

Abstract
The resurgence of immunotherapy as an effective anticancer strategy has been coupled with more mature understandings of the underlying immune pathways and the development of novel immune checkpoint targets. The clinical development of antibodies first directed against cytotoxic T-lymphocyte–associated antigen 4, and later against program death 1, achieved durable disease control in a subset of patients across a large number of tumor types. Previous work demonstrates that targeting the programmed death 1 pathway alone does not result in complete restoration of T cell function and in some cancers, targeting this axis does not restore T cell function at all, suggesting a need to identify other molecules and inhibitory pathways that are involved in T cell exhaustion. In a comprehensive immune profiling study of patients with bladder cancer, we demonstrate T-cell immunoglobulin domain and mucin domain-containing molecule and T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain as possible targets as perhaps monotherapy or in combination with other immune checkpoint inhibitors.
1. Introduction
Blocking immune checkpoints as a potential anticancer strategy was initially proposed in 1996 when Leach and colleagues demonstrated that antibodies to cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) resulted in the rejection of tumors in preclinical models [1]. The clinical development of antibodies directed first against CTLA-4, and later against program death 1/programmed death ligand 1 (PD-1/PD-L1), achieved durable disease control in a subset of patients across a large number of different tumor types, opening an entirely new era in cancer immunotherapy. The introduction of these immune checkpoint inhibitors represented a paradigm shift in the clinical management of patients with urothelial carcinoma of the bladder (UCB). However, the observed successes in targeting PD-1 and CTLA-4 in UCB has proven beneficial in only a small subset of patients, likely due to the complexity associated with the various immune inhibitory pathways, underscoring the need to identify resistance mechanisms and novel checkpoint targets.
1.1. TIM-3 and TIGIT: novel checkpoint targets
