

Abstract
Recent molecular genetic findings on endometriosis and normal endometrium suggest a modified model in which circulating epithelial progenitor or stem cells intended to regenerate uterine endometrium after menstruation may become overreactive and trapped outside the uterus. These trapped epithelium-committed progenitor cells form nascent glands through clonal expansion and recruit polyclonal stromal cells, leading to the establishment of deep infiltrating endometriosis. Once formed, the ectopic tissue becomes subject to immune surveillance, resulting in chronic inflammation. The inflammatory response orchestrated by nuclear factor-κB signaling is exacerbated by aberrations in the estrogen receptor-β and progesterone receptor pathways, which are also affected by local inflammation, forming a dysregulated inflammation–hormonal loop. Glandular epithelium within endometriotic tissue harbors cancer-associated mutations that are frequently detected in endometriosis-related ovarian cancers. In this review, we summarize recent advances that have illuminated the origin and pathogenesis of endometriosis and have provided new avenues for research that promise to improve the early diagnosis and management of endometriosis.
Keywords: endometriosis, ovarian cancer, stem cell, genetic and epigenetic alterations, chronic inflammation, immune dysregulation, endocrine dysregulation
INTRODUCTION
Endometriosis is a reproductive disorder in which endometrial tissue is aberrantly located outside the uterus. Endometriosis affects nearly 10% of women of reproductive age, and 30% to 50% of those with the condition suffer from chronic pelvic pain and/or infertility, the two major clinical symptoms (1, 2). Endometriosis causes a significant economic burden, costing $70 billion dollars annually in the United States alone (3). Endometriosis can be classified into three subtypes based on its histopathology and anatomical locations: superficial endometriosis, deep infiltrating endometriosis (DIE), and ovarian endometriotic cysts (known as endometriomas, or so-called chocolate cysts) (2) (Figure 1). Superficial endometriosis usually appears on the surface or in the subserosal soft tissue of the peritoneum or visceral organs, whereas DIE involves lesions that extend deep into the muscular layer of the intestine, bladder wall, diaphragm, or other organs (Figure 2). Ovarian endometriotic cysts are found on the ovary, usually forming a large cystic structure (clinically interpreted as an adnexal mass). Different types of endometriosis are characterized by distinct biological and clinical features. Unlike superficial endometriosis, DIE frequently causes severe clinical symptoms, including significant pain and gastrointestinal and urological tract abnormalities. For women with DIE who do not respond to medical therapy, surgical removal can be extensive, requiring segmental bowel resection and extirpative resection of pelvic tissue. Ovarian endometriotic cysts, however, are commonly associated with infertility and carry an increased risk for the development of endometriosis-related ovarian cancer.
Figure 1.
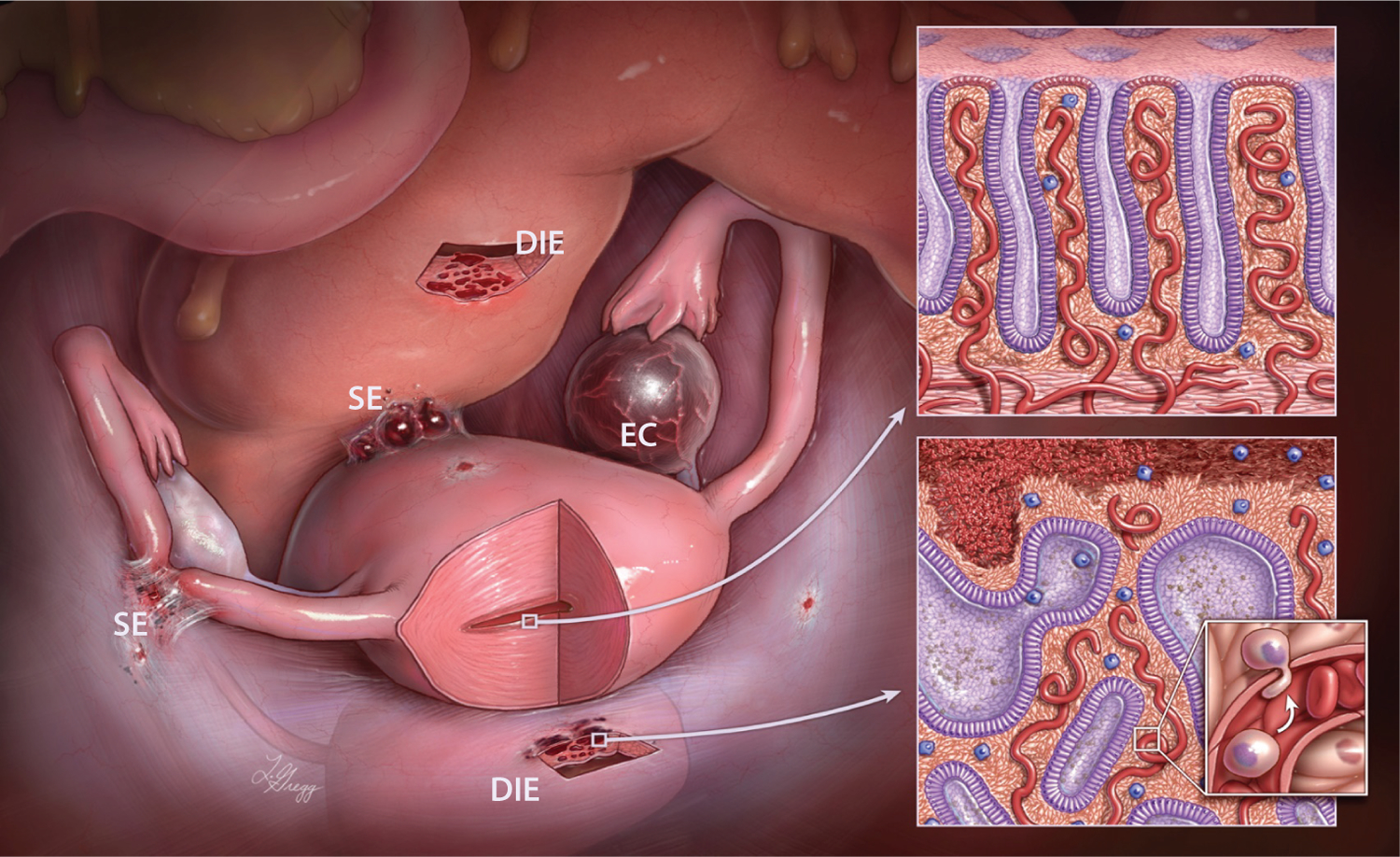
Different types of endometriosis. Endometriosis can occur in several forms: deep infiltrating endometriosis (DIE), ovarian endometriotic cyst (EC), and superficial endometriosis (SE). The insets illustrate the similar histology of (top) eutopic endometrium and (bottom) endometriosis, which is composed of both glandular epithelium and stroma. The small inset illustrates the transdifferentiation of a single circulating progenitor or stem epithelial cell or a group of such progenitor or stem epithelial cells responsible for establishing deep endometriosis. Figure copyright Ie-Ming Shih, Johns Hopkins University.
Figure 2.
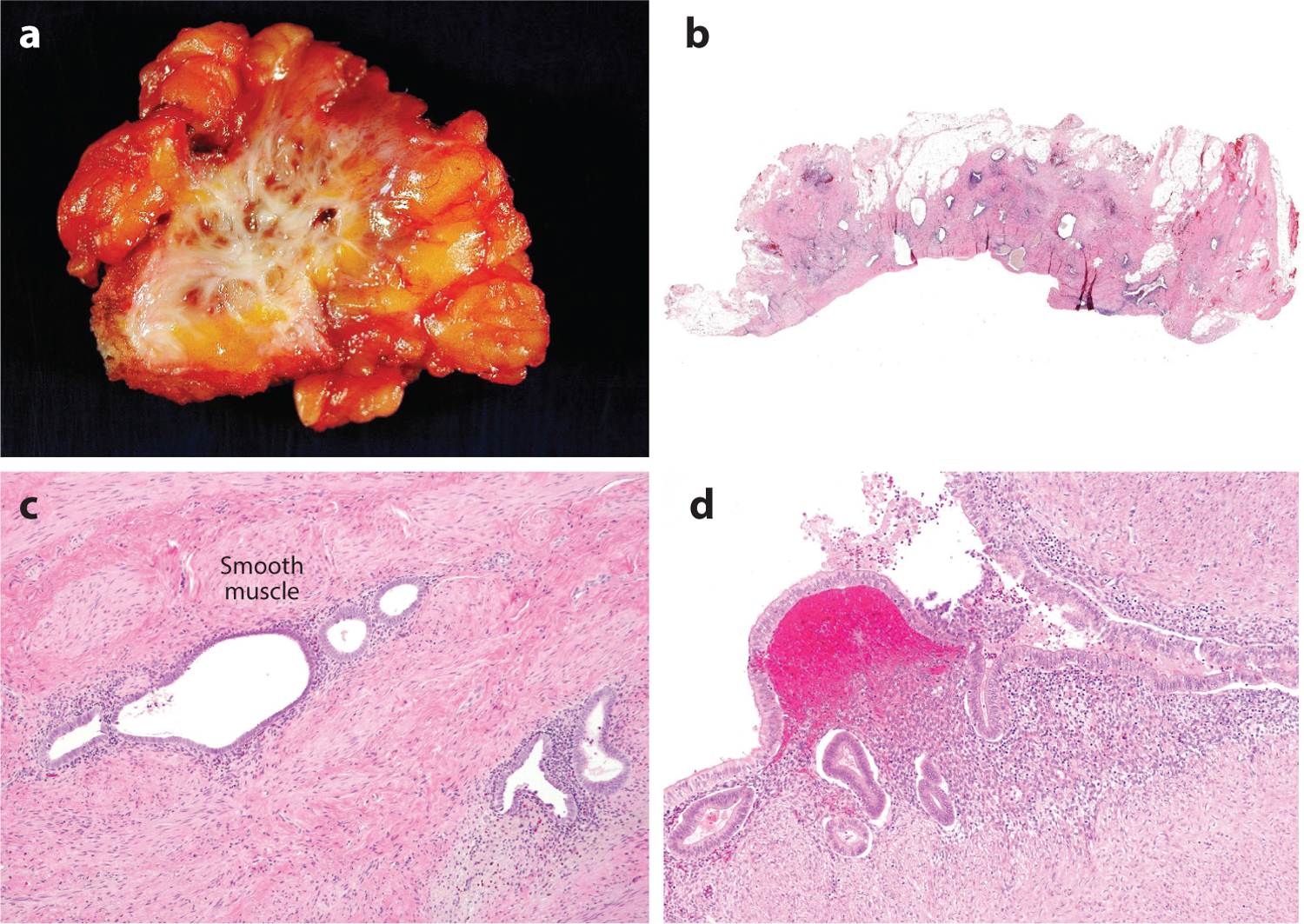
Gross appearance and histology of deep infiltrating endometriosis (DIE). (a) DIE involves fibro-adipose tissue, resulting in extensive scarring. (b) Discrete endometrial glands surrounded by fibrosis are evident in sections from the DIE shown in panel a. (c) Hematoxylin and eosin–stained section of DIE shows the presence of glands and endometrial-type stroma in soft tissue. (d) An example of superficial endometriosis showing involvement of the surface of a fallopian tube.
Current understanding of the pathogenesis and etiology of endometriosis is limited, even though significant effort has been devoted to their study for several decades. Several important questions remain to be answered. Among them, what is the origin of the different endometriosis species? Why does benign DIE behave like disseminated cancer? Why do lesions with normal-appearing histology always induce chronic inflammation, whereas their uterine counterparts do not? Researchers, gynecologists, and patients alike have puzzled over these questions since the disease was first characterized almost a century ago. Despite the expanding inventory of gonadotropin-releasing hormone (GnRH) antagonists or agonists, aromatase inhibitors, selective estrogen receptor (ER) and progesterone receptor (PR) modulators, and anti-inflammatory drugs, in some patients the disease becomes refractory to these medical treatments, and patients experience recurrence. They require multiple laparoscopic-assisted surgical interventions, leading to a reduced quality of life. The development of new drugs [e.g., antioxidants, kinase inhibitors, epigenetic modifiers, microRNAs (miRNAs)] to target specific pathways in individual patients offers hope for personalized treatment. This entails further understanding of the pathobiology of endometriosis, including the compensatory networks that remain active after specific hormonal pathways are blocked in different molecular types of endometriosis.
In this review, we summarize recent advances in understanding the pathogenesis of endometriosis and discuss how new and emerging knowledge will inform our future research, with the ultimate goal of reducing disease-associated symptoms through early diagnosis, the molecular classification of disease, and the development of new treatments.
