

Abstract
Buccinum undatum is a subtidal gastropod that exhibits clear spatial variation in several phenotypic shell traits (color, shape, and thickness) across its North Atlantic distribution. Studies of spatial phenotypic variation exist for the species; however, population genetic studies have thus far relied on a limited set of mitochondrial and microsatellite markers. Here, we greatly expand on previous work by characterizing population genetic structure in B. undatum across the North Atlantic from SNP variation obtained by RAD sequencing. There was a high degree of genetic differentiation between Canadian and European populations (Iceland, Faroe Islands, and England) consistent with the divergence of populations in allopatry (F ST > 0.57 for all pairwise comparisons). In addition, B. undatum populations within Iceland, the Faroe Islands, and England are typified by weak but significant genetic structuring following an isolation‐by‐distance model. Finally, we established a significant correlation between genetic structuring in Iceland and two phenotypic traits: shell shape and color frequency. The works detailed here enhance our understanding of genetic structuring in B. undatum and establish the species as an intriguing model for future genome‐wide association studies.
Keywords: divergence, FST, genetics, phylogeography, speciation
The current study described novel trends in population genetic structure of Common Whelk, Buccinum undatum, from across the North Atlantic. We described strong genetic structuring between Canadian and European populations concurrent with the divergence in allopatry, as well as associations between genotypic and phenotypic structuring in Iceland. The works detailed here enhance our understanding of genetic structuring in B. undatum across multiple geographical scales and establish the species as an intriguing model for future genotype–phenotype associative studies.

1. INTRODUCTION
The reconstruction of spatial genetic structure can provide valuable insights into the evolutionary processes affecting species—such as genetic drift, adaptive selection, and gene flow between populations (Crawford & Oleksiak, 2016; Funk et al., 2012; Kohn et al., 2006)—while facilitating the characterization of historical demographic events and ongoing evolutionary trajectories (Emerson et al., 2010). Marine invertebrate phylogeography in the north Atlantic Ocean has been shaped by climate variation during the Pleistocene Epoch; glacial cycles have repeatedly altered species' distributions over the last two million years. Several marine invertebrate species have gone extinct in North America and have subsequently been recolonized from Europe (Ingolfsson, 1992; Vermeij, 1991; Wares & Cunningham, 2001). Expansion from southern refugia since the last glacial maximum 18–25 k years ago is generally believed to account for present‐day distributions. However, some species survived in glacial refugia and have recolonized from more northerly latitudes (Coyer et al., 2011; Maggs et al., 2008; Panova et al., 2011), undergoing genetic bottlenecks that resulted in signatures of low genetic diversity as found in recently expanded populations.
The common whelk, Buccinum undatum (Linnaeus, 1758), is a commercially valuable neogastropod species characterized by slow maturation rates, a mostly sedentary adult lifestyle, internal fertilization, and direct development of larvae (Himmelman, 1988; Himmelman & Hamel, 1993; Jalbert et al., 1989; Martel et al., 1986). Common whelk is broadly distributed at high latitudes across the North Atlantic Ocean, from eastern North America to western Europe, as well as Greenland, Iceland, and the Norwegian Sea (Gendron, 1992; Golikov, 1968; Taylor & Taylor, 1977). Phylogenetic reconstructions utilizing mitochondrial (mt)DNA cytochrome oxidase subunit I (COI) markers identified two monophyletic lineages (eastern and western North Atlantic: ENA and WNA, respectively), hypothesized to have diverged early during the Pleistocene approximately 2.1 mya (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Pálsson et al., 2014). Molecular indices, based on COI, indicate that the WNA and ENA lineages should be considered cryptic species that have diverged in allopatry (Magnúsdóttir et al., 2019). Canadian and ENA whelk also display variation in shell morphology (Magnúsdóttir, Pálsson, Westfall, Jónsson, & Örnólfsdóttir, 2019), as well as temporal spawning differences (Canadian whelk breed in May–July while ENA whelk in September–February) (Kideys et al., 1993; Magnúsdóttir et al., 2010; Martel et al., 1986), further supporting the diversification of the Canadian and ENA lineages. Within the ENA lineage, multiple studies of B. undatum have reported patterns of local population structure following an isolation‐by‐distance model (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Mariani et al., 2012; Pálsson et al., 2014; Weetman et al., 2006) at distances as short as 30 km in Icelandic waters (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Pálsson et al., 2014). However, despite repeated observations of fine‐scale population structure, genetic divergence (as measured by F ST) across the entire ENA lineage is uncharacteristically low (Weetman et al., 2006). Large effective population size and recent divergence may explain the limited genetic differentiation. Semi‐continuity has also been proposed as a potential mechanism maintaining gene flow among the European populations (Mariani et al., 2012; Weetman et al., 2006).
Comparing intraspecific patterns of genotypic and phenotypic variation may also elucidate concordant or distinctive responses to environmental landscapes (Zamudio et al., 2016). Common whelk displays discordant relationships between genotypic and phenotypic variation across small geographic scales in Iceland (Magnúsdóttir et al., 2010) and Ireland (Mariani et al., 2012). In Breiðafjörður (western Iceland), shell traits such as shape and color are highly differentiated among geographically proximate populations (20–30 km), exhibiting small but significant amounts of neutral genetic variation (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Pálsson et al., 2014). Fine‐scale phenotypic patterns have been documented, with shell shape and color exhibiting gradients from the inner to the outer bay that correlated with environmental variables (Magnúsdóttir et al., 2018). Limited demographic connectivity was apparent from the patterns of phenotypic variation; still, with only low levels of neutral genetic differentiation among the same populations, it was hypothesized that plastic responses to environmental variation are driving the observed phenotypic divergence (Mariani et al., 2012).
Although phenotypic variation may reflect population differentiation at the molecular level, a certain level of discordance can be expected due to sampling of markers, selection on functional traits, or plasticity. Previous studies on common whelk were based on a handful of loci (microsatellite and mtDNA COI) (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Mariani et al., 2012; Pálsson et al., 2014; Weetman et al., 2006). Studies utilizing genome‐wide genetic datasets such as RAD sequencing (Davey & Blaxter, 2010) may be required to delineate concordant responses between the genotypic and phenotypic variations observed in common whelk. RAD sequencing has been used to resolve fine‐scale population structure in several marine invertebrates species, including the American and European lobster (Benestan et al., 2015; Jenkins et al., 2019), great and Mediterranean scallop (Vendrami et al., 2017, 2019), sea scallop (Van Wyngaarden et al., 2016), and staghorn corals (Drury et al., 2016). Additionally, RAD sequencing has the potential to detect uncharacterized cryptic species complexes within known species distributions (e.g., in the River Limpet (Weiss et al., 2017)). As such, utilization of large‐scale RAD sequencing datasets may help clarify the low overall genetic differentiation across the ENA lineage and provide further evidence of the cryptic species found within common whelk.
The current study utilizes new and extensive geographic sampling of common whelk across the North Atlantic combined with RAD sequencing technologies to assess the genetic structure within the species at varying geographic scales, ranging from a broad‐scale analysis of genetic divergence across the North Atlantic to a more targeted fine‐scale analysis of divergence within Iceland. In addition, broad‐scale analyses (herein referred to as North Atlantic‐specific analyses) aimed to evaluate the existence of cryptic species in Canada and Europe and the low genetic divergence within the ENA lineage (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Pálsson et al., 2014; Weetman et al., 2006). Within Iceland, individuals from Breiðafjörður Bay (western Iceland) display exceptionally high levels of shell color variation (Magnúsdóttir et al., 2018). Detailed analyses of geographical patterns of phenotypic shell traits in Breiðafjörður have been described by Magnúsdóttir et al. (2018). Therefore, in addition to describing fine‐scale population trends in Breiðafjörður using a RAD sequencing approach, we aim to examine further the relationship between population genetic differentiation and existing phenotypic profiling data (shell color and shape) within the bay.
2. MATERIALS AND METHODS
2.1. Sample collection
Live B. undatum were collected at seven sites in Breiðafjörður, Iceland (IS, n = 295, Figure 1): Bjarneyjaráll (BJA, n = 31), Brjánslækur (BRJ, n = 60), Hvammsfjörður (HVA, n = 58), Hrútey (HRU, n = 66), Oddbjarnarsker (ODD, n = 40), Skor (SKO, n = 32), and Rauðasandur (RAU, n = 8). Across the North Atlantic (Figure 2), whelk were sampled from Canada (CAN, Quebec, Gulf of St. Lawrence, n = 29), England (ENG, English Channel, n = 30), and Faroe Islands (FAR, n = 29). Geographical coordinates for all sample sites are listed in Table 1. For all individuals, foot tissue was sampled via dissection and fixed immediately in 96% ethanol. Samples were stored at 4°C for 2 days before being exchanged with fresh ethanol and stored at –30°C.
Figure 1.