Recent advances have yielded a better understanding of anticancer immune pathways and novel immune checkpoint targets including T-cell immunoglobulin domain and mucin domain-containing molecule (TIM-3). Commonly targeted and newly emerging immune checkpoints in UCB are demonstrated in Fig. 1. The TIM protein family, composed of 3 members in humans (hTIM1, 3, and 4), are type-1 cell surface glycoproteins involved in apoptosis and the regulation of innate and adaptive immune responses. Specifically, TIM-3, unlike the most well characterized negative regulators PD-1 and CTLA-4, has been postulated to have more complex regulatory functions in immune response [2,3].
Fig. 1.
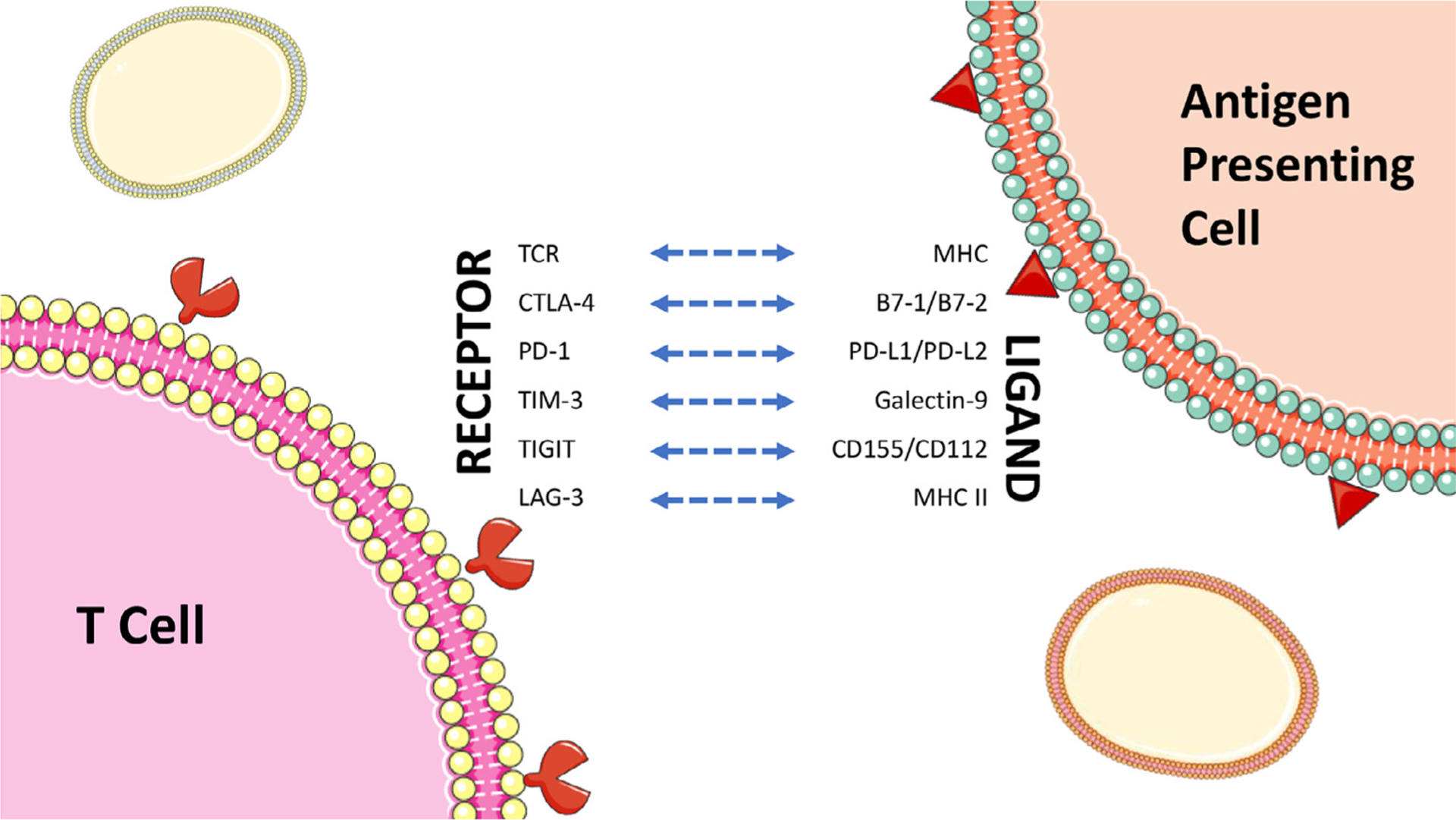
Commonly targeted and newly emerging immune checkpoints in bladder cancer. Note: CTLA-4, PD-1, TIM-3, TIGIT, and LAG-3 are inhibitory receptors.
TIM-3 is expressed on CD4+ and CD8+ T cells, Treg, and innate immune cells (DC, NK cells, and monocytes). Furthermore, it has been proposed as a regulator of the Th1 type immune response [4]. TIM-3 has been found to be upregulated on tumor antigen-specific CD8+ T cells and CD8+ tumor infiltrating lymphocytes (TILs), with increased expression of TIM-3 having an association with disease progression and shorter survival across a variety of distinct tumors including lung, gastric, cervical as well as sarcomas, and myeloid leukemias [5].
Preclinical models suggest the upregulation of TIM-3 on TILs and the synergistic potential of TIM-3 blockade in combination with other immune checkpoints. Alone, only a modest antitumor effect was seen across several tumor types in murine models; but a more robust, significant suppression of tumor growth was observed in combination with anti PD-1/PD-L1 monoclonal antibodies [6]. In preclinical studies across a variety of mouse tumor models, including prostate, a slowed tumor progression with TIM-3 blockade and a greater antitumor effect in combination with PD-1 and CTLA-4 blockade was observed [7,8]. Targeting the PD-1/PD-L1 pathway alone does not result in complete restoration of T-cell function as suggested by Blackburn et al. [9] and in some cancers, targeting this axis does not restore T cell function at all [10], suggesting a need to identify other molecules and inhibitory pathways that are involved in T cell exhaustion. Prior work demonstrates other markers of T cell exhaustion that, when inhibited, did not completely restore T cell function alone but rather synergized with PD-1 blockade to improve T cell responses [9]. As such, it appears that targeting multiple pathways may be an effective strategy in reversing T cell exhaustion.