RISK FACTORS AND ASSOCIATED DISORDERS
A number of genetic, endocrine, immunological, microbiotic, and environmental factors (4, 5) have been reported to be both positively and negatively associated with the development of endometriosis. Among factors associated with increased risk are Asian ethnicity (6), prolonged estrogen exposure (e.g., early age at menarche, shorter menstrual cycles, nulliparity) (7), low body mass index (8), and uterine outlet obstruction (8). However, cigarette smoking and greater parity are associated with reduced risk (9). Molecularly, endometriosis is associated with specific genetic changes. A genome-wide association study from a recent meta-analysis identified nine previously reported and five novel loci significantly associated with endometriosis risk. Interestingly, all loci harbor genes that function in sex steroid hormone pathways (FN1, CCDC170, ESR1, SYNE1, and FSHB), emphasizing the role of these pathways in the development of endometriosis (10).
After retrospective review of medical and pathology information from 1,000 women with endometriosis (excluding those with ovarian endometriotic cysts), Matalliotaki et al. (11) reported that endometriosis is associated with an increased risk of developing other benign gynecological disorders, especially ovarian endometriotic cysts and adenomyosis (Figure 3). This finding suggests that endometriosis of different types share a developmental pathobiology. While the most common endometriosis-associated symptoms, such as pelvic pain, dysmenorrhea, and infertility, involve the gynecological tract, endometriosis also has systemic effects on multiple organ systems. Women with endometriosis have a higher risk of infection, allergy, autoimmune disease, psychiatric conditions, preterm birth, metabolic syndrome, coronary heart disease, and cancer, especially ovarian and breast cancers, and melanoma (12–16). The cause of systemic comorbidities is unknown, but the production of cytokines, miRNA, and, perhaps, stem cell migration and dissemination have been implicated. Nevertheless, the broad systemic effects of endometriosis and its comorbidities deserve more clinical attention with respect to the management of patients with endometriosis.
Figure 3.
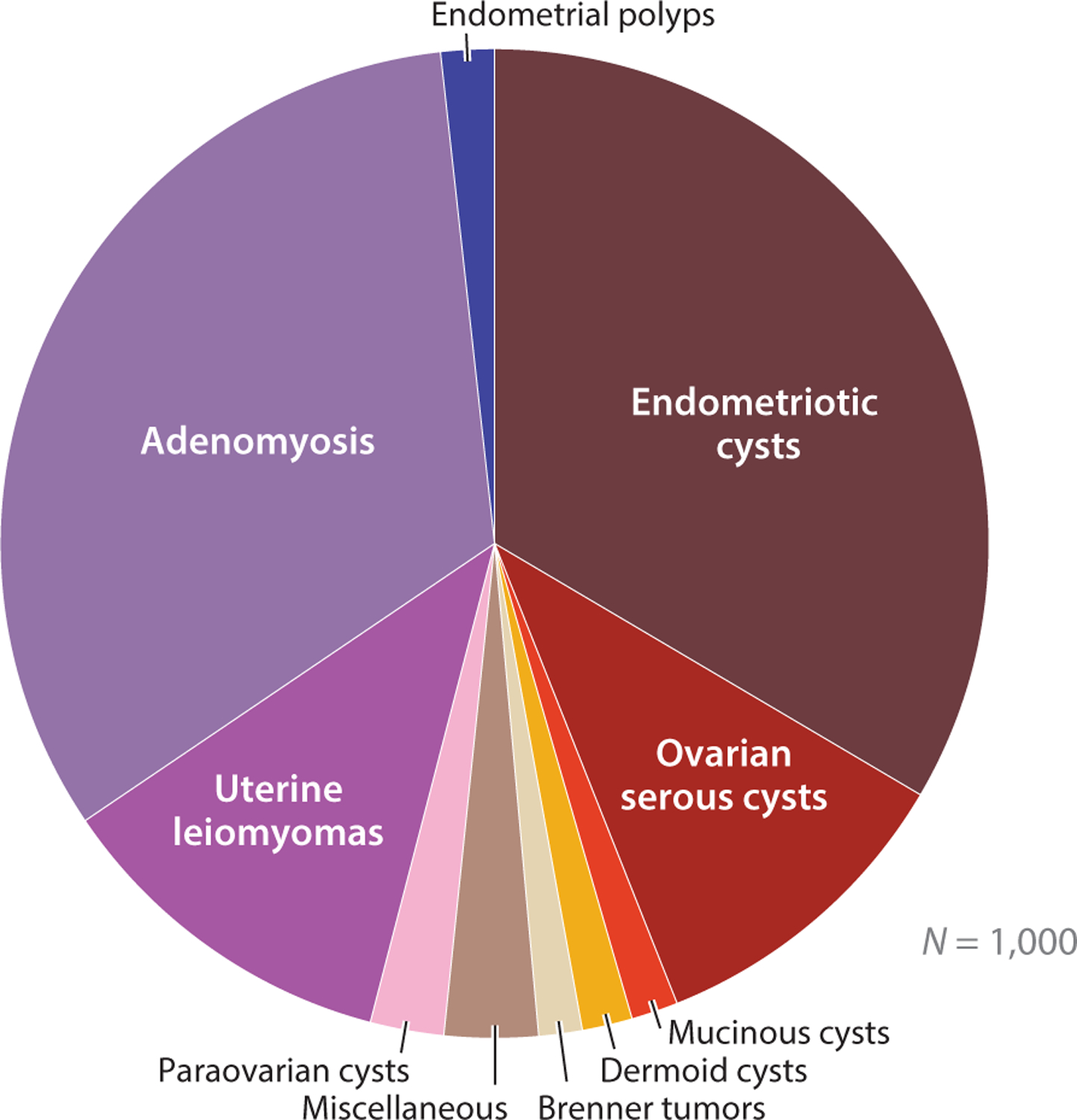
Disorders of female reproductive organs that are associated with endometriosis. Endometriosis is linked with increased risk of benign gynecological diseases, such as endometriotic cysts, uterine leiomyomas, and adenomyosis. Data collected from 1,000 women with endometriosis (11).
THE ORIGIN OF ENDOMETRIOSIS
The normal-appearing ectopic endometrium that becomes established and survives outside the uterus is one of the most intriguing features of endometriosis. Evolutionarily, multicellular organisms have established basic principles of differentiation governed by expectations for cells to follow on their path toward tissue formation: (a) repair their DNA when damaged by either intrinsic or extrinsic factors; (b) wait for signals from neighbors to divide; (c) reside legitimately in their appointed location. Apparently, endometriosis does not obey the third basic evolutionary rule. Whether driven by intrinsic or environmental needs or forces, endometriosis cells seem to have lost connection with their homeland—a cellular diaspora, perhaps.
There are several theories to account for the origin of endometriosis and to explain how tissue can be scattered throughout the abdominal cavity. Among the most-examined and most popular theories are retrograde menstruation and, more recently, extrauterine-sourced stem cells, but other theories have also been proposed, including hematogenous or lymphatic spread, coelomic metaplasia, and Müllerian rest induction (2). However, there is no single theory that explains all of the different clinical presentations and pathological features in endometriosis and the rare cases in which endometriosis is present in the thoracic cavity in women with Mayer-Rokitansky-Küster-Hauser syndrome and in men. Moreover, it is possible that superficial endometriosis, DIE, and ovarian endometriotic cysts develop via different mechanisms, and they invoke different theories. Unlike cancer, in which the epithelial cell is usually the focus of study, efforts to determine the evolutionary trajectories and clonal development of endometriosis are often confounded by the fact that endometrium-specific stromal cells are an inherent component of endometriosis in addition to glandular epithelium. The clonal relationship between epithelial cells and endometrium-like stromal components must be elucidated in various models to account for the origin of endometriosis. In the next sections, we elaborate on the retrograde menstruation and stem cell theories and examine their validity in different types of endometriosis, with special emphasis on assessing the clonality between epithelium and stroma within the same endometriotic lesions.
RETROGRADE MENSTRUATION MODEL
Sampson’s retrograde menstruation theory is widely accepted, and it is supported by the fact that women with uterine outflow obstruction have a higher risk of endometriosis (14, 17). However, this theory does not explain the low incidence of endometriosis relative to retrograde menstruation (18), nor does it explain some cases in which lesions are found deep in abdominal organs or even outside of the peritoneal cavity (18). Nonetheless, the retrograde theory well explains the superficial endometriosis found on the mucosa of fallopian tubes and on the subserosa of the fallopian tube, visceral organs, and peritoneal wall, as well as ovarian endometriotic cysts. For superficial endometriosis to develop, a few retrograde menstrual tissue fragments must first employ a molecular strategy to adhere to the serosal surface. Once attached and surviving on the surface, the ischemic endometrial tissue needs to grow into the superficial soft tissues where angiogenesis takes place to sustain their development.
It has been proposed that the rupture of an enlarged corpus luteum (or cyst) during the menstrual period may allow the retrograde menstrual endometrium to have access to and enter the disrupted ovarian tissue. After closure of the wound created by the rupture of the corpus luteum, the retained endometrial tissues are temporally and spatially nourished by the corpus luteal cells to form foci of ovarian endometriosis. As a result of local estrogen stimulation and the enriched blood supply unique to the ovary, these foci, as compared with their superficial counterparts, continue to grow as a blood-containing endometriotic cyst, grossly recognizable as a tumor mass on the ovary. In both the scenario of superficial endometriosis and that of the ovarian endometriotic cyst, it is most likely that the stromal and epithelial cells within the retrograde menstrual endometrium comigrate and can form either superficial endometriosis or an ovarian endometrioma. Recently, Suda et al. (19) observed cancer-associated mutations in epithelial cells from ovarian endometriotic cysts. Interestingly, they also detected a similar repertoire of somatic cancer-associated mutations in individual endometrial glands isolated from normal uterine endometrium. The investigators suggested that clonal expansion of epithelial cells with cancer-associated mutations occurs in normal endometrial glands, and these cells can leave the uterine cavity through retrograde menstruation, leading to the development of ovarian endometriotic cysts. This argument would be strengthened if there were evidence of a clonal relationship between individual glands and an endometriotic cyst in the same woman.
Applying the retrograde menstruation model to explain the occurrence of DIE appears challenging. Unlike superficial endometriosis, DIE is located deep in the organ structure, usually in the muscular layers of the gastrointestinal tract, urinary bladder, and ureter. In fact, the pathology of DIE is similar to cancer metastasis, in which a single or minute cluster of tumor cells containing stem-like cells is responsible for establishing new foci through angiolymphatic routes. If retrograde menstruation is the mechanism, one must assume that the menstrual endometrium containing both epithelial and stromal cells can enter into angiolymphatic circulation without disruption and has the ability to co-invade and undergo extravasation from the vessels in order to reside within the muscular layers of organs. However, there is no evidence that menstrual endometrium arising from benign endometrium is able to accomplish these demanding cancer-like tasks. Furthermore, the retrograde menstruation model cannot explain endometriosis found outside the abdominal cavity, such as in the thoracic cavity or brain, and at other sites. Thus, the long-held model of retrograde menstruation faces a crisis, and such a crisis leads to changes in models and changes in paradigms. The stem cell theory of endometriosis represents one of the new paradigms.
ENDOMETRIAL STEM CELL RECRUITMENT THEORY
The stem cell origin theory of endometriosis has gained considerable attention in recent years. The two main variants of the theory are based on the tissue origin of the stem cells, which are thought to arise either from the uterine endometrium or from bone marrow. Irrespective of the site of stem cell origin, hormones and other factors in the tissue microenvironment contribute to the adhesion, invasion, inflammation, angiogenesis, and evasion of immunosurveillance required for the establishment of endometriosis. In normal endometrium, there are several populations of multipotent endometrial stem cells, including epithelial derived, mesenchymal, and a mixed side population (20). The strength of this endometrial stem cell theory is that it not only fits the retrograde menstruation model but also explains the pathogenesis of DIE and endometriosis outside the abdominal cavity because stem cells of endometrial origin may enter the angiolymphatic space passively during menstruation and gain entry into the circulation system to find environmentally friendly “soil” for seeding.
Epithelial stem cells are hypothesized to localize in the basalis layer of the endometrium near the functionalis layer, where they are geographically protected from regular menstruation. These stem cells are thought to be responsible for regenerating the epithelium of the functionalis layer during the proliferative phase in response to estrogen. To date, there are no specific markers available for isolating endometrial epithelial stem cells. Markers deserving of further study include stage specific embryonic antigen 1 (SSEA-1), a marker highly expressed in the endometrium basalis; leucine repeat–containing G protein–coupled receptor 5 (LGR5), a marker dynamically expressed in functionalis epithelium closest to the basalis layer; and N-cadherin, a potential marker of epithelial progenitor cells (21–23). N-cadherin expression is high in the glandular epithelium in the basalis and is gradually lost as the gland extends into the functionalis. N-cadherin-positive basalis epithelial cells in human endometrium have high clonogenicity, proliferative potential, and self-renewing capacity, properties that are lost concomitant with the loss of N-cadherin. These cells can differentiate into cytokeratin-positive gland-like structures in three-dimensional cultures, consistent with their stem cell–like properties (23).
In contrast to endometrial epithelial stem cells, endometrial mesenchymal stem cells can be readily isolated and enriched (24), and, consequently, they have been intensively studied. These cells are localized in the perivascular area of both the basalis and functionalis, and they are responsible for generating functionalis stroma. They can be efficiently isolated from endometrial biopsies or from shed menstrual blood using combinations of the markers CD146, PDGFR-B, and SUSD2 (25–27).