Sampling sites across the Bay of Breiðafjörður, west Iceland. Buccinum undatum were collected from the following sites: Rauðasandur (RAU), Skor (SKO), Bjarneyjaráll (BJA), Oddbjarnarsker (ODD), Brjánslækur (BRJ), Hrútey (HRU), and Hvammsfjörður (HVA). GPS coordinates for each sample site can be found in Table 1
Figure 2.
Distribution of Buccinum undatum sample sites across the North Atlantic. Individuals sampled are representative of populations from Canada, the Gulf of Saint Lawrence (CAN), Faroe Islands (FAR), England (ENG), and Breiðafjörður in west Iceland. GPS coordinates for each sample site can be found in Table 1
Table 1.
Sample locations, depths, GPS coordinates, and sample numbers (pre‐ and post‐data filtering) for Buccinum undatum sampled within Breiðafjörður (Iceland) and across the North Atlantic (Canada, England, Faroe Islands)
| Location | Abbreviation | Depth (m) | Latitude | Longitude | Year Sampled | Captured (n) | Analyzed (n) |
|---|---|---|---|---|---|---|---|
| Iceland | IS | – | – | – | 2014 | 295 | 104 |
| Bjarneyjaráll | BJA | 125 | 65° 08′27″ N | 23° 35′04″ W | 2014 | 31 | 9 |
| Brjánslækur | BRJ | 37 | 65° 29′52″ N | 23° 08′24″ W | 2014 | 60 | 9 |
| Hvammsfjörður | HVA | 15 | 65° 07′36″ N | 22° 22′37″ W | 2014 | 58 | 34 |
| Hrútey | HRU | 36 | 65° 01′34″ N | 22° 56′20″ W | 2014 | 66 | 18 |
| Oddbjarnarsker | ODD | 43 | 65° 18′50″ N | 23° 14′01″ W | 2014 | 40 | 4 |
| Skor | SKO | 53 | 65° 20′26″ N | 23° 55′28″ W | 2014 | 32 | 24 |
| Rauðasandur | RAU | 31 | 65° 27′59″ N | 24° 09′05″ W | 2014 | 8 | 6 |
| Canada | CAN | 18 | 50° 02′24″ N | 66° 25′48″ W | 2015 | 29 | 28 |
| England | ENG | 10 | 50° 23′24″ N | 1° 22′12″ W | 2007 | 30 | 14 |
| Faroe Islands | FAR | 40 | 61° 46′48″ N | 7° 36′00″ W | 2008 | 29 | 18 |
2.2. DNA extraction, RAD library preparation, and sequencing
Fixed foot tissue was dried briefly (to evaporate residual ethanol) and cut into small pieces that underwent DNA isolation using an Omega Bio‐Tek E.Z.N.A. Mollusc DNA Kit (as per manufacturer's instructions, https://www.omegabiotek.com/product/e‐z‐n‐a‐mollusc‐dna‐kit/). Double digest RAD sequencing libraries were constructed by digesting whole genomic DNA with Sau3AI and ApeKI and selecting fragments 390 bp to 430 bp using a Pippin Prep (Sage Science). Individuals were assigned a molecular identification tag using combinatorial barcoding of forward (5bp) and reverse (6bp) reads, respectively, then pooled into four separate libraries before size selection and amplification using 10 PCR cycles. Resulting library concentrations were quantified using SYBR Gold double‐stranded DNA assay measured on a TECAN GENios plate reader (TECAN™, www.tecan.com), and the sizes estimated by 2% agarose gel. Library quality was assessed using two pilot sequencing runs on Illumina MiSeq (paired‐end 2 × 150 bp). Libraries were sequenced using Illumina HiSeq 2500 (paired‐end 2 × 125 bp) across four lanes, generating an average of 4 ± 3.5 SD million paired raw reads per individual.
2.3. Data assessment, quality filtering, and stacks assembly
Raw reads were demultiplexed in paired mode (‐‐paired) using the process_radtags program in Stacks v.2.0b (Catchen et al., 2011, 2013), with restriction enzymes specified (‐‐renz_1 apeKI; renz_2 sau3AI) and the ‐‐inline_inline, –r, –c, –q, –t = 119 options enabled (demultiplexed reads available from Goodall, et al., 2020). Note that reads were truncated to 119 bp to ensure equal length following the removal of molecular identification tags. Demultiplexed reads (see Table S1), which included both Illumina MiSeq and HiSeq data (average of 3,499,689 (±3,131,707 standard deviation) reads per sample), were processed using the de novo stacks pipeline (i.e., ustacks, cstacks, sstacks, tsv2bam, gstacks, and populations).
The initial pass through the de novo stacks pipeline was designed to optimize the sample selection of downstream primary analyses by identifying and removing low‐quality individuals within the dataset. Overall, Stacks parameters were selected with the aim of retaining a high number of SNPs, with more rigorous filtering of SNPs and individuals occurring downstream. Initially, trimmed reads were processed in ustacks with the –M 4, –m 3 options enabled. To generate cstacks catalogs, sample inputs were limited to n = 200 individuals, following the recommendations of Rochette and Catchen (2017). To avoid the inclusion of over‐ or under‐sequenced individuals in cstacks assemblies, the median total read count (as derived from FastQC (Andrews, 2010)) was calculated from all n = 383 individuals. The n = 200 individuals used constituted those with a total read count closest to this median value, which included n individuals from each of the four North Atlantic sample locations: Canada(n) = 19; England(n) = 14; Faroe Island(n) = 17; Iceland(n) = 150. Following cstacks catalog construction (–n 6 option enabled), the remaining de novo stacks pipeline proceeded using the original n = 383 samples. Sstacks and tsv2bam steps proceeded using default settings, while gstacks was run in de novo mode (‐P option enabled). Finally, populations analyses were run with –p 3 and –r 0.5 options enabled, where the ‐‐popmap designated individuals based on their country of origin (i.e., Canada, England, Faroe Islands, and Iceland).
The resulting populations .vcf file was imported into Radiator (Gosselin, 2017) for quality analysis. Radiator's filter_rad pipeline was run in interactive mode, with the aim of blacklisting poor‐quality markers and individuals. Filtering parameters and outputs are listed in Table S2. After Radiator filtering, the following samples were considered as whitelisted and used for all primary analyses downstream: Canada(n) = 28; England(n) = 14; Faroe Island(n) = 18; and Iceland(n) = 117.
Primary analyses were initiated by undertaking a second pass through the de novo stacks pipeline using only the optimized n = 177 whitelisted sample set. Again, Stacks parameters were selected with the aim of retaining high numbers of SNPs, with more rigorous filtering occurring downstream in Radiator. Initially, the cstacks catalog was reassembled (–n 6 option enabled) using all n = 177 individuals. Sstacks and tsv2bam steps were run using default settings, and gstacks run in de novo mode (–P option enabled), yielding 900,825 loci, with an effective per‐sample coverage of 17.7 (± 6.8 standard deviation). Populations analyses were again run with –p 3 and –r 0.5 options enabled, and ‐‐popmap designating individuals based on their country of origin. Low‐quality individuals and markers were again filtered using Radiator (parameters and outputs listed in Table S3). The number of individuals retained, postfiltering, across all geographic scales of analysis is listed in Table 1. The average coverage per marker following Radiator was 18.6 (±6.3 standard deviation).
Departure from Hardy–Weinberg equilibrium (HWE) was assessed using a chi‐squared test (χ2) of expected genotype frequencies in the R‐package Pegas (Paradis, 2010), where the false discovery rate of p was controlled for using Benjamini and Yekutieli (2001) procedure for multiple testing. HWE tests were undertaking in a population‐wise manner, where population status was assigned based on country of origin, except for Icelandic individuals who were assigned population status relative to their sample site of origin in Breiðafjörður. In addition to population‐wise chi‐squared values, combined probabilities across all populations were calculated using Fisher's combined probability (Sokal & Rohlf, 1969) and adjusted using Benjamini‐Yekutieli's procedure. Loci were removed from the dataset when either two or more populations significantly deviated from HWE, or significant Fisher's combined probability values (p < .05) were observed (these parameters are not mutually exclusive). Notably, departure from HWE was assessed on a scale‐specific manner; firstly, at the North Atlantic scale where all individuals from all populations (with all sites from Breiðafjörður representing Iceland) were included and, secondly, at the Icelandic scale where only individuals from Iceland were assessed (again with population designated relative to sample site in Breiðafjörður). Adaptive SNPs (putative loci under selection) were identified either at the North Atlantic or Icelandic scale using BayeScan (Foll & Gaggiotti, 2008), with parameters set for 50,000 iterations with prior odds of 100, following 20 pilot runs of 5,000 interactions each, a thinning interval of 10, and a burn‐in length of 50,000. Confirmation of Markov chain convergence was conducted using Gelman and Rubin multiple sequence diagnostic and Heidelberger and Welch's Convergence diagnostic tests within the R‐package CODA (Plummer et al., 2006). Putative adaptive SNPs were removed from the North Atlantic or Icelandic datasets using Adegenet (Jombart & Ahmed, 2011) (loci were considered outliers under a false discovery rate (FDR) of 0.05). All downstream analysis of the quality filtered North Atlantic‐ and Iceland‐specific datasets were conducted independently.