To date, 3 TIM-3 antagonistic monoclonal antibodies are in early-phase clinical development. The safety and efficacy of MGB453 (Novartis, Basel, Switzerland) is being evaluated in a Phase I-Ib/II open-label multicenter study as a single agent and in combination with PD-1 monoclonal antibodies in patients with advanced malignancies including renal cell carcinoma, among others (NCT02608268). TSR-022 (TESARO, Waltham, USA) is being evaluated as a single agent in a Phase I trial in patients with advanced solid tumors (NCT02817633), which aims to explore the safety and clinical activity of TSR-022 as monotherapy and in combination with an anti-PD-1 antibody in patients with selected tumor types. Finally, Sym023 (Symphogen A/S, Ballerup, Denmark) is a Phase I trial investigating the safety, tolerability, and antineoplastic activity of this anti-TIM 3 in patients with advanced solid tumor malignancies and lymphomas (NCT03489343).
Another marker of T cell “exhaustion,” T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT) or is expressed on both T cells and NK cells [11,12]. In preclinical models, blockade of TIGIT can reverse the exhaustion of cytotoxic T lymphocyte (CTL)-mediated antitumor immunity and inhibit tumor growth, and its expression is associated with tumor progression [13]. The mechanism by which immunotolerance is achieved with TIGIT blockade has not been completely elucidated; the potential of TIGIT blockade as a therapeutic target, however, appears promising. TIGIT expression on tumor-infiltrating NK cells has been associated with tumor progression and was linked to functional exhaustion of NK cells. Blockade of TIGIT reversed the exhaustion of antitumor NK cells in multiple tumor models, demonstrating that the NK cell–associated TIGIT signaling pathway has a role in tumor evasion of the immune system and that reversing NK cell exhaustion is critical for the therapeutic effects of antitumor immunotherapy [14].
1.2. Implications of TIM-3 and TIGIT expression in UCB
Given the potential single or combinational therapeutic benefits of these novel immune checkpoint targets we sought to better understand their role in patients with UCB. We further sought to perform a comprehensive immune profiling of patients with UCB. Most studies to date have focused on the effect of blocking signaling through TIM-3 and TIGIT in the context of T cell exhaustion, despite significant promiscuity in their expression between both T and NK cells [15–19]. To determine if expression of TIM-3 and TIGIT changes on NK as well as T cells, and to determine if there are differences in expression between the PMBC and primary tumor, we analyzed the peripheral blood mononuclear cells (PBMC) (non-muscle invasive (NMI), n = 29, muscle invasive (MI), n = 16, healthy-donor (HD) = 17) and primary tumors (NMI, n = 13, MI, n = 16) of a cohort of UCB patients that included 24 individuals from whom matched blood and tumor tissue were available. Informed consent was obtained from all patients prior to collection of whole blood and/or primary tumor tissue under an IRB-approved, genitourinary biorepository protocol at the Tisch Cancer Institute at the Icahn School of Medicine at Mount Sinai. Cells were stained with a 14 color FACS panel to identify immune subsets and exhaustion markers (PD-1, TIM-3 and TIGIT). PBMC from UCB patients showed significant upregulation of TIM-3 on NK cells (2.8–4.6-fold), CD4+ T cells (9.8–17-fold), and CD8+ T cells (10.4–36.6-fold) compared to healthy controls. TIM-3 expression was higher on NK and T cells from the PBMC of individuals with MI compared to NMI disease, identifying TIM-3 expression as a barometer of primary tumor-invasiveness that is detectable in the peripheral blood. Importantly, the frequency of TIM-3+ peripheral blood NK cells was greater than that of TIM-3+ T cells, suggesting that NK cells are more sensitive to perturbations induced by UCB. TIGIT was also significantly increased in UCB patient PBMC relative to HD PBMC by NK cells (10.8–11.8-fold), CD4+ T cells (3.2–3.3-fold), and CD8+ T cells (1.9–2.1-fold). Of note, as TIM-3 expression increased on tumor NK cells, we observed a similar increase by peripheral blood NK cells, suggesting that perturbations within tumor tissue are recapitulated, and can be monitored, in the periphery.