One previous study has shown the presence of very small embryonic stem cell–like cells in adult mouse endometrium (25, 26). It was proposed that these cells might be a source of the endometrial stem cell population. Another study supported the presence of such cells, showing expression of human endometrium glandular and stromal markers in mice in early differentiating embryonic stem cells, and the study later identified CD146+PDGFR-B+ mesenchymal stem cells in the culture. This unique cell population deserves further characterization and investigation (27). Despite significant work in this area, there is no direct evidence that endometrial stem cells are involved in the pathogenesis of endometriosis.
BONE MARROW–DERIVED STEM CELL THEORY
Several bone marrow–derived stem cell populations (BMDSCs), including mesenchymal stem cells, hematopoietic stem cells, and endothelial progenitor cells, are suggested to contribute to the physiological regeneration of endometrium (28). The numbers of these cells in circulation may increase during the menstrual cycle and the proliferative phase of the endometrium. BMDSCs have been reported to directly contribute to the generation of both epithelial and stromal cells in human and mouse endometrium, while having a slight preference for stroma (29–31). This conclusion is supported by the distribution of human leukocyte antigen mismatched cells in the endometrium of women receiving bone marrow transplantation (32). Circulating CD45+ blood cells have been shown to colonize and regenerate the mouse uterine epithelium (33), and BMDSCs have been shown to engraft in transplanted endometrium in the mouse peritoneum (32). However, the efficiency with which stem cells have been found to regenerate uterine epithelial cells has been modest, raising questions about their physiological role in this process.
According to the theory, if BMDSCs go astray in soft tissue rather than homing back to the endometrium, endometriosis can then develop. Once formed, ectopic endometriotic tissue competes with eutopic endometrium for a limited number of circulating BMDSCs. Treating mice with bazedoxifene, an ER modulator, effectively suppresses endometriotic cell proliferation, decreases lesion size and number, and increases BMDSC recruitment to eutopic tissue. The decreased size and function of endometriotic lesions is likely due to decreasing ER-α and proliferating cell nuclear antigen (PCNA) in ectopic endometrium (34, 35).
The recruitment factors for this phenomenon remain unclear. Previously, granulocyte colony–stimulating factor (also known as colony stimulating factor 3) and interleukin (IL)-1B have been implied, but neither has been shown to significantly increase BMDSC recruitment. Recent evidence suggests that the CXCL12/CXCR4 axis is involved in recruiting BMDSCs to both eutopic and ectopic endometrial tissues. In cancer, activation of CXCL12/CXCR4 has been demonstrated to increase the expression of metalloproteinase, promote angiogenesis, and recruit endothelial cells to facilitate tumor progression and metastasis (36, 37). CXCR4 is a chemokine receptor expressed on the surface of stem cells (38), and CXCL12 is its ligand. CXCL12 is expressed by stromal and epithelial cells in various tissues, particularly from inflamed or injured tissue (39). An in vitro study showed that physiological estradiol (E2) increases the expression of CXCL12 and CXCR4 by mouse bone marrow stem cells, while progesterone (P4) increases their expression by human endometrial stromal cells. Expression levels of both CXCR4 and CXCL12 are higher in endometriotic epithelial cells than in normal uterine endometrium (40, 41). Conditioned culture medium from primary cultures of human endometriotic cells induces greater migration of BMDSCs than does conditioned medium from primary culture of normal endometrium (40).
BMDSCs are thought to be the major source of stem cells that give rise to endometriosis outside of the peritoneal cavity, and they could be a source of the rare endometriosis cases in men (42, 43). A recent study in mice found that mesenchymal stem cells from peritoneal endometriosis contribute to lesion vascularization and are capable of dissemination to the lungs, where they differentiate into cells expressing alveolar cell markers, suggesting multipotency (44). Interestingly, endometriosis-derived cells can return to the endometrium. Compared with eutopic endometrium, these endometriosis-derived cells express higher levels of cytokeratin, Wnt, and proteins involved in epithelial–mesenchymal transition. Endometriotic epithelium is hypothesized to undergo epithelial–mesenchymal transition and to return to the uterus through the venous circulation (45). It remains uncertain whether these phenomena can be confirmed in mice and extrapolated to humans.
THE NEW POSSIBLE UNIFIED THEORY
Central to validating the stem cell theory of endometriosis is accounting for the evolution of distinct tissue types, that is, glandular epithelium and endometrium-specific stroma, within an endometriotic lesion. Specifically, are both epithelial and stromal components of an endometriotic lesion clonally related or do they derive from independent stem or progenitor cells? In the retrograde menstruation model, fragments of epithelial tissue (presumably originating from multiple glands) and adjacent stromal cells implant onto the peritoneal surface; therefore, populations of epithelial and endometrium-specific stromal cells, which are presumably polyclonal, comigrate from the beginning of their journey. In the stem cell theory, which posits clonal proliferation and differentiation, one must assume that one stem cell is responsible for producing the progeny of differentiated epithelial and stromal cells to establish the entire focus of DIE lesions. Thus, among the various theories, there are five possible scenarios to account for DIE lesions: (a) a multipotent stem cell differentiates into both epithelial and stromal cell types simultaneously (i.e., clonally related epithelium and stroma); (b) an independent epithelial stem cell and an independent stromal stem cell colocalize, and they differentiate into their respective progeny (i.e., epithelium and stroma are clonal but unrelated); (c) epithelial stem or progenitor cells clonally differentiate into epithelial cells, which recruit multiple polyclonal independent stromal cells (i.e., only epithelium is clonal); (d) stromal stem or progenitor cells clonally differentiate into endometrium-specific stromal cells that recruit multiple polyclonal independent epithelial cells (i.e., only stroma is clonal); (e) different clones of epithelial and stromal stem or progenitor cells arrive at the same tissue locations (both components are polyclonal). To determine which of the above scenarios actually occurs in endometriosis, molecular genetic tests of clonality should be applied both to epithelial and stromal components. Both components might be separately enriched on tissue sections, and an analysis of somatic mutations, as has been widely employed to assess tumor clonality in cancer biology, could be applied.
Anglesio et al. (46) have recently applied next-generation sequencing to allow for discovery of cancer-associated driver mutations that initiate tumor development, as well as missense and synonymous passenger mutations that do not increase the selective growth advantage of the cells harboring them. Over time, these harmless passenger mutations accumulate during tissue self-renewal and tumorigenic clonal proliferation. These private passenger mutations provide the best barcode for tracing clonality because they are inherited, random, and functionally neutral. Although cancer-associated mutations can also be used as clonality markers, they are not ideal for two reasons. First, cancer-associated mutations are not neutral with respect to clonal selection, and, second, these mutations are found in transformed epithelial cells but not in stromal neoplasms among a wide variety of human cancers. To determine the clonality of DIE lesions, Noë et al. (47) took advantage of synonymous or missense passenger mutations and used laser-capture microdissection to isolate epithelial and stromal cells independently from within the same DIE lesions. They applied droplet digital polymerase chain reaction followed by nucleotide sequencing to determine mutant allele frequencies of individual mutations, from which they inferred their clonality status. The investigators found that only the epithelial component contained these synonymous or missense passenger mutations, while the stromal component from the same lesions did not. This suggests that endometriosis follows a complex evolutionary path, in which the epithelium is clonally and developmentally distinct from the stroma, supporting scenario c above.
Importantly, this finding by Noë et al. (47) does not support the view that endometriosis originates from a single stem or progenitor cell that differentiates into both epithelial and stromal cells or that epithelial cells differentiate into stromal cells through epithelial–mesenchymal transition, or vice versa. In these scenarios, stromal cells would acquire the same somatic mutations as the epithelial cells. This is not, however, the case. Rather, their data support a modified hypothesis that a single circulating endometrial epithelial progenitor cell undergoes a transient clonal expansion to form a nascent endometrial gland at the prospective endometriotic site (Figure 4). Subsequently, different circulating mesenchymal stem cells or endometrial stroma progenitor cells are recruited by the endometriotic lesion (gland only, at this moment) to establish the endometriosis, which is composed of both epithelial and stromal cells. Since the stromal cells are derived from different progenitor cells, the stromal component, unlike the epithelial counterpart, will be polyclonal.
Figure 4.
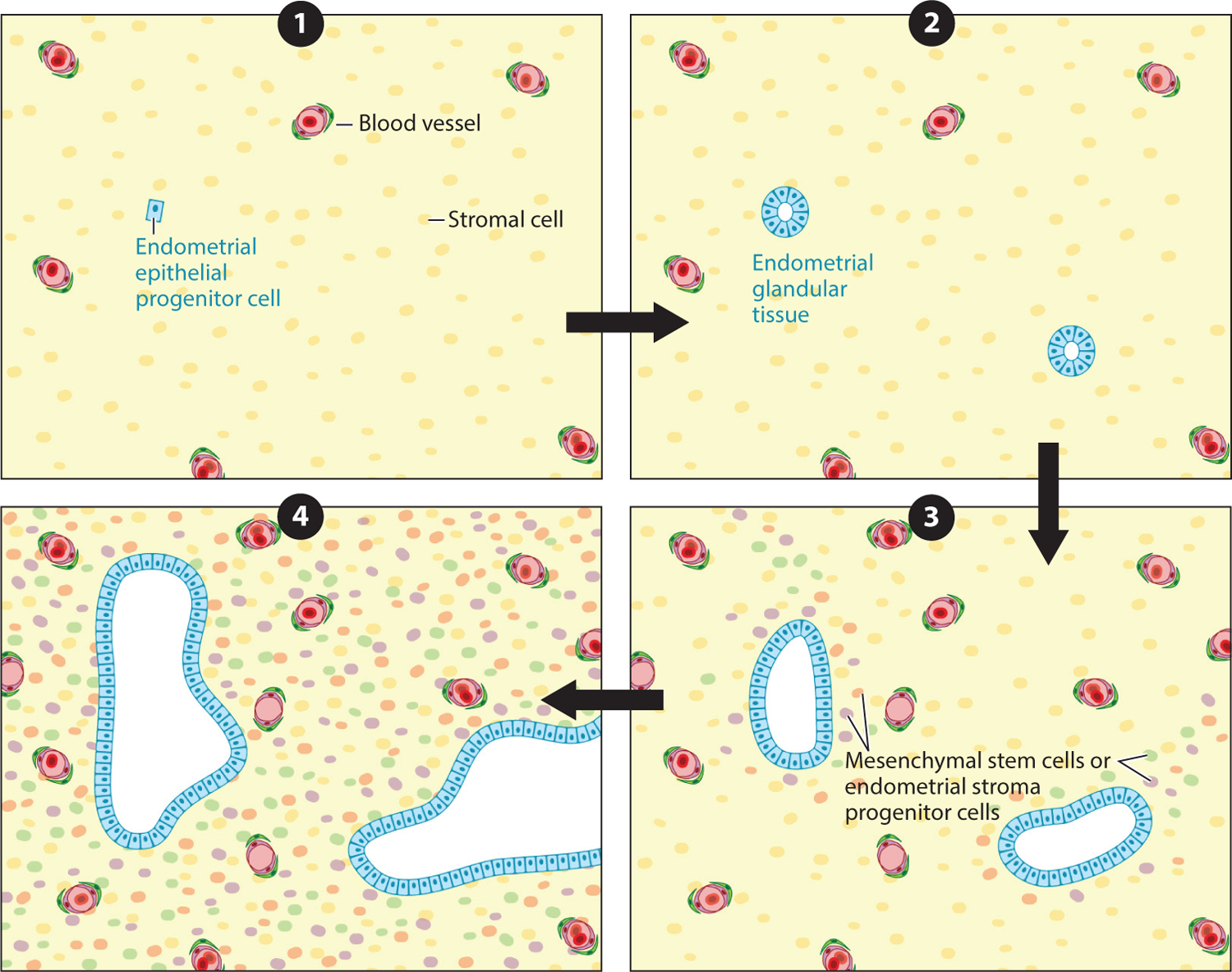
The modified stem cell hypothesis for deep infiltrating endometriosis proposes (❶) that a single circulating endometrial epithelial progenitor cell (blue), presumably already carrying certain somatic mutations, originating from the endometrium or elsewhere, (❷) undergoes transient clonal expansion to form endometrial glandular tissue at the prospective endometriotic site. Then, (❸) circulating mesenchymal stem cells or endometrial stroma progenitor cells are recruited by the nascent endometriosis (glands only, at this moment) to (❹) establish endometriosis composed of both epithelial and stromal cells.
Since human endometrial epithelium appears to be mutation prone—probably due to a mutagenic environment endowed by rapid cellular proliferation during the proliferative phase and primed by estrogen stimulation—the endometrium needs to undergo continued cycles of growth and shedding to suppress cancer development by preventing neoplastic clones from expanding and to provide freshly regenerated endometrium for embryonic implantation. From this perspective, menstruation is evolutionarily advantageous. How can this regenerative process become highly efficient in women? This modified stem cell theory may offer an explanation—that is, the abundant circulating epithelial progenitor or stem cells continue homing to the uterus to establish clonal endometrial glands.