2.4. Analysis of genetic variation and population divergence
The estimation of genetic variation and divergence was undertaken in both North Atlantic and Icelandic datasets using the neutral SNP dataset exclusively. Mean observed heterozygosity (H O), expected heterozygosity (H E), and inbreeding coefficient (F IS) per population were calculated using Hierfstat (Goudet, 2005). All pairwise F ST calculations were computed in DartR following 1,000 permutations (Gruber et al., 2018). The presence of putative population clusters (K) within the genomic data was estimated using the find.clusters function of Adegenet (Jombart, 2008; Jombart & Ahmed, 2011). The optimal number of clusters (K) was selected by computing the value of K with the lowest associated goodness of fit value estimated with BIC.
Population genetic structure was visualized by using Gower PCoA ordination (using Euclidean distance) in DartR. Population structure was visualized using PCoA both when considering population assignments as sample site of origin or as a function of the putative population clusters (K) assignments. Population admixture was estimated using the Discriminant Analysis of Principal Components (DAPC) in Adegenet (Jombart et al., 2010). The number of principal components retained within DAPC was optimized using the a‐score optimization test prior to analysis. Admixture was expressed both when considering population assignments as sample site of origin or as a function of the putative population clusters (K) assignments. Isolation‐by‐distance was assessed by analyzing the association of genetic and geographic distances for all individuals using a Mantel test in Ade4 (Dray & Dufour, 2007; Mantel, 1967). Relative migration rates for Icelandic individuals were calculated on a per‐sample site basis using the divMigrate function in diveRsity (Keenan et al., 2013), where relative migration was derived from Nei's GST statistic.
Finally, genomic divergence across the North Atlantic was investigated by assaying haplotype coancestry. Populations derived haplotype.tsv files were converted to a fineRADstructure (Malinsky et al., 2018) compatible format using fineRADstructure_tools (https://github.com/edgardomortiz/fineRADstructure‐tools). Haplotype coancestry of North Atlantic populations was calculated using fineRADstructure and visualized in R using the scripts provided in the fineRADstructure manual (http://cichlid.gurdon.cam.ac.uk/fineRADstructure.html).
2.5. Comparison of genetic and phenotypic trends in Breiðafjörður, Iceland
Comparisons of genetic and phenotypic distances were performed for the Icelandic dataset only. Phenotypic data corresponding to each of the Icelandic sites was sourced from Magnúsdóttir et al. (2018). No phenotypic data could be sourced from RAU (Rauðasandur), and therefore, individuals from this site were excluded from phenotype association analyses. Phenotypic data corresponding to the following measures were sourced: (a) shell shape, based on eleven geometric morphometric landmarks on the ventral surface of the shell that combine information on shell spire/body whorl ratio, aperture shape, shell shape, and shell lip thickness; and (b) shell color frequency, based on the primary color of the shell (See Table S4).
Correlation of genetic distances (euclidean) with phenotypic distances was tested on a per‐sample site basis, using Mantel's test, with 1,000 permutations in the R‐package vegan (Oksanen et al., 2017). Euclidean distances were calculated for variation in shell shape between sites, while differences in shell color composition at each site were assessed using Bray–Curtis distances (Oksanen et al., 2015). To account for spatial autocorrelation due to either geographic distance or differences in depth, a partial Mantel test was implemented (Smouse et al., 1986) whereby either the effect of geographic distance or depth differences was kept constant.
The relationship between population genetic and phenotypic structuring was tested using cluster (K = 2) population assignments. Correlation between putative population clusters and shell shape differences were assessed using permutational multivariate analysis of variance of shape distances, implemented using the adonis() function in vegan (Oksanen et al., 2017) with the assigned putative genetic clusters as the explanatory variable. Similarly, correlation between putative population clusters and shell color were assessed by comparing differences in shell color frequencies between putative population clusters, using Fisher's exact test (Fisher, 1934).
3. RESULTS
3.1. Population genetic structure and divergence across the North Atlantic
Following genotype calling (populations) and Radiator quality filtering, North Atlantic‐specific analyses (n = 164 individuals) yielded a total of 35,836 common putative single locus SNP across Canada (n = 28), Iceland (n = 104), Faroe Islands (n = 18), and England (n = 14). A further n = 314 loci significantly deviated from HWE and were removed from the dataset, as were n = 2 loci flagged as putative adaptive SNP candidates (qval ≤ 0.05). All downstream analyses were performed on the remaining n = 35,518 putative neutral loci (total missing data 18.4%) retained postfiltering.
Genetic diversity was similar in all ENA populations (Iceland, Faroe Islands, England), with observed heterozygosity ranging from 0.146 to 0.158 (Table 2), while gene diversity was lowest for Canadian individuals (H O = 0.107). None of the North Atlantic populations displayed signals of inbreeding with F IS ranging from 0.043 to 0.068. At the North Atlantic scale, calculations of pairwise F ST identified significant genetic divergence for all pairwise comparisons (p < .001, Table 3). F ST values for all pairwise comparisons with Canadian exceeded 0.57, while F ST values within the ENA ranged from 0.059 to 0.098. Overall, genetic divergence followed a model of isolation‐by‐distance (r = 0.884, p = .001), with the Faroe Islands and England whelk displaying the greatest degree of genetic similarity, followed by Iceland, while Canada represented the most genetically divergent population relative to ENA populations (Figure 3a). No signals of population admixture (DAPC optimized to retain n = 23 principal components) were observed between Canada, Iceland, Faroe Islands, and England (Figure 3b).
Table 2.
Summary of average observed heterozygosity (HO), expected heterozygosity (HE), and inbreeding coefficient (FIS) for Buccinum undatum populations sampled from across the North Atlantic. Populations are designated relative to the country of origin with countries denoted as CAN (Canada), ICE (Iceland), FAR (Faroe Island), and ENG (England). The standard error is denoted as SE
| Population | Individuals | H O | ±SE | H E | ±SE | F IS | ±SE |
|---|---|---|---|---|---|---|---|
| CAN | 28 | 0.107 | 0.001 | 0.108 | 0.001 | 0.047 | 0.001 |
| ICE | 104 | 0.157 | 0.001 | 0.167 | 0.001 | 0.068 | 0.001 |
| FAR | 18 | 0.158 | 0.001 | 0.159 | 0.001 | 0.043 | 0.001 |
| ENG | 14 | 0.146 | 0.001 | 0.148 | 0.001 | 0.050 | 0.001 |
| Overall | 164 | 0.147 | 0.001 | 0.214 | 0.001 | 0.053 | 0.001 |
Table 3.
Pairwise F ST values for Buccinum undatum sampled across the North Atlantic. Values of F ST are listed above the diagonal, while values below represent significance values (p) of pairwise comparisons. Significance values (p) of pairwise comparisons were obtained following 1,000 permutations. Populations are listed based on country of origin, with abbreviations corresponding to the following: CAN (Canada); ICE (Iceland); ENG (England); and FAR (Faroe Islands)
| CAN | ICE | FAR | ENG | |
|---|---|---|---|---|
| CAN | – | 0.572 | 0.603 | 0.619 |
| ICE | <0.001 | – | 0.061 | 0.098 |
| FAR | <0.001 | <0.001 | – | 0.059 |
| ENG | <0.001 | <0.001 | <0.001 | – |
Figure 3.
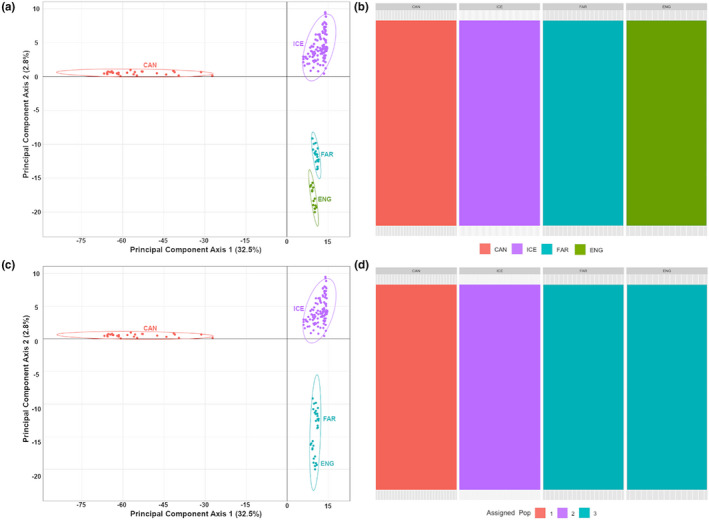
Summary of inferred population genetic structure for Buccinum undatum (n = 164) sampled from across the North Atlantic. All plots are derived from 35,518 putatively neutral SNP. Plots corresponding to the following: (a) Gower PCoA ordination (using euclidean distance) plot where population is assigned relative country of origin; (b) Admixture plot where population is assigned relative country of origin; (c) Gower PCoA ordination plot where population is assigned relative to optimal cluster (K = 3) assignments; (d) Admixture plot where population is assigned relative to optimal cluster (K = 3) assignments. Individuals are labeled relative country of origin: Canada (CAN, orange), Iceland (ICE, purple), Faroe Island (FAR, blue), and England (ENG, green)
The Bayesian information criterion estimated optimal goodness of fit value at K = 3 population clusters. Canadian and Icelandic populations formed distinct putative populations relative to the Faroe Islands and England whelk (Figure 3c). No signals of population admixture were detected between putative population assignments at the North Atlantic (DAPC optimized to retain n = 23 principal components).