These data are unpublished, thus limiting full disclosure of the results. However in sum, the presence of bladder cancer leads to an upregulation of the inhibitory receptors TIM-3 and TIGIT by NK and T cells in a tissue- and disease-stage-specific manner in both PBC and the TME of UCB patients. Upregulation of TIM-3 and TIGIT are correlated with the inhibition of NK cell effector functions. Our data shows that NK cell inhibition can be reversed with TIM-3 blockade in the PBMC of UCB patients causing enhanced cytokine production and the release of cytolytic effector molecules. NK dysfunction appears to be more prominent in the tumor, with no ability to reverse this function immediately ex vivo, but tumor-resident NK cells are not terminally dysfunctional because physical disruption of the TME and ex vivo culture restores competent function. TIM-3 blockade also improved IFNy production by T cells highlighting the potential synergy and strategies that target receptors expressed by multiple cell types. Our data suggest that overexpression of TIM-3 and TIGIT by peripheral blood NK cells of UCB patients may be useful as biomarkers to assess disease severity and monitor treatment as well as TIM-3 inhibition as a possible novel target in patients with UCB. Limitations to this work exist which parallel the limitations of using the most widely studied immune checkpoint target, PD-1, as a biomarker and/or therapeutic target. Clinically, our work requires validation through aforementioned clinical trials. As biomarkers, the optimal threshold for TIM-3 and TIGIT positivity is ill-defined, and the heterogeneity which exists between primary (including tumoral vs. tumoral and immune infiltrate expression) and metastatic sites lends clinical dilemma as to which sites should be sampled for TIM-3 and TIGIT expression. Variability in expression on the basis of specimen age, assay and vendor choice is likewise important considerations. As TIM-3 and TIGIT are dynamic markers, expression may be altered on the basis of prior therapy.
2. Conclusion
Although great strides have been made in the treatment of UCB in recent years in the form of immunotherapeutics, a large proportion of patients are yet to derive benefit from current treatment strategies, emphasizing a critical need to identify novel checkpoint molecules. Ongoing cycles of bench to bedside and bedside to bench research are required to further accelerate progress in enabling effective targeting and reliable combination strategies of these novel checkpoint molecules for tumor immunotherapy. Our work has identified TIM-3 and TIGIT as possible targets, as perhaps monotherapy or in combination with other immune checkpoint inhibitors, in patients with UCB.
Footnotes
Disclosures
The authors have no disclosures relevant to this study.
References
- [1].Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271(5256):1734–6. [DOI] [PubMed] [Google Scholar]
- [2].Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 2007;27(6):927–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol 2010;184(4):1918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol 2011;31(19):3963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ocana-Guzman R, Torre-Bouscoulet L, Sada-Ovalle I. TIM-3 regulates distinct functions in macrophages. Front Immunol 2016;7:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010; 207(10):2187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res 2013;19(18): 4917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ngiow SF, Teng MW, Smyth MJ. Prospects for TIM3-targeted anti-tumor immunotherapy. Cancer Res 2011;71(21):6567–71. [DOI] [PubMed] [Google Scholar]
- [9].Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A 2008;105(39):15016–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gehring AJ, Ho ZZ, Tan AT, Aung MO, Lee KH, Tan KC, et al. Profile of tumor antigen-specific CD8 T cells in patients with hepatitis B virus-related hepatocellular carcinoma. Gastroenterology 2009;137(2):682–90. [DOI] [PubMed] [Google Scholar]
- [11].Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol 2009;10(1):48–57. [DOI] [PubMed] [Google Scholar]
- [12].Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A 2009;106(42): 17858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014;26(6):923–37. [DOI] [PubMed] [Google Scholar]
- [14].Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol 2018;19(7):723–32. [DOI] [PubMed] [Google Scholar]
- [15].PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8⁺ T cells induced by melanoma vaccines., (2014). [DOI] [PMC free article] [PubMed]
- [16].Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018;558(7710):454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Audenet F, Farkas AM, Anastos H, Galsky MD, Bhardwaj N, Sfakianos JP. Immune phenotype of peripheral blood mononuclear cells in patients with high-risk non-muscle invasive bladder cancer. World J Urol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer cell 2014;26(6):923–37. [DOI] [PubMed] [Google Scholar]
- [19].Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol 2018. [DOI] [PubMed] [Google Scholar]