From where do these epithelial stem or progenitor cells originate? Most likely, these epithelial progenitor cells are derived from the endometrium. This notion is based on the fact that glands within normal endometrium have been reported to harbor cancer-associated mutations in endometriosis-associated genes. Thus, epithelial stem or progenitor cells may enter the circulation, home to endometrium during the regeneration of the proliferative phase, and go astray ectopically to form DIE. Alternatively, BMDSCs may be the source of epithelial stem or progenitor cells, although this seems less likely because mesenchymal stem cells from bone marrow need to be committed to become epithelial cells, an observation that has never been proven. More importantly, there is no evidence that BMDSCs harbor somatic mutations in the cancer-associated genes that are commonly detected in endometriosis.
MOLECULAR GENETIC CHANGES IN ENDOMETRIOSIS
The most exciting molecular genetic finding in recent years is that somatic mutations of cancer-associated genes are commonly found in different types of endometriosis, including DIE (46), endometriotic cysts (19), and iatrogenic endometriosis (48). This is surprising given that endometriotic tissue appears normal and is histopathologically indistinguishable from eutopic endometrium. In addition, unlike cancer cells, endometriotic cells have only limited proliferative activity in general. Nevertheless, the original study by Anglesio et al. in 2017 (46) reported that 83% of benign DIE lesions contained somatic mutations, and 26% harbored cancer driver mutations, including in KRAS, PIK3CA, ARID1A, and PPP2R1A, all of which were confined to the epithelium (Table 1). These well-known genes are also frequently mutated in uterine endometrioid carcinomas and endometriosis-related ovarian cancers (19, 46, 49) (Figure 4). This unexpected observation prompts new avenues of investigation in the pathogenesis of endometriosis, and it may lead to the generation of a biologically informed classification scheme that ultimately improves prognostication and personalized treatment.
Table 1.
Cancer-associated genes reported in endometriosis cases
| Gene symbol | Gene name | Localization | Function | Role in cancer |
|---|---|---|---|---|
| KRAS | Kirsten rat sarcoma viral oncogene homolog | Chromosome 12: p12.1; plasma membrane | GTPase activity, positive control of cell growth and division, cell response to stimuli, cellular metabolism, regulator of signal transduction | Oncogene |
| ARID1A | AT-rich interaction domain 1A (SWI-like) | Chromosome 1: p35.3; nucleus | ATP-dependent chromatin remodeling factor, DNA binding, transcription regulator, DNA damage response | Tumor suppressor |
| PIK3CA | Phosphatidylinositol- 4,5-bisphosphate 3-kinase, catalytic subunit alpha | Chromosome 3: q26.3; cytosol | AKT pathway activator; serine/threonine protein kinase; cell growth, proliferation, differentiation, motility, survival; intracellular trafficking | Oncogene |
| PPP2R1A | Protein phosphatase 2, regulatory subunit Aalpha | Chromosome 19: q13.41; cytosol, mitochondria, nucleus | Serine/threonine protein phosphatase, negative control of cell growth and division, regulation of gene transcription | Oncogene or tumor suppressor, depending on mutation type |
It is also important to determine whether the cancer-associated mutations are biologically important for endometriosis development. The answer to this is confounded by recent sequencing data showing that some normal endometrial glands harbor somatic mutations in cancer-associated genes, such as KRAS (19). Thus, although these mutations may occur in cancer driver genes, they may simply serve as clonality markers (discussed previously) that are useful for relating the endometriosis to mutated stem cells in the endometrium but are otherwise of little biological significance. Generally speaking, carcinogenesis requires multiple (at least three) cancer driver mutations (50). Among all cases examined to date, no more than one mutation of a cancer driver gene has been reported in endometriosis. Thus, the three-mutation rule is not met, and the single mutation in cancer-associated genes per lesion is insufficient for cancer development. To determine whether KRAS mutations in DIE lesions are associated with increased proliferation and increased levels of phosphorylated AKT, we performed immunohistochemistry on a cohort of DIE lesions with or without KRAS mutations. We found that there was no difference in the Ki-67 proliferative index or in phosphorylated AKT levels between KRAS-mutated and wild-type lesions (S-F Lin, unpublished data). These findings are consistent with the observation that malignant transformation of endometriosis to carcinoma is exceedingly rare, although large ovarian endometriotic cysts may carry an increased, albeit still modest, risk because of their large number of constituent epithelial cells (approximately 1 million–fold more than discrete DIE lesions) (51, 52).
However, somatic mutations of a specific cancer-associated gene may contribute to phenotypes of the disease other than carcinogenesis. In fact, several reports have shown that increased KRAS activity (through either genetic or epigenetic mechanisms) may contribute to the survival of ectopic endometrium and to resistance to P4 treatment–induced apoptosis. For example, activation of KRAS in mice was associated with endometriosis-like lesions on the peritoneum and ovaries (53). Endometriotic lesions derived from mice with KRAS activating mutations survive longer than do those from wild-type mice (54). Another study demonstrated that KRAS activation led to aberrant overexpression of SIRT1, which colocalizes with BCL6, contributing to P4 resistance through inactivation of the GLI1 promoter (55). Let-7 miRNA normally binds to the 3′ untranslated region of KRAS transcripts, resulting in KRAS mRNA degradation and suppression of its expression. Interestingly, a specific germline KRAS polymorphic variant involving the Let-7 miRNA binding site is associated with increased KRAS expression levels. This variant is enriched in women who have endometriosis (56).
ARID1A is another gene that is mutated or its expression is lost in endometriosis. ARID1A encodes a protein that participates in SWI/SNF-mediated chromatin remodeling. As a remodeler for regulating local chromatin openness, ARID1A is known to play a critical role in many biological processes, including transcription, DNA methylation, and DNA damage repair (57). ARID1A is classified as a tumor suppressor gene, and inactivating mutations of ARID1A are detected in many human carcinomas, most commonly in endometrium-derived and -related cancers, including uterine endometrioid carcinoma, ovarian clear cell carcinoma, and ovarian endometrioid carcinoma (58–62). ARID1A may contribute to the clinical phenotype of endometriosis by increasing invasiveness and affecting the transforming growth factor (TGF)-β signaling pathway, which, in turn, affects P4 signaling. It will be important to determine whether mutations of cancer-associated genes are related to clinical P4 resistance since 40% of women with endometriosis exhibit P4 resistance (i.e., failure to respond to P4 therapy).
EPIGENETIC AND microRNA ALTERATIONS IN ENDOMETRIOSIS
Despite the similarity of genetic changes between eutopic and ectopic endometrial tissues, several studies report that they are different in epigenetic alterations and have different miRNA profiles (1, 63, 64). By analyzing global promoter methylation patterns, investigators have demonstrated that differentially methylated genes are associated with immune surveillance, inflammatory response, cell adhesion, negative regulation of apoptosis, response to steroid hormones, and activation of mitogen-activated protein kinase (MAPK) activity. It is uncertain whether these differences in methylation are biologically significant, as few of them have been shown to be associated with changes in gene expression. Nevertheless, some differentially methylated genes have been suggested to play a role in the development of endometriosis. Studies have reported aberrant methylation in promoters of ESR1, ESR2, PGR, NR5A1, CYP19A1, HOX gene clusters, and GATA family genes, with corresponding changes in gene and protein expression (65–68). It is likely that these genes form a complex signaling network acting in the pathogenesis of endometriosis. As an example, hypermethylation of GATA2 and hypomethylation of GATA6 are known to promote endometriotic phenotypes, including decreased expression of ESR1, PGR, MMP11, and ALD1A2 and increased expression of ESR2, NR5A1, HOXC6, and CYP19A1 (1).
Overexpression of NR5A1 and GATA6 in endometrial stromal cells may also affect spontaneous E2 biosynthesis, an important mechanism promoting endometriosis (69). DNMT3b, one of three DNA methyltransferases, has been reported to be responsible for the changes in methylation patterns. The failure of endometriotic stromal cells to downregulate DNMT3b in response to decidualization has been suggested to lead to changes in promoter methylation and gene expression in endometriosis (70, 71). It is apparent that studies are needed to further elucidate the mechanisms by which specific epigenetic alterations associated with endometriosis establish and promote lesion growth and survival. It is especially important to know how epigenetic alterations reprogram the endometriosis microenvironment and how these alterations account for clinical phenotypes. It is also important to determine how epigenetic changes develop separately in glandular epithelium and stroma within the same endometriotic lesions.
miRNAs are small noncoding RNAs of 19 to 25 nucleotides. They can regulate gene and protein expression at the posttranscriptional level by degrading mRNA or interfering with mRNA translation (72–74). Traveling via exosome transportation or microvesicular secretion, these miRNAs with other vesicular contents, such as enzymes, can affect cell functions, including the survival, differentiation, migration, and immune response of cells at other body sites, through paracrine and endocrine intercellular communication (75–78). Because endometriosis is a disease involving chronic inflammation and angiogenesis, it is conceivable that miRNAs from the blood and peritoneal fluid could influence endometriotic initiation and progression. Several studies have indicated that miRNA expression profiles differ between endometriosis and eutopic endometrial tissue (79, 80). For example, upregulation of miR-20a in endometriosis decreases target protein DUSP2, which subsequently results in aberrant activation of extracellular signal-regulated kinase (ERK)- and prostaglandin E2 (PGE2)-induced FGF9 expression (81, 82). Downregulation of miRNA-200b and upregulation of its targets ZEB1 and ZEB2 in endometriotic tissue are also implicated in promoting invasive and proliferative behavior (83). Polymorphisms in the targets of differentially expressed miRNAs may also play a role in the pathogenesis of endometriosis (56). As previously discussed, an inherited polymorphism of a Let-7 miRNA binding site in KRAS may be associated with abnormal endometrial growth and endometriosis (56). This finding resonates with the molecular genetic result showing KRAS as the gene most commonly mutated in endometriosis (19, 46, 48). Let-7 miRNA has also been proposed as a potential local treatment for endometriosis (84). We note that a consistent map of miRNA in endometriosis is difficult to draw because the findings have not been reproducible, presumably due to different methods used and different types of endometriosis included in these studies. Moreover, no evidence exists to show that a combination of miRNAs meets the necessary criteria for use as a diagnostic tool (82, 85). Further systemic study with uniformly well-controlled approaches will be required to establish miRNAs as diagnostic or prognostic biomarkers.
ENDOMETRIOSIS IS AN INFLAMMATORY DISORDER
It has been well established that local inflammation and immune dysregulation characterize endometriosis, but whether they are the cause or the consequence of the disease remains to be elucidated (86–88). Thus, understanding why ectopic but not eutopic endometrium tends to be inflammatory is fundamental to developing new treatment options for this disease, as inflammation and subsequent fibrosis account for the major clinical symptoms, including pelvic pain, bowel and urinary problems, and infertility. One possible explanation is that inflammation is induced by hemorrhage and tissue injury. Although endometriosis can respond to the cyclic changes of estrogen and P4, degenerating endometriotic tissue, unlike normal endometrium, cannot be shed during menstruation. Blood accumulates inside endometriotic lesions, especially in ovarian endometriotic cysts, which then undergoes degradation and induces the Fenton reaction. This results in the production of reactive oxygen species that initiate cell death, elicit inflammation, and prime the peritoneal surface for adherence by retrograde endometrium or progenitor or stem cells. This process is best evidenced by the presence of numerous hemosiderin-laden macrophages within endometriotic lesions, especially in the wall of ovarian endometriotic cysts.
There is abundant evidence to demonstrate that endometriosis is inflammatory. Several key inflammatory mediators, including COX-2, IL-1β, IL-8, tumor necrosis factor (TNF)-α, PGE2, and E2, are elevated in endometriotic lesions compared with eutopic endometrium (89–94). These mediators work synergistically to sustain and aggravate inflammation. In contrast to normal endometrium from women without associated endometriosis, uterine endometrium from women with endometriosis exhibits increased levels of COX-2 expression, and the endometriotic tissue has even higher COX-2 levels. Moreover, IL-1β also increases COX-2 expression and, therefore, upregulates PGE2 (95). As a result, endometriotic stromal cells have elevated PGE2 levels, which induces the production of E2, and this, in turn, promotes local inflammation (96). Excessive E2 and PGE2 thus form a positive feedback loop and promote persistent inflammation, immune responses, angiogenesis, and survival of endometriotic tissue (97–99). Several types of immune cells, including B lymphocytes (100), macrophages (101), CD1a+ dendritic cells (102), natural killer (NK) cells (103), T regulatory cells (104), and monocytic myeloid-derived suppressor cells (105), are prevalent in the inflamed microenvironment of endometriosis. These immune cells are also present in peritoneal fluid, and they interact with each other and with the epithelial and stromal cells of the lesions. Interestingly, the increase in immune cell infiltration, especially infiltration of macrophages, is also found in eutopic endometrium in patients with endometriosis (106), supporting the view that eutopic endometrium in endometriosis patients is different from endometrium in the absence of the disease, although follow-up studies are needed to validate these findings.