Haplotype coancestry analyses indicated a lack of shared genetic ancestry between CAN and all ENA individuals, with the split of CAN and the ENA lineages occurring before any subsequent ENA divergence (Figure 4). Regarding the ENA clade, Icelandic individuals formed an outgroup distinct from the Faroe Islands and English haplotypes. Within the Iceland‐specific branch, haplotypes were primarily structured into two further subgroups. For the Faroe Island‐ and England‐specific clade, individual haplotypes were structured by country of origin, with Faroe Islands and English individuals forming two distinct sister groups.
Figure 4.
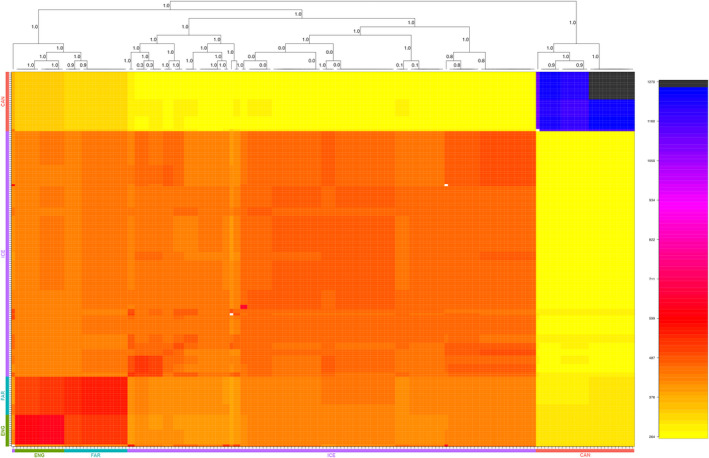
Haplotype coancestry matrix inferred for North Atlantic Buccinum undatum populations. Each pixel within the heatmap represents the inferred coancestry coefficient for a given individual based on haplotype loci. Colored bars adjacent to the heatmap represent individuals' country of origin, including England (green), the Faroe Islands (blue), Iceland (purple), and Canada (red). Within the heatmap, individuals with the highest shared coancestry are depicted in darker colors, while lesser values of coancestry are depicted in brighter shades. Bootstrap support is listed for each branch divergence for the inferred phylogeny
3.2. Population genetic structure and divergence in Iceland
Quality filtering reduced sample representation across all sample sites (particularly for ODD), with Iceland‐specific analyses being conducted across BJA (n = 9), BRJ (n = 9), HVA (n = 34), HRU (n = 18), ODD (n = 4), SKO (n = 24), and RAU (n = 6). Following the preparation of the Icelandic‐specific dataset (35,836 common putative single locus SNP across all individuals), quality filtering removed a further n = 268 loci, which significantly deviated from HWE, as well as a further n = 175 putatively adaptive SNP candidates (qval ≤ 0.05). All downstream Iceland‐specific analyses proceeded with the remaining n = 35,393 putative neutral loci (total percentage of missing data = 19.4%).
Genetic diversity was similar across all Icelandic populations, with observed heterozygosity ranging from 0.149 to 0.165 and inbreeding coefficient (F IS) ranging from −0.019 to 0.040 (Table 4). Calculations of pairwise F ST identified significant genetic divergence between all pairwise comparisons (p < .001, Table 5). Genetic divergence followed a model of isolation‐by‐distance (r = 0.203, p = .001), with individuals within the outermost (SKO, RAU, BJA) and innermost (BRJ, HRU, HVA, ODD) regions of Breiðafjörður, generally, displaying the greatest degree of genetic similarity (Figure 5a) and admixture (Figure 5b), respectively. Estimations of clusters (K) in adegenet indicated further regional segregation of population genetic variation, predicting an optimal BIC goodness of fit value at K = 2, with clusters segregating outer‐ (SKO, RAU, BJA) and innermost (BRJ, HRU, HVA, ODD) bay sites (Figure 5c). When considering the optimal cluster (K) population assignments, admixture plots indicated minimal gene flow between the putative outer‐ and innermost bay populations (see Figure 5d). Migration analyses indicated that coastal sites generally share the greatest degree of migration (Figure 6), particularly for the inner bay sites BRJ, HRU, and HVA. For both mid‐bay sites (BJA and ODD), which occur at the greatest depth, unidirectional migration patterns were observed, with migration occurring exclusively from deep site to proximal coastal sites.
Table 4.
Summary of average observed heterozygosity (H O), expected heterozygosity (H E), and inbreeding coefficient (F IS) for Buccinum undatum populations sampled from Breiðafjörður Bay, Iceland. Populations are designated relative to their sample site of origin with sites denoted as BJA (Bjarneyjaráll), BRJ (Brjánslækur), HVA (Hvammsfjörður), HRU (Hrútey), ODD (Oddbjarnarsker), SKO (Skor), and RAU (Rauðasandur). The standard error is denoted as SE
| Population | Individuals | H O | ±SE | H E | ±SE | F IS | ±SE |
|---|---|---|---|---|---|---|---|
| BJA | 9 | 0.149 | 0.001 | 0.140 | 0.001 | 0.026 | 0.001 |
| BRJ | 9 | 0.157 | 0.001 | 0.148 | 0.001 | 0.016 | 0.001 |
| HRU | 18 | 0.157 | 0.001 | 0.157 | 0.001 | 0.040 | 0.001 |
| HVA | 34 | 0.160 | 0.001 | 0.162 | 0.001 | 0.034 | 0.001 |
| ODD | 4 | 0.165 | 0.001 | 0.143 | 0.001 | −0.019 | 0.002 |
| RAU | 6 | 0.161 | 0.001 | 0.150 | 0.001 | 0.017 | 0.002 |
| SKO | 24 | 0.159 | 0.001 | 0.161 | 0.001 | 0.036 | 0.001 |
| Overall | 104 | 0.158 | 0.001 | 0.167 | 0.001 | 0.024 | 0.001 |
Table 5.
Pairwise F ST values for Buccinum undatum sampled within Breiðafjörður Bay, Iceland. Values of F ST are listed above the diagonal, while values below represent significance values (p) of pairwise comparisons. Significance values (p) of pairwise comparisons were obtained following 1,000 permutations. Populations are listed relative to individuals sample site of origin, with abbreviations corresponding to the following: BJA (Bjarneyjaráll), BRJ (Brjánslækur), HVA (Hvammsfjörður), HRU (Hrútey), ODD (Oddbjarnarsker), SKO (Skor), and RAU (Rauðasandur)
| BJA | BRJ | HRU | HVA | ODD | RAU | SKO | |
|---|---|---|---|---|---|---|---|
| BJA | – | 0.068 | 0.067 | 0.068 | 0.051 | 0.057 | 0.053 |
| BRJ | <0.001 | – | 0.009 | 0.007 | 0.015 | 0.035 | 0.038 |
| HRU | <0.001 | <0.001 | – | 0.007 | 0.012 | 0.036 | 0.040 |
| HVA | <0.001 | <0.001 | <0.001 | – | 0.012 | 0.035 | 0.038 |
| ODD | <0.001 | <0.001 | <0.001 | <0.001 | – | 0.018 | 0.026 |
| RAU | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | – | 0.003 |
| SKO | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | – |
Figure 5.
