

Abstract
Use of non-steroidal anti-inflammatory drugs (NSAIDs) is associated with reduced risk of colorectal cancer (CRC). However, the mechanism by which NSAIDs suppress colorectal tumorigenesis remains unclear. We previously showed that NSAIDs selectively kill emerging tumor cells via death receptor (DR) signaling and a synthetic lethal interaction mediated by the proapoptotic Bcl-2 family protein BID. In this study, we found NSAIDs induce endoplasmic reticulum (ER) stress to activate DR signaling and BID in tumor suppression. Importantly, our results unveiled an ER stress- and BID-dependent immunogenic effect of NSAIDs, which may be critical for tumor suppression. NSAID treatment induced hallmarks of immunogenic cell death (ICD) in CRC cells and colonic epithelial cells upon loss of APC tumor suppressor, and elevated tumor-infiltrating lymphocytes (TILs) in the polyps of APCMin/+ mice. ER stress inhibition or BID deletion abrogated the antitumor and immunogenic effects of NSAIDs. Furthermore, increased ER stress and TILs were detected in human advanced adenomas from NSAID-treated patients. Together, our results suggest that NSAIDs induce ER stress- and BID-mediated ICD to restore immunosurveillance and suppress colorectal tumor formation.
Keywords: Colorectal cancer, NSAIDs, Chemoprevention, DR5, ER Stress, Immunogenic cell death, Tumor infiltrating lymphocytes
Introduction
Colorectal cancer (CRC) remains the second leading cause of cancer-related death in the United States despite recent successes in early detection and therapeutic intervention [1]. Prevention of CRC is a promising approach for reducing the mortality and morbidity of this disease. The widely used non-steroidal anti-inflammatory drugs (NSAIDs) have been shown to reduce CRC incidence in epidemiological studies, clinical trials, and animal model studies [2, 3]. It is well known that a key effect of NSAIDs is inhibition of cyclooxygenases [2, 3]. However, the antitumor mechanisms of NSAIDs and other chemopreventive agents are not well understood.
Colorectal tumorigenesis often initiates with the formation of adenomatous precursor lesions. This process is driven by gatekeeper alterations in the Adenomatous Polyposis Coli (APC) tumor suppressor pathway, leading to aberrant Wnt signaling, accumulation and nuclear translocation of β-catenin, and subsequent activation of c-Myc and other oncogenes [4]. A critical activity of NSAIDs in chemoprevention is selective killing of intestinal stem cells acquiring the gatekeeper alterations [5, 6]. Our previous studies showed that NSAIDs activate death receptor (DR) signaling to trigger a synthetic lethal interaction in intestinal epithelial cells with APC loss and c-Myc accumulation [7]. This selective killing effect of NSAIDs is mediated by apoptosis through the mitochondrial pathway [8–10]. Blocking this effect by deleting the proapoptotic Bcl-2 family member BID abrogates the antitumor activity of NSAIDs such as sulindac in APCMin/+ mice, a commonly used genetic model of intestinal tumorigenesis and chemoprevention [7]. However, the upstream events leading to DR signaling and BID-mediated cell death in response NSAIDs remain unclear.
Previous work on CRC prevention has mostly focused on tumor cell intrinsic alterations. Recent studies suggest antitumor immunity also plays a critical role in CRC prevention [11]. Emerging tumor cells must overcome immunosurveillance to sustain the course of development by accumulating additional oncogenic alterations [12, 13]. Chemopreventive agents such as NSAIDs are known to have immune-modulatory effects [14]. However, the origin and functional role of immune responses in NSAID-mediated chemoprevention are poorly understood. It is known that different cell death pathways have diverse immunological consequences [15, 16]. For example, apoptosis mediated by endoplasmic reticulum (ER) stress promotes a robust immune response and is therefore considered as immunogenic cell death (ICD) [17]. NSAIDs can induce various cellular responses including ER stress, which has not been carefully examined in previous studies.
In this study, we further investigated the selective cell-killing effect of NSAIDs and its immunogenic consequences using cultured cells, animal models, and human adenoma specimens. Our results reveal the antitumor activity of NSAIDs is mediated by ER stress, which not only leads to death of emerging tumor cells, but also triggers an antitumor immune response that may be critical for tumor suppression.
Results
Induction of ER stress mediates apoptosis induced by sulindac sulfide in HCT116 cells
We previously showed that NSAIDs upregulate DR5 to engage the extrinsic apoptotic pathway in CRC cells [7]. Upon examining DR5 upstream pathways, we found ER stress is responsible for DR5 upregulation in response to NSAIDs. Treating HCT116 CRC cells with sulindac sulfide (SUS), an active metabolite of the NSAID sulindac, markedly induced ER stress markers in a dose- and time-dependent manner, including phosphorylation of PERK (pPERK; T981) and EIF2α (pEIF2α; S51), and increased expression of BiP (GRP78), ATF4, and CHOP (Fig. 1, A and B). It is worth noting that SUS at 30 μM, which is close to its plasma concentration range in humans [18], was sufficient to induce DR5, pPERK and pEIF2α (Fig. 1A), suggesting relevance of ER stress to its anticancer activity. The induction of the ER stress markers coincided with induction of DR5 mRNA and protein (Fig. 1A, 1B and S1A). Transmission electron microscopy (TEM) detected rough ER with much elongated membrane structures in SUS-treated HCT116 cells compared to untreated cells (Fig. 1C). This abnormal ER morphology is distinct from swollen ER (up to five times the volume) in response to other ER stress inducers as described previously [19, 20]. Similar to swollen ER, the elongated ER should also create additional ER space to accommodate increased protein folding and allow cells to cope with increased protein load [19, 20]. In addition, immunostaining and immunogold TEM revealed cytoplasmic and cell surface enrichment of BiP (Fig. S1, B and C), which has not been described in previous ER stress studies, suggesting atypical ER stress in response to SUS.
Fig. 1. ER stress mediates the killing effect of sulindac sulfide in HCT116 cells.