Like cancer, endometriosis resembles a chronic wound that never heals. In cancer, NF-κB, one of the most extensively studied inflammatory pathways, orchestrates many aspects of cancer-associated inflammatory phenotypes by coordinating cross talk between NF-κB and other signaling pathways, including the STAT3, p53, IRF, NRF2, JNK, Notch, and WNT/β-catenin pathways (107). Biologically, NF-κB participates in a multitude of cancer-promoting phenotypes, such as metastasis, angiogenesis, immunosuppression, tumor cell survival and proliferation, cancer stem cell maintenance, and therapy resistance (107, 108). NF-κB also appears to play a critical role in the development of endometriosis (109–111). In endometrial stromal cells, iron overload activates IKKβ and results in the production of abundant reactive oxygen species that stimulate the NF-κB pathway. Lipopolysaccharide promotes the development of murine endometriosis-like lesions via NF-κB pathway activation (112), and disulfiram, a candidate NF-κB inhibitor, prevents endometriotic implant growth in a rat endometriosis model (113).
However, increased anti-inflammatory cytokines, such as IL-6, IL-10, IL-15, and TGF-β, together with the soluble NKG2D ligands, MICA and MICB, in peritoneal fluid may mitigate the proinflammatory effects of PGE2 and NF-κB (114–119). Several immunosuppressive and immunoevasive mechanisms also regulate the immune response in endometriosis. A recent study has demonstrated that both PD-L1 and PD-1 are upregulated in endometriotic tissue relative to healthy controls, and the in vitro expression of PD-L1 on endometrial epithelium is induced by E2 (120). These data suggest that the PD-1/PD-L1 pathway may be involved in immune evasion in endometriosis. Thus, it appears that the fine balance between proinflammatory and anti-inflammatory mechanisms dictates the actual inflammatory phenotypes of individual endometriotic lesions.
It is intriguing why endometriosis is always associated with chronic inflammation. One explanation is that ectopic endometrium, although benign in nature, is considered abnormal by the body due to its aberrant location. Similar to an inflammatory response that clears infecting microorganisms, the inflammatory response to endometriosis may represent a host effort to eliminate an intruder. From this perspective, inflammatory cells are recruited to the site of endometriosis, and they secrete mediators (discussed above) to amplify or sustain the inflammation. It is possible that some, perhaps many, subclinical inflammatory events are actually physiological, and they may effectively prevent or abort the formation of some endometriotic lesions or eradicate preexisting endometriosis, or both. Then, fibrosis may clear the earlier active endometriosis and associated inflammation and replace the entire lesion with fibrotic tissue, giving rise to the so-called white lesions that cause adhesion. Lesions that are not eliminated by inflammatory clearance (due to ineffective anti-inflammatory activities or impaired immune surveillance) may survive, causing the clinical symptoms of endometriosis.
HORMONAL DYSREGULATION IN ENDOMETRIOSIS
One of the documented differences in endometrium between patients with and without endometriosis is that patients with endometriosis exhibit lower PR pathway activity and higher ER pathway activity. The difference is even greater when endometriotic tissues are compared with eutopic endometrium. This finding is important, as P4-based and antiestrogen-based therapies are the mainstay therapeutic modalities in the current medical management of endometriosis. Not all patients respond to P4-based therapy, and overcoming P4 resistance has become an unmet need for improving the quality of life in patients. The molecular basis of P4 resistance in endometriosis is emerging (121), and a reduction in the levels of PRs, especially the lack of the PR-B isoform, appears to be the main culprit. Similarly, decreased PR pathway activity promotes inflammation in endometriosis because the reduced PR activity can no longer effectively antagonize the proinflammatory ER pathway.
However, the ER pathway is highly active in endometriosis because of increased local E2 production and intrinsic ER-β pathway activation. A recent report describes possible mechanisms by which this could occur (122). It was found that suppressing elevated ER-β activity using selective ER-β antagonists inhibits the growth of endometriosis in mice. ER-β increases IL-1β, which enhances the cellular adhesion and proliferation of endometriosis by interacting with components of the cytoplasmic inflammasome. The report also showed that ER-β inhibits TNF-α-induced apoptosis, and it suggested that endometriosis survives by employing this mechanism to escape from endogenous immune surveillance. The ER-β pathway also enhances epithelial–mesenchymal transition, thus facilitating invasive activity, which is a characteristic feature of endometriotic tissue. Increased ER-β activity further downregulates PR expression by negatively acting on its promoter region. Thus, it appears that ER-β plays a central role in hormonal dysregulation in endometriosis.
In addition, increased E2 concentrations in endometriotic tissue further activate the ER pathway. First, the reduced PR activity promotes E2 synthesis. It has been established that endometrial stromal cells in normal endometrium respond to P4 by secreting paracrine factors, which, in turn, act on nearby epithelial cells to induce the expression of 17β-hydroxysteroid dehydrogenase type 2 (17β-HSD2). This enzyme effectively metabolizes the biologically active E2 to almost inactive estrone (E1). Thus, a decrease in 17β-HSD2 in the epithelial cells of endometriosis tissue due to reduced PR expression indirectly leads to increased E2 levels. Second, compared with eutopic endometrium, endometriotic tissue has higher aromatase activity, which results in increased E2 production. As a result, E2 accumulates locally and enhances ER-β activity to promote survival, disease progression, and P4 resistance in endometriosis (93, 123).
The above findings may have implications for the medical management of women with endometriosis. The initial recommended medical treatment is nonsteroidal anti-inflammatory drugs in combination with oral contraceptives (124, 125). The goal of hormone therapy is to maintain a local hypoestrogenic state, and the purpose of oral contraceptives is to inhibit ovarian E2 production and ovulation. Second-line medical treatment includes progestin monotherapy and a GnRH agonist. Progestin monotherapy is used in patients who are resistant to oral contraceptives and who are older than 35 years or who are susceptible to the side effects of antiestrogen therapy (e.g., deep vein thrombosis). In addition to inhibiting ovarian steroidogenesis, progestin also induces decidualization and apoptosis of ectopic endometrial tissue. GnRH agonists induce an initial stimulation of the hypothalamic–pituitary–gonadal axis, which is followed by inhibition due to desensitization. Most of the side effects of this conventional therapy come from the hypoestrogenic state, and they include breakthrough bleeding, lipid profile changes, osteoporosis, weight gain, and mood change. Future research focused on targeting specific abnormalities unique to endometriosis (such as increased ER-β activity) would be useful to help minimize the systemic side effects of current hormone therapy.
ENDOMETRIOSIS-ASSOCIATED OVARIAN CANCERS
Despite generally being considered a benign condition, histopathological and epidemiological studies have indicated that endometriotic tissue is the origin of ovarian clear cell carcinomas and ovarian endometrioid carcinomas, which are collectively known as endometriosis-associated cancers. These are type I ovarian cancers characterized by unique molecular genetic alterations that are different from type II ovarian cancers (which are high-grade serous carcinomas) (126). The risk of eventual ovarian cancer diagnosis in women with long-standing endometriosis is two- to three-fold higher than in women without endometriosis, but those reports mostly assess the risk in ovarian endometriotic cysts rather than DIE or superficial endometriosis. The best evidence for an endometriotic origin of some type I ovarian cancers comes from features seen during examination of resected endometriotic cysts. Different stages of tumor progression—ranging from normal-appearing epithelium lining the endometriotic cyst to atypical endometriosis to borderline tumor to in situ carcinoma to invasive carcinoma—have been observed in resected endometriotic cysts, providing unequivocal evidence that endometriotic cysts can develop into endometriosis-associated cancers (52, 127) (Figure 5). Multifocal endometriotic lesions can be clonally related, and endometriosis-associated ovarian neoplasms can develop from endometriotic lesions that carry a sufficient number of cancer-associated mutations, a circumstance seen in large ovarian endometriotic cysts (46, 49). It is likely that the somatic mutations are acquired during the expansion of endometriotic cysts. The original cells that establish the cyst must undergo many divisions during the formation of the cyst, and the number of epithelial cells may be significantly greater than in superficial or deep infiltrating lesions. Except in rare case reports (128), discrete small endometriotic lesions, such as superficial endometrioses and DIE, do not carry an increased risk of progression to endometriosis-associated cancer because they have a very low mutation burden.
Figure 5.
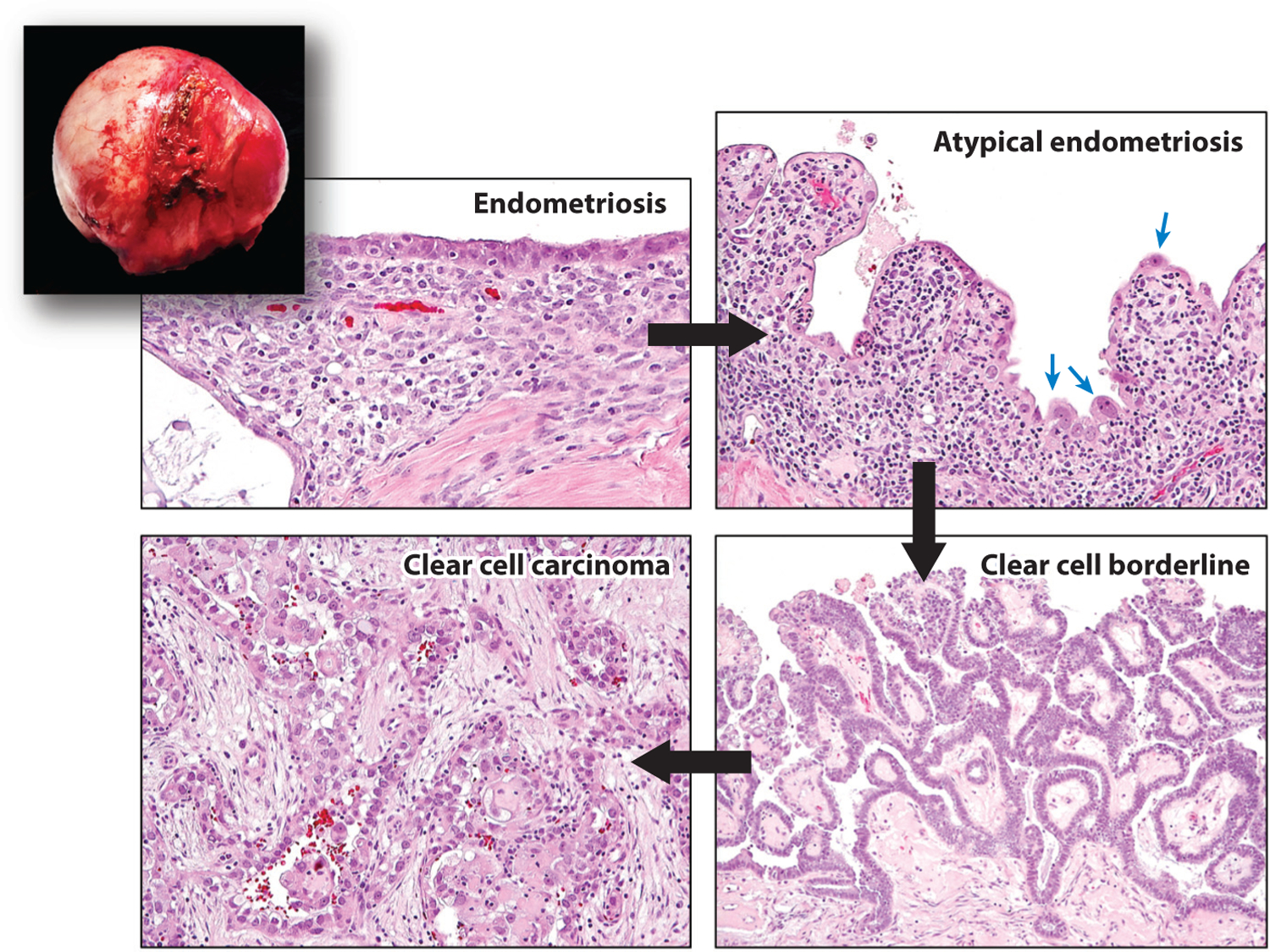
An ovarian endometriotic cyst (inset) containing different stages of tumor progression, from normal-appearing endometriosis to atypical endometriosis with highly atypical epithelial cells (blue arrows) to clear cell borderline tumor to invasive clear cell carcinoma. Black arrows indicate the sequence in tumor progression.
Somatic ARID1A inactivating mutations characterize endometriosis-associated ovarian neoplasms (60, 61, 127). Importantly, the loss of ARID1A expression, most likely due to somatic inactivating mutations—is an early molecular event in the development of most ovarian clear cell and endometrioid carcinomas arising in endometriomas (127, 129). Despite their shared origin, clear cell carcinoma and endometrioid carcinoma of the ovary may develop via different molecular genetic and epigenetic mechanisms. For example, somatic mutations of the TERT promoter and DNA copy number gain and upregulation of CCNE1 are frequent in ovarian clear cell carcinomas but not in ovarian endometrioid carcinomas (130, 131).