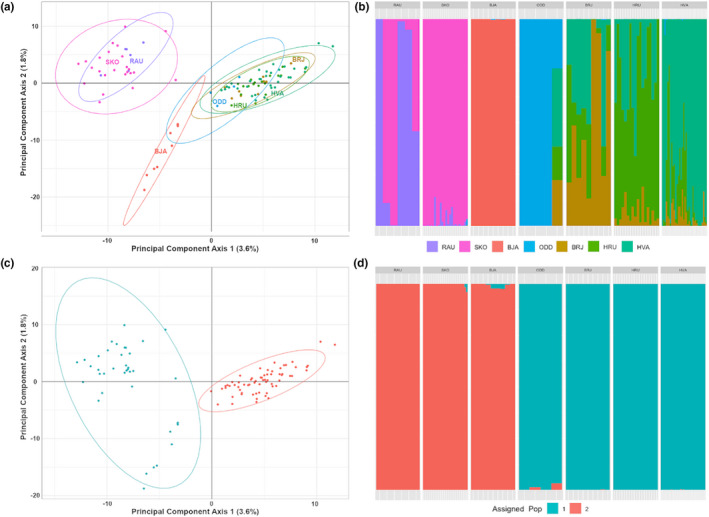
Summary of inferred population genetic structure for Buccinum undatum (n = 104) sampled from Breiðafjörður, Iceland. All plots are derived from 35,836 putatively neutral SNP. Plots corresponding to the following: (a) Gower PCoA ordination (using euclidean distance) plot where individual population is assigned relative to capture site in Breiðafjördur; (b) Admixture plot where individual population is assigned relative to capture site in Breiðafjördur; (c) Gower PCoA ordination plot where individual population is assigned relative to optimal cluster (K = 2) assignments; (d) Admixture plot where individual population is assigned relative to optimal cluster (K = 2) assignments. Individuals are labeled relative to capture site in Breiðafjördur: Rauðasandur (RAU, purple), Skor (SKO, pink), Bjarneyjaráll (BJA, red), Oddbjarnarsker (ODD, blue), Brjánslækur (BRJ, brown), Hrútey (HRU, green), and Hvammsfjörður (HVA, blue‐green)
Figure 6.
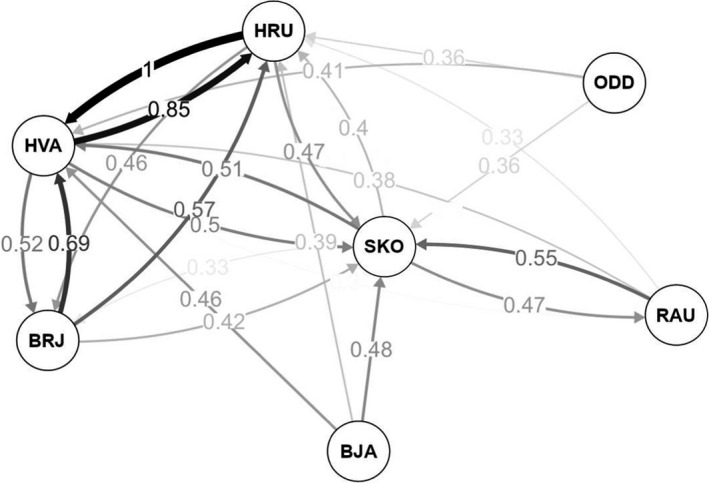
Relative migration network of sample sites within Breiðafjörður, Iceland. Migration rates are calculated from Nei's GST. Filter thresholds were set to exclude weak migration patterns at values < 0.3. Sample sites within Breiðafjörður are abbreviated as follows: Rauðasandur (RAU), Skor (SKO), Bjarneyjaráll (BJA), Oddbjarnarsker (ODD), Brjánslækur (BRJ), Hrútey (HRU), and Hvammsfjörður (HVA)
3.3. Comparison of genetic and phenotypic variation in Iceland
No significant correlation between shell shape distances or color dissimilarity were observed (p > .05) when considering individuals on a site‐specific basis, regardless of correction for spatial autocorrelation (geographic distance or depth). Sites were grouped into putative population clusters based on K‐means clustering (K = 2) and analyzed for correlations between genetic distance and phenotypic traits, shell shape distances, and color frequency (Magnúsdóttir et al., 2018). Significant differences in both color frequency (Fisher's test: p = .0005) and shape distances (F′ = 15.95, p = .001) were observed between the two putative genetic clusters. The outer bay cluster consisted primarily of lighter shell colors, with orange and white being the most common (Table S4), while the inner bay cluster consisted predominantly of green and brown shells. With regard to shell shape, the outer bay cluster was correlated with rounder shells, a proportionally shorter spire (compared with the body whorl), and a more elongate aperture (Magnúsdóttir et al., 2018), whereas the inner bay cluster was correlated with more elongate shells, with a comparatively taller spire, and smaller rounder apertures.
4. DISCUSSION
The current study undertook a comprehensive analysis of North Atlantic population genetic structure in the common whelk utilizing a RAD sequencing approach. WNA (Canada) and ENA (England, Faroe Islands, and Iceland) populations were highly divergent, with an F ST ≥ 0.57 relative to within‐ENA divergence (F ST ≤ 0.1), bolstering previous claims that the two lineages constitute cryptic species that diverged under allopatric speciation (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Pálsson et al., 2014). Smaller but significant differentiation was also observed between all ENA sites (Iceland, Faroe Islands, England), with putative cluster assignments suggesting genetic substructuring within the ENA likely exists, particularly regarding Icelandic populations being differentiated from both Faroe Islands and English populations. Haplotype coancestry analyses further support the characterization of ENA populations as a distinct lineage, typified by moderate to low intra‐lineage population divergence. Limited but significant population structure following an isolation‐by‐distance model was also detected in Icelandic populations, suggesting that limited dispersal in B. undatum constrains gene flow at localized and broad geographic scales across the ENA. Analyses of genetic variation in the bay of Breiðafjörður (Iceland) identified two putative population clusters composed exclusively of individuals sampled from the bay's outer‐ and innermost regions. Significant correlation between shell shape variation and shell color frequency were observed in Iceland, with phenotypic traits showing concordance with the denotation of Breiðafjörður whelk as distinct outer‐ and innermost bay population clusters.
Direct‐developing, benthic marine species are hypothesized to exhibit greater population genetic structure, relative to broadcast spawners, due to their limited dispersal capabilities. While rafting of egg masses may confer some means of dispersal for direct‐developing gastropods (see Johannesson, 1988; Kyle & Boulding, 2000; Marko, 2004), the frequency of such events is unlikely to maintain gene flow to an extent which counteracts the effects of high self‐recruitment. Existing population genetic studies of B. undatum described genetic structuring following an isolation‐by‐distance model across the North Atlantic (Magnúsdóttir, Pálsson, Westfall, Jónsson, Goodall, et al., 2019; Mariani et al., 2012; Pálsson et al., 2014; Weetman et al., 2006); however, it remained unclear whether the variation observed from mitochondrial/microsatellite markers was representative of broader genomic trends. In the present study, trends in population structure both within Iceland and across the North Atlantic (as scored from a set of RAD sequencing SNPs) were commensurate with existing mitochondrial/microsatellite works.
Our results reaffirm mtDNA data at the North Atlantic scale, indicating the Canadian and ENA populations constitute two distinct genetic lineages, which diverged some 2.1 million years ago (Magnúsdóttir, Pálsson, Westfall, Jónsson, & Örnólfsdóttir, 2019). Haplotype coancestry estimates indicated Canadian and ENA whelk represent two distinct genetic lineages, with subsequent divergence within the ENA lineage occurring more recently. For all pairwise comparisons, F ST and DAPC analyses indicated significant genetic divergence with restricted gene flow between Canadian and ENA populations. Recent work focused on the B. undatum mtDNA gene, COI, indicated the presence of cryptic species or clear allopatric divergence between Canadian and ENA whelk based on multiple species screening indices (Magnúsdóttir, Pálsson, Westfall, Jónsson, & Örnólfsdóttir, 2019). Regarding SNP‐based population genetic comparisons, Hey and Pinho (2012) proposed an F ST = 0.35 as the upper threshold for intra‐species population comparisons, whereby comparisons exceeding this value likely constitute interspecies comparisons. Under this scheme, Canadian and the ENA whelk exceed Hey and Pinho's (2012) F ST threshold at ≥0.57 and would thus be considered separate species. Nevertheless, while our data further consolidate evidence of substantial genetic divergence between Canadian and ENA lineages, breeding studies are ultimately required to determine whether the two lineages represent reproductively isolated species conforming to the biological species concept. However, due to asynchronous breeding times, these populations are unlikely to interbreed in nature.
Our results support claims of low, but significant, genetic divergence as a persistent feature within the ENA lineage. Further research is required to derive the mechanisms driving low divergence within the ENA; however, our data suggest that maintenance of gene flow under a stepping stone model is unlikely given little to no admixture is observed between Icelandic, Faroe Islands, and English whelk. At the Icelandic scale, our estimates of F ST (ranging from 0.003 to 0.068) fall within the ranges previously estimated for Breiðafjörður using mtDNA (ranging from 0.0003 to 0.145) (Pálsson et al., 2014). On a regional basis, global F ST for Iceland (global F ST = 0.029) is similar, albeit higher on average, to those derived for British (global F ST = 0.014) and Irish (global F ST = 0.019) B. undatum populations (Mariani et al., 2012; Weetman et al., 2006).
