.7 ± 0.5 | 96.1 ± 0.4 | 82.9 ± 0.6 | 74.2 ± 0.9 | 89.9 ± 0.3 |
| RPITER | 86.0 ± 1.0 | 75.1 ± 1.1 | 95.6 ± 1.9 | 95.3 ± 1.8 | 82.9 ± 1.1 | 74.0 ± 1.9 | 88.5 ± 0.7 | |
| IPMinter | 79.9 ± 0.8 | 84.6 ± 0.9 | 78.6 ± 1.9 | 79.4 ± 1.3 | 79.5 ± 0.9 | 63.7 ± 1.6 | 81.6 ± 0.9 | |
| CFRP | 79.9 ± 2.0 | 75.3 ± 1.5 | 85.3 ± 2.7 | 82.2 ± 2.8 | 77.1 ± 2.3 | 60.8 ± 4.3 | 86.0 ± 1.8 |
The values in bold indicate this performance metric is the best among the four methods
The mathematical notation (±) represents standard deviation
From the Fig. 1a, EDLMFC achieves the highest ACC, TNR, PPV, F1, and MCC. As shown in Table 1, we can see that EDLMFC yielded an ACC of 93.8%, which is 0.3%, 0.3%, and 1.0% higher than that of RPITER, IPMiner, and CFRP, respectively. The standard deviation of ACC under 5CV is smaller than RPITER and CFRP. The TNR of EDLMFC is 84.5%, which is 1.8%, 7.7%, and 7.1% higher than that of RPITER, IPMiner, and CFRP, respectively. The PPV of EDLMFC is 94.9%, which is 0.6%, 2.2%, and 2.2% higher than that of RPITER, IPMiner, and CFRP, respectively. F1 of EDLMFC is 95.9%, which is 0.2%, 0.1% and 0.7% higher than that of RPITER, IPMiner and CFRP, respectively. MCC of EDLMFC is 83.3%, which is 0.9%, 0.7%, and 3.6% higher than that of RPITER, IPMiner, and CFRP, respectively. Although the TPR of EDLMFC is 2.3% lower than the IPMinter, the AUC is 1.0% lower than RPITER, EDLMFC method performs better than the two methods in general. Therefore, compared with the above three methods, our method EDLMFC has superior performance in predicting ncRPIs on RPI1807 dataset.
From the Fig. 1b, EDLMFC is superior to all the methods on seven performance metrics on NPInter v2.0 dataset. From the Fig. 1c, EDLMFC achieves the highest ACC, TNR, PPV, F1, MCC, and AUC on RPI488 dataset. It suggests that the method relied on integrated deep learning with a combination of multi-scale features presented in this work is an effective and efficient way to predict ncRPIs.
Performance of EDLMFC in independent test
To further validate the ability of EDLMFC in distinguishing whether ncRNAs interact with proteins or not. We used the RPI1807 dataset to train our model and verified it on NPInter v2.0 dataset. There is no overlap between the two processed datasets. The processed NPInter v2.0 dataset contains 1943 interaction pairs, which can be divided into 6 organisms: Homo sapiens, Mus musculus, Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and Escherichia coli with the number of interaction pairs of 740, 229, 693, 33, 46, and 202, respectively, which were tested by EDLMFC separately. As shown in Table 2, EDLMFC predicted the correct number of interacted pairs on the 6 organisms for 631, 217, 632, 31, 41, and 188, with ACC rates of 85%, 95%, 91%, 94%, 89%, and 93%, respectively. On the independent NPInter v2.0 dataset, we finally predicted the correct number of ncRNA–protein pairs to be 1740, with an overall ACC of 90%.
Table 2.
Independent testing results of EDLMFC on six organisms from NPInter v2.0
| Organism | Total ncRNA–protein pairs in NPInter v2.0 | EDLMFC performance |
|---|---|---|
| Homo sapiens | 740 | 631 (85%) |
| Mus musculus | 229 | 217 (95%) |
| Saccharomyces cerevisiae | 693 | 632 (91%) |
| Caenorhabditis elegans | 33 | 31 (94%) |
| Drosophila melanogaster | 46 | 41 (89%) |
| Escherichia coli | 202 | 188 (93%) |
| Total | 1943 | 1742 (90%) |
Analyses of different feature combination strategies
We adopted three kinds of feature of ncRNAs and proteins to construct EDLMFC model, including sequence features, secondary structure features, and tertiary structure features. To analyse the contributions of the three kinds of feature, seven different feature combinations: sequence, secondary structure, tertiary structure, sequence together with secondary structure, sequence together with tertiary structure, secondary structure together with tertiary structure, and all features were used as inputs to experiment the classification performance of the model. The ROC curves of seven different feature combinations as inputs tested on RPI1807 and NPInter v2.0 were shown in Fig. 2a and Fig. 2b, respectively. The results of seven performance metrics under 5CV are all listed in Table 3.
Fig. 2.

AUC comparison among different feature combination strategies. a AUC comparison on RPI1807 dataset. b AUC comparison on NPInter v2.0 dataset. The ROC curves of all feature, sequence together with secondary structure, sequence together with tertiary structure, only sequence, secondary structure together with tertiary structure, only secondary structure, and only tertiary structure as inputs were expressed in red, blue, green, magenta, orange, purple and black, respectively. The maximum AUC represents the best performance of the model
Table 3.