Given the potential for an ovarian endometriotic cyst to develop into endometriosis-associated ovarian cancer, Wang et al. (132) studied whether the mutations found in ovarian cancers could be identified in ovarian cyst fluids. They found that tumor-specific mutations were detectable in the cyst fluids of 77% of endometriosis-associated ovarian cancers, but no mutations were found in the cyst fluids of patients with benign cysts. This finding warrants thorough follow-up to determine the utility of this analysis for the early diagnosis of endometriosis-associated ovarian cancer.
CONCLUSIONS AND DIRECTIONS FOR FUTURE RESEARCH
Endometriosis is an estrogen-dependent inflammatory disorder. The ectopic residency of otherwise normal-appearing uterine tissues may result from an overreaction during endometrial regeneration in the uterus. Endometrial progenitor and stem cells are enriched in circulation during the proliferative phase of endometrial regeneration. These cells may have a high capacity to survive at an extrauterine site, and, thus, they may be able to establish endometriosis in addition to their obligatory function of regenerating the endometrium after menstruation. Additionally, local immune surveillance and inflammatory networks normally function to clear these ectopic lesions, and the outcome of the battle between endometriosis and local inflammation dictates the fate of endometriosis, which can be either fibrotic or active. For active lesions, dysregulation in local hormonal homeostasis and aberrant ER and PR signaling conspire with local inflammation to promote the survival of endometriotic tissue and cause symptoms and resistance to hormonal therapy (Figure 6). More interestingly, recent advances in the molecular genetic analysis of clonal trajectories in developing endometriosis have necessitated revisions to our model of the genesis of endometriosis, especially of DIE lesions.
Figure 6.
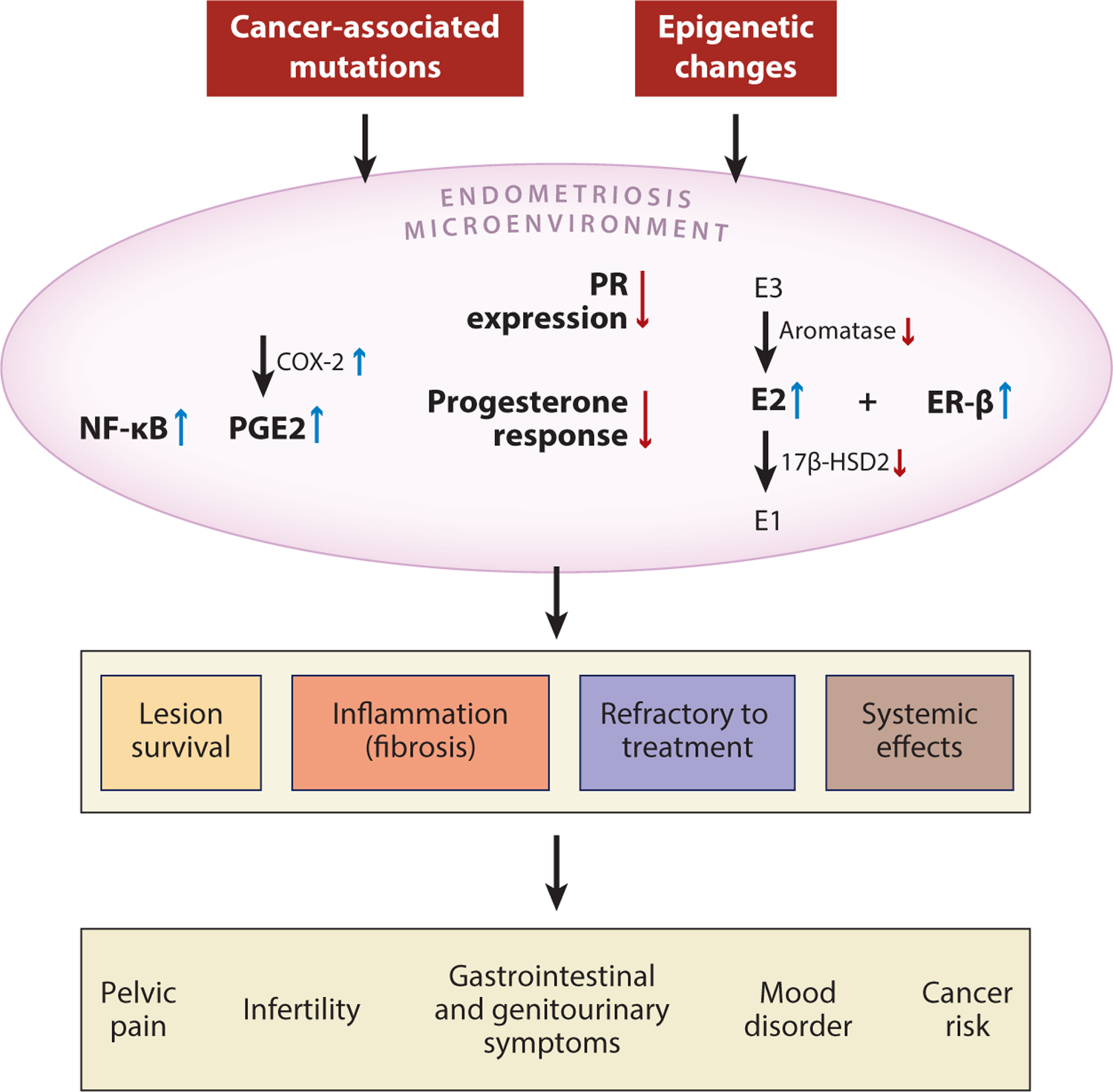
Summary of molecular changes involving inflammation and aberrant hormonal signaling in the pathogenesis of endometriosis. Both genetic and epigenetic alterations are associated with the development of endometriosis; these may include somatic mutations in cancer-associated genes or promoter methylation that alters the expression of genes involved in ER and PR signaling. As a result, the endometriosis microenvironment demonstrates numerous key molecular changes, including increased activity of NF-κB, PGE2, E2, and ER-β, that lead to inflammation and local dysregulation of hormonal pathways. As the chronic inflammation persists, it supports survival of the endometriotic lesion and induces local fibrosis. Persistent inflammation causes several systemic clinical effects and contributes to the refractory response to hormone-based treatment. Abbreviations: 17β-HSD2, 17β-hydroxysteroid dehydrogenase type 2; E1, estrone; E2, estradiol; E3, estriol; ER, estrogen receptor; NF-κB, nuclear factor-κB; PGE2, prostaglandin E2; PR, progesterone receptor.
Despite recent advances, several fundamental issues about endometriosis remain to be resolved. The first is to fully elucidate how endometriosis develops. The evidence is inconsistent with any single model, and it will likely be necessary to integrate different models for different types of endometriosis. A key step will be to identify the as-yet-unknown circulating epithelial progenitor or stem cells that are responsible for regenerating epithelium in both the endometrium and endometriotic lesions. Equally important will be to determine the origins of these progenitors and to explore the possibility of using them as biomarkers for predicting endometriosis risk and treatment response. Second, multispectral immunoprofiling will be needed to assess immune cell populations and their geographical distribution within individual endometriotic lesions. In vitro and in vivo models similar to those commonly used in cancer immunology can be applied to determine how distinct immune cell types contribute to the pathogenesis of endometriosis and how the NF-κB and COX-2 pathways, in particular, are activated to induce inflammation in endometriosis. Molecular pathology studies will be needed to determine whether different types of endometriosis (i.e., superficial, DIE, endometriotic cysts) are characterized by different inflammatory milieus or are functions of distinct immune checkpoints. Third, an emerging research direction is the analysis of exosome-mediated intracellular signaling, which may impact the development of endometriosis (133). Fourth, new classifications of endometriosis may be proposed if molecular genetic alterations, such as KRAS mutations, are found to correlate with clinical severity or treatment response. Finally, further efforts are required to assess the safety and efficacy of emerging target-based therapies intended to maximize therapeutic effects and minimize systemic side effects. Evaluating these new strategies in more relevant and larger animal models will likely be necessary in the course of translating results obtained in mouse models to humans. The recent progress in studying endometriosis has provided insight into this mysterious but common reproductive disorder, and it promises to lead to an improved quality of life for women suffering from endometriosis.
ACKNOWLEDGMENTS
This review was supported by a grant from the US National Institutes of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01HD096147) and the Richard W. TeLinde Gynecologic Pathology Research Program.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Dyson MT, Roqueiro D, Monsivais D, Ercan CM, Pavone ME, et al. 2014. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLOS Genet 10:e1004158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burney RO, Giudice LC. 2012. Pathogenesis and pathophysiology of endometriosis. Fertil. Steril 98:511–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simoens S, Dunselman G, Dirksen C, Hummelshoj L, Bokor A, et al. 2012. The burden of endometriosis: costs and quality of life of women with endometriosis and treated in referral centres. Hum. Reprod 27:1292–99 [DOI] [PubMed] [Google Scholar]
- 4.Olovsson M 2011. Immunological aspects of endometriosis: an update. Am. J. Reprod. Immunol 66(Suppl. 1):101–4 [DOI] [PubMed] [Google Scholar]
- 5.Baranov VS, Ivaschenko TE, Liehr T, Yarmolinskaya MI. 2015. Systems genetics view of endometriosis: a common complex disorder. Eur. J. Obstet. Gynecol. Reprod. Biol 185:59–65 [DOI] [PubMed] [Google Scholar]
- 6.Williams C, Long AJ, Noga H, Allaire C, Bedaiwy MA, et al. 2018. East and South East Asian ethnicity and moderate-to-severe endometriosis. J. Minim. Invasive Gynecol 26:507–15 [DOI] [PubMed] [Google Scholar]
- 7.Missmer SA, Hankinson SE, Spiegelman D, Barbieri RL, Malspeis S, et al. 2004. Reproductive history and endometriosis among premenopausal women. Obstet. Gynecol 104:965–74 [DOI] [PubMed] [Google Scholar]
- 8.McLeod BS, Retzloff MG. 2010. Epidemiology of endometriosis: an assessment of risk factors. Clin. Obstet. Gynecol 53:389–96 [DOI] [PubMed] [Google Scholar]
- 9.Shafrir AL, Farland LV, Shah DK, Harris HR, Kvaskoff M, et al. 2018. Risk for and consequences of endometriosis: a critical epidemiologic review. Best Pract. Res. Clin. Obstet. Gynaecol 51:1–15 [DOI] [PubMed] [Google Scholar]
- 10.Sapkota Y, Steinthorsdottir V, Morris AP, Fassbender A, Rahmioglu N, et al. 2017. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat. Commun 8:15539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matalliotaki C, Matalliotakis M, Ieromonachou P, Goulielmos GN, Zervou MI, et al. 2018. Co-existence of benign gynecological tumors with endometriosis in a group of 1,000 women. Oncol. Lett 15:1529–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris HR, Costenbader KH, Mu F, Kvaskoff M, Malspeis S, et al. 2016. Endometriosis and the risks of systemic lupus erythematosus and rheumatoid arthritis in the Nurses’ Health Study II. Ann. Rheum. Dis 75:1279–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen NM, Jorgensen KT, Pedersen BV, Rostgaard K, Frisch M. 2011. The co-occurrence of endometriosis with multiple sclerosis, systemic lupus erythematosus and Sjögren syndrome. Hum. Reprod 26:1555–59 [DOI] [PubMed] [Google Scholar]
- 14.Barbieri RL. 1998. Stenosis of the external cervical os: an association with endometriosis in women with chronic pelvic pain. Fertil. Steril 70:571–73 [DOI] [PubMed] [Google Scholar]
- 15.Ness RB, Cramer DW, Goodman MT, Kjaer SK, Mallin K, et al. 2002. Infertility, fertility drugs, and ovarian cancer: a pooled analysis of case-control studies. Am. J. Epidemiol 155:217–24 [DOI] [PubMed] [Google Scholar]
- 16.Alderman MH 3rd, Yoder N, Taylor HS. 2017. The systemic effects of endometriosis. Semin. Reprod. Med 35:263–70 [DOI] [PubMed] [Google Scholar]
- 17.Sanfilippo JS, Wakim NG, Schikler KN, Yussman MA. 1986. Endometriosis in association with uterine anomaly. Am. J. Obstet. Gynecol 154:39–43 [DOI] [PubMed] [Google Scholar]
- 18.D’Hooghe TM, Debrock S. 2002. Endometriosis, retrograde menstruation and peritoneal inflammation in women and in baboons. Hum. Reprod. Update 8:84–88 [DOI] [PubMed] [Google Scholar]
- 19.Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, et al. 2018. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Rep 24:1777–89 [DOI] [PubMed] [Google Scholar]
- 20.Tampaki EC, Tampakis A, Kontzoglou K, Kouraklis G. 2017. Commentary: somatic stem cells and their dysfunction in endometriosis. Front. Surg 4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valentijn AJ, Palial K, Al-Lamee H, Tempest N, Drury J, et al. 2013. SSEA-1 isolates human endometrial basal glandular epithelial cells: phenotypic and functional characterization and implications in the pathogenesis of endometriosis. Hum. Reprod 28:2695–708 [DOI] [PubMed] [Google Scholar]
- 22.Gil-Sanchis C, Cervello I, Mas A, Faus A, Pellicer A, et al. 2013. Leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5) as a putative human endometrial stem cell marker. Mol. Hum. Reprod 19:407–14 [DOI] [PubMed] [Google Scholar]
- 23.Nguyen HPT, Xiao L, Deane JA, Tan KS, Cousins FL, et al. 2017. N-cadherin identifies human endometrial epithelial progenitor cells by in vitro stem cell assays. Hum. Reprod 32:2254–68 [DOI] [PubMed] [Google Scholar]
- 24.Chan RW, Schwab KE, Gargett CE. 2004. Clonogenicity of human endometrial epithelial and stromal cells. Biol. Reprod 70:1738–50 [DOI] [PubMed] [Google Scholar]
- 25.Gunjal P, Bhartiya D, Metkari S, Manjramkar D, Patel H. 2015. Very small embryonic-like stem cells are the elusive mouse endometrial stem cells—a pilot study. J. Ovarian Res 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhartiya D, James K. 2017. Very small embryonic-like stem cells (VSELs) in adult mouse uterine perimetrium and myometrium. J. Ovarian Res 10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parasar P, Sacha CR, Ng N, McGuirk ER, Chinthala S, et al. 2017. Differentiating mouse embryonic stem cells express markers of human endometrium. Reprod. Biol. Endocrinol 15:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker CM, Beaudry P, Funakoshi T, Benny O, Zaslavsky A, et al. 2011. Circulating endothelial progenitor cells are up-regulated in a mouse model of endometriosis. Am. J. Pathol 178:1782–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor HS. 2004. Endometrial cells derived from donor stem cells in bone marrow transplant recipients. JAMA 292:81–85 [DOI] [PubMed] [Google Scholar]
- 30.Mints M, Jansson M, Sadeghi B, Westgren M, Uzunel M, et al. 2008. Endometrial endothelial cells are derived from donor stem cells in a bone marrow transplant recipient. Hum. Reprod 23:139–43 [DOI] [PubMed] [Google Scholar]
- 31.Ikoma T, Kyo S, Maida Y, Ozaki S, Takakura M, et al. 2009. Bone marrow–derived cells from male donors can compose endometrial glands in female transplant recipients. Am. J. Obstet. Gynecol 201:608.e1–e8 [DOI] [PubMed] [Google Scholar]
- 32.Du H, Taylor HS. 2007. Contribution of bone marrow–derived stem cells to endometrium and endometriosis. Stem Cells 25:2082–86 [DOI] [PubMed] [Google Scholar]
- 33.Bratincsak A, Brownstein MJ, Cassiani-Ingoni R, Pastorino S, Szalayova I, et al. 2007. CD45-positive blood cells give rise to uterine epithelial cells in mice. Stem Cells 25:2820–26 [DOI] [PubMed] [Google Scholar]
- 34.Sakr S, Naqvi H, Komm B, Taylor HS. 2014. Endometriosis impairs bone marrow–derived stem cell recruitment to the uterus whereas bazedoxifene treatment leads to endometriosis regression and improved uterine stem cell engraftment. Endocrinology 155:1489–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulak J Jr., Fischer C, Komm B, Taylor HS. 2011. Treatment with bazedoxifene, a selective estrogen receptor modulator, causes regression of endometriosis in a mouse model. Endocrinology 152:3226–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh S, Singh UP, Grizzle WE, Lillard JW Jr. 2004. CXCL12–CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Investig 84:1666–76 [DOI] [PubMed] [Google Scholar]
- 37.Chu CY, Cha ST, Chang CC, Hsiao CH, Tan CT, et al. 2007. Involvement of matrix metalloproteinase-13 in stromal-cell-derived factor 1α-directed invasion of human basal cell carcinoma cells. Oncogene 26:2491–501 [DOI] [PubMed] [Google Scholar]
- 38.Lai CY, Yamazaki S, Okabe M, Suzuki S, Maeyama Y, et al. 2014. Stage-specific roles for Cxcr4 signaling in murine hematopoietic stem/progenitor cells in the process of bone marrow repopulation. Stem Cells 32:1929–42 [DOI] [PubMed] [Google Scholar]
- 39.Hattori K, Heissig B, Rafii S. 2003. The regulation of hematopoietic stem cell and progenitor mobilization by chemokine SDF-1. Leuk. Lymphoma 44:575–82 [DOI] [PubMed] [Google Scholar]
- 40.Moridi I, Mamillapalli R, Cosar E, Ersoy GS, Taylor HS. 2017. Bone marrow stem cell chemotactic activity is induced by elevated CXCl12 in endometriosis. Reprod. Sci 24:526–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leconte M, Chouzenoux S, Nicco C, Chereau C, Arkwright S, et al. 2014. Role of the CXCL12–CXCR4 axis in the development of deep rectal endometriosis. J. Reprod. Immunol 103:45–52 [DOI] [PubMed] [Google Scholar]
- 42.Figueira PG, Abrao MS, Krikun G, Taylor HS. 2011. Stem cells in endometrium and their role in the pathogenesis of endometriosis. Ann. N. Y. Acad. Sci 1221:10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nezhat C, King LP, Paka C, Odegaard J, Beygui R. 2012. Bilateral thoracic endometriosis affecting the lung and diaphragm. JSLS 16:140–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li F, Alderman MH 3rd, Tal A, Mamillapalli R, Coolidge A, et al. 2018. Hematogenous dissemination of mesenchymal stem cells from endometriosis. Stem Cells 36:881–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santamaria X, Massasa EE, Taylor HS. 2012. Migration of cells from experimental endometriosis to the uterine endometrium. Endocrinology 153:5566–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noë M, et al. 2017. Cancer-associated mutations in endometriosis without cancer. N. Engl. J. Med 376:1835–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noë M, Ayhan A, Wang TL, Shih IM. 2018. Independent development of endometrial epithelium and stroma within the same endometriosis. J. Pathol 245:265–69 [DOI] [PubMed] [Google Scholar]
- 48.Lac V, Verhoef L, Aguirre-Hernandez R, Nazeran TM, Tessier-Cloutier B, et al. 2019. Iatrogenic endometriosis harbors somatic cancer-driver mutations. Hum. Reprod 34:69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anglesio MS, Bashashati A, Wang YK, Senz J, Ha G, et al. 2015. Multifocal endometriotic lesions associated with cancer are clonal and carry a high mutation burden. J. Pathol 236:201–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogelstein B, Kinzler KW. 2015. The path to cancer—three strikes and you’re out. N. Engl. J. Med 373:1895–98 [DOI] [PubMed] [Google Scholar]
- 51.Chui MH, Wang TL, Shih IM. 2017. Endometriosis: benign, malignant, or something in between? Oncotarget 8:78263–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilbur MA, Shih IM, Segars JH, Fader AN. 2017. Cancer implications for patients with endometriosis. Semin. Reprod. Med 35:110–16 [DOI] [PubMed] [Google Scholar]
- 53.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, et al. 2005. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med 11:63–70 [DOI] [PubMed] [Google Scholar]
- 54.Cheng CW, Licence D, Cook E, Luo F, Arends MJ, et al. 2011. Activation of mutated K-ras in donor endometrial epithelium and stroma promotes lesion growth in an intact immunocompetent murine model of endometriosis. J. Pathol 224:261–69 [DOI] [PubMed] [Google Scholar]
- 55.Yoo JY, Kim TH, Fazleabas AT, Palomino WA, Ahn SH, et al. 2017. KRAS activation and overexpression of SIRT1/BCL6 contributes to the pathogenesis of endometriosis and progesterone resistance. Sci. Rep 7:6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grechukhina O, Petracco R, Popkhadze S, Massasa E, Paranjape T, et al. 2012. A polymorphism in a let-7 microRNA binding site of KRAS in women with endometriosis. EMBO Mol. Med 4:206–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu RC, Wang TL, Shih IM. 2014. The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther 15:655–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guan B, Mao TL, Panuganti PK, Kuhn E, Kurman RJ, et al. 2011. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am. J. Surg. Pathol 35:625–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guan B, Wang TL, Shih IM. 2011. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res 71:6718–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones S, Wang TL, Shih IM, Mao TL, Nakayama K, et al. 2010. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330:228–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, et al. 2010. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med 363:1532–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maeda D, Mao TL, Fukayama M, Nakagawa S, Yano T, et al. 2010. Clinicopathological significance of loss of ARID1A immunoreactivity in ovarian clear cell carcinoma. Int. J. Mol. Sci 11:5120–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borghese B, Barbaux S, Mondon F, Santulli P, Pierre G, et al. 2010. Genome-wide profiling of methylated promoters in endometriosis reveals a subtelomeric location of hypermethylation. Mol. Endocrinol 24:1872–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L, Zhao J, Li Y, Wang Z, Kang S. 2019. Genome-wide analysis of DNA methylation in endometriosis using Illumina Human Methylation 450 K BeadChips. Mol. Reprod. Dev 86:491–501 [DOI] [PubMed] [Google Scholar]
- 65.Xue Q, Lin Z, Cheng YH, Huang CC, Marsh E, et al. 2007. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol. Reprod 77:681–87 [DOI] [PubMed] [Google Scholar]
- 66.Xue Q, Lin Z, Yin P, Milad MP, Cheng YH, et al. 2007. Transcriptional activation of steroidogenic factor-1 by hypomethylation of the 5r CpG island in endometriosis. J. Clin. Endocrinol. Metab 92:3261–67 [DOI] [PubMed] [Google Scholar]
- 67.Izawa M, Taniguchi F, Terakawa N, Harada T. 2013. Epigenetic aberration of gene expression in endometriosis. Front. Biosci 5:900–10 [DOI] [PubMed] [Google Scholar]
- 68.Wu Y, Halverson G, Basir Z, Strawn E, Yan P, et al. 2005. Aberrant methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis. Am. J. Obstet. Gynecol 193:371–80 [DOI] [PubMed] [Google Scholar]
- 69.Bernardi LA, Dyson MT, Tokunaga H, Sison C, Oral M, et al. 2018. The essential role of GATA6 in the activation of estrogen synthesis in endometriosis. Reprod. Sci 26:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamagata Y, Asada H, Tamura I, Lee L, Maekawa R, et al. 2009. DNA methyltransferase expression in the human endometrium: down-regulation by progesterone and estrogen. Hum. Reprod 24:1126–32 [DOI] [PubMed] [Google Scholar]
- 71.Dyson MT, Kakinuma T, Pavone ME, Monsivais D, Navarro A, et al. 2015. Aberrant expression and localization of deoxyribonucleic acid methyltransferase 3B in endometriotic stromal cells. Fertil. Steril 104:953–63.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ibrahim SA, Hassan H, Gotte M. 2014. MicroRNA-dependent targeting of the extracellular matrix as a mechanism of regulating cell behavior. Biochim. Biophys. Acta Gen. Subj 1840:2609–20 [DOI] [PubMed] [Google Scholar]
- 73.Ambros V 2004. The functions of animal microRNAs. Nature 431:350–55 [DOI] [PubMed] [Google Scholar]
- 74.Ebert MS, Sharp PA. 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149:515–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raposo G, Stoorvogel W. 2013. Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol 200:373–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Braicu C, Tomuleasa C, Monroig P, Cucuianu A, Berindan-Neagoe I, et al. 2015. Exosomes as divine messengers: Are they the Hermes of modern molecular oncology? Cell Death Differ. 22:34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Finn NA, Eapen D, Manocha P, Al Kassem H, Lassegue B, et al. 2013. Coronary heart disease alters intercellular communication by modifying microparticle-mediated microRNA transport. FEBS Lett 587:3456–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kosaka N, Yoshioka Y, Hagiwara K, Tominaga N, Katsuda T, et al. 2013. Trash or treasure: extracellular microRNAs and cell-to-cell communication. Front. Genet 4:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ibrahim SA, Yip GW, Stock C, Pan JW, Neubauer C, et al. 2012. Targeting of syndecan-1 by microRNA miR-10b promotes breast cancer cell motility and invasiveness via a Rho-GTPase- and E-cadherin-dependent mechanism. Int. J. Cancer 131:E884–96 [DOI] [PubMed] [Google Scholar]
- 80.Ohlsson Teague EM, Van der Hoek KH, Van der Hoek MB, Perry N, Wagaarachchi P, et al. 2009. MicroRNA-regulated pathways associated with endometriosis. Mol. Endocrinol 23:265–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsiao KY, Lin SC, Wu MH, Tsai SJ. 2015. Pathological functions of hypoxia in endometriosis. Front. Biosci 7:309–21 [DOI] [PubMed] [Google Scholar]
- 82.Lin SC, Wang CC, Wu MH, Yang SH, Li YH, et al. 2012. Hypoxia-induced microRNA-20a expression increases ERK phosphorylation and angiogenic gene expression in endometriotic stromal cells. J. Clin. Endocrinol. Metab 97:E1515–23 [DOI] [PubMed] [Google Scholar]
- 83.Eggers JC, Martino V, Reinbold R, Schafer SD, Kiesel L, et al. 2016. microRNA miR-200b affects proliferation, invasiveness and stemness of endometriotic cells by targeting ZEB1, ZEB2 and KLF4. Reprod. Biomed. Online 32:434–45 [DOI] [PubMed] [Google Scholar]
- 84.Sahin C, Mamillapalli R, Yi KW, Taylor HS. 2018. microRNA Let-7b: a novel treatment for endometriosis. J. Cell. Mol. Med 22:5346–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Agrawal S, Tapmeier T, Rahmioglu N, Kirtley S, Zondervan K, et al. 2018. The miRNA mirage: How close are we to finding a non-invasive diagnostic biomarker in endometriosis? A systematic review. Int. J. Mol. Sci 19:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zondervan KT, Becker CM, Koga K, Missmer SA, Taylor RN, Viganò P. 2018. Endometriosis. Nat. Rev. Dis. Primers 4:9. [DOI] [PubMed] [Google Scholar]
- 87.Giudice LC, Kao LC. 2004. Endometriosis. Lancet 364:1789–99 [DOI] [PubMed] [Google Scholar]
- 88.Grund EM, Kagan D, Tran CA, Zeitvogel A, Starzinski-Powitz A, et al. 2008. Tumor necrosis factor-α regulates inflammatory and mesenchymal responses via mitogen-activated protein kinase kinase, p38, and nuclear factor κB in human endometriotic epithelial cells. Mol. Pharmacol 73:1394–404 [DOI] [PubMed] [Google Scholar]
- 89.Tseng JF, Ryan IP, Milam TD, Murai JT, Schriock ED, et al. 1996. Interleukin-6 secretion in vitro is up-regulated in ectopic and eutopic endometrial stromal cells from women with endometriosis. J. Clin. Endocrinol. Metab 81:1118–22 [DOI] [PubMed] [Google Scholar]
- 90.Noble LS, Takayama K, Zeitoun KM, Putman JM, Johns DA, et al. 1997. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. J. Clin. Endocrinol. Metab 82:600–6 [DOI] [PubMed] [Google Scholar]
- 91.Noble LS, Simpson ER, Johns A, Bulun SE. 1996. Aromatase expression in endometriosis. J. Clin. Endocrinol. Metab 81:174–79 [DOI] [PubMed] [Google Scholar]
- 92.Juhasz-Boss I, Fischer C, Lattrich C, Skrzypczak M, Malik E, et al. 2011. Endometrial expression of estrogen receptor β and its splice variants in patients with and without endometriosis. Arch. Gynecol. Obstet 284:885–91 [DOI] [PubMed] [Google Scholar]
- 93.Han SJ, Hawkins SM, Begum K, Jung SY, Kovanci E, et al. 2012. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat. Med 18:1102–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu MH, Wang CA, Lin CC, Chen LC, Chang WC, et al. 2005. Distinct regulation of cyclooxygenase-2 by interleukin-1β in normal and endometriotic stromal cells. J. Clin. Endocrinol. Metab 90:286–95 [DOI] [PubMed] [Google Scholar]
- 95.Tamura M, Sebastian S, Yang S, Gurates B, Fang Z, et al. 2002. Interleukin-1β elevates cyclooxygenase-2 protein level and enzyme activity via increasing its mRNA stability in human endometrial stromal cells: an effect mediated by extracellularly regulated kinases 1 and 2. J. Clin. Endocrinol. Metab 87:3263–73 [DOI] [PubMed] [Google Scholar]
- 96.Bulun SE, Lin Z, Imir G, Amin S, Demura M, et al. 2005. Regulation of aromatase expression in estrogen-responsive breast and uterine disease: from bench to treatment. Pharmacol. Rev 57:359–83 [DOI] [PubMed] [Google Scholar]
- 97.Tsai SJ, Wu MH, Lin CC, Sun HS, Chen HM. 2001. Regulation of steroidogenic acute regulatory protein expression and progesterone production in endometriotic stromal cells. J. Clin. Endocrinol. Metab 86:5765–73 [DOI] [PubMed] [Google Scholar]
- 98.Sun HS, Hsiao KY, Hsu CC, Wu MH, Tsai SJ. 2003. Transactivation of steroidogenic acute regulatory protein in human endometriotic stromal cells is mediated by the prostaglandin EP2 receptor. Endocrinology 144:3934–42 [DOI] [PubMed] [Google Scholar]
- 99.Bulun SE, Monsavais D, Pavone ME, Dyson M, Xue Q, et al. 2012. Role of estrogen receptor-β in endometriosis. Semin. Reprod. Med 30:39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ahn SH, Khalaj K, Young SL, Lessey BA, Koti M, et al. 2016. Immune-inflammation gene signatures in endometriosis patients. Fertil. Steril 106:1420–31.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Capobianco A, Rovere-Querini P. 2013. Endometriosis, a disease of the macrophage. Front. Immunol 4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schulke L, Berbic M, Manconi F, Tokushige N, Markham R, et al. 2009. Dendritic cell populations in the eutopic and ectopic endometrium of women with endometriosis. Hum. Reprod 24:1695–703 [DOI] [PubMed] [Google Scholar]
- 103.Matarese G, De Placido G, Nikas Y, Alviggi C. 2003. Pathogenesis of endometriosis: natural immunity dysfunction or autoimmune disease? Trends Mol. Med 9:223–28 [DOI] [PubMed] [Google Scholar]
- 104.Berbic M, Hey-Cunningham AJ, Ng C, Tokushige N, Ganewatta S, et al. 2010. The role of Foxp3+ regulatory T-cells in endometriosis: a potential controlling mechanism for a complex, chronic immunological condition. Hum. Reprod 25:900–7 [DOI] [PubMed] [Google Scholar]
- 105.Chen Y, Wang K, Xu Y, Guo P, Hong B, et al. 2019. Alteration of myeloid-derived suppressor cells, chronic inflammatory cytokines, and exosomal miRNA contribute to the peritoneal immune disorder of patients with endometriosis. Reprod. Sci 26:1130–38 [DOI] [PubMed] [Google Scholar]
- 106.Berbic M, Schulke L, Markham R, Tokushige N, Russell P, et al. 2009. Macrophage expression in endometrium of women with and without endometriosis. Hum. Reprod 24:325–32 [DOI] [PubMed] [Google Scholar]
- 107.Taniguchi K, Karin M. 2018. NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol 18:309–24 [DOI] [PubMed] [Google Scholar]
- 108.Jinawath N, Vasoontara C, Jinawath A, Fang X, Zhao K, et al. 2010. Oncoproteomic analysis reveals co-upregulation of RELA and STAT5 in carboplatin resistant ovarian carcinoma. PLOS ONE 5:e11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lebovic DI, Mueller MD, Taylor RN. 2001. Immunobiology of endometriosis. Fertil. Steril 75:1–10 [DOI] [PubMed] [Google Scholar]
- 110.Gonzalez-Ramos R, Defrere S, Devoto L. 2012. Nuclear factor-κB: a main regulator of inflammation and cell survival in endometriosis pathophysiology. Fertil. Steril 98:520–28 [DOI] [PubMed] [Google Scholar]
- 111.Gonzalez-Ramos R, Van Langendonckt A, Defrere S, Lousse JC, Colette S, et al. 2010. Involvement of the nuclear factor-κB pathway in the pathogenesis of endometriosis. Fertil. Steril 94:1985–94 [DOI] [PubMed] [Google Scholar]
- 112.Azuma Y, Taniguchi F, Nakamura K, Nagira K, Khine YM, et al. 2017. Lipopolysaccharide promotes the development of murine endometriosis-like lesions via the nuclear factor-κB pathway. Am. J. Reprod. Immunol 77:e12631. [DOI] [PubMed] [Google Scholar]
- 113.Celik O, Ersahin A, Acet M, Celik N, Baykus Y, et al. 2016. Disulfiram, as a candidate NF-κB and proteasome inhibitor, prevents endometriotic implant growing in a rat model of endometriosis. Eur. Rev. Med. Pharmacol. Sci 20:4380–89 [PubMed] [Google Scholar]
- 114.Salih HR, Goehlsdorf D, Steinle A. 2006. Release of MICB molecules by tumor cells: mechanism and soluble MICB in sera of cancer patients. Hum. Immunol 67:188–95 [DOI] [PubMed] [Google Scholar]
- 115.Salih HR, Rammensee HG, Steinle A. 2002. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol 169:4098–102 [DOI] [PubMed] [Google Scholar]
- 116.Guo SW, Du Y, Liu X. 2016. Platelet-derived TGF-β1 mediates the down-modulation of NKG2D expression and may be responsible for impaired natural killer (NK) cytotoxicity in women with endometriosis. Hum. Reprod 31:1462–74 [DOI] [PubMed] [Google Scholar]
- 117.Kang YJ, Jeung IC, Park A, Park YJ, Jung H, et al. 2014. An increased level of IL-6 suppresses NK cell activity in peritoneal fluid of patients with endometriosis via regulation of SHP-2 expression. Hum. Reprod 29:2176–89 [DOI] [PubMed] [Google Scholar]
- 118.Yu JJ, Sun HT, Zhang ZF, Shi RX, Liu LB, et al. 2016. IL15 promotes growth and invasion of endometrial stromal cells and inhibits killing activity of NK cells in endometriosis. Reproduction 152:151–60 [DOI] [PubMed] [Google Scholar]
- 119.Yang HL, Zhou WJ, Chang KK, Mei J, Huang LQ, et al. 2017. The crosstalk between endometrial stromal cells and macrophages impairs cytotoxicity of NK cells in endometriosis by secreting IL-10 and TGF-beta. Reproduction 154:815–25 [DOI] [PubMed] [Google Scholar]
- 120.Wu L, Lv C, Su Y, Li C, Zhang H, et al. 2018. Expression of programed death-1 (PD-1) and its ligand PD-L1 is upregulated in endometriosis and promoted by 17β-estradiol. Gynecol. Endocrinol 35:251–56 [DOI] [PubMed] [Google Scholar]
- 121.Al-Sabbagh M, Lam EW, Brosens JJ. 2012. Mechanisms of endometrial progesterone resistance. Mol. Cell. Endocrinol 358:208–15 [DOI] [PubMed] [Google Scholar]
- 122.Han SJ, Jung SY, Wu SP, Hawkins SM, Park MJ, et al. 2015. Estrogen receptor β modulates apoptosis complexes and the inflammasome to drive the pathogenesis of endometriosis. Cell 163:960–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bulun SE, Cheng YH, Yin P, Imir G, Utsunomiya H, et al. 2006. Progesterone resistance in endometriosis: link to failure to metabolize estradiol. Mol. Cell. Endocrinol 248:94–103 [DOI] [PubMed] [Google Scholar]
- 124.Am. Coll. Obstet. Gynecol. 2010. Practice bulletin no. 114: management of endometriosis. Obstet. Gynecol 116:223–36 [DOI] [PubMed] [Google Scholar]
- 125.Pract. Comm. Am. Soc. Reprod. Med. 2014. Treatment of pelvic pain associated with endometriosis: a committee opinion. Fertil. Steril 101:927–35 [DOI] [PubMed] [Google Scholar]
- 126.Kurman RJ, Shih IM. 2016. The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am. J. Pathol 186:733–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maeda D, Shih IM. 2013. Pathogenesis and the role of ARID1A mutation in endometriosis-related ovarian neoplasms. Adv. Anat. Pathol 20:45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wu RC, Veras E, Lin J, Gerry E, Bahadirli-Talbott A, et al. 2017. Elucidating the pathogenesis of synchronous and metachronous tumors in a woman with endometrioid carcinomas using a whole-exome sequencing approach. Cold Spring Harb. Mol. Case Stud 3:a001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ayhan A, Mao TL, Seckin T, Wu CH, Guan B, et al. 2012. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int. J. Gynecol. Cancer 22:1310–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu RC, Ayhan A, Maeda D, Kim KR, Clarke BA, et al. 2014. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J. Pathol 232:473–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ayhan A, Kuhn E, Wu RC, Ogawa H, Bahadirli-Talbott A, et al. 2017. CCNE1 copy-number gain and overexpression identify ovarian clear cell carcinoma with a poor prognosis. Mod. Pathol 30:297–303 [DOI] [PubMed] [Google Scholar]
- 132.Wang Y, Sundfeldt K, Mateoiu C, Shih IM, Kurman RJ, et al. 2016. Diagnostic potential of tumor DNA from ovarian cyst fluid. eLife 5:e15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schjenken JE, Panir K, Robertson SA, Hull ML. 2019. Exosome-mediated intracellular signalling impacts the development of endometriosis—new avenues for endometriosis research. Mol. Hum. Reprod 25:2–4 [DOI] [PubMed] [Google Scholar]