While previous studies have speculated on the occurrence of multiple spatially differentiated B. undatum populations within Breiðafjörður (Magnúsdóttir et al., 2010, 2018; Woods & Jonasson, 2017), our study is the first to characterize fine‐scale genetic structure in the region. Interestingly, profiles of heterozygosity and genetic divergence in Iceland are dissimilar to those described for Britain. Weetman et al. (2006) characterized reduced genetic diversity and greater genetic differentiation in inshore, relative to offshore populations, resulting from unidirectional migration by British whelk. Breiðafjörður's populations exhibit similar observed heterozygosity across all site‐specific populations, with populations from the outermost regions of Breiðafjörður (BJA, RAU, and SKO) displaying the greatest degree of genetic differentiation. To summarize, trends in genetic diversity and divergence in Iceland do not indicate unidirectional offshore migration, as seen in Britain. Instead, modeling of migration patterns indicated that some degree of migration likely occurs between all sites; however, said migration is primarily restricted to shallower more coastal sites and, even then, occurs mostly within the putatively assigned outermost and innermost bay population clusters (a trend also reflected by estimations of admixture).
Interestingly, both BJA and ODD, which represent the deepest sites for each putative population cluster, respectively, displayed unidirectional migration patterns toward shallower more coastal sites. Signatures of restricted gene flow in both BJA and ODD were evident in site‐specific admixture plots, even within their putatively assigned population clusters. Shallower, more coastal sites may be more susceptible to disturbance (i.e., in the case of storms), increasing the frequency of rafting events, and thus, admixture between coastal sites. Surface waters in Breiðafjörður are considered to be well mixed, with nearshore currents flowing in a continuous clockwise fashion (Logemann et al., 2013) across Breiðafjörður. Taken together, deeper sites such as BJA and ODD are likely more stable (with respect to storm events) and less influenced by coastal currents, resulting in reduced gene flow from adjacent sites (a trend that is particularly evident from the high divergence and limited admixture of BJA). As such, gene flow from deep water sites in Breiðafjörður may be primarily dictated by patterns of vertical migration (as seen in the migration pattern analyses), while coastal sites experience more complex gene flow patterns resulting from more dynamic hydrological conditions. Ultimately, the impact of hydrological conditions on benthic communities in Breiðafjörður has not been quantified, and therefore, more targeted studies are required to determine and quantify factors driving the migration of B. undatum in Breiðafjörður. Still, based on this study, population genetic structuring in Breiðafjördur is broadly stratified into two distinct population clusters corresponding to the bay's outermost and innermost geographical regions, with both clusters displaying independent signatures of inshore migration and increased migration and gene flow in the shallower, more coastal sites.
Magnúsdóttir et al. (2018) described a trend of thinner, rounder shells with relatively shorter spires, as well as increasing color diversity (and striping) from the innermost to outmost regions of Breiðafjörður. Our study showed that both color frequency distribution and shell shape distance were strongly correlated with putative population cluster (K) assignments. Divergence in phenotypic shell traits between putative populations can reflect random fluctuations (i.e., genetic drift) in separate populations with limited demographic connectivity, which may be further augmented by selection due to different environmental settings. Previous genetic data are congruent with a model of recent population expansion into Breiðafjörður following the last glacial maximum (Pálsson et al., 2014), where the ice sheet is considered to have extended 130 km west of the coast, as witnessed by findings of glacial moraines (Norddahl et al., 2008). Given the species' limited dispersal capabilities, the formation of multiple genetically isolated populations diverging under a genetic drift model is possible. Similar concepts have been proposed to explain the low F ST values recorded for the ENA lineage, whereby population bottlenecks would have facilitated genetic drift, leading to low but significant population differentiation (Weetman et al., 2006). However, the inner regions of Breiðafjörður, which are typically shallower, also contain a variety of predatory crab species such as the Spider crab (Hyas araneus), Green crab (Carcinus maenas), and Atlantic Rock crab (Cancer irroratus). Thicker, more elongate shells with smaller apertures were hypothesized to occur in association with the predatory crab species in the innermost regions of Breiðafjörður (Bourdeau & Johansson, 2012; Magnúsdóttir et al., 2018; Thomas & Himmelman, 1988). At greater depths, negative selection by visual predators may be less intense, allowing for greater shell color diversity to be represented by individuals inhabiting the outermost regions of Breiðafjörður. In the presence of visual predators, negative selection may also constrain the frequency of particular color morphs within Breiðafjörður while selecting for specific shell shapes. Future studies should seek to characterize the relationship between genetic variation and shell shape and color in Breiðafjörður using genome‐wide association studies. In particular, GWAS studies should seek to link genetic variation with specific color morphs in a bid to characterize the distribution and frequency of color‐linked genes across Breiðafjörður.
The current study undertook the first genome‐wide analysis of population structure and divergence in B. undatum. Our genetic works reinforce the need to re‐examine B. undatum as a singular trans North Atlantic species, with Canadian and ENA whelk likely representing a potentially cryptic species complex. A clear pattern of fine‐scale population genetic structuring following a model of isolation‐by‐distance was established as a persistent trend across both small‐ (Breiðafjörður) and large‐scale (North Atlantic) geographical distances. Clear population genetic structure was defined for B. undatum within Breiðafjörður, Iceland, constituting two distinct putative genetic clusters. The frequency distribution of shell color and shell shape distance was significantly correlated with the two putative genetic clusters in Breiðafjörður. The work detailed here provides a rigid framework for ongoing associative studies of genetic variation in the subtidal mollusk, B. undatum, while laying the foundations for future comparative studies aimed at characterizing genotype–phenotype associations in common whelk.
CONFLICT OF INTEREST
None to declare.
AUTHOR CONTRIBUTION
Jake Goodall: Conceptualization (equal); Data curation (equal); Formal analysis (lead); Methodology (equal); Project administration (equal); Validation (lead); Visualization (lead); Writing‐original draft (lead); Writing‐review & editing (lead). Kristen M. Westfall: Conceptualization (equal); Data curation (equal); Formal analysis (supporting); Funding acquisition (supporting); Investigation (equal); Methodology (equal); Project administration (equal); Validation (supporting); Visualization (supporting); Writing‐original draft (supporting); Writing‐review & editing (supporting). Hildur Magnúsdóttir: Conceptualization (equal); Data curation (equal); Formal analysis (supporting); Funding acquisition (supporting); Investigation (equal); Methodology (equal); Project administration (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting). Snæbjörn Pálsson: Conceptualization (equal); Formal analysis (supporting); Funding acquisition (supporting); Methodology (equal); Project administration (equal); Resources (supporting); Supervision (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting). Erla Björk Örnólfsdóttir: Conceptualization (equal); Funding acquisition (lead); Project administration (equal); Resources (lead); Supervision (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting). Zophonías O. Jónsson: Conceptualization (equal); Data curation (supporting); Formal analysis (supporting); Funding acquisition (supporting); Investigation (equal); Methodology (equal); Project administration (equal); Resources (supporting); Supervision (equal); Writing‐original draft (supporting); Writing‐review & editing (supporting).
Supporting information
Supinfo
ACKNOWLEDGEMENTS
The project was funded by research grants #141302‐051 and #196417‐051 from the Icelandic Centre for Research (RANNÍS). We want to thank Símon Sturluson, the captain of Ronja SH‐53, and Will Butler for their assistance in sampling in Breiðafjörður. We are also immensely grateful to Bernard Sainte‐Marie (Maurice Lamontagne Institute, Department of Fisheries and Oceans, Canada), Una Matras (Havstovan in the Faroe Islands), and Kathryn E. Smith (University of Exeter, England) for providing us with samples from their respective regions. We are very grateful to Ólafur Th. Magnússon at deCODE Genetics for advice and the Illumina sequencing. We greatly appreciate valuable input from Hörður Guðmundsson, Kalina Kapralova, Denis Warshan, Charles Hansen, Marcos Lagunas, and Han Xiao, who participated in several discussions on population genetic theory and methodologies during the development of this project.
Goodall J, Westfall KM, Magnúsdóttir H, Pálsson S, Örnólfsdóttir EB, Jónsson ZO. RAD sequencing of common whelk, Buccinum undatum, reveals fine‐scale population structuring in Europe and cryptic speciation within the North Atlantic. Ecol Evol. 2021;11:2616–2629. 10.1002/ece3.7219
DATA AVAILABILITY STATEMENT
Demultiplexed Illumina sequences are available from the European Nucleotide Archive (ENA) via study accession: PRJEB37370 (ERP120679).
REFERENCES
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Google Scholar]
- Benestan, L. , Gosselin, T. , Perrier, C. , Sainte‐Marie, B. , Rochette, R. , & Bernatchez, L. (2015). RAD genotyping reveals fine‐scale genetic structuring and provides powerful population assignment in a widely distributed marine species, the American lobster (Homarus americanus). Molecular Ecology, 24(13), 3299–3315. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , & Yekutieli, D. (2001). The control of the false discovery rate in multiple testing under dependency. Annals of Statistics, 29(4), 1165–1188. [Google Scholar]
- Bourdeau, P. E. , & Johansson, F. (2012). Predator‐induced morphological defences as by‐products of prey behaviour: A review and prospectus. Oikos, 121(8), 1175–1190. 10.1111/j.1600-0706.2012.20235.x [DOI] [Google Scholar]
- Catchen, J. M. , Amores, A. , Hohenlohe, P. , Cresko, W. , & Postlethwait, J. H. (2011). Stacks: Building and genotyping loci de novo from short‐read sequences. G3: Genes, Genomes, Genetics, 1(3), 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: An analysis tool set for population genomics. Molecular Ecology, 22(11), 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyer, J. A. , Hoarau, G. , Van Schaik, J. , Luijckx, P. , & Olsen, J. L. (2011). Trans‐Pacific and trans‐Arctic pathways of the intertidal macroalga Fucus distichus L. reveal multiple glacial refugia and colonizations from the North Pacific to the North Atlantic. Journal of Biogeography, 38(4), 756–771. 10.1111/j.1365-2699.2010.02437.x [DOI] [Google Scholar]
- Crawford, D. L. , & Oleksiak, M. F. (2016). Ecological population genomics in the marine environment. Briefings in Functional Genomics, 15(5), 342–351. 10.1093/bfgp/elw008 [DOI] [PubMed] [Google Scholar]
- Davey, J. W. , & Blaxter, M. L. (2010). RADSeq: Next‐generation population genetics. Briefings in Functional Genomics, 9(5–6), 416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray, S. , & Dufour, A.‐B. (2007). The ade4 Package: Implementing the duality diagram for ecologists. Journal of Statistical Software, 1(4), 1–20. [Google Scholar]
- Drury, C. , Dale, K. E. , Panlilio, J. M. , Miller, S. V. , Lirman, D. , Larson, E. A. , Bartels, E. , Crawford, D. L. , & Oleksiak, M. F. (2016). Genomic variation among populations of threatened coral: Acropora cervicornis. BMC Genomics, 17(1), 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson, K. J. , Merz, C. R. , Catchen, J. M. , Hohenlohe, P. A. , Cresko, W. A. , Bradshaw, W. E. , & Holzapfel, C. M. (2010). Resolving postglacial phylogeography using high‐throughput sequencing. Proceedings of the National Academy of Sciences, 107(37), 16196–16200. 10.1073/pnas.1006538107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A. (1934). Statistical methods for research workers. Edinburgh, Oliver and Boyd. [Google Scholar]
- Foll, M. , & Gaggiotti, O. (2008). A genome‐scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics, 180(2), 977–993. 10.1534/genetics.108.092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk, W. C. , McKay, J. K. , Hohenlohe, P. A. , & Allendorf, F. W. (2012). Harnessing genomics for delineating conservation units. Trends in Ecology and Evolution, 27(9), 489–496. 10.1016/j.tree.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron, L. (1992). Determination of the size at sexual maturity of the waved whelk Buccinum undatum Linnaeus, 1758, in the Gulf of St. Lawrence, as a basis for the establishment of a minimum catchable size. Journal of Shellfish Research, 11(1), 1–7. [Google Scholar]
- Golikov, A. N. (1968). Distribution and variability of long‐lived benthic animals as indicators of currents and hydrological conditions. Sarsia, 34(1), 199–208. 10.1080/00364827.1968.10413382 [DOI] [Google Scholar]
- [dataset] Goodall, J. , Westfall, K. M. , Magnúsdóttir, H. , Pálsson, S. , Örnólfsdóttir, E. B. , & Jónsson, Z. O. (2020). ddRAD sequencing of Common Whelk, Buccinum undatum; European Nucleotide Archive; Accession: P RJEB37370 (ERP120679).
- Gosselin, T. (2017). radiator: RADseq data exploration, manipulation and visualization using R. R package version 0.0 5. [Google Scholar]
- Goudet, J. (2005). hierfstat, a package for r to compute and test hierarchical F‐statistics. Molecular Ecology Notes, 5(1), 184–186. 10.1111/j.1471-8286.2004.00828.x [DOI] [Google Scholar]
- Gruber, B. , Unmack, P. J. , Berry, O. F. , & Georges, A. (2018). dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Molecular Ecology Resources, 18(3), 691–699. [DOI] [PubMed] [Google Scholar]
- Hey, J. , & Pinho, C. (2012). Population genetics and objectivity in species diagnosis. Evolution, 66(5), 1413–1429. 10.1111/j.1558-5646.2011.01542.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelman, J. H. (1988). Movement of whelks (Buccinum undatum) towards a baited trap. Marine Biology, 97(4), 521–531. 10.1007/BF00391048 [DOI] [Google Scholar]
- Himmelman, J. H. , & Hamel, J. R. (1993). Diet, behaviour and reproduction of the whelk Buccinum undatum in the northern Gulf of St. Lawrence, eastern Canada. Marine Biology, 116(3), 423–430. 10.1007/BF00350059 [DOI] [Google Scholar]
- Ingolfsson, A. (1992). The Origin of the Rocky Shore Fauna of Iceland and the Canadian Maritimes. Journal of Biogeography, 19(6), 705–712. 10.2307/2845711 [DOI] [Google Scholar]
- Jalbert, P. , Himmelman, J. , Béland, P. , & Thomas, B. (1989). Whelks (Buccinum undatum) and other subtidal invertebrate predators in the northern Gulf of St. Lawrence. Le Naturaliste Canadien, 116(1), 1–15. [Google Scholar]
- Jenkins, T. L. , Ellis, C. D. , Triantafyllidis, A. , & Stevens, J. R. (2019). Single nucleotide polymorphisms reveal a genetic cline across the north‐east Atlantic and enable powerful population assignment in the European lobster. Evolutionary Applications, 12(10), 1881–1899. 10.1111/eva.12849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesson, K. (1988). The paradox of Rockall: Why is a brooding gastropod (Littorina saxatilis) more widespread than one having a planktonic larval dispersal stage (L. littorea)? Marine Biology, 99(4), 507–513. 10.1007/BF00392558 [DOI] [Google Scholar]
- Jombart, T. (2008). adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics, 24(11), 1403–1405. 10.1093/bioinformatics/btn129 [DOI] [PubMed] [Google Scholar]
- Jombart, T. , & Ahmed, I. (2011). adegenet 1.3‐1: New tools for the analysis of genome‐wide SNP data. Bioinformatics, 27(21), 3070–3071. 10.1093/bioinformatics/btr521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. , Devillard, S. , & Balloux, F. (2010). Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genetics, 11(1), 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan, K. , McGinnity, P. , Cross, T. F. , Crozier, W. W. , & Prodöhl, P. A. (2013). diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods in Ecology and Evolution, 4(8), 782–788. [Google Scholar]
- Kideys, A. E. , Nash, R. D. M. , & Hartnoll, R. G. (1993). Reproductive cycle and energetic cost of reproduction of the neogastropod Buccinum undatum in the Irish Sea. Journal of the Marine Biological Association of the United Kingdom, 73(2), 391–403. [Google Scholar]
- Kohn, M. H. , Murphy, W. J. , Ostrander, E. A. , & Wayne, R. K. (2006). Genomics and conservation genetics. Trends in Ecology and Evolution, 21(11), 629–637. 10.1016/j.tree.2006.08.001 [DOI] [PubMed] [Google Scholar]
- Kyle, C. J. , & Boulding, E. G. (2000). Comparative population genetic structure of marine gastropods (Littorina spp.) with and without pelagic larval dispersal. Marine Biology, 137(5), 835–845. 10.1007/s002270000412 [DOI] [Google Scholar]
- Logemann, K. , Ólafsson, J. , Snorrason, Á. , Valdimarsson, H. , & Marteinsdóttir, G. (2013). The circulation of Icelandic waters – a modelling study. Ocean Science, 9(5), 931–955. 10.5194/os-9-931-2013 [DOI] [Google Scholar]
- Maggs, C. A. , Castilho, R. , Foltz, D. , Henzler, C. , Jolly, M. T. , Kelly, J. , Olsen, J. , Perez, K. E. , Stam, W. , Väinölä, R. , Viard, F. , & Wares, J. (2008). Evaluating signatures of glacial refugia for north Atlantic benthic marine taxa. Ecology, 89(sp11), S108–S122. [DOI] [PubMed] [Google Scholar]