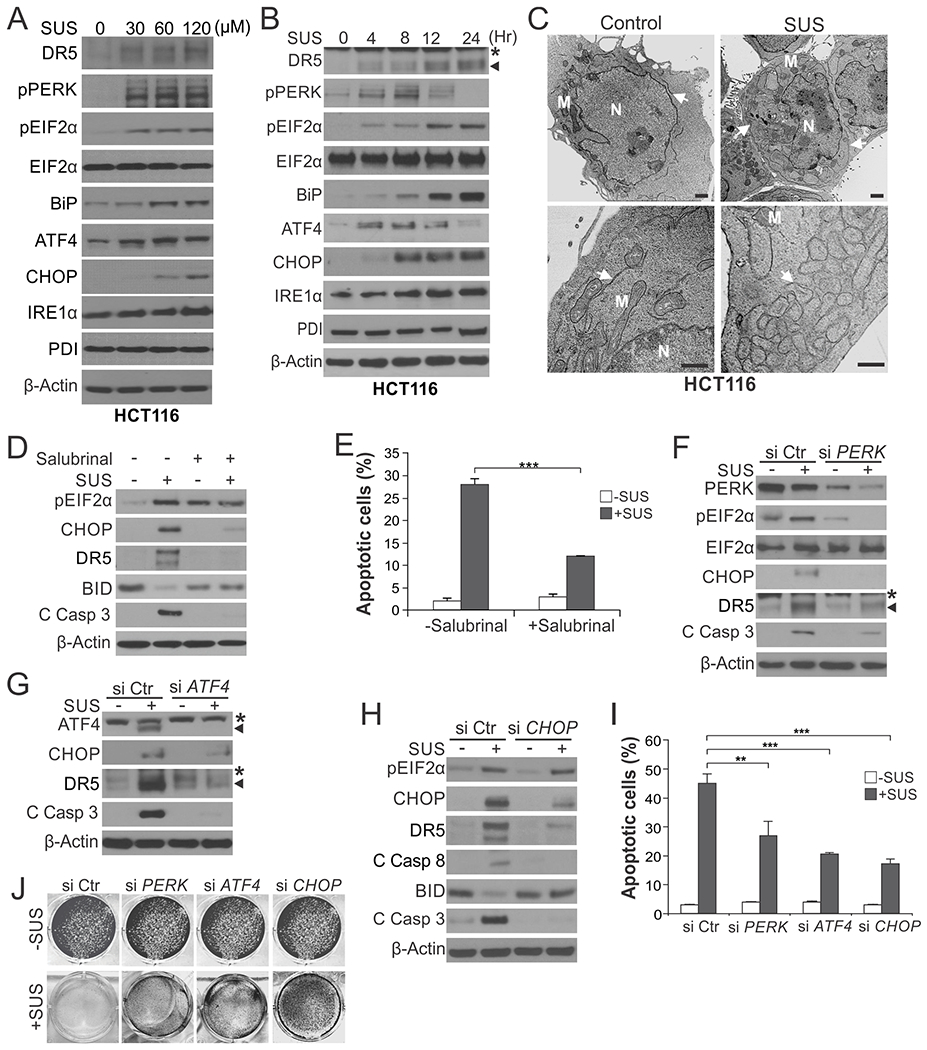
(A) Western blotting of indicated ER stress markers in HCT116 cells treated with sulindac sulfide (SUS) at indicated concentrations for 24 hr. (B) Western blotting of indicated ER stress markers in HCT116 cells treated with 120 μM SUS at indicated time points. *, non-specific bands. (C) HCT116 colon cancer cells treated with 120 μM SUS for 24 hr were analyzed by transmission electron microscopy (TEM). Arrows: endoplasmic reticulum; M: Mitochondria; N: nucleus. Scale bars: 1.0 µm. (D) Western blotting of indicated proteins in HCT116 cells treated with 120 μM SUS for 24 hr with or without pre-treatment with the ER stress inhibitor Salubrinal (1.0 μM) for 1.5 hr. C Casp 3, cleaved caspase 3. (E) Apoptosis in HCT116 cells treated with SUS for 48 hr with or without Salubrinal pre-treatment as in (D) was analyzed by counting condensed and fragmented nuclei after nuclear staining with Hoechst 33258. (F)-(J) HCT116 cells transfected with control scrambled, PERK, ATF4 or CHOP siRNA were treated with 120 μM SUS. (F)-(H) Western blotting of indicated proteins after SUS treatment for 24 hr. (I) Analysis of apoptosis after SUS treatment for 48 hr as in (E). (J) Crystal violet staining of viable cells after SUS treatment for 48 hr. Results in (E) and (I) were expressed as means + SD of three independent experiments. **P < 0.01; ***P < 0.001.
We used pharmacological and genetic approaches to investigate the functional role of ER stress in SUS-induced apoptosis. Inhibiting ER stress by salubrinal, which indirectly suppresses eIF2α by preventing its dephosphorylation [21], abrogated SUS-induced CHOP and DR5 expression, as well as caspase 3 and BID cleavage and nuclear fragmentation (Fig. 1, D and E). Knockdown of PERK, ATF4, or CHOP by small-interfering RNA (siRNA) in HCT116 cells suppressed SUS-induced DR5 expression and apoptosis and rescued cell viability (Fig. 1, F–J and S1D). In contrast, BID and DR5 knockout (KO) did not affect CHOP induction, despite blocking apoptosis and caspase activation (Fig. S1, E–G), consistent with ER stress as the key upstream event leading to DR5 induction and subsequent BID-dependent apoptosis [7].
Induction of ER stress mediates the killing activity of other NSAIDs in different CRC cells
The induction of ER stress markers was detected in HCT116 cells undergoing apoptosis induced by indomethacin (Indo) (Fig. 2A), which is an NSAID with a distinct chemical structure compared to sulindac [8]. Indomethacin-induced apoptosis and caspase 3 activation was also suppressed by ER stress inhibition by salubrinal (Fig. 2, B and C), and knockdown of PERK, ATF4, or CHOP (Fig. 2, D–F). Furthermore, other NSAIDs including diclofenac, naproxen, sodium salicylate, and celecoxib, as well as commonly used drugs acetaminophen and metformin, also induced pEIF2α and/or CHOP at a concentration that caused apoptosis in HCT116 cells (Fig. 2G). Apoptosis induced by these drugs in HCT116 cells was inhibited by DR5 KO and CHOP knockdown (Fig. 2, H and I).
Fig. 2. ER stress mediates the killing effect of indomethacin and other NSAIDs in HCT116 cells.
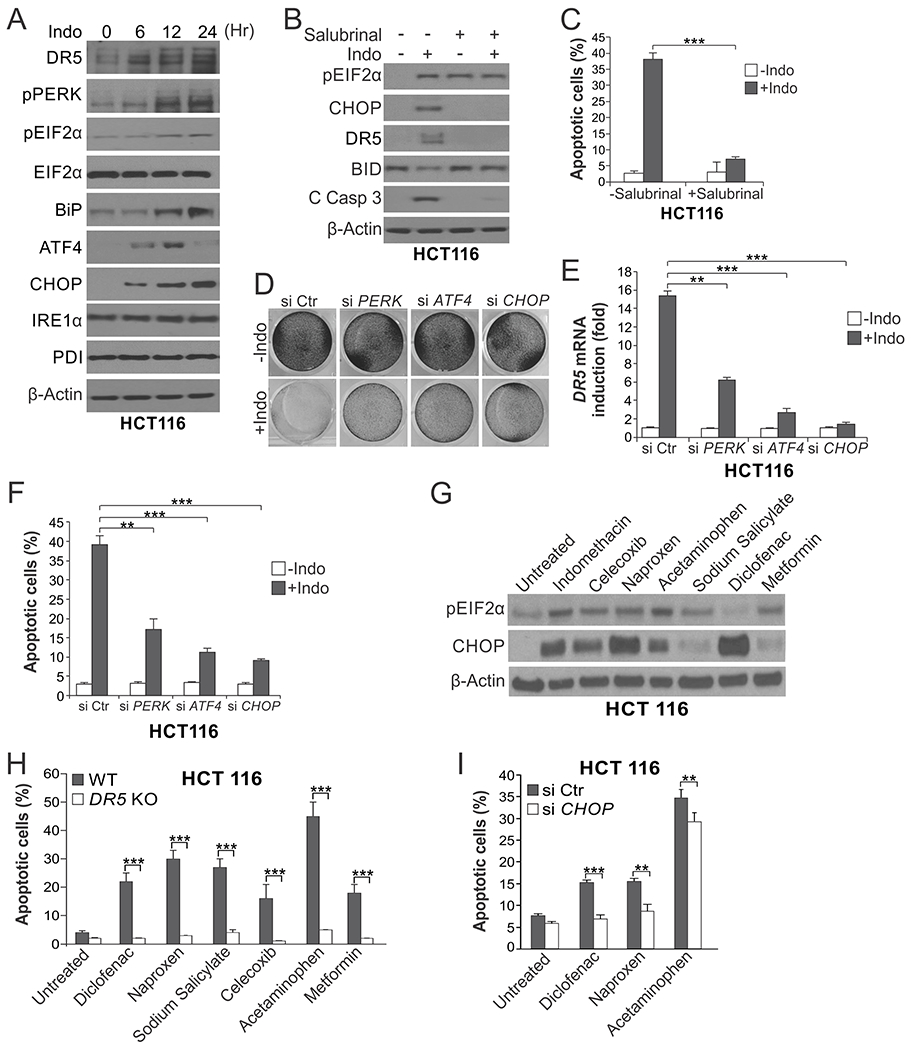
(A) Western blotting of indicated ER stress markers in HCT116 cells treated with 500 μM indomethacin (Indo) at indicated time points. (B) Western blotting of indicated proteins in HCT116 cells treated with Indo as in (A) for 24 hr with or without pre-treatment with the ER stress inhibitor Salