

Abstract
Heritable variation in gene expression is common within and between species. This variation arises from mutations that alter the form or function of molecular gene regulatory networks that are then filtered by natural selection. High-throughput methods for introducing mutations and characterizing their cis- and trans-regulatory effects on gene expression (particularly, transcription) are revealing how different molecular mechanisms generate regulatory variation, while studies comparing these mutational effects to variation seen in the wild are teasing apart the role of neutral and non-neutral evolutionary processes. This integration of molecular and evolutionary biology allows us to understand how the variation in gene expression we see today came to be and to predict how it is most likely to evolve in the future.
Keywords: gene regulatory network, transcription, evolution, mutation, cis-regulation, trans-regulation
Introduction
The regulation of gene expression is a critical step in translating genotypes into phenotypes. Variation in this regulation is common within and between species1, and contributes to trait diversity. For example, changes in the regulation of gene expression have been shown to contribute to divergent pigmentation in plants and animals2,3, polymorphic body size in mice4, sporulation rate in domesticated yeast5, and many other morphological, physiological, and behavioural traits6,7, including disease states in humans8. Understanding how regulatory variation arises and evolves is thus critical for understanding many aspects of biology.
Genetic variation that affects the activity of regulatory networks underlies variation in gene expression. These networks include interactions among proteins, RNAs, and DNA sequences. Transcription factor proteins and DNA sequences such as enhancers and promoters are most often considered to define the structure of gene regulatory networks9,10, but protein-protein interactions, signaling pathways, and even metabolic states can also impact their activity11. Mutations that alter any of these elements can give rise to variation in gene expression. Such mutations can be classified as either cis-acting or trans-acting12: cis-acting mutations alter expression of a gene located on the same chromosome and tend to be located close to the affected gene, whereas trans-regulatory mutations have effects on gene expression that are mediated by diffusible molecules (such as RNAs and proteins) and can be located anywhere in the genome. Both types of mutations contribute to variation in gene expression, but differences in their molecular mechanisms suggest that they might contribute unequally to regulatory variation over evolutionary time.
Genomic studies describing variation in gene expression and the relative contributions of cis- and trans-acting variants have now been performed for diverse plant, animal, and microbial species13. As with all traits, this variation reflects the introduction of new genetic variants by mutation, the filtering of these variants by natural selection, and the chance survival of variants mediated by genetic drift. The extent to which each of these processes shapes the variation we see in wild populations, however, remains difficult to discern. For example, if one gene shows more variation in its expression than another, it might be because expression of the first gene is under less selective constraint or because a greater fraction of new mutations alters its expression (among other possibilities). Studies investigating the role of selection in shaping regulatory variation have thus far relied heavily on assumptions about the effects of new mutations because little empirical data was available14–17. However, this knowledge gap is beginning to close as recent advances in DNA synthesis, genome editing, and high-throughput expression analysis allow regulatory mutations to be generated and characterized on a large scale18.
Here, we examine our current understanding of the molecular and evolutionary processes generating variation in gene expression. We focus on variation in RNA expression because this is where the most data are available; quantifying variation in protein expression levels remains much more technically challenging. We begin by briefly reviewing studies describing the relative contributions of cis- and trans-regulatory variation to variation in gene expression. We then discuss the molecular sources of this regulatory variation, including studies describing the effects of mutations in these sequences as well as their contributions to expression differences within and between species. Finally, we close by showing how contrasting the effects of new mutations and genetic variants segregating in natural populations reveals the evolutionary processes responsible for the evolution of gene expression.
Partitioning cis- and trans- regulatory variation
Distinguishing between cis- and trans-regulatory variation reveals the relationship between mutations and their effects on gene expression. Two general strategies have primarily been used to disentangle the effects of cis- and trans-regulatory variants on a genomic scale. The first approach uses allele-specific expression (ASE) in F1 hybrids to compare activity of cis-regulatory alleles in a common trans-regulatory background to expression in the parents of the F1 hybrid19. The second strategy uses statistical associations between genetic variants and gene expression to identify quantitative trait loci affecting gene expression (eQTL)20,21. These two approaches provide complementary information about cis- and trans- regulatory variation, with the first capturing the net effect of all cis- and trans- regulatory variants, and the second providing information about the effects of individual loci.
Studies using ASE to estimate the relative contributions of cis- and trans-regulatory variants to variation in gene expression have been conducted in a variety of taxa, including plants22–25, yeast26–29, mice30,31, birds32,33, wasps34, and flies35–38. These studies include analysis of gene expression among individuals from outbred populations, between more isolated strains of the same species, and between species, each of which captures the evolution of gene expression at a different stage in the evolutionary process. Within species, trans-regulatory variants seem to contribute more to variation in gene expression than cis-regulatory variants13,28,29,39,40. This pattern has been suggested to be due to a larger mutational target size for trans-regulatory variants41: that is, there are more places in the genome where a mutation can affect a gene’s expression in trans than in cis. Trans-acting variants are also often assumed to affect expression of more genes on average than cis-acting variants. However, cis-regulatory variants often make similar24,37,38,42 or greater24,31,35 contributions to gene expression divergence between species. Studies directly comparing the relative contributions of cis- and trans-regulatory variants to expression divergence suggest that the relative contribution of cis-regulatory variants increases with divergence time29,37 (Figure 1A, B). This increasing cis-regulatory contribution can be explained by cis-regulatory variants being either more beneficial28,43 and/or less deleterious39 than trans-regulatory variants, which might result from differences in their average pleiotropy, as discussed more below.
Figure 1: cis- and trans-regulatory contributions to expression differences between and within species.
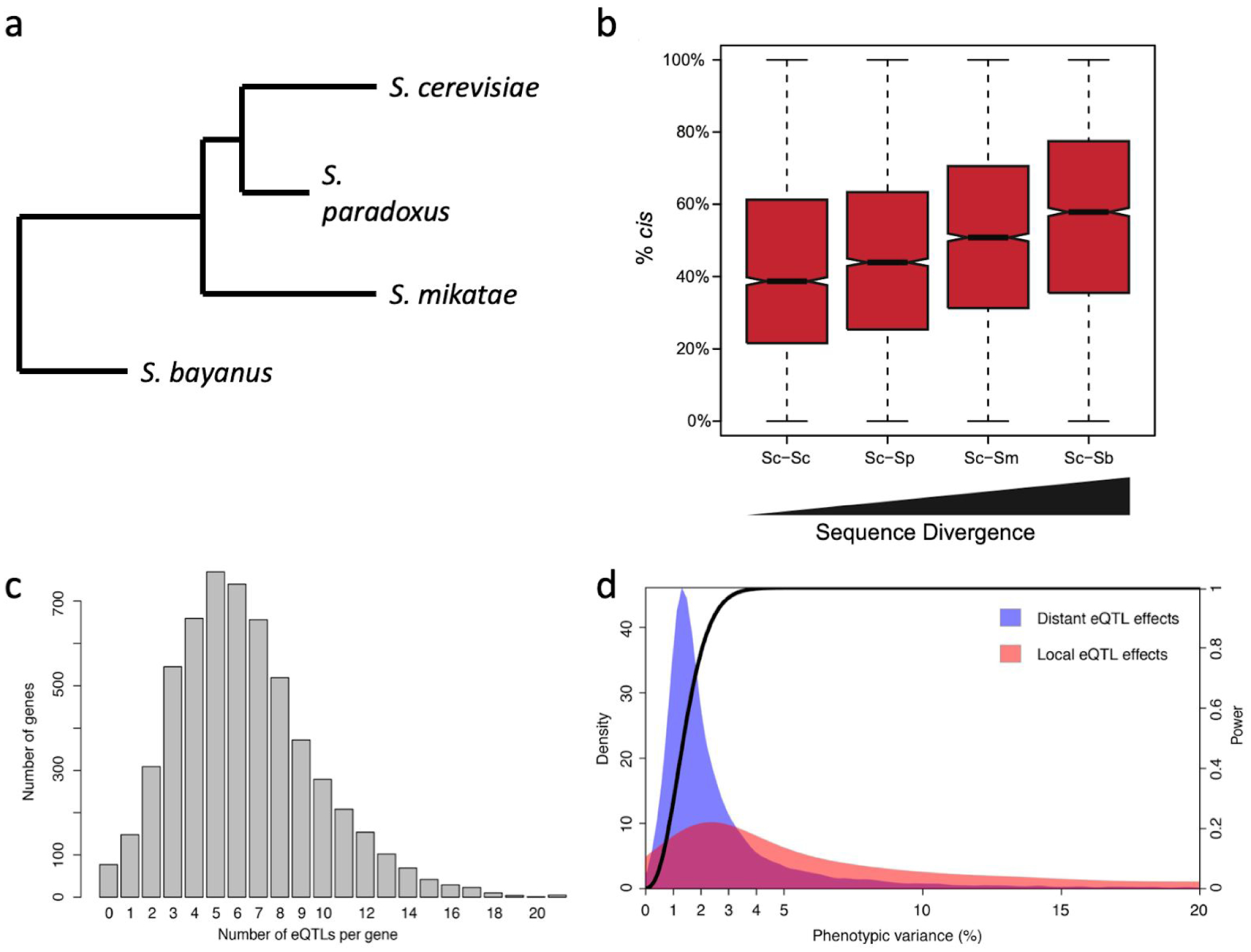
(a,b) An analysis of allele-specific expression in hybrid yeast (Saccharomyces) species with a range of divergence times (a, branch lengths reflect relative divergence times) showed increasing contributions of cis-regulatory variation to expression differences with increasing divergence time (b, notches in the boxplot indicate 95% CI of the median). (c,d) A highly-powered study of eQTL in Saccharomyces cerevisiae shows how the number of eQTL affecting expression varies among genes (c) and that putatively cis-acting eQTL end to have larger effects than trans-regulatory eQTL (d). Panel (b) reproduced with permission from Coolon et al.37, and panels (c) and (d) reproduced with permission from Albert et al.47
Studies identifying eQTL contributing to variation in gene expression have been conducted in a similarly diverse array of taxa12,44–46. Data from such studies provide insight into the number, location, and effects of regulatory variants within the genome and have shown that variation in gene expression is typically polygenic, with multiple variants contributing to variation in expression of most genes. For example, a study of the baker’s yeast, Saccharomyces cerevisiae, with 90% power to identify eQTL explaining 2.5% or more of the variation in a gene’s expression, found a median of 6 eQTL affecting expression of individual genes, with a max of 21 eQTL47 (Figure 1C). eQTL often span relatively large genomic regions and may contain multiple genetic variants, making identifying causal variants difficult. Approaches that increase the number of recombination breakpoints can be used to obtain higher resolution48. For example, eQTL mapping experiments that incorporate more than one generation of recombination to break up linked sites followed by bulk segregant analysis of individuals with extreme phenotypes49 have found even more eQTL, with over 100 eQTL affecting expression of a single gene (TDH3) in S. cerevisiae50.
eQTL located close to the affected gene (that is, proximal) are often considered cis-acting whereas eQTL located further from the affected gene (that is, distal) are often considered trans-acting20. Consistent with this assumption, proximal eQTL often have allele-specific effects on gene expression51. Indeed, the largest study of eQTL to date, which was conducted by the Genotype-Tissue Expression (GTEx) consortium and surveyed gene expression in cells derived from 49 tissues from up to 838 humans, has shown a strong correlation between the estimated effect of eQTL designated as cis-acting and allele-specific measures of expression in heterozygous individuals52. Several eQTL studies have reported that the majority of heritable expression variation is explained by trans-acting eQTL53–55, some of which affect the expression of many genes and are known as “hotspots”47,56–58 the GTEx study detected at least one cis-acting eQTL for nearly 95% of protein coding genes, whereas, trans-acting eQTL were detected for only 121 protein coding genes. The number of individuals surveyed for each tissue was a strong predictor of the number of trans-acting eQTL detected, however, underscoring the importance of taking statistical power into account when comparing the number of trans-acting eQTL reported among studies52. The unequal power for detecting cis- and trans-regulatory variants must also be considered when comparing eQTL: systematically testing for trans-regulatory variants requires many more statistical tests and thus a greater multiple testing burden than cis-regulatory variants. For this reason, some eQTL studies have focused solely on identifying cis-eQTL48,59.
Relative effect sizes of putatively cis- and trans-eQTL can be more fairly compared. Such comparisons tend to show that cis-eQTL have larger effects on gene expression than trans-eQTL46,57. For example, in the GTEx study, more cis- than trans-acting eQTL caused a two-fold or greater change in gene expression52. Similarly, in a recent, highly powered eQTL mapping study between two strains of S. cerevisiea the average cis-eQTL also explained more of the expression variation than the average trans-eQTL47. But genes are often regulated by multiple trans-regulatory variants, and sets of trans-eQTL affecting expression of the same gene tend to explain more of that gene’s expression variation than its cis-eQTL47,53,54 (Figure 1D). This observation is consistent with the greater combined contribution of trans-regulatory variation to polymorphic gene expression inferred using ASE.
Although ASE and eQTL studies reveal the relative contributions of cis- and trans-regulatory variation, they provide little insight into the specific genetic changes and molecular mechanisms altered by this variation. Only when such studies reach single variant resolution can they provide this type of insight, which is necessary for a complete understanding of why the patterns of regulatory variation we see today exist60. In the next two sections, we examine the molecular processes that give rise to cis- and trans-regulatory variation in more detail, highlighting studies examining the impact of mutations on these sequences as well as those that investigate their contribution to the evolution of gene expression.
Mechanisms generating cis-regulatory variation
cis-regulatory variation arises from genetic changes affecting sequences controlling expression of a particular allele of a gene. These sequences include the core promoter and enhancers of the gene, which both contain binding sites for transcription factors, chromatin structure influencing the accessibility of DNA to transcription factors, and sequences in the RNA transcript that affect its structure, stability, or translation. Below we discuss each of these components as a source of cis-regulatory variation.
Core promoters
At the most proximal level, a gene’s expression is controlled by its core promoter sequence, which contains binding sites for the general transcription factors necessary for transcription (Figure 2). Core promoter sequences typically lie close to the transcription start site, for example within 300 bp in humans61. Some of these core promoters contain discrete sequences with consistent positioning such as the TATA box or the downstream core promoter element, whereas others are enriched for sequence motifs such as CpG islands that are distributed over a broader region61,62.
Figure 2. Sources of cis-regulatory variation in eukaryotes.
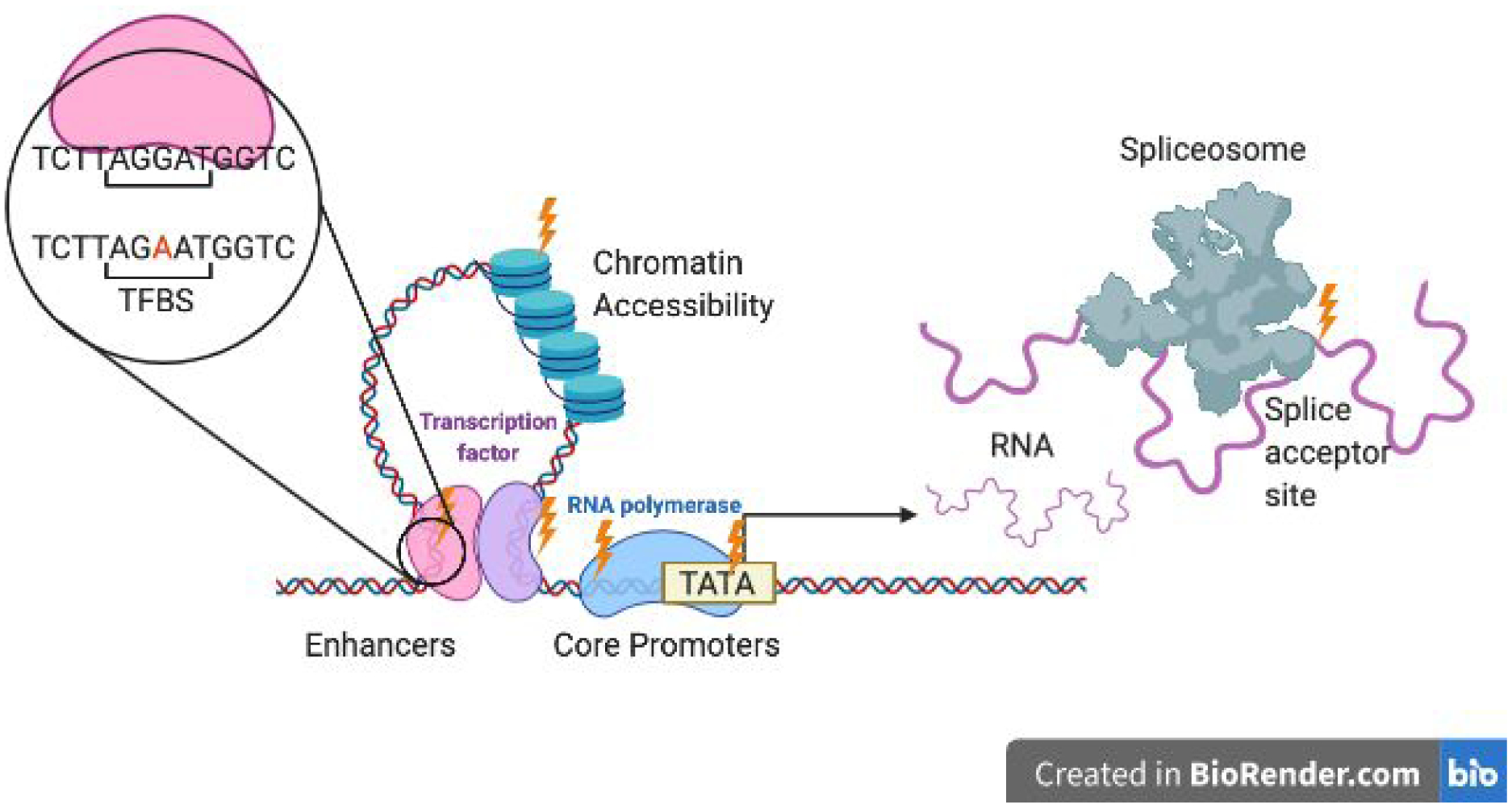
Mutations (indicated with lightning bolts) affecting the core promoter (including in motifs such as the TATA box used to assemble the transcription machinery activating RNA polymerase), enhancers (whose functional units are transcription factor binding sites (TFBS)), chromatin accessibility (altered by nucleosome placement and stability) can have cis-regulatory effects on gene expression. Mutations that affect the splicing, stability, and/or translation of mRNA in an allele-specific manner can also be sources of cis-regulatory variation.
High-throughput mutagenesis studies assaying the effects of thousands of single-nucleotide changes on activity of core promoters show how variation in these sequences might contribute to regulatory variation within and among species. One of the first such studies63 used a massively parallel reporter assay (Box 3) to assess the impact of cis-regulatory mutations in core promoters from bacteriophage and humans, with activity determined using in vitro transcription assays. The largest effect mutations were located within TATA boxes and initiator regions overlapping the transcription start site. Outside of these motifs, most mutations had no statistically significant effect. However, a more recent, more highly-powered, study of core promoters in humans assayed the activity of various promoter alleles after integration into the genome of a human cell line and found that sequences outside of these key regions can also harbor genetic variation impacting promoter activity64. Studies of mutations in core promoters of the baker’s yeast Saccharomyces cerevisiae have also described a broader distribution of mutations within the promoter that have significant effects65.
Box 3. Surveying effects of cis-regulatory mutations.
Determining the distribution of mutational effects for a cis-regulatory sequence requires generating many alleles of the cis-regulatory element (ideally with each allele carrying a single mutation) and then assaying the ability of each allele to drive gene expression in a cell. Mutant alleles can be generated by programmable DNA synthesis on microarrays179, synthesis of DNA fragments with degenerate positions, error-prone PCR, or site-directed mutagenesis. After cloning these fragments upstream of a reporter gene or DNA barcode, and introducing these alleles into a cell (either in cell culture or by injecting into living organisms), expression of the reporter gene or barcode is measured. If the reporter gene is fluorescent, expression can be measured using flow cytometry or microscopy. If a barcode is used, expression is quantified based on the number of copies of each barcode observed in an RNA-seq experiment180.
Experiments coupling the high-throughput production of mutant alleles with a high-throughput readout of expression using barcodes are often referred to as massively parallel reporter assays181. Briefly, as shown in the figure, a library of regulatory element alleles is synthesized on an array, these DNA sequences are integrated into plasmids bearing unique DNA barcodes, and then these trasmids are transformed into cells. Finally, RNA-seq is used to measure expression of the barcode driven by each allele of the regulatory element. Using this technique, thousands of mutant alleles for one or multiple cis-regulatory elements can be assayed simultaneously. However, because alleles are not integrated into the genome, this experiment might not accurately predict the effects of cis-regulatory mutations in their native genomic contexts63,85. By contrast, studies using reporter genes are more likely to integrate cis-regulatory alleles into the genome and tend to have greater power to detect small changes in expression, but typically survey fewer cis-regulatory elements and mutations. Reporter genes that can be assayed in many single cells also make it easier to examine the impact of mutations on expression noise88. The next frontiers for this work are increasing the scale of reporter-gene experiments, increasing the sensitivity of single-cell bar-coding strategies, and adding spatial information for expression in multicellular organisms182.
Despite the potential for core promoters to contribute to expression divergence, key elements of their sequence61, histone marks66, and function65 are often highly conserved among species. This conservation is presumably driven by the requirement for a functional promoter to express a gene as well as the strong functional constraints on proteins that bind to these sequences because they regulate so many different genes. Indeed, sequences within promoters that serve as binding sites for general transcription factors, such as TATA boxes, are the most highly conserved portions of mammalian core promoters61. However, a comparison of core promoter sequences between human and rhesus macaque suggested that core promoters for a small number of genes might be diverging due to positive selection67, and other work has shown that the gain and loss of core promoters contributes to expression divergence between mouse and human68. Furthermore, even if variation in the core promoter itself is not the source of expression divergence, the structure of the core promoter can still influence expression divergence. For example, the presence of a TATA box69,70, nucleosome positioning in the core promoter71, and tandem repeats in the core promoter sequence have all been shown to correlate with expression divergence in yeast72.
Enhancers
Compared to core promoters, enhancers are typically located further from the transcription start site in either upstream (5’), downstream (3’) or intronic regions73 (Figure 2) and seem to more often be the source of cis-regulatory variation affecting gene expression74–76. Because enhancers regulate gene expression in a more time-, tissue-, or environment-specific manner than core promoters, they are expected to be subject to less functional constraint due to pleiotropy77 and thus more evolvable78. Indeed, histone marks commonly associated with enhancers show greater divergence among mammalian species than histone marks associated with core promoters66. Although single cell organisms such as S. cerevisiae lack enhancers, they have upstream activating and repressing sequences that often work in a similarly context-dependent manner79.
Transcription factor binding sites
The primary functional units within all of these cis-regulatory DNA sequences are binding sites for transcription factors, which can activate or repress transcription80. These sequences are short, degenerate, and able to evolve relatively quickly, even from random sequences81,82. Mutations that change the identity, affinity, orientation, number, and/or spacing of transcription factor binding sites (TFBSs) can alter cis-regulatory activity75,83,84. Large-scale mutagenesis studies of enhancers and other similar cis-regulatory elements have shown that although many mutations in these sequences can alter gene expression, mutations in TFBSs tend to have the largest effects85–88. Although TFBSs are often among the most highly conserved sequences within an enhancer89–92, they can also harbor genetic changes responsible for variation in gene expression within93,94 and between species95,96,97. However, in most cases where functional changes have been mapped to enhancers or similar cis-regulatory sequences, the specific genetic changes responsible for altering their function have not yet been identified6,98–100.
Chromatin accessibility
For a TFBS to regulate expression of a gene, the transcription factor it binds must be able to access the DNA sequence. In eukaryotes, DNA is packaged into chromatin by wrapping it around a complex of histone proteins known as a nucleosome, which can interfere with this access (Figure 2). Compeitition between nucleosomes and transcription factors for interactions with cis-regulatory DNA sequences can thus affect gene expression101–103, making genetic differences affecting chromatin structure another potential source of cis-regulatory variation24. Indeed, different patterns of nucleosome positioning at promoters have been shown to correlate with expression plasticity, species-level expression divergence, and the effects of new mutations on gene expression71,104, indicating that the pattern of nucleosome occupancy and stability at the promoter could play an important role in shaping evolutionary trajectories72.
Direct evidence of changes in chromatin structure contributing to the evolution of gene expression remains scarce, but is starting to accumulate. For example, in flies, combining information about chromatin accessibility and TFBSs explained expression divergence between Drosophila melanogaster and Drosophila virilis better than considering TFBSs alone105. In yeast, divergent chromatin structure has also been shown to correlate with divergent gene expression106,107, but most differences in nucleosome positioning between species are outside of regulatory regions and do not correlate with expression divergence108. However, in at least some cases, changes in chromatin structure seemed to have been offset by compensatory changes in TFBS exposed by the change in nucleosome position108,109.
Post-transcriptional sources of cis-regulatory variation
Although core promoters, enhancers, and chromatin accessibility are the most often discussed sources of cis-regulatory variation, they are not the only ways by which allele-specific variation in gene expression arises110. For example, variation in splice sites can have allele-specific effects on splicing of mRNA111–114; variation in polyadenylation signals can alter mRNA stability, translation, and location within the cell115; and variation in the 3’ UTR can affect mRNA degradation rates116 as well as regulation by microRNAs117. Sequence variation within the mRNA can also affect ribosome occupancy and translation efficiency118. Future work focusing on these post-transcriptional mechanisms is needed to more fully evaluate their relative contributions to regulatory evolution.
Mechanisms generating trans-regulatory variation
Whereas cis-regulatory variants tend to lie near the affected gene, trans-regulatory variants affecting a gene’s expression can be located virtually anywhere in the genome. These potential sites of trans-regulatory variants include both coding and non-coding sequences that affect expression or activity of gene products that regulate the focal gene’s expression either directly (by binding to its cis-acting sequences) or indirectly (by influencing the activity of direct regulators)58 (Figure 3). This large potential target size for trans-regulatory variants makes it difficult to interrogate them by targeted analysis of candidate regions. Rather, genome-wide mutagenesis and mapping strategies are needed to introduce and characterize trans-regulatory variants, often requiring follow-up experiments to separate the effects of causal variants from linked loci11,47.
Figure 3. Sources of trans-regulatory variation.
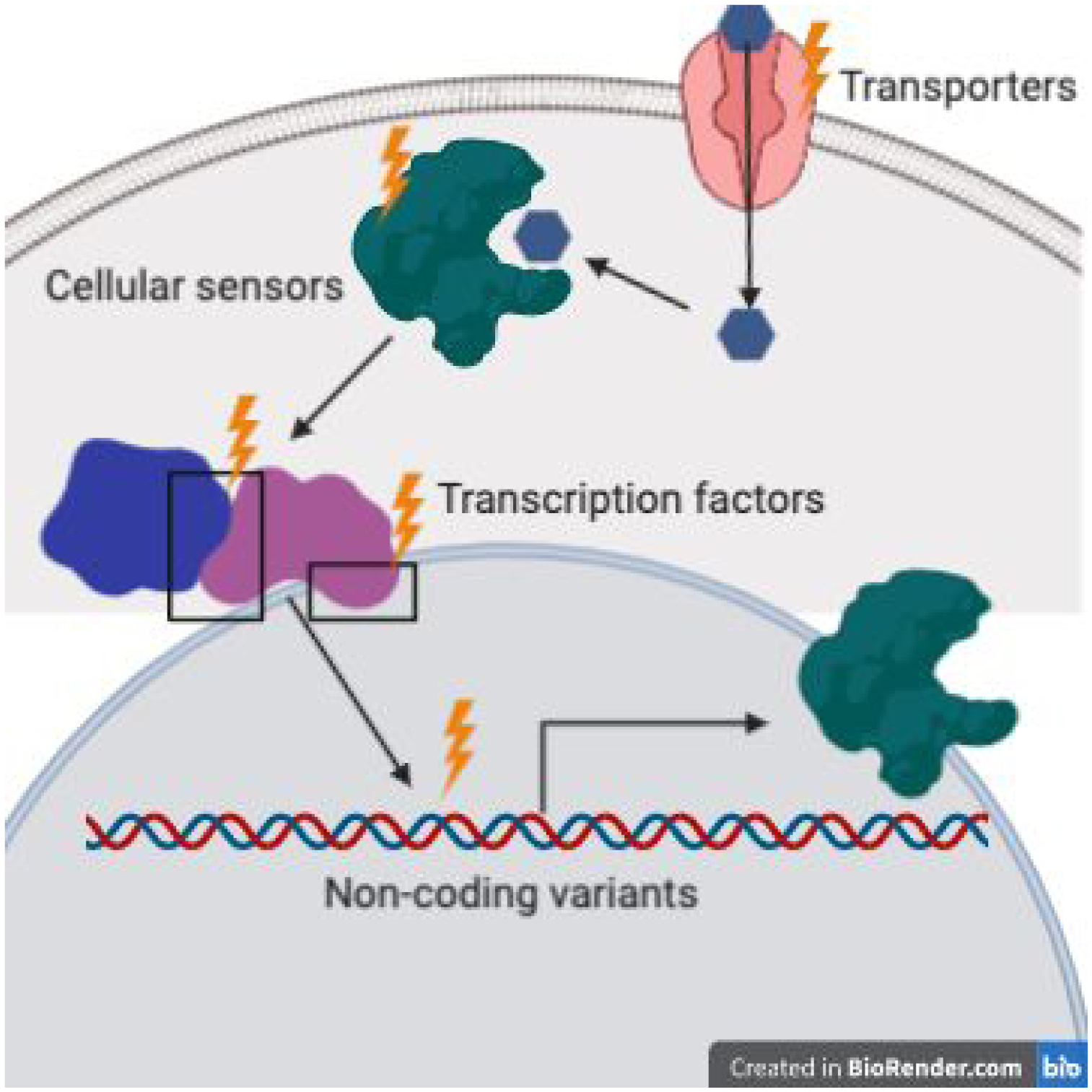
Mutations (indicated by lightning bolts) that can affect expression of a gene via diffusible molecules are trans-acting. These mutations can occur in non-coding or coding sequences of transcription factors, cellular sensors, transporters, and other molecules that influence transcription of many genes via effects on the many interconnected cellular networks.
Coding and non-coding sequences
Although the effects of trans-regulatory variants are mediated by diffusible molecules such as RNAs or proteins, studies of regulatory variation segregating in humans suggest that most trans-acting variants are not located within the sequences encoding these molecules114. Instead, in large-scale genome-wide association studies (Box 2), the majority of trans-regulatory variants have been found in non-coding, putatively cis-regulatory sequences controlling the gene’s expression52,114,119,120. By changing expression of the gene they affect in cis, such variants can impact the expresssion of other genes in trans53,56,114,121. For example, a cis-acting eQTL located near the gene encoding lysozyme (an enzyme that breaks down bacterial cell walls) has been shown to also act as a trans-acting eQTL for expression of other genes in monocytes122. Similarly, a cis-acting eQTL near the transcription factor KLF14, which regulates expression of genes in adipose tissue, explains trans-acting effects observed on expression of other genes123.
Box 2. Using genetic associations to localize cis- and trans-regulatory variants.
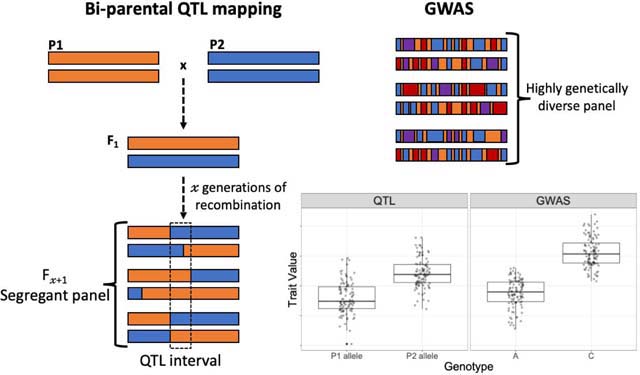
Specific genetic changes impacting gene expression can be localized within the genome using genetic mapping approaches with gene expression phenotypes20,21. These strategies rely on statistical associations with the effects of variants in different parts of the genome separated from each other by recombination. This recombination can come from two (or more) parental strains (e.g. P1 & P2 in figure) being crossed in a controlled manner (QTL mapping) to produce heterozygous F1 progeny which are then further crossed to produce a segregant panel. Alternatively, instead of experimentally generating recombinants, and thus capturing allelic and phenotypic variation between two strains, one can instead rely on existing genetic diversity within a population sample and perform a Genome Wide Association Study (GWAS, see figure). In each case, individuals within the segregant panel or population sample are genotyped and phenotyped allowing the detection of statistical associations between genetic variants and quantitative traits (in this case gene expression). Variants with statistically significant effects are called expression quantitative trait loci (eQTL). eQTL studies have been used to provide insight into the relative contributions of cis- and trans-regulatory variants to expression variation by designating each eQTL as (putatively) cis- or trans-acting based on its physical proximity to the gene whose expression it affects. Thus, associated variants proximal to the affected gene are commonly considered cis-eQTL and associated variants outside of a given cis- window are considered trans-eQTL. While this assumption often holds, it is possible for proximal variants to regulate the affected gene through a diffusible product (such as an RNA or protein) and for cis- acting variants to be located in distal enhancers, far from the gene they regulate. Because tests for cis-eQTL are typically restricted to variants in a small region of the genome close to the focal gene, and tests for trans-eQTL include all variants outside of this putatively cis-acting region, there is a much larger multiple testing burden, and thus lower statistical power, for identifying trans-eQTL. Despite these limitations, eQTL mapping is currently the best approach available for localizing regulatory variants within the genome.
However, studies of the baker’s yeast S. cerevisiae suggest that this species might have a different distribution of trans-regulatory variants in coding and non-coding sequences. As in humans, hotspot genes with trans-regulatory eQTL affecting expression of many genes are more likely to have a local, putatively cis-acting eQTL than expected by chance47, but the functional trans-regulatory variants mapped and validated in S. cerevisiae so far have primarily, although not exclusively, been in coding regions20,47,56,58,124,125. S. cerevisiae might have a higher proportion of trans-regulatory variants in coding sequences than humans because so much less of their genome is non-coding (27% in S. cerevisiae vs 97% in humans126); however, the higher proportion of coding variants might also be a consequence of often using a lab-adapted strain that carries many variants absent from wild populations127. Determining the true relative contributions of coding and non-coding variants to trans-regulatory variation in yeast (and other species) will require much more extensive mapping and functional testing of variants from natural populations.
If trans-regulatory variants generally do map to non-coding sequences more often than coding sequences, it might be because mutations in non-coding sequences tend to be less pleiotropic. For example, non-coding mutations that affect activity of a tissue-specific enhancer are expected to impact fewer traits than coding mutations altering the same gene’s protein sequence everywhere it is expressed16,76,78,128. Indeed, most trans-acting eQTL in human non-coding sequences seem more likely to affect enhancers than core promoters114, and often have tissue-specific effects53,114. Because mutations that are more pleiotropic are expected to typically be more deleterious than less pleiotropic mutations129, coding mutations might be selected against more strongly than non-coding mutations, reducing their frequency in natural populations. However, this paradigm is challenged by data showing that cis-regulatory sequences are more pleiotropic130, and protein sequences more modular131,132, than generally appreciated. Indeed, a recent study has shown how modularity in the yeast MATalpha2 transcription factor protein facilitated its divergence, which was then followed by changes in cis-regulatory, non-coding sequences of the genes it regulates133.
Transcription factors
Transcription factors (TFs) are proteins that bind to short sequences within cis-acting promoters and enhancers to regulate expression of a gene. They are often considered the most likely source of trans-regulatory variation, especially for hotspot eQTL, because most TFs regulate expression of many target genes134–138. Indeed, TFs do often seem to be responsible for hotspot eQTL in both humans120,139,140 and S. cerevisiae47,141. However, the ability of TFs to affect expression of multiple downstream target genes also results in functional constraint on their variation. Indeed, their protein coding sequences, DNA binding specificities, and general physiological roles are often conserved over long evolutionary timescales142. Despite these general trends of conservation, TFs can and do diverge in function, as changes in TF protein sequences, including those that affect their DNA binding specificity, have been reported for TFs controlling mating type in yeast,143,144, flower development and cell division in plants145, and body patterning in insects146,147, among others.
Sources of trans-regulatory variation other than transcription factors
Variants affecting genes not encoding TFs are also important sources of trans-regulatory variation. For example, chromatin regulators can have widespread effects on gene expression148, and an eQTL study in S. cerevisiae suggests that genes encoding these types of proteins harbor trans-acting eQTL affecting expression of many genes149. Functional studies in S. cerevisiae have also demonstrated trans-regulatory effects of variants in co-factors that modulate the activity of TFs150 as well as genes that influence metabolism such as the glucose receptor RGT258 and a membrane protein, SSY1, that senses the concentration of extracellular amino acids151. In humans, trans-eQJL have also been shown to map to genes that do not encode TFs, such as the Slco1a6 gene, in which a genetic variant was shown to alter expression of many genes by altering the transport of bile acids in pancreatic islets152. The diverse sources of trans-regulatory variation illustrated by these and other studies result from the interconnectedness of transcriptional, structural, signaling, and metabolic networks, and underscore the challenge of predicting and identifying trans-regulatory variants with our current understanding of systems biology11. They are also consistent with the proposed ‘omnigenic’ model of heritability, in which every gene expressed has the potential to influence every trait153. Ultimately, more functional tests of candidate trans-regulatory variants will be needed to fully understand the sources of trans-regulatory variation.
Surveying the effects of trans-regulatory mutations
Targetted mutagenesis strategies like those used to elucidate the effects of cis-regulatory mutations cannot be used for unbiased surveys of trans-regulatory mutations because trans-regulatory mutations can be located anywhere in the genome. trans-regulatory mutations are thus best surveyed by introducing mutations randomly throughout the genome and measuring their effects on gene expression. Two general strategies have been used to isolate the mutations needed to characterize the effects of trans-regulatory mutations: mutation accumulation and random mutagenesis (Box 4). Neither of these approaches distinguishes between mutations that act in cis or trans, but the vast majority of randomly introduced mutations affecting expression of a focal gene are expected to act in trans41, suggesting that cis-regulatory mutations captured in these studies are negligible. Indeed, studies of the TDH3 gene in S. cerevisiae have estimated that a random mutation is at least 265 times more likely to affect expression of this gene in trans than in cis154,155.
Box 4. Surveying effects of trans-regulatory mutations.
Because a trans-acting mutation can reside virtually anywhere in the genome, effects of trans-regulatory mutations are most efficiently surveyed by examining the effects of mutations introduced randomly genome-wide. Such mutations are generally collected using one of two strategies: (1) mutation accumulation or (2) random mutagenesis. With either strategy, effects of the mutations captured can be assayed for single genes using reporter genes or for the entire genome using RNA-seq.
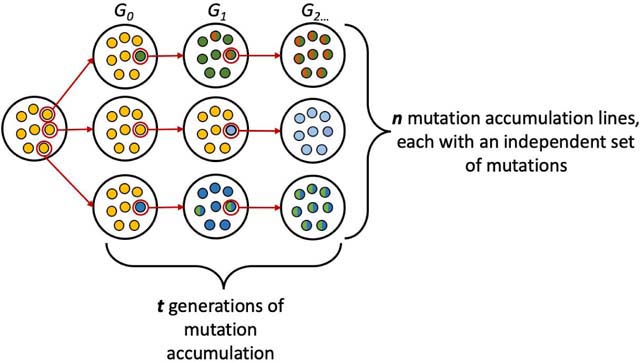
Mutation accumulation studies collect spontaneous mutations arising over many generations in the near absence of natural selection156,157. Multiple independent lines are initiated from a single starting population (highly inbred, if not isogenic) and propagated with bottlenecks of 1 asexual or 2 sexual individuals each generation (see figure). These extreme bottlenecks allow selection to remove only lethal or sterile mutations. This strategy captures the full range of spontaneous mutations, but requires many generations of mutation accumulation to capture even a small number of mutations given that per base mutation rates are typically in the range of 10−8 to 10−10 per generation183mutation accumulation experiments tend to provide only sparse sampling of trans-regulatory mutations affecting expression of any given gene.
By contrast, random mutagenesis can introduce tens to hundreds of new mutations per cell in a single generation184,185. These mutations can be introduced by using chemical mutagenesis, DNA repair deficient strains, or activation of transposons. Mutations introduced by these methods, however, reflect only a subset of the types of mutations that arise spontaneously. For example, ethyl methanesulfonate (EMS), perhaps the most widely-used chemical mutagen, introduces almost exclusively G-to-A and C-to-T transitions186. Random mutagenesis approaches are thus an important complement to, rather than a replacement for, studying the effects of spontaneous mutations.
Mutation accumulation studies typically summarize the effects of new mutations on gene expression by estimating the mutational variance (Vm), which describes the increase in expression variance caused by new mutations each generation156,157. This parameter has been estimated genome-wide for two Drosophila species158–160, S. cerevisiae70 and the nematode Caenorhabditis elegans161,162. These data suggest that new mutations often have widespread effects on gene expression. For example, a 200 generation mutation accumulation experiment in D. melanogaster examined about 360 mutations in each of 12 independent strains and found that ~39% of genes showed significant expression variance among the mutation accumulation lines158. About one third of the genes in S. cerevisiae were also found to have significant expression variance among 4 independent lines from a mutation accumulation study lasting 4000 generations70. In general, mutation accumulation studies suggest that many mutations affect expression of multiple genes158–161, consistent with them often having trans-regulatory effects.
Mutagenesis studies that specifically examine a set of mutations affecting expression of a single gene are an important complement to mutation accumulation studies because they provide much deeper sampling of trans-regulatory mutations affecting the gene’s expression. (Mutation accumulation lines generally recover only a few mutations affecting expression of any particular gene.) Thus far, this mutagenesis approach has been used most extensively to study the distribution of mutational effects for trans-regulatory mutations altering expression driven by the promoter of the S. cerevisiae TDH3 gene154,155. These studies have shown, for example, that even though TDH3 is one of the most highly expressed genes in the genome, mutations increasing its expression are at least as common as mutations decreasing its expression. Using this same approach to characterize the effects of thousands of mutations on expression driven by promoters from 9 other S. cerevisiae genes showed how gene-specific distributions of mutational effects can differ in terms of skew, kurtosis, and dispersion, none of which are captured by Vm163.
These more focused studies of predominantly trans-acting mutations affecting expression of a particular gene also allow direct comparisons between the effects of cis- and trans-regulatory mutations affecting expression of the same gene. For example, a study comparing the effects of 235 cis-regulatory mutations in the S. cerevisiae TDH3 promoter to the effects of ~47,000 mutations spread throughout the genome showed that cis-regulatory mutations tended to have larger average effects on expression driven by the TDH3 promoter than trans-regulatory mutations154 These cis-regulatory mutations were also more likely than trans-regulatory mutations to decrease expression of this gene154 and to have dominant effects in diploid cells155,164. To the best of our knowledge, TDH3 is the only gene for which such comparable information on cis- and trans-regulatory mutations currently exists; however, if other genes show similar trends, these differences between cis- and trans-regulatory mutations, combined with the expected differences in pleiotropy described above, might explain the unequal contributions of cis- and trans-regulatory variants to the evolution of gene expression.
Mechanisms of evolutionary change
Understanding how new mutations generate variation in gene expression is critical for understanding how gene expression evolves because it allows us to predict how much variation in gene expression we should see after different amounts of evolutionary time due to neutral processes alone. That is, when a gene’s expression is evolving neutrally, mutations introduce new variants that can affect its expression and genetic drift fixes and eliminates these variants by chance, effectively sampling randomly from the distribution of mutational effects. However, when natural selection is acting on a gene’s expression, some regulatory variants are more likely to fix or be eliminated than others based on their effects, causing the distribution of mutational effects to differ from the distribution of effects observed for polymorphisms segregating within a species or divergent sites that differ between species (Figure 4). Comparing the effects of mutations to the effects of polymorphic and/or divergent sites is thus a powerful way to infer the effects of natural selection165. This general strategy has been used to infer the role of selection in generating variation in gene expression within and between species, first using mutational effects inferred from mutation accumulation studies158,161 and more recently using mutational effects derived from studies interrogating cis- and trans-regulatory mutations affecting expression of a particular gene more deeply50,88,166.
Figure 4: Using mutational effects to infer the action of natural selection.
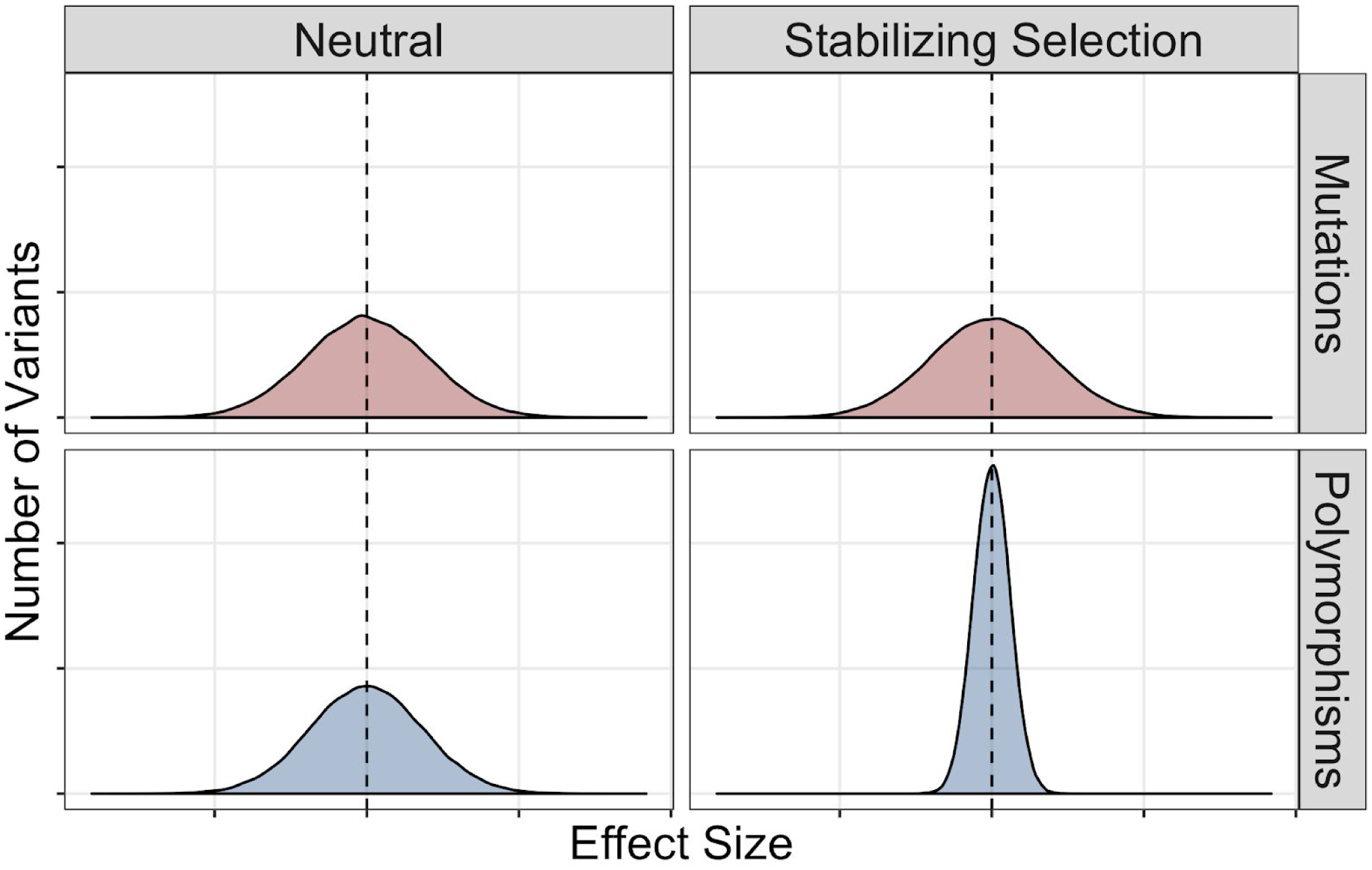
Distinguishing between neutral and adaptive explanations for gene expression variation can be achieved by contrasting the effects of mutations (red, which shows the amount of expression variation expected to result from the accumulation of mutations in the absence of selection) and polymorphisms (blue, which shows expression variation affected by both neutral processes and selection). Dashed lines represent an effect size of zero (that is, no change in expression). If a gene’s expression is evolving neutrally (left panels), the effects of polymorphisms are expected to be consistent with a random sampling of effects from the mutational distribution: there should be no statistically significant difference between the distributions of effects for mutations (red) and polymorphisms (blue). By contrast, if expression of a gene is under stabilizing or directional selection, for example, the distribution of effects for polymorphisms will have lower variance than the distribution of mutational effects. The example shown here (right panels) is consistent with stabilizing selection, which maintains expression at its current level (that is, selection disfavors variants that either decrease or increase expression). Directional selection would also shift the mean effect of polymorphisms to higher or lower expression than the mean effect of mutations.
As described above, mutation accumulation studies typically measure the effects of mutations on gene expression in terms of mutational variance, Vm. For Drosophila spp., this estimate of how expression variance increases each generation was used to calculate the variance in gene expression expected to evolve under mutation-drift equilibrium for three pairs of Drosophila species158. Comparing the observed expression differences between these three pairs of species to this neutral expectation showed the expression divergence was substantially lower than predicted by the neutral model, suggesting that stabilizing selection had acted to reduce variation in gene expression levels158. A study comparing Vm for D. melanogaster to expression variation among strains of D. meianogaster reached the same conclusion160. Similarly, Vm estimated from four mutation accumulation lines of C. elegans maintained for 280 generations predicted more expression variance than was observed among five recently isolated lines of C. elegans separated by many thousands of generations161. These findings, combined with other types of analyses, have led to the prevailing view that stabilizing selection typically constrains variation in gene expression on a genomic scale17,167.
Gene-specific distributions of mutational effects are beginning to refine these analyses, allowing more specific questions to be addressed about the impact of selection on variation in gene expression. For example, effects of mutations in two human enhancers and one mouse enhancer assayed in mice85 were used to predict the effects of divergent sites in other rodent and primate lineages, showing evidence of different types of selection acting on each enhancer168. More direct comparisons between the effects of mutations and polymorphisms assayed in their native species have been performed for the S. cerevisiae TDH3 gene. Specifically, effects of cis-regulatory mutations in the TDH3 promoter on both gene expression level and gene expression noise were compared to the effects of polymorphisms in the TDH3 promoter observed among 85 strains of S. cerevisiae88 These data showed no evidence of selection acting on mean expression level, but did show evidence of stabilizing selection constraining expression noise88. Comparing the effects of these cis-regulatory mutations and polymorphisms in multiple environments also showed evidence of stabilizing selection acting to maintain a particular degree of expression plasticity for TDH3166. Finally, evidence of stabilizing selection was also seen when the effects of trans-regulatory mutations determined using mutagenesis were compared to the effects of polymorphisms affecting TDH3 expression inferred from eQTL mapping50.
Future directions
Molecular biology explains how new mutations give rise to variation in gene expression whereas population genetics explains how these new mutations might contribute to evolutionary divergence once they arise. We believe that both perspectives must be considered together to understand why we see the expression variation we see in the wild. Moving forward, we think it is important for the field to grow in at least three critical directions.
First, we think that more gene-specific distributions of mutational effects are needed for cis- and trans-regulatory mutations. Such work is required because new mutations are expected to have effects on gene expression that vary from gene to gene and between cis- and trans-acting mutations, but we have only begun to discover the range of these differences and do not yet know which properties of mutational effects are most important for accurately predicting polymorphism and divergence. New techniques such as saturation mutagenesis of regulatory elements169 and massively parallel genome editing to functionally validate trans-regulatory variants170 are making collection of such data more feasible at the scales necessary to answer these questions.
Despite these advances, it will likely never be practical to survey all genes and regulatory elements in all species. Consequently, the second critical direction is to understand how properties of regulatory networks shape distributions of mutational effects. We anticipate that such properties exist because the effects of new mutations on gene expression are determined by how they impact the structure and function of regulatory networks9. Indeed, a study comparing patterns of expression polymorphism and divergence to regulatory network structure in Drosophila spp found that genes regulated by a greater number of transcription factors were less likely to show variation in expression within and between species, presumably because the coordinate control of gene expression by sets of regulators tends to buffer the effects of mutations impacting activity of individual regulators171. This pattern might not be general though, as it was not observed among yeast species172 and no relationship was detected between loci harboring eQTL hotspots and network connectivity in S. cerevisiae47. Many questions remain, however, about the form and function of regulatory networks that might obscure these relationships11. The context-dependency of regulatory networks further adds to this challenge, as regulatory networks are expected to differ between cell types, genetic backgrounds, sexes, and environments. Yet here too, technical advances such as single-cell RNA-sequencing hold great promise for elucidating temporal and tissue-specific regulatory networks, and how they are impacted by new mutations173.
Once the effects of new mutations on gene expression are known or can be predicted, a third challenge is linking the changes in gene expression caused by these mutations to fitness and using the existing theoretical framework of population genetics to predict the evolutionary fate of different types of regulatory mutations. Fitness curves describing the relationship between expression of a gene and relative fitness are available for a few genes in S. cerevisiae174–176, but remain unknown for most genes in most species. Filling this knowledge gap will require more efficient ways to both modify gene expression and quantify fitness in many species. Despite this challenge, such data are key for connecting the too often disparate fields of molecular and evolutionary biology, which is essential for understanding the biological world as it exists now and how it is most likely to be in the future.
Box 1. Using allele-specific expression to disentangle cis- and trans-regulatory variation.
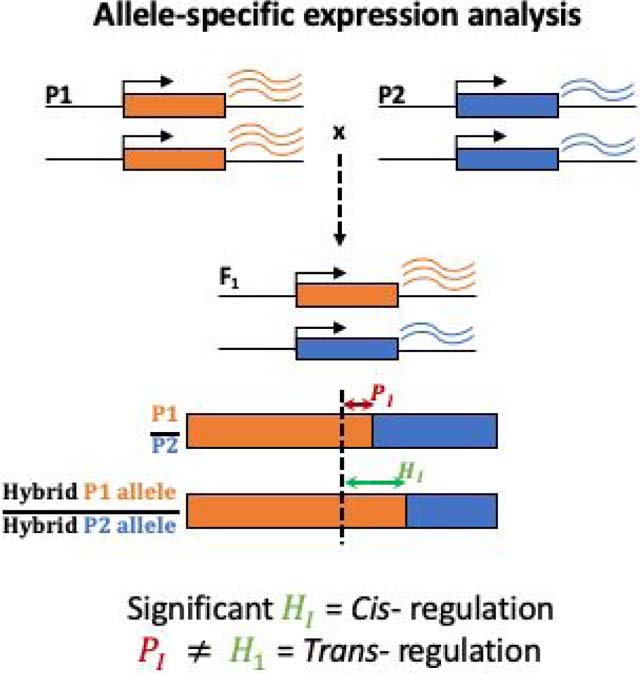
By definition, cis-regulatory variants have an allele-specific effect on gene expression, with a cis-regulatory variant altering expression of only the transcribed sequence located on the same chromosome. Consequently, when expression of two alleles of the same gene is compared in a single trans-regulatory environment - as is the case for two alleles within an F1 hybrid - differences in the abundance of RNA transcripts produced from the two alleles captures their relative cis-regulatory activity177. Comparing this relative cis-regulatory activity in F1 hybrids to the relative expression of the same alleles in the parental genotypes (P1 & P2) crossed to produce the F1 hybrid allows the effects of trans-regulatory variation to also be inferred19. Thus, using this approach (see figure), cis effects are detected when there is a significant difference in expression between the two alleles in the F1 hybrid (quantity H1), and trans effects are detected when the ratios of allelic expression in the parental (P1) and hybrid strains (H1) differ (P1 ≠ H1). With the advent of RNA-seq, allele-specific expression can be quantified genome-wide and the relative contribution of cis- and trans-regulatory variation to differences in gene expression assessed on a gene-by-gene basis. This general strategy can be used to characterize regulatory variation both within and between species, as long as there is allelic variation and the two parental genotypes can produce viable F1 hybrids.
The most significant limitation of this approach is that it is blind to the identity and genomic location of the cis- and trans-regulatory variants causing the observed regulatory effects. In addition, tests for cis-regulatory variation are typically more highly powered than tests for trans-regulatory variation because the former relies only on the measurements of allele-specific expression in the F1 hybrids whereas the latter compares this expression ratio in the hybrids to that between the parental genotypes. Thus, the number of parameters that can vary across biological replicates is higher when testing the effects of trans- than cis-regulatory variation. Care must also be taken to ensure independent estimates of the effects of cis- and trans-regulatory variation when testing for evidence of compensatory evolution28,178.
Acknowledgments
We thank members of the Wittkopp laboratory for helpful discussions during the drafting of the manuscript. Support for this work was provided by the John Simon Guggenheim Memorial Foundation, Alexander von Humboldt Foundation, National Science Foundation (DEB-1911322), and National Institutes of Health (R35GM118073) to P.J.W. and the National Institutes of Health Training Grant T32GM007544 to P.V.Z.
Glossary
- Pleiotropy
The phenomenon whereby a single genetic variant affects multiple independent traits.
- Genetic Drift
Variation in allele frequencies caused by random sampling of individuals.
- Bulk segregant analysis
A technique used to associate genetic markers with trait variation by contrasting allele frequencies between two groups of individuals defined by differences in trait values.
- TATA box
An element of some promoter sequences that serves as a binding site for certain general transcription factors and is rich in T/A nucleotides.
- Core promoter element
Functional sequences proximal to the transcription start site that are sufficient to initiate transcription.
- CpG island
A region of the genome containing a large number of CpG dinucleotide repeats, found in the promoters of many mammalian genes.
- Initiator region
An element of core promoter sequences downstream of the TATA box which overlaps with the transcription start site.
- Gene expression noise
The variability of expression level among genetically identical cells in the same environment.
- Skew
A measure of the asymmetry of a distribution about its mean.
- Kurtosis
A measure of how much weight is concentrated in the tails of a distribution, relative to its center.
- Dispersion
The extent to which a set of values is clustered or dispersed, often measured by the variance or standard deviation of a distribution.
References cited
- 1.Zheng W, Gianoulis TA, Karczewski KJ, Zhao H & Snyder M Regulatory variation within and between species. Annu. Rev. Genomics Hum. Genet 12, 327–346 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Kronforst MR et al. Unraveling the thread of nature’s tapestry: the genetics of diversity and convergence in animal pigmentation. Pigment Cell Melanoma Res. 25, 411–433 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Wessinger CA & Rausher MD Lessons from flower colour evolution on targets of selection. J. Exp. Bot 63, 5741–5749 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Oliver F et al. Regulatory variation at glypican-3 underlies a major growth QTL in mice. PLoS Biol. 3, e135 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deutschbauer AM & Davis RW Quantitative trait loci mapped to single-nucleotide resolution in yeast. Nat. Genet 37, 1333–1340 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Martin A & Orgogozo V The Loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution 67, 1235–1250 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Courtier-Orgogozo V, Arnoult L, Prigent SR, Wiltgen S & Martin A Gephebase, a database of genotype-phenotype relationships for natural and domesticated variation in Eukaryotes. Nucleic Acids Res. 48, D696–D703 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albert FW & Kruglyak L The role of regulatory variation in complex traits and disease. Nat. Rev. Genet 16, 197–212 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Babu MM, Luscombe NM, Aravind L, Gerstein M & Teichmann SA Structure and evolution of transcriptional regulatory networks. Curr. Opin. Struct. Biol 14, 283–291 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Yu H & Gerstein M Genomic analysis of the hierarchical structure of regulatory networks. Proc. Natl. Acad. Sci. U. S. A 103, 14724–14731 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flint J & Ideker T The great hairball gambit. PLoS Genet. 15, e1008519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rockman MV & Kruglyak L Genetics of global gene expression. Nat. Rev. Genet 7, 862–872 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Signor SA & Nuzhdin SV The Evolution of Gene Expression in cis and trans. Trends Genet. 34, 532–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fay JC & Wittkopp PJ Evaluating the role of natural selection in the evolution of gene regulation. Heredity 100, 191–199 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Streisfeld MA & Rausher MD Population genetics, pleiotropy, and the preferential fixation of mutations during adaptive evolution. Evolution 65, 629–642 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Stern DL & Orgogozo V Is genetic evolution predictable? Science 323, 746–751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bedford T & Hartl DL Optimization of gene expression by natural selection. Proc. Natl. Acad. Sci. U. S. A 106, 1133–1138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinney JB & McCandlish DM Massively Parallel Assays and Quantitative Sequence-Function Relationships. Annu. Rev. Genomics Hum. Genet 20, 99–127 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Wittkopp PJ, Haerum BK & Clark AG Evolutionary changes in cis and trans gene regulation. Nature 430, 85–88 (2004). [DOI] [PubMed] [Google Scholar]; This study describes the use of allele-specific RNA expression in F1 hybrids to estimate the relative contributions of cis- and trans-regulatory variation to expression differences between species.
- 20.Brem RB, Yvert G, Clinton R & Kruglyak L Genetic dissection of transcriptional regulation in budding yeast. Science 296, 752–755 (2002). [DOI] [PubMed] [Google Scholar]; This study was the first to couple linkage analysis with genome-wide gene expression data, identifying eQTL likely to act in cis and in trans. Expression of 570 genes was statistically linked to one or more loci, indicating complex inheritance patterns for most gene expression levels.
- 21.Schadt EE et al. Genetics of gene expression surveyed in maize, mouse and man. Nature 422, 297–302 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Springer NM & Stupar RM Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell 19, 2391–2402 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X & Borevitz JO Global analysis of allele-specific expression in Arabidopsis thaliana. Genetics 182, 943–954 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi X et al. Cis- and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun 3, (2012). [DOI] [PubMed] [Google Scholar]
- 25.Bell GDM, Kane NC, Rieseberg LH & Adams KL RNA-Seq Analysis of Allele-Specific Expression, Hybrid Effects, and Regulatory Divergence in Hybrids Compared with Their Parents from Natural Populations. Genome Biol. Evol 5, 1309–1323 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D et al. Expression evolution in yeast genes of single-input modules is mainly due to changes in trans-acting factors. Genome Res. 17, 1161–1169 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sung H-M et al. Roles of Trans and Cis Variation in Yeast Intraspecies Evolution of Gene Expression. Mol. Biol. Evol 26, 2533–2538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emerson JJ et al. Natural selection on cis and trans regulation in yeasts. Genome Res. 20, 826–836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzger BPH, Wttkopp PJ & Coolon JD Evolutionary Dynamics of Regulatory Changes Underlying Gene Expression Divergence among Saccharomyces Species. Genome Biol. Evol 9, 843–854 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goncalves A et al. Extensive compensatory cis-trans regulation in the evolution of mouse gene expression. Genome Res. 22, 2376–2384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mack KL, Campbell P & Nachman MW Gene regulation and speciation in house mice. Genome Res. 26, 451–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson JH & Balakrishnan CN Gene regulatory evolution during speciation in a songbird. G3: Genes, Genomes, Genetics 6, 1357–1364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M, Uebbing S & Ellegren H Bayesian Inference of Allele-Specific Gene Expression Indicates Abundant Cis-Regulatory Variation in Natural Flycatcher Populations. Genome Biol. Evol 9, 1266–1279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Werren JH & Clark AG Allele-Specific Transcriptome and Methylome Analysis Reveals Stable Inheritance and Cis-Regulation of DNA Methylation in Nasonia. PLoS Biol. 14, e1002500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wttkopp PJ, Haerum BK & Clark AG Regulatory changes underlying expression differences within and between Drosophila species. Nat. Genet 40, 346–350 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Suvorov A et al. Intra-Specific Regulatory Variation in Drosophila pseudoobscura. PLoS One 8, e83547 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coolon JD, McManus CJ, Stevenson KR, Graveley BR & Wittkopp PJ Tempo and mode of regulatory evolution in Drosophila. Genome Res. 24, 797–808 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McManus CJ et al. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res. 20, 816–825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaefke B et al. Inheritance of gene expression level and selective constraints on trans-and cis-regulatory changes in yeast. Mol. Biol. Evol 30, 2121–2133 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Nolte V & Schlötterer C Temperature stress mediates decanalization and dominance of gene expression in Drosophila melanogaster. PLoS Genet. 11, e1004883 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wttkopp PJ Genomic sources of regulatory variation in cis and in trans. Cell. Mol. Life Sci 62, 1779–1783 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guerrero RF, Posto AL, Moyle LC & Hahn MW Genome-wide patterns of regulatory divergence revealed by introgression lines. Evolution 70, 696–706 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Coolon JD et al. Molecular mechanisms and evolutionary processes contributing to accelerated divergence of gene expression on the Drosophila X chromosome. Mol. Biol. Evol 32, 2605–2615 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nica AC & Dermitzakis ET Expression quantitative trait loci: Present and future. Philosophical Transactions of the Royal Society B: Biological Sciences vol. 368 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson G & Weir B The quantitative genetics of transcription. Trends Genet. 21, 616–623 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Gilad Y, Rifkin SA & Pritchard JK Revealing the architecture of gene regulation: the promise of eQTL studies. Trends Genet. 24, 408–415 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Albert FW, Bloom JS, Siegel J, Day L & Kruglyak L Genetics of trans-regulatory variation in gene expression. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This highly-powered study of eQTL in S. cerevisiae explained 70% of variation in gene expression and found that the combined effects of trans-regulatory eQTL were greater than the effects of cis-regulatory eQTL for most genes. More than 90% of trans-eQTL were clustered into 102 hotspot regions, with some hotspots contributing to expression variation for thousands of genes.