- Magnúsdóttir, H. , Olsen, K. , Matras, U. , & Örnólfsdóttir, E. (2010). Biology and distribution of the common whelk (Buccinum undatum) in Icelandic and Faroese waters/Konksneglens (Buccinum undatum) biologi og udbredelse i farvandet ved Island og Faerøerne Project No. 282, VÖR Marine Research Institute. http://www.hav.fo/PDF/Ritgerdir/2011/Smarit1107.pdf [Google Scholar]
- Magnúsdóttir, H. , Pálsson, S. , Westfall, K. M. , Jónsson, Z. O. , Goodall, J. , & Örnólfsdóttir, E. B. (2019). Revised phylogeography of the common whelk Buccinum undatum (Gastropoda: Buccinidae) across the North Atlantic. Biological Journal of the Linnean Society, 127(4), 890–899. 10.1093/biolinnean/blz060 [DOI] [Google Scholar]
- Magnúsdóttir, H. , Pálsson, S. , Westfall, K. M. , Jónsson, Z. O. , & Örnólfsdóttir, E. B. (2018). Shell morphology and color of the subtidal whelk Buccinum undatum exhibit fine‐scaled spatial patterns. Ecology and Evolution, 8(9), 4552–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir, H. , Pálsson, S. , Westfall, K. M. , Jónsson, Z. O. , & Örnólfsdóttir, E. B. (2019). Morphological variation in genetically divergent populations of the common whelk, Buccinum undatum (Gastropoda: Buccinidae), across the North Atlantic. Biological Journal of the Linnean Society, 128(1), 93–106. 10.1093/biolinnean/blz095 [DOI] [Google Scholar]
- Malinsky, M. , Trucchi, E. , Lawson, D. J. , & Falush, D. (2018). RADpainter and fineRADstructure: Population Inference from RADseq Data. Molecular Biology and Evolution, 35(5), 1284–1290. 10.1093/molbev/msy023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Research, 27(2), 209–220. [PubMed] [Google Scholar]
- Mariani, S. , Peijnenburg, K. , & Weetman, D. (2012). Independence of neutral and adaptive divergence in a low dispersal marine mollusc. Marine Ecology Progress Series, 446, 173–187. [Google Scholar]
- Marko, P. B. (2004). 'What's larvae got to do with it?' Disparate patterns of post‐glacial population structure in two benthic marine gastropods with identical dispersal potential. Molecular Ecology, 13(3), 597–611. 10.1046/j.1365-294X.2004.02096.x [DOI] [PubMed] [Google Scholar]
- Martel, A. , Larrivée, D. H. , & Himmelman, J. H. (1986). Behaviour and timing of copulation and egg‐laying in the neogastropod Buccinum undatum L. Journal of Experimental Marine Biology and Ecology, 96(1), 27–42. 10.1016/0022-0981(86)90011-0 [DOI] [Google Scholar]
- Norddahl, H. , Ingólfsson, Ó. , Pétursson, H. , & Hallsdottir, M. (2008). Late Weichselian and Holocene environmental history of Iceland. Jökull, 58, 343–364. [Google Scholar]
- Oksanen, J. , Blanchet, F. , Kindt, R. , Legendre, P. , Ohara, R. , Simpson, G. , Solymos, P. , Stevens, M. , & Wagner, H. (2015). Multivariate analysis of ecological communities. Vegan Tutorial, 1–40. [Google Scholar]
- Oksanen, J. , Blanchet, G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, R. , O'Hara, R. B. , Simpson, G. , Solymos, P. , Stevens, H. , Szoecs, E. , & Wagner, H. (2017). vegan: Community Ecology Package. [Google Scholar]
- Pálsson, S. , Magnúsdóttir, H. , Reynisdóttir, S. , Jónsson, Z. O. , & Örnólfsdóttir, E. B. (2014). Divergence and molecular variation in common whelk Buccinum undatum (Gastropoda: Buccinidae) in Iceland: A trans‐Atlantic comparison. Biological Journal of the Linnean Society, 111(1), 145–159. [Google Scholar]
- Panova, M. , Blakeslee, A. M. H. , Miller, A. W. , Mäkinen, T. , Ruiz, G. M. , Johannesson, K. , & André, C. (2011). Glacial History of the North Atlantic Marine Snail, Littorina saxatilis, inferred from distribution of mitochondrial DNA lineages. PLoS One, 6(3), e17511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis, E. (2010). pegas: An R package for population genetics with an integrated–modular approach. Bioinformatics, 26(3), 419–420. 10.1093/bioinformatics/btp696 [DOI] [PubMed] [Google Scholar]
- Plummer, M. , Best, N. , Cowles, K. , & Vines, K. (2006). CODA: Convergence diagnosis and output analysis for MCMC. R News, 6(1), 7–11. [Google Scholar]
- Rochette, N. C. , & Catchen, J. M. (2017). Deriving genotypes from RAD‐seq short‐read data using Stacks. Nature Protocols, 12(12), 2640–2659. 10.1038/nprot.2017.123 [DOI] [PubMed] [Google Scholar]
- Smouse, P. , Long, J. , & Sokal, R. (1986). Multiple regression and correlation extensions of the mantel test of matrix correspondence. Systematic Zoology, 35(4), 627–632. 10.2307/2413122 [DOI] [Google Scholar]
- Sokal, R. R. , & Rohlf, F. J. (1969). Biometry: The principles and practice of statistics in biological research. W.H. Freeman. [Google Scholar]
- Taylor, J. D. , & Taylor, C. N. (1977). Latitudinal distribution of predatory gastropods on the Eastern Atlantic Shelf. Journal of Biogeography, 4(1), 73–81. 10.2307/3038130 [DOI] [Google Scholar]
- Thomas, M. L. H. , & Himmelman, J. H. (1988). Influence of predation on shell morphology of Buccinum undatum L. on Atlantic coast of Canada. Journal of Experimental Marine Biology and Ecology, 115(3), 221–236. 10.1016/0022-0981(88)90156-6 [DOI] [Google Scholar]
- Van Wyngaarden, M. , Snelgrove, P. V. R. , DiBacco, C. , Hamilton, L. C. , Rodríguez‐Ezpeleta, N. , Jeffery, N. W. , Stanley, R. R. E. , & Bradbury, I. R. (2016). Identifying patterns of dispersal, connectivity and selection in the sea scallop, Placopecten magellanicus, using RADseq‐derived SNPs. Evolutionary Applications, 10(1), 102–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrami, D. L. J. , De Noia, M. , Telesca, L. , Handal, W. , Charrier, G. , Boudry, P. , Eberhart‐Phillips, L. , & Hoffman, J. I. (2019). RAD sequencing sheds new light on the genetic structure and local adaptation of European scallops and resolves their demographic histories. Scientific Reports, 9(1), 7455–7455. 10.1038/s41598-019-43939-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrami, D. L. , Telesca, L. , Weigand, H. , Weiss, M. , Fawcett, K. , Lehman, K. , Clark, M. S. , Leese, F. , McMinn, C. , Moore, H. , & Hoffman, J. I. (2017). RAD sequencing resolves fine‐scale population structure in a benthic invertebrate: Implications for understanding phenotypic plasticity. Royal Society Open Science, 4(2), 160548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij, G. J. (1991). Anatomy of an Invasion: The Trans‐Arctic Interchange. Paleobiology, 17(3), 281–307. 10.1017/S0094837300010617 [DOI] [Google Scholar]
- Wares, J. P. , & Cunningham, C. W. (2001). Phylogeography and historical ecology of the North Atlantic intertidal. Evolution, 55(12), 2455–2469. 10.1111/j.0014-3820.2001.tb00760.x [DOI] [PubMed] [Google Scholar]
- Weetman, D. , Hauser, L. , Bayes, M. K. , Ellis, J. R. , & Shaw, P. (2006). Genetic population structure across a range of geographic scales in the commercially exploited marine gastropod Buccinum undatum. Marine Ecology Progress Series, 317, 157–169. [Google Scholar]
- Weiss, M. , Weigand, H. , Weigand, A. M. , & Leese, F. (2017). Genome‐wide single‐nucleotide polymorphism data reveal cryptic species within cryptic freshwater snail species‐The case of the Ancylus fluviatilis species complex. Ecology and Evolution, 8(2), 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods, P. , & Jonasson, J. P. (2017). Bayesian hierarchical surplus production model of the common whelk Buccinum undatum in Icelandic waters. Fisheries Research, 194, 117–128. [Google Scholar]
- Zamudio, K. , Bell, R. , & Mason, N. (2016). Phenotypes in phylogeography: Species’ traits, environmental variation, and vertebrate diversification. Proceedings of the National Academy of Sciences, 113(29), 8041–8048. 10.1073/pnas.1602237113 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- [dataset] Goodall, J. , Westfall, K. M. , Magnúsdóttir, H. , Pálsson, S. , Örnólfsdóttir, E. B. , & Jónsson, Z. O. (2020). ddRAD sequencing of Common Whelk, Buccinum undatum; European Nucleotide Archive; Accession: P RJEB37370 (ERP120679).
Supplementary Materials
Supinfo
Data Availability Statement
Demultiplexed Illumina sequences are available from the European Nucleotide Archive (ENA) via study accession: PRJEB37370 (ERP120679).