Results under 5CV of different feature combinations considered on RPI1807 and NPInter v2.0
| Dataset | Combinations of features | ACC (%) | TPR (%) | TNR (%) | PPV (%) | F1 (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|---|---|
| RPI1807 | Sequence | 92.1 ± 1.3 | 94.5 ± 2.7 | 85.1 ± 2.9 | 94.9 ± 0.8 | 94.7 ± 0.9 | 79.5 ± 2.9 | 96.2 ± 0.8 |
| Secondary structure | 92.8 ± 1.2 | 96.6 ± 1.9 | 81.5 ± 6.6 | 94.0 ± 2.1 | 95.2 ± 0.7 | 80.7 ± 3.5 | 96.1 ± 1.1 | |
| Tertiary structure | 72.9 ± 9.6 | 85.7 ± 21.7 | 28.7 ± 28.7 | 82.9 ± 6.9 | 79.7 ± 12.7 | 26.9 ± 21.5 | 81.6 ± 8.7 | |
| Sequence + secondary structure | 93.8 ± 0.6 | 96.6 ± 1.2 | 85.5 ± 2.5 | 95.2 ± 0.8 | 95.9 ± 0.4 | 83.5 ± 1.5 | 96.5 ± 0.8 | |
| Sequence + tertiary structure | 92.6 ± 1.6 | 95.1 ± 2.2 | 85.1 ± 2.9 | 95.0 ± 0.9 | 95.0 ± 1.1 | 80.5 ± 3.9 | 96.3 ± 0.8 | |
| Secondary structure + tertiary structure | 92.4 ± 0.4 | 96.5 ± 1.5 | 80.6 ± 5.6 | 93.7 ± 1.6 | 95.0 ± 0.2 | 79.7 ± 1.3 | 96.2 ± 0.9 | |
| All features | 94.3 ± 0.2 | 97.4 ± 1.0 | 85.1 ± 1.9 | 95.1 ± 0.6 | 96.2 ± 0.2 | 84.7 ± 0.7 | 96.7 ± 0.8 | |
| NPInter v2.0 | Sequence | 87.7 ± 0.8 | 89.7 ± 1.1 | 85.7 ± 2.4 | 86.3 ± 1.9 | 87.9 ± 0.7 | 75.5 ± 1.6 | 94.6 ± 0.3 |
| Secondary structure | 78.8 ± 1.4 | 87.5 ± 1.4 | 70.1 ± 2.4 | 74.6 ± 1.6 | 80.5 ± 1.2 | 58.5 ± 2.7 | 88.1 ± 1.0 | |
| Tertiary structure | 54.7 ± 3.8 | 68.1 ± 27.0 | 41.4 ± 33.6 | 58.7 ± 8.7 | 56.3 ± 10.6 | 10.9 ± 8.7 | 59.9 ± 2.7 | |
| Sequence + secondary structure | 89.1 ± 0.9 | 91.2 ± 1.1 | 86.9 ± 1.5 | 87.5 ± 1.3 | 89.3 ± 0.8 | 78.3 ± 1.7 | 95.4 ± 0.3 | |
| Sequence + tertiary structure | 88.9 ± 0.8 | 91.1 ± 1.1 | 86.8 ± 1.3 | 87.3 ± 1.1 | 89.2 ± 0.7 | 77.9 ± 1.5 | 95.2 ± 0.5 | |
| Secondary structure + tertiary structure | 83.5 ± 1.0 | 88.6 ± 1.5 | 78.5 ± 2.6 | 80.5 ± 1.8 | 84.3 ± 0.7 | 67.5 ± 1.9 | 92.0 ± 0.4 | |
| All features | 90.0 ± 0.7 | 92.2 ± 1.1 | 87.6 ± 0.9 | 88.2 ± 0.8 | 90.2 ± 0.7 | 80.0 ± 1.4 | 96.2 ± 0.3 |
The values in bold indicate this performance metric is the best among the three methods
The mathematical notation (±) represents standard deviation
From the Fig. 2, on RPI1807 and NPInter v2.0 datasets, only secondary structure as input have a slightly lower AUC than only sequence as input and notably higher AUC than only tertiary structure as input. Thus, the sequence is the most important feature in ncRPIs; the following is the predicted secondary structure, and then is the tertiary structure. When any combination of two features was sent into the model, we find that its AUC value is higher than that of one of the two features. Moreover, the AUC value of the model is the highest when all features were entered. Therefore, we can conclude that all the features contain useful information, and at the same time, as inputs, they complement each other to give the model a better predictive performance.
Application of EDLMFC for ncRNA–protein network construction
To visualize how many interactions have been correctly predicted, we further used the independent test results of EDLMFC to construct the ncRNA–protein networks. Here, we adopted a software named Cytoscape [47–49] for Mus musculus networks clustering. For Mus musculus in the NPInter v2.0 dataset, we correctly predicted the 217 of 229 interactions, the ACC up to 95%. As is shown in Fig. 3, we found that the ncRPIs of Mus musculus contain both hub proteins (a protein interacts with multiple RNAs) and hub ncRNAs (an RNA interacts with multiple proteins) [50]. P84104 and Q8VE97 hub proteins have the largest number of interactions and are both considered to be serine or arginine with rich splicing factor 3 [51]. Especially, P84104 hub protein is the splicing factor that specifically promotes exon-inclusion during alternative splicing. Interaction with YTHDC1, an RNA-binding protein that recognizes and binds N6-methyladenosine (m6A)-containing RNAs, promotes recruitment of SRSF3 to its mRNA-binding elements adjacent to m6A sites, leading to exon-inclusion during alternative splicing [52, 53]. Q8VE97 hub protein plays a role in alternative splice site selection during pre-mRNA splicing. Repressing the splicing of MAPT/Tau exon 10 as well [54]. Therefore, constructing ncRNA–protein networks help identify the important functions and pathways of key proteins and ncRNAs, which will facilitate various medical and pharmaceutical studies [55].
Fig. 3.

The Mus musculus networks constructed based on interaction pairs predicted by the EDLMFC. The oval nodes with green and blue represent the ncRNA and proteins, respectively. The gray and red edges indicate correctly and wrongly predicted ncRPIs, respectively
Discussion
In this work, we proposed a multi-scale features combination-based computational method, EDLMFC, to predict ncRPIs through an ensemble deep learning combined CNN and BLSTM. Compared with the other three state-of-the-art methods on RPI1807, NPInter v2.0, and RPI488 datasets, comprehensive experimental results indicate our method EDLMFC has the best classification performance for ncRPI predictions. This is mainly because of the following reasons:
The multi-scale features were used, which includes not only sequence features information but also structural information. The results of different feature combinations show that sequence features are the most important, followed by secondary structure features and tertiary structure features. All features contain useful information, so the classification performance of the model was best when all features were used as input for prediction.
Using conjoint k-mer method to encode sequence features of ncRNAs and proteins, a variety of k-mer features are considered so that proteins and ncRNAs can be represented more accurately and comprehensively.
CNN was used to dig the hidden abstract high-level features of proteins and ncRNAs, then feeding into BLSTM to capture their long-range dependencies, and a three-layer fully-connected layer was employed to predict the ncRPIs.
Although EDLMFC achieves a better performance in ncRPI predictions, there are still some limits that need to be noticed. Like other deep learning-based approaches, it's like a black box that automatically learns the features of proteins and ncRNAs and makes predictions that we can't understand biologically. Besides, the method of ncRNA secondary structure prediction, SPOT-RNA, can only predict RNAs with a length of no more than 500 nucleotides. Therefore, our work mainly predicts the interaction between ncRNAs with a length of fewer than 500 nucleotides and proteins. In future work, we will consider designing more advanced neural network models to learn high-level abstract features with biological insights and choosing a more accurate prediction method of secondary structure to predict ncRPIs more accurately and efficiently.
Conclusions
The prediction of ncRPIs contributes to understand the molecular mechanism within various fundamental biological processes and diseases. Many computational methods have been proposed for ncRPI predictions. However, only a small number of previous studies considered high-order structural features of ncRNAs and proteins, and overwhelming majority of them only used shallow machine learning to build classifiers for prediction. In this work, we presented a computational method based on CNN and BLSTM to predict ncRPIs through learning high-level abstract features from multi-scale features. To gain as much information of proteins and ncRNAs as possible, we employed not only primary sequence features, secondary structure sequence features but also tertiary structure features, and adopted a conjoint k-mer method to extract multiple-mers features by extending the range of k. Then, we adopted BLSTM to capture long-range dependencies between dominating features of ncRNAs/proteins learned by CNN, and send them to the full connection layer to predict whether they have the interaction relationship. Compared with the other three state-of-the-art methods under 5CV on RPI1807, NPInter v2.0, and RPI488 datasets, EDLMFC improved the performance with an increase of roughly 0.1%-7.7%. And the independent test between 6 organisms divided from NPInter v2.0 has an overall ACC of 90%, indicating that the ensemble deep learning framework can reveal and learn the high-level hidden information to improve prediction performance. Besides, according to the analyses of different feature combination strategies, we can conclude that all the features contain useful information. When multiple features were fed into the model, they complemented each other to make the model achieve a better prediction performance. In conclusion, EDLMFC method can be a useful tool for predicting unknown ncRPIs.
Methods
Benchmark datasets
Primary sequence data of paired samples, ncRNAs, and proteins in RPI1807, NPInter v2.0, and RPI488 were downloaded from the previous study [40]. RPI1807 has extracted the possible interaction pairs by parsing a nucleic acid database (NAD) that provides RNA protein complex and protein–RNA interface, consisting of 1078 RNA chains and 3131 protein chains in total [31]. In data preprocessing, the EMBOSS needle program has used to remove protein and RNA chains with high sequence similarity (cutoff ≥ 30%), then further distinguishing the atomic interactions with a distance threshold (cutoff = 3.40 Å), which was reasonable and sufficient to cover ‘strong’ and ‘moderate’ hydrogen bonds and energy-rich van der Waals contacts [56, 57]. It contains 1807 positive pairs and 1436 negative pairs after deleting the RNA sequences length of fewer than 15 nucleotides and the protein sequences of less than 25 amino acids. NPInter v2.0 was obtained from NPInter database, which documents functional interactions between noncoding RNAs (except tRNAs and rRNAs) and biomolecules (proteins, RNAs, and DNAs) verified by experiments [58]. In addition, as NPInter database only contains interaction (primarily physical interactions) pairs, and lack non-interaction pairs to work as negative samples in the training model, the same number of non-interaction pairs were generated by randomly pairing the ncRNAs and proteins in positive samples and further discarding similar known interaction pairs [26, 27] (a randomly generated pair R2–P2 was discarded if there has existed an interaction pair R1–P1 of P2 shared ≥ 40% sequence identity with P1 and R2 shared ≥ 80% sequence identity with R1). RPI488 is a lncRNA–protein interaction dataset, which was obtained from 18 ncRNA–protein complexes downloaded from the PDB database [27]. The atomic interactions were distinguished with a distance threshold (5 Å). CD-HIT tool [59] was used to remove protein and RNA chains with high sequence similarity (cut-off ≥ 90%). After redundancy removal, RPI488 dataset contains 488 lncRNA–protein pairs, including 243 interacting pairs and 245 non-interacting pairs.
Additionally, we used ncRNA secondary structure prediction method, SPOT-RNA, which was trained via RNAs with a maximum length of 500 nucleotides. Thus, ncRNAs with more than 500 nucleotides in primary sequence were deleted. ncRNAs-proteins paired samples of more than 500 nucleotides were further deleted based on the deleted ncRNA samples. Then, the protein primary sequences that were not paired with ncRNAs were deleted based on the deleted paired samples. Finally, RPI1807 contains 652 positive pairs and 221 negative pairs, NPInter v2.0 contains 1943 positive pairs and 1943 negative pairs. RPI488 contains 43 positive pairs and 233 negative pairs. The sample information of the original and processed set is shown in Table 4. Due to the large gap between the number of positive and negative samples in RPI488 dataset after processing. The negative samples were randomly divided into 5 groups to form 5 subsets with the positive samples. The average results of the 5 subsets were taken as the result of RPI488. The details of the 5 subsets are listed in Additional file 1: Table S1.
Table 4.
The three original and processed ncRPI datasets used in this study
| Dataset | Positive pairs | Negative pairs | RNAs | Proteins | |
|---|---|---|---|---|---|
| Original set | RPI1807 | 1807 | 1436 | 1078 | 3131 |
| NPInter v2.0 | 10,412 | 10,412 | 4636 | 449 | |
| RPI488 | 243 | 245 | 25 | 247 | |
| Processed set | RPI1807 | 652 | 221 | 646 | 868 |
| NPInter v2.0 | 1943 | 1943 | 513 | 448 | |
| RPI488 | 43 | 233 | 13 | 155 |
Features extraction
SPOT-RNA based features
The secondary structure of ncRNA was predicted by SPOT-RNA [60, 61]. We localized their work by downloading it from https://github.com/jaswindersingh2/SPOT-RNA/. SPOT-RNA represented RSS with a macroscopic secondary structure, which is seven single character identifiers for the structure types of each nucleotide in the primary sequence. In this representation, S = stem, H = hairpin loop, M = multi-loop, I = internal loop, B = bulge, X = external loop, and E = end. Thus, each secondary structure sequence of ncRNAs can be represented by the seven-letter alphabet.
SPIDER3 based features
For protein secondary structure prediction, we localized SPIDER3 from the server http://www.sparks-lab.org/server/spider3/ [62], in which three classical protein secondary structures (α-helix, β-sheet, and coil) were used to represent each amino acid in the protein primary sequence. Besides, SPIDER3 also can be used to predict tertiary structures: solvent accessible surface area (ASA), contact number (CN), the upper half sphere exposure (HSEα-up), and the down half sphere exposure (HSEα-down) [62]. We calculated the average value of these tertiary structures for all amino acids in each protein sample.
Interface propensity
For interface propensity (IP) between a residue and nucleotide [63], we used an improved work by our team [63], which got the residue-nucleotide propensities (60 × 8) with secondary structure information of RNAs and proteins considered by scoring. Here, we calculated the average value of the binding preferences of all nucleotides to amino acids in a paired sample.
Sequence coding
To input ncRNA and protein sequences into deep learning or conventional machine learning models, the sequence data need to be encoded as numeric vectors. Most existing studies extracted ncRNA and protein sequence features by using a simple k-mer: 3-mer frequency feature for proteins and 4-mer frequency feature for ncRNAs [27, 30, 32, 35, 39]. For protein, 20 amino acids can be classified into seven groups based on their dipole moments and side-chain volume:={A, G, V}, ={I, L, F, P},={Y, M, T, S}, ={H, N, Q, W}, ={R, K}, ={D, E} and ={C} [39]. Then, each protein sequence can be represented by the seven-letter alphabet. Thus, a protein sequence can be represented as a numeric vector with 343 () elements by calculating the 3-mer frequency. For ncRNA, using four ribonucleotides (A, U, G, C), an ncRNA sequence can be represented as a numeric vector of 256() elements.
We adopted a conjoint k-mer method to extract more feature information by extending the range of k to 1–4 in the k-mer frequency coding process for a ncRNA and 1–3 for a protein. That is to say, for ncRNA, we considered not only the 4-mer frequency information but also the 1-mer, 2-mer, and 3-mer. Similar to 4-mer, 3-mer of ncRNAs can be represented as a numeric vector with 64() elements; 2-mer of ncRNAs can be represented as a numeric vector with 16() elements; 1-mer of ncRNAs can be represented as a numeric vector with 4() elements. As shown in Fig. 4a, the rows and columns are corresponding to all kinds of k-mer comprised of four ribonucleotides (A, U, G, C) and the primary sequence of each ncRNA. Then, a primary sequence of ncRNA can be represented by a binary matrix, which was then transformed into a numeric vector with 340 () elements by calculating each kind of k-mer frequency. Similar to Fig. 4b, using seven structure types (S, H, M, I, B, X, E), a secondary structure sequence of ncRNAs can be represented as a numeric vector with 2800 () elements. Therefore, integrating IP would produce the ncRNAs coding vector with 3141 () elements. For protein, we considered the 1-mer, 2-mer, and 3-mer frequency information, combining primary sequence represented by the reduced seven-letter alphabet, secondary structure sequence represented by three classical secondary structures (α-helix, β-sheet, and coil) with tertiary structures (IP, ASA, CN, HSEα-up and HSEα-down) would produce the proteins coding vector with 443 () elements.
Fig. 4.

a The primary sequence of ncRNAs representation by conjoint k-mer. b The secondary structure sequence of ncRNAs representation by conjoint k-mer
Performance metrics
We adopted 5CV to evaluate the performance of EDLMFC and other methods by seven widely used metrics. Due to the random effects of the training procedure, the 5CV was repeated 10 times. The average of the performance metrics predicted from the 10 times was used as the final prediction, and the 10 results of EDLMFC on the three datasets are listed in Additional file 1: Tables S2–S4. The formulas of ACC, TPR, TNR, PPV, F1, MCC, and AUC of the ROC are as follows:
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
where TP, FP, TN, and FN denote the number of true positive, false positive, true negative, and false negative, respectively. ACC reflects the ability of the classifier to discriminate against the whole sample. TPR reflects the ability to predict positive samples. TNR reflects the ability to predict negative samples. PPV represents the ability to discriminate positive samples that are actually positive samples. MCC reflects the classification performance of the classification model when the number of positive and negative samples are not balanced. F1 is a comprehensive index that considers TPR and PPV. And AUC is used to evaluate the performance of a classification model.
Model design
We adopted a conjoint k-mer to encode the primary sequence and secondary structure sequence features, merging IP and IP, ASA, HSEα-up, HSEα-down, CN for ncRNAs, and proteins, respectively, forming 3141 and 443-dimensional feature column vectors. Then the ensemble deep learning framework did the rest of the work automatically. Specifically, the two encoded feature column vectors of ncRNAs and proteins were separately fed into layered networks, including CNN and BLSTM layers. Then, a concatenated vector of the two outputs from the BLSTM layer was wired as the input of the fully connected layer. Finally, the ensemble module used the softmax activation function at the last layer to make binary predictions. The details of the proposed framework are shown in Fig. 5.
Fig. 5.

The workflow of the proposed EDLMFC
CNN consists of several layers, including the input layer, convolution layer, max-pooling layer, full connection layer, and output layer [64]. Among these, the convolution layer includes activation operation and the max-pooling layer includes batch normalization operation. In the convolution layer, assume that is the feature map of the layer, which can be described as:
| 7 |
where is the weight matrix of the convolution kernel of layer, operator represents convolution operation, is the offset vector, and is the activation function.
After convolution operation, a commonly used activation function rectified linear unit (ReLU) was applied to sparse the output of the convolution layer, which can be used to speed up the supervised train process and maintain the rate of convergence at a steady state to avoid the vanishing gradient problem [65]. Suppose that is the activating layer, its formula defined as:
| 8 |
Followed by the convolution layer, the max-pooling layer was used to sample the feature graph according to certain rules to reduce the parameters and calculation while maintaining the main features. suppose that is the pooling layer, its formula is:
| 9 |
After the max-pooling operation, batch normalization (BN) [66] operation was employed to reduce internal covariate shift and help train the designed deep network.
LSTM is a widely used RNN with the memory cells [67], which store information over an arbitrary time allowing the network to learn long dependencies in the sequential data. Three non-linear gating units (input, output and forget) control the information flow through the time steps. Each gate gets a similar input as the input neuron. Moreover, each gate has an activation function [68], which forward mechanism expressed by the following equation:
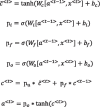 |
10 |
where W, b denote the weights and bias, respectively, denotes the Logistic Sigmoid function, denotes pointwise multiplication, ![]() represent the input gate, forget gate, and output gate, respectively. input data at current step t, is hidden state at previous step t − 1. is cell state at previous step t − 1, is cell state at current step t, is the hidden state at the current step t, which equal to the output at current.
represent the input gate, forget gate, and output gate, respectively. input data at current step t, is hidden state at previous step t − 1. is cell state at previous step t − 1, is cell state at current step t, is the hidden state at the current step t, which equal to the output at current.
We used the variant BLSTM, which consists of two parallel LSTMs: one input sequence forward and the other input sequence inverted [69], to capture long-range dependencies between high-level abstract features extracted from primary sequence, secondary structure sequence, and tertiary structure by CNN.
To predict ncRPIs effectively, we designed a training model based on a three-layer CNN combining BLSTM. Two similar ensemble neural network parts analyzed the ncRNA and protein input vectors separately, and two feature vectors were formed by using a one-layer fully-connected layer. Then, a three-layer fully-connected concatenated the two feature vectors as input and made the interaction prediction. The main parameters in the ensemble deep learning framework, including the number of layers, filter size, kernel size, learning rate, dropout rate, BLSTM hidden size, and fully-connection size, were tuned to maximize the MCC on a validation set randomly selected from the training set. For ensemble neural network of analyzing proteins, the values of the parameters are as follows: number of layers: 3; filter size: 45, 64, and 86; kernel size: 6, 6, and 6; and dropout rate: 0.2, 0.2, and 0.2; BLSTM hidden size: 45; fully-connection size: 64; For ensemble neural network of analyzing ncRNAs, the values of parameters are the same as the ones for analyzing proteins, except for kernel sizes which are 6, 5, and 5. In the end, the three-layer fully-connected with 128, 64, and 2 neurons, respectively, and the dropout with 0.25 and 0.3. Adam [70] and stochastic gradient descent (SGD) [71] were employed successively to train each part, among which Adam with a learning rate 0.001 first gave the module a quick converge and then SGD with a learning rate 0.005 was used to fine tune the module after. Besides, we used the back-propagation algorithm [72] to minimize the loss function of binary cross entropy, also used regularization [73] and early stopping [74] algorithms to avoid overfitting. Our model was implemented by the Keras2.2.5 library.
Supplementary Information
Additional file 1. Table S1: The 5 subsets divided from the processed RPI488 dataset. Table S2: The results of EDLMFC under 5CV after running 10 times on RPI488 dataset. Table S3: The results of EDLMFC under 5CV after running 10 times on RPI1807 dataset. Table S4: The results of EDLMFC under 5CV after running 10 times on NPInter v2.0 dataset.
Acknowledgements
We thank all the staff of the journal for their efforts and those who have helped us in the course of our work.
Abbreviations
- RBPs
RNA-binding proteins
- ncRNA
Non-coding RNA
- ncRPI
Non-coding RNA protein interaction
- ncRNAs
Non-coding RNAs
- ncRPIs
Non-coding RNA protein interactions
- X-ray
X-ray crystal diffraction
- NMR
Nuclear magnetic resonance
- cryo-EM
Electron cryo-microscopy
- CNN
Convolutional neural network
- BLSTM
Bi-directional long short-term memory network
- RNN
Recurrent neural networks
- RSS
RNA secondary structures
- IP
Interface propensity
- ASA
Solvent accessible surface area
- CN
Contact number
- HSEα-up
A upper half sphere exposure
- HSEα-down
A down half sphere exposure
- RF
Random forest
- SVM
Support vector machine
- ELM
Extreme learning machine
- lncRNAs
Long non-coding RNAs
- NAD
Nucleic acid database
- 5CV
Five-fold cross-validation
- ACC
Accuracy
- TPR
True positive rate
- TNR
True negative rate
- PPV
Positive predictive value
- F1
F1-score
- MCC
Matthews correlation coefficient
- AUC
Area under the receiver operating characteristic curve
- ROC
Receiver operation characteristic
- ReLU
Rectified linear unit
- BN
Batch normalization
- SGD
Stochastic gradient descent
Authors’ contributions
JJW designed the method, prepared the datasets, implemented the experiment, and wrote the manuscript; YPZ conceived the algorithm; WKG solved the work technical problems; YL revised the manuscript; MW adjusted the manuscript format; XQH sorted out the references; JJT guided the work ideas and revised the manuscript; everyone finally reviewed the manuscript.
Funding
We thank the Beijing Natural Science Foundation (No. 2202002) and the Chinese Natural Science Foundation project (21173014) for financial supports.
Availability of data and materials
The source code of EDLMFC and the datasets used in this work are available at https://github.com/JingjingWang-87/EDLMFC.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Knowling S, Morris KV. Non-coding RNA and antisense RNA. Nature's trash or treasure? Biochimie. 2011;93(11):1922–1927. doi: 10.1016/j.biochi.2011.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaikkonen MU, Lam MTY, Glass CK. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res. 2011;90(3):430–440. doi: 10.1093/cvr/cvr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rinn JL, Ule J. 'Oming in on RNA–protein interactions. Genome Biol. 2014;15(1):401. doi: 10.1186/gb4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramanathan M, Porter DF, Khavari PA. Methods to study RNA–protein interactions (vol. 16, p. 225, 2019) Nat Methods. 2019;16(4):351. doi: 10.1038/s41592-019-0366-2. [DOI] [PubMed] [Google Scholar]
- 5.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143(1):46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142(3):409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim G, Zhu S, Zhang K, Hoey T, Deragon J, Kachroo A, Kachroo P. The analogous and opposing roles of double-stranded RNA-binding proteins in bacterial resistance. J Exp Bot. 2019;70(5):1627–1638. doi: 10.1093/jxb/erz019. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Wen L, Zhu H. Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Biosci. 2015;5(1):59. doi: 10.1186/s13578-015-0050-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan L, Zhu L, Guo W, Zhou X, Zhang Y, Huang Z, Huang D. Nonconvex penalty based low-rank representation and sparse regression for eQTL mapping. IEEE ACM Trans Comput Biol. 2017;14(5):1154–1164. doi: 10.1109/TCBB.2016.2609420. [DOI] [PubMed] [Google Scholar]
- 12.Yuan L, Huang D. A network-guided association mapping approach from DNA methylation to disease. Sci Rep Uk. 2019;9(1):5601. doi: 10.1038/s41598-019-42010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitagawa M, Kotake Y, Ohhata T. Long non-coding RNAs involved in cancer development and cell fate determination. Curr Drug Targets. 2012;13(13):1616–1621. doi: 10.2174/138945012803530026. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Y, Bian X, Ye D, Yao X, Zhang S, Dai B, Zhang H, Shen Y. Long noncoding RNA expression signatures of bladder cancer revealed by microarray. Oncol Lett. 2014;7(4):1197–1202. doi: 10.3892/ol.2014.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Yan CC, Zhang X, You Z. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2017;18(4):558–576. doi: 10.1093/bib/bbw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, Laurent GSI, Kenny PJ, Wahlestedt C. Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14(7):723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng S, Zhu L, Huang D. Mining the bladder cancer-associated genes by an integrated strategy for the construction and analysis of differential co-expression networks. BMC Genomics. 2015;163:S4. doi: 10.1186/1471-2164-16-S3-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan L, Guo L, Yuan C, Zhang Y, Han K, Nandi AK, Honig B, Huang D. Integration of multi-omics data for gene regulatory network inference and application to breast cancer. IEEE ACM Trans Comput Biol. 2019;16(3):782–791. doi: 10.1109/TCBB.2018.2866836. [DOI] [PubMed] [Google Scholar]
- 19.Ray D, Kazan H, Chan ET, Castillo LP, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol. 2009;27(7):135–667. doi: 10.1038/nbt.1550. [DOI] [PubMed] [Google Scholar]
- 20.Keene JD, Komisarow JM, Friedersdorf MB. RIP-chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc. 2006;1(1):302–307. doi: 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
- 21.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and MicroRNA target sites by PAR-CLIP. Cell. 2010;141(1):129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ke A, Doudna JA. Crystallization of RNA and RNA–protein complexes. Methods. 2004;34(3):408–414. doi: 10.1016/j.ymeth.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 23.Scott LG, Hennig M. RNA structure determination by NMR. Methods Mol Biol. 2008;452:29–61. doi: 10.1007/978-1-60327-159-2_2. [DOI] [PubMed] [Google Scholar]
- 24.Jin P, Bulkley D, Guo Y, Zhang W, Guo Z, Huynh W, Wu S, Meltzer S, Cheng T, Jan LY, et al. Electron cryo-microscopy structure of the mechanotransduction channel NOMPC. Nature. 2017;547(7661):118–122. doi: 10.1038/nature22981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu L, Guo W, Deng S, Huang D. ChIP-PIT: enhancing the analysis of ChIP-Seq data using convex-relaxed pair-wise interaction tensor decomposition. IEEE ACM Trans Comput Biol. 2016;13(1):55–63. doi: 10.1109/TCBB.2015.2465893. [DOI] [PubMed] [Google Scholar]
- 26.Muppirala UK, Honavar VG, Dobbs D. Predicting RNA–protein interactions using only sequence information. BMC Bioinform. 2011;12:489. doi: 10.1186/1471-2105-12-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan X, Fan Y, Yan J, Shen H. IPMiner: hidden ncRNA–protein interaction sequential pattern mining with stacked autoencoder for accurate computational prediction. BMC Genomics. 2016;17(1):582. doi: 10.1186/s12864-016-2931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai Q, Guo M, Duan X, Teng Z, Fu Y. Construction of complex features for computational predicting ncRNA–protein interaction. Front Genet. 2019;10:18. doi: 10.3389/fgene.2019.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, You Z, Huang D, Zhou F. Combining high speed ELM learning with a deep convolutional neural network feature encoding for predicting protein–RNA interactions. IEEE ACM Trans Comput Biol. 2020;17(3):972–980. doi: 10.1109/TCBB.2018.2874267. [DOI] [PubMed] [Google Scholar]
- 30.Cheng S, Zhang L, Tan J, Gong W, Li C, Zhang X. DM-RPIs: predicting ncRNA–protein interactions using stacked ensembling strategy. Comput Biol Chem. 2019;83:107088. doi: 10.1016/j.compbiolchem.2019.107088. [DOI] [PubMed] [Google Scholar]
- 31.Zhan Z, You Z, Li L, Zhou Y, Yi H. Accurate prediction of ncRNA–protein interactions from the integration of sequence and evolutionary information. Front Genet. 2018;9:458. doi: 10.3389/fgene.2018.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akbaripour-Elahabad M, Zahiri J, Rafeh R, Eslami M, Azari M. rpiCOOL: a tool for in silico RNA–protein interaction detection using random forest. J Theor Biol. 2016;402:1–8. doi: 10.1016/j.jtbi.2016.04.025. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Yan X, Liu M, Song K, Sun X, Pan W. Prediction of RNA–protein interactions by combining deep convolutional neural network with feature selection ensemble method. J Theor Biol. 2019;461:230–238. doi: 10.1016/j.jtbi.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Zhang X, Fan X, Li W. LPI-CNNCP: prediction of lncRNA–protein interactions by using convolutional neural network with the copy-padding trick. Anal Biochem. 2020;601:113767. doi: 10.1016/j.ab.2020.113767. [DOI] [PubMed] [Google Scholar]
- 35.Yi H, You Z, Huang D, Li X, Jiang T, Li L. A deep learning framework for robust and accurate prediction of ncRNA–protein interactions using evolutionary information. Mol Ther Nucl Acids. 2018;11:337–344. doi: 10.1016/j.omtn.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bellucci M, Agostini F, Masin M, Tartaglia GG. Predicting protein associations with long noncoding RNAs. Nat Methods. 2011;8(6):444–445. doi: 10.1038/nmeth.1611. [DOI] [PubMed] [Google Scholar]
- 37.Agostini F, Zanzoni A, Klus P, Marchese D, Cirillo D, Tartaglia GG. catRAPID omics: a web server for large-scale prediction of protein–RNA interactions. Bioinformatics. 2013;29(22):2928–2930. doi: 10.1093/bioinformatics/btt495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Q, Ren S, Lu M, Zhang Y, Zhu D, Zhang X, Li T. Computational prediction of associations between long non-coding RNAs and proteins. BMC Genomics. 2013;14:651. doi: 10.1186/1471-2164-14-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suresh V, Liu L, Adjeroh D, Zhou X. RPI-Pred: predicting ncRNA–protein interaction using sequence and structural information. Nucl Acids Res. 2015;43(3):1370–1379. doi: 10.1093/nar/gkv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng C, Han S, Zhang H, Li Y. RPITER: a hierarchical deep learning framework for ncRNA–protein interaction prediction. Int J Mol Sci. 2019;20(5):1070. doi: 10.3390/ijms20051070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan X, Zhang S. LPI-BLS: Predicting lncRNA–protein interactions with a broad learning system-based stacked ensemble classifier. Neurocomputing. 2019;370:88–93. doi: 10.1016/j.neucom.2019.08.084. [DOI] [Google Scholar]
- 42.Liu Y, Gong W, Zhao Y, Deng X, Li C. aPRBind: protein–RNA interface prediction by combining sequence and I-TASSER model-based structural features learned with convolutional neural networks. Bioinformatics. 2020;2020:a747. doi: 10.1093/bioinformatics/btaa747. [DOI] [PubMed] [Google Scholar]
- 43.Alipanahi B, Delong A, Weirauch MT, Frey BJ. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol. 2015;33(8):831. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- 44.Quang D, Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucl Acids Res. 2016;44(11):e107. doi: 10.1093/nar/gkw226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan X, Shen H. Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics. 2018;34(20):3427–3436. doi: 10.1093/bioinformatics/bty364. [DOI] [PubMed] [Google Scholar]
- 46.Ben-Bassat I, Chor B, Orenstein Y. A deep neural network approach for learning intrinsic protein–RNA binding preferences. Bioinformatics. 2018;34(17):638–646. doi: 10.1093/bioinformatics/bty600. [DOI] [PubMed] [Google Scholar]
- 47.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2(10):2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shannon P, Markeil A, Ozier O, Baliga NS, Wang J. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otasek D, Morris JH, Bouas J, Pico AR, Demchak B. Cytoscape automation: empowering workflow-based network analysis. Genome Biol. 2019;20(1):185. doi: 10.1186/s13059-019-1758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu L, Deng S, Huang D. A two-stage geometric method for pruning unreliable links in protein–protein networks. IEEE Trans Nanobiosci. 2015;14(5SI):528–534. doi: 10.1109/TNB.2015.2420754. [DOI] [PubMed] [Google Scholar]
- 51.DeLigio JT, Stevens SC, Nazario-Munoz GS, MacKnight HP, Doe KK, Chalfant CE, Park MA. Serine/arginine-rich splicing factor 3 modulates the alternative splicing of cytoplasmic polyadenylation element binding protein 2. Mol Cancer Res. 2019;17(9):1920–1930. doi: 10.1158/1541-7786.MCR-18-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansen GM, Markesich DC, Burnett MB, Zhu Q, Dionne KM, Richter LJ, Finnell RH, Sands AT, Zambrowicz BP, Abuin A. Large-scale gene trapping in C57BL/6N mouse embryonic stem cells. Genome Res. 2008;18(10):1670–1679. doi: 10.1101/gr.078352.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manley JL, Krainer AR. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins) Gene Dev. 2010;24(11):1073–1074. doi: 10.1101/gad.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo H, Li Y, Luo M, Lin S, Chen J, Ma Q, Gu Y, Jiang Z, Gui Y. Androgen receptor binding to an androgen-responsive element in the promoter of the Srsf4 gene inhibits its expression in mouse sertoli cells. Mol Reprod Dev. 2015;82(12):976–985. doi: 10.1002/mrd.22576. [DOI] [PubMed] [Google Scholar]
- 55.Deng S, Zhu L, Huang D. Predicting hub genes associated with cervical cancer through gene co-expression networks. IEEE ACM Trans Comput Biol. 2016;13(1):27–35. doi: 10.1109/TCBB.2015.2476790. [DOI] [PubMed] [Google Scholar]
- 56.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 57.Rajagopal S, Vishveshwara S. Short hydrogen bonds in proteins. Febs J. 2005;272(8):1819–1832. doi: 10.1111/j.1742-4658.2005.04604.x. [DOI] [PubMed] [Google Scholar]
- 58.Teng X, Chen X, Xue H, Tang Y, Zhang P, Kang Q, Hao Y, Chen R, Zhao Y, He S. NPInter v4.0: an integrated database of ncRNA interactions. Nucl Acids Res. 2019;48(D1):D160–D165. doi: 10.1093/nar/gkz969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26(5):680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh J, Hanson J, Paliwal K, Zhou Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat Commun. 2019;10(1):5407. doi: 10.1038/s41467-019-13395-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Danaee P, Rouches M, Wiley M, Deng D, Huang L, Hendrix D. bpRNA: large-scale automated annotation and analysis of RNA secondary structure. Nucl Acids Res. 2018;46(11):5381–5394. doi: 10.1093/nar/gky285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heffernan R, Yang Y, Paliwal K, Zhou Y. Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics. 2017;33(18):2842–2849. doi: 10.1093/bioinformatics/btx218. [DOI] [PubMed] [Google Scholar]
- 63.Li CH, Cao LB, Su JG, Yang YX, Wang CX. A new residue-nucleotide propensity potential with structural information considered for discriminating protein–RNA docking decoys. Proteins. 2012;80(1):14–24. doi: 10.1002/prot.23117. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J, Chen Q, Liu B. iDRBP_MMC: identifying DNA-binding proteins and RNA-binding proteins based on multi-label learning model and motif-based convolutional neural network. J Mol Biol. 2020;432(22):5860–5875. doi: 10.1016/j.jmb.2020.09.008. [DOI] [PubMed] [Google Scholar]
- 65.Glorot X, Bordes A, Bengio Y. Deep sparse rectifier neural networks. J Mach Learn Res. 2011;15:315–323. [Google Scholar]
- 66.Wang SH, Muhammad K, Hong J, Sangaiah AK, Zhang YD. Alcoholism identification via convolutional neural network based on parametric ReLU, dropout, and batch normalization. Neural Comput Appl. 2018;32(3SI):665–680. [Google Scholar]
- 67.Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12(10):931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo Y, Li W, Wang B, Liu H, Zhou D. DeepACLSTM: deep asymmetric convolutional long short-term memory neural models for protein secondary structure prediction. BMC Bioinformatics. 2019;20(1):341. doi: 10.1186/s12859-019-2940-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X, Li B, Zeng G, Liu Q, Ai D. Prediction of long non-coding RNAs based on deep learning. Genes Basel. 2019;10(4):273. doi: 10.3390/genes10040273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kingma D, Ba J. Adam: a method for stochastic optimization. 2014. arXiv:1412.6980
- 71.Monro RS. A stochastic approximation method. Ann Math Stat. 1951;22(3):400–407. doi: 10.1214/aoms/1177729586. [DOI] [Google Scholar]
- 72.De R, Ge H, Rj W. Learning representations by back-propagating errors. Nature. 1986;323(6088):533–536. doi: 10.1038/323533a0. [DOI] [Google Scholar]
- 73.Srivastava N, Hinton G, Krizhevsky A, Sutskever I, Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15:1929–1958. [Google Scholar]
- 74.Lecun Y, Bottou L, Orr GB. Neural networks: tricks of the trade. Can J Anaesth. 2012;41(7):658. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Table S1: The 5 subsets divided from the processed RPI488 dataset. Table S2: The results of EDLMFC under 5CV after running 10 times on RPI488 dataset. Table S3: The results of EDLMFC under 5CV after running 10 times on RPI1807 dataset. Table S4: The results of EDLMFC under 5CV after running 10 times on NPInter v2.0 dataset.
Data Availability Statement
The source code of EDLMFC and the datasets used in this work are available at https://github.com/JingjingWang-87/EDLMFC.