- 48.Kita R, Venkataram S, Zhou Y & Fraser HB High-resolution mapping of cis-regulatory variation in budding yeast. Proc. Natl. Acad. Sci. U. S. A 114, E10736–E10744 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ehrenreich IM et al. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature 464, 1039–1042 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metzger BPH & Wittkopp PJ Compensatory trans-regulatory alleles minimizing variation in TDH3 expression are common within Saccharomyces cerevisiae. Evolution letters 3, 448–461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mohammadi P, Castel SE, Brown AA & Lappalainen T Quantifying the regulatory effect size ofcis-acting genetic variation using allelic fold change. Genome Research vol. 27 1872–1884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aguet F et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. bioRxiv 787903 (2019) doi: 10.1101/787903. [DOI] [PMC free article] [PubMed] [Google Scholar]; This large eQTL study examined regulatory variation across 49 tissues from 838 human donors. They identified thousand of cis- and trans-eQTL and were further able to examine the tissue-specificity of regulatory effects.
- 53.Grundberg E et al. Mapping cis-and trans-regulatory effects across multiple tissues in twins. Nat. Genet 44, 1084–1089 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wright FA et al. Heritability and genomics of gene expression in peripheral blood. Nat. Genet 46, 430–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Li YI & Pritchard JK Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 177, 1022–1034.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yvert G et al. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet 35, 57–64 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Kliebenstein D Quantitative genomics: analyzing intraspecific variation using global gene expression polymorphisms or eQTLs. Annu. Rev. Plant Biol 60, 93–114 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Lutz S, Brion C, Kliebhan M & Albert FW DNA variants affecting the expression of numerous genes in trans have diverse mechanisms of action and evolutionary histories. PLoS Genet. 15, e1008375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies the specific nucleotide changes responsible for three eQTL impacting expression of a large number of genes and demonstrates the distinct mechanisms by which they have their effects.
- 59.Lappalainen T et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lemos B et al. Evolution of genomic expression. Evolutionary genomics and proteomics. Sunderland (MA): Sinauer Associates; 81–118 (2008). [Google Scholar]
- 61.Carninci P et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet 38, 626–635 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Juven-Gershon T & Kadonaga JT Regulation of gene expression via the core promoter and the basal transcriptional machinery. Dev. Biol 339, 225–229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patwardhan RP et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol 27, 1173–1175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes a method for high throughput analysis of all possible point mutations in regulatory elements using massively parallel DNA synthesis and sequencing.
- 64.Weingarten-Gabbay S et al. Systematic interrogation of human promoters. Genome Res. 29, 171–183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lubliner S et al. Core promoter sequence in yeast is a major determinant of expression level. Genome Res. 25, 1008–1017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents the first comprehensive analysis of yeast core promoters, finding that core promoter activity is highly correlated with total promoter activity and that sequence variation can tune expression in predictable ways.
- 66.Villar D et al. Enhancer evolution across 20 mammalian species. Cell 160, 554–566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liang H, Lin Y-S & Li W-H Fast evolution of core promoters in primate genomes. Mol. Biol. Evol 25, 1239–1244 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Young RS et al. The frequent evolutionary birth and death of functional promoters in mouse and human. Genome Res. 25, 1546–1557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tirosh I, Weinberger A, Carmi M & Barkai N A genetic signature of interspecies variations in gene expression. Nat. Genet 38, 830–834 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Landry CR, Lemos B, Rifkin SA, Dickinson WJ & Hartl DL Genetic properties influencing the evolvability of gene expression. Science 317, 118–121 (2007). [DOI] [PubMed] [Google Scholar]
- 71.Hornung G, Oren M & Barkai N Nucleosome organization affects the sensitivity of gene expression to promoter mutations. Mol. Cell 46, 362–368 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study examines the sensitivity of yeast promoters to mutation and finds a relationship between nucleosome organization and sensitivity to mutation.
- 72.Tirosh I, Barkai N & Verstrepen KJ Promoter architecture and the evolvability of gene expression. J. Biol 8, 95 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andersson R & Sandelin A Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet 21, 71–87 (2020). [DOI] [PubMed] [Google Scholar]
- 74.Wittkopp PJ & Kalay G Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet 13, 59–69 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Long HK, Prescott SL & Wysocka J Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wray GA The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet 8, 206–216 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Paaby AB & Rockman MV The many faces of pleiotropy. Trends Genet. 29, 66–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wray GA et al. The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol 20, 1377–1419 (2003). [DOI] [PubMed] [Google Scholar]
- 79.Hahn S & Young ET Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189, 705–736 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spitz F & Furlong EEM Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626 (2012). [DOI] [PubMed] [Google Scholar]
- 81.de Boer CG et al. Deciphering eukaryotic gene-regulatory logic with 100 million random promoters. Nat. Biotechnol 38, 56–65 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rockman MV & Wray GA Abundant raw material for cis-regulatory evolution in humans. Mol. Biol. Evol 19, 1991–2004 (2002). [DOI] [PubMed] [Google Scholar]
- 83.Sharon E et al. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol 30, 521–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Swanson CI, Schwimmer DB & Barolo S Rapid evolutionary rewiring of a structurally constrained eye enhancer. Curr. Biol 21, 1186–1196 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Patwardhan RP et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol 30, 265–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kwasnieski JC, Mogno I, Myers CA, Corbo JC & Cohen BA Complex effects of nucleotide variants in a mammalian cis-regulatory element. Proc. Natl. Acad. Sci. U. S. A 109, 19498–19503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Melnikov A et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol 30, 271–277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Metzger BPH, Yuan DC, Gruber JD, Duveau F & Wittkopp PJ Selection on noise constrains variation in a eukaryotic promoter. Nature 521, 344–347 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Glenwinkel L, Wu D, Minevich G & Hobert O TargetOrtho: a phylogenetic footprinting tool to identify transcription factor targets. Genetics 197, 61–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burgess D & Freeling M The most deeply conserved noncoding sequences in plants serve similar functions to those in vertebrates despite large differences in evolutionary rates. Plant Cell 26, 946–961 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cooper SJ, Trinklein ND, Anton ED, Nguyen L & Myers RM Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res. 16, 1–10 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Z & Gerstein M Of mice and men: phylogenetic footprinting aids the discovery of regulatory elements. J. Biol 2, 11 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Claussnitzer M et al. Leveraging cross-species transcription factor binding site patterns: from diabetes risk loci to disease mechanisms. Cell 156, 343–358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Corradin O & Scacheri PC Enhancer variants: evaluating functions in common disease. Genome Med. 6, 85 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lewinsky RH et al. T-13910 DNA variant associated with lactase persistence interacts with Oct-1 and stimulates lactase promoter activity in vitro. Hum. Mol. Genet 14, 3945–3953 (2005). [DOI] [PubMed] [Google Scholar]
- 96.Chang J et al. The Molecular Mechanism of a Cis-Regulatory Adaptation in Yeast. PLoS Genet. 9, e1003813 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arnoult L et al. Emergence and diversification of fly pigmentation through evolution of a gene regulatory module. Science 339, 1423–1426 (2013). [DOI] [PubMed] [Google Scholar]
- 98.Stern DL & Orgogozo V The loci of evolution: how predictable is genetic evolution? Evolution 62, 2155–2177 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rebeiz M & Williams TM Using Drosophila pigmentation traits to study the mechanisms of cis-regulatory evolution. Curr Opin Insect Sci 19, 1–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Klein JC, Keith A, Agarwal V, Durham T & Shendure J Functional characterization of enhancer evolution in the primate lineage. Genome Biol. 19, 99 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lickwar CR, Mueller F, Hanlon SE, McNally JG & Lieb JD Genome-wide protein-DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature 484, 251–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klemm SL, Shipony Z & Greenleaf WJ Chromatin accessibility and the regulatory epigenome. Nature Reviews Genetics vol. 20 207–220 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Raveh-Sadka T et al. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. (2012) doi: 10.1038/ng.2305. [DOI] [PubMed] [Google Scholar]
- 104.Tirosh I & Barkai N Two strategies for gene regulation by promoter nucleosomes. Genome Res. 18, 1084–1091 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peng P-C et al. The Role of Chromatin Accessibility in cis-Regulatory Evolution. Genome Biol. Evol 11, 1813–1828 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Field Y et al. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nat. Genet 41, 438–445 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chapal M, Mintzer S, Brodsky S, Carmi M & Barkai N Resolving noise-control conflict by gene duplication. PLoS Biol. 17, e3000289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tirosh I, Sigal N & Barkai N Divergence of nucleosome positioning between two closely related yeast species: genetic basis and functional consequences. Mol. Syst. Biol 6, 365 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tsankov AM, Thompson DA, Socha A, Regev A & Rando OJ The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol. 8, e1000414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schaefke B, Sun W, Li Y-S, Fang L & Chen W The evolution of posttranscriptional regulation. Wiley Interdiscip. Rev. RNA e1485 (2018). [DOI] [PubMed] [Google Scholar]
- 111.McManus CJ, Coolon JD, Eipper-Mains J, Wittkopp PJ & Graveley BR Evolution of splicing regulatory networks in Drosophila. Genome Res. 24, 786–796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang X et al. Cis-regulated alternative splicing divergence and its potential contribution to environmental responses in Arabidopsis. Plant J. 97, 555–570 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Gao Q, Sun W, Ballegeer M, Libert C & Chen W Predominant contribution of cis-regulatory divergence in the evolution of mouse alternative splicing. Mol. Syst. Biol 11, 816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.GTEx Consortium et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xiao M-S et al. Global analysis of regulatory divergence in the evolution of mouse alternative polyadenylation. Mol. Syst. Biol 12, 890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pai AA et al. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 8, e1003000 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yuan Y & Weidhaas JB Functional microRNA binding site variants. Mol. Oncol 13, 4–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McManus CJ, May GE, Spealman P & Shteyman A Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res. 24, 422–430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Battle A et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 24, 14–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yao C et al. Dynamic Role of trans Regulation of Gene Expression in Relation to Complex Traits. Am. J. Hum. Genet 100, 571–580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brynedal B et al. Large-Scale trans-eQTLs Affect Hundreds of Transcripts and Mediate Patterns of Transcriptional Co-regulation. Am. J. Hum. Genet 100, 581–591 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fairfax BP et al. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet 44, 502–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Small KS et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat. Genet 43, 561–564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sudarsanam P & Cohen BA Single nucleotide variants in transcription factors associate more tightly with phenotype than with gene expression. PLoS Genet. 10, e1004325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ronald J, Brem RB, Whittle J & Kruglyak L Local regulatory variation in Saccharomyces cerevisiae. PLoS Genet. 1, e25 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Alexander RP, Fang G, Rozowsky J, Snyder M & Gerstein MB Annotating non-coding regions of the genome. Nat. Rev. Genet 11, 559–571 (2010). [DOI] [PubMed] [Google Scholar]
- 127.Doniger SW et al. A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet. 4, e1000183 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Carroll SB Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134, 25–36 (2008). [DOI] [PubMed] [Google Scholar]
- 129.Kimura M & Ohta T On some principles governing molecular evolution. Proc. Natl. Acad. Sci. U. S. A 71, 2848–2852 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sabarís G, Laiker I, Preger-Ben Noon E & Frankel N Actors with Multiple Roles: Pleiotropic Enhancers and the Paradigm of Enhancer Modularity. Trends Genet. 35, 423–433 (2019). [DOI] [PubMed] [Google Scholar]
- 131.Lynch VJ & Wagner GP Resurrecting the role of transcription factor change in developmental evolution. Evolution 62, 2131–2154 (2008). [DOI] [PubMed] [Google Scholar]
- 132.Wagner GP & Lynch VJ The gene regulatory logic of transcription factor evolution. Trends Ecol. Evol 23, 377–385 (2008). [DOI] [PubMed] [Google Scholar]
- 133.Britton CS, Sorrells TR & Johnson AD Protein-coding changes preceded cis-regulatory gains in a newly evolved transcription circuit. Science 367, 96–100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yue F et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gerstein MB et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330, 1775–1787 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.modENCODE Consortium et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kemmeren P et al. Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell 157, 740–752 (2014). [DOI] [PubMed] [Google Scholar]
- 139.Cesar ASM et al. Identification of putative regulatory regions and transcription factors associated with intramuscular fat content traits. BMC Genomics 19, 499 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bryois J et al. Cis and trans effects of human genomic variants on gene expression. PLoS Genet. 10, e1004461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lee E & Bussemaker HJ Identifying the genetic determinants of transcription factor activity. Mol. Syst. Biol 6, 412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lambert SA et al. The Human Transcription Factors. Cell 175, 598–599 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Gerke J, Lorenz K & Cohen B Genetic interactions between transcription factors cause natural variation in yeast. Science 323, 498–501 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baker CR, Tuch BB & Johnson AD Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc. Natl. Acad. Sci. U. S. A 108, 7493–7498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sayou C et al. A promiscuous intermediate underlies the evolution of LEAFY DNA binding specificity. Science 343, 645–648 (2014). [DOI] [PubMed] [Google Scholar]
- 146.Galant R & Carroll SB Evolution of a transcriptional repression domain in an insect Hox protein. Nature 415, 910–913 (2002). [DOI] [PubMed] [Google Scholar]
- 147.Ronshaugen M, McGinnis N & McGinnis W Hox protein mutation and macroevolution of the insect body plan. Nature 415, 914–917 (2002). [DOI] [PubMed] [Google Scholar]
- 148.Choi JK & Kim YJ Epigenetic regulation and the variability of gene expression. Nat. Genet 40, 141–147 (2008). [DOI] [PubMed] [Google Scholar]
- 149.Lee S-I, Pe’er D, Dudley AM, Church GM & Koller D Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification. Proc. Natl. Acad. Sci. U. S. A 103, 14062–14067 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fazlollahi M, Muroff I, Lee E, Causton HC & Bussemaker HJ Identifying genetic modulators of the connectivity between transcription factors and their transcriptional targets. Proc. Natl. Acad. Sci. U. S. A 113, E1835–E1843 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Brown KM, Landry CR, Hartl DL & Cavalieri D Cascading transcriptional effects of a naturally occurring frameshift mutation in Saccharomyces cerevisiae. Mol. Ecol 17, 2985–2997 (2008). [DOI] [PubMed] [Google Scholar]
- 152.Tian J et al. Identification of the Bile Acid Transporter Slco1a6 as a Candidate Gene That Broadly Affects Gene Expression in Mouse Pancreatic Islets. Genetics 201, 1253–1262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Boyle EA, Li YI & Pritchard JK An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Metzger BPH et al. Contrasting Frequencies and Effects of cis- and trans-Regulatory Mutations Affecting Gene Expression. Mol. Biol. Evol 33, 1131–1146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study measured the effects of hundreds of cis-regulatory mutations and thousands of trans-regulatory mutations on expression level and expression noise of the TDH3 gene in S. cerevisiae. cis-regulatory mutations tended to have larger effects on TDH3 expression than trans-regulatory mutations.
- 155.Gruber JD, Vogel K, Kalay G & Wittkopp PJ Contrasting properties of gene-specific regulatory, coding, and copy number mutations in Saccharomyces cerevisiae: frequency, effects, and dominance. PLoS Genet. 8, e1002497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Katju V & Bergthorsson U Old Trade, New Tricks: Insights into the Spontaneous Mutation Process from the Partnering of Classical Mutation Accumulation Experiments with High-Throughput Genomic Approaches. Genome Biol. Evol 11, 136–165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Halligan DL & Keightley PD Spontaneous Mutation Accumulation Studies in Evolutionary Genetics. Annu. Rev. Ecol. Evol. Syst 40, 151–172 (2009). [Google Scholar]
- 158.Rifkin SA, Houle D, Kim J & White KP A mutation accumulation assay reveals a broad capacity for rapid evolution of gene expression. Nature 438, 220–223 (2005). [DOI] [PubMed] [Google Scholar]; This study measured genome-wide transcript abundance among 12 independent Drosophila melanogaster mutation accumulation lines that had been evolving for over 200 generations. They used these data to estimate mutational variance in gene expression and contrasted these variances with neutral expectations of expression divergence between species, finding that gene expression does not appear to be evolving neutrally.
- 159.McGuigan K et al. The Nature and Extent of Mutational Pleiotropy in Gene Expression of MaleDrosophila serrata. Genetics vol. 196 911–921 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huang W et al. Spontaneous mutations and the origin and maintenance of quantitative genetic variation. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Denver DR et al. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat. Genet 37, 544–548 (2005). [DOI] [PubMed] [Google Scholar]
- 162.Zalts H & Yanai I Developmental constraints shape the evolution of the nematode mid-developmental transition. Nature Ecology & Evolution 1, 0113 (2017). [DOI] [PubMed] [Google Scholar]
- 163.Hodgins-Davis A, Duveau F, Walker EA & Wittkopp PJ Empirical measures of mutational effects define neutral models of regulatory evolution in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A 116, 21085–21093 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lemos B, Araripe LO, Fontanillas P & Hartl DL Dominance and the evolutionary accumulation of cis- and trans-effects on gene expression. Proc. Natl. Acad. Sci. U. S. A 105, 14471–14476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rice DP & Townsend JP A test for selection employing quantitative trait locus and mutation accumulation data. Genetics 190, 1533–1545 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes a framework for using empirical data describing effects of new mutations on quantitative traits to determine neutral models of trait evolution and subsequently infer whether and to what extent selection has shaped existing trait variation.
- 166.Duveau F, Yuan DC, Metzger BPH, Hodgins-Davis A & Wttkopp PJ Effects of mutation and selection on plasticity of a promoter activity in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A 114, E11218–E11227 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gilad Y, Oshlack A & Rifkin SA Natural selection on gene expression. Trends Genet. 22, 456–461 (2006). [DOI] [PubMed] [Google Scholar]
- 168.Smith JD, McManus KF & Fraser HB A novel test for selection on cis-regulatory elements reveals positive and negative selection acting on mammalian transcriptional enhancers. Mol. Biol. Evol 30, 2509–2518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kircher M et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sharon E et al. Functional Genetic Variants Revealed by Massively Parallel Precise Genome Editing. Cell 175, 544–557.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang B & Wittkopp PJ Structure of the Transcriptional Regulatory Network Correlates with Regulatory Divergence in Drosophila. Mol. Biol. Evol 34, 1352–1362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kopp A & McIntyre LM Transcriptional network structure has little effect on the rate of regulatory evolution in yeast. Mol. Biol. Evol 29, 1899–1905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jackson CA, Castro DM, Saldi G-A, Bonneau R & Gresham D Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. Elife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rest JS et al. Nonlinear fitness consequences of variation in expression level of a eukaryotic gene. Mol. Biol. Evol 30, 448–456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Duveau F, Toubiana W & Wittkopp PJ Fitness Effects of Cis-Regulatory Variants in the Saccharomyces cerevisiae TDH3 Promoter. Mol. Biol. Evol 34, 2908–2912 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Keren L et al. Massively Parallel Interrogation of the Effects of Gene Expression Levels on Fitness. Cell 166, 1282–1294.e18 (2016). [DOI] [PubMed] [Google Scholar]
- 177.Cowles CR, Hirschhorn JN, Altshuler D & Lander ES Detection of regulatory variation in mouse genes. Nat. Genet 32, 432–437 (2002). [DOI] [PubMed] [Google Scholar]
- 178.Fraser HB Improving Estimates of Compensatory cis-trans Regulatory Divergence. Trends Genet. 35, 3–5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.LeProust EM et al. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Res. 38, 2522–2540 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Inoue F & Ahituv N Decoding enhancers using massively parallel reporter assays. Genomics 106, 159–164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.White MA Understanding how cis-regulatory function is encoded in DNA sequence using massively parallel reporter assays and designed sequences. Genomics 106, 165–170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fuqua T et al. Dense encoding of developmental regulatory information may constrain evolvability. bioRxiv 2020.04.17.046052 (2020) doi: 10.1101/2020.04.17.046052. [DOI] [Google Scholar]
- 183.Sung W, Ackerman MS, Miller SF, Doak TG & Lynch M Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl. Acad. Sci. U. S. A 109, 18488–18492 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Duveau F et al. Mapping small effect mutations in Saccharomyces cerevisiae: impacts of experimental design and mutational properties. G3 4, 1205–1216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Halligan DL, Peters AD & Keightley PD Estimating numbers of EMS-induced mutations affecting life history traits in Caenorhabditis elegans in crosses between inbred sublines. Genet. Res 82, 191–205 (2003). [DOI] [PubMed] [Google Scholar]
- 186.Greene EA et al. Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics 164, 731–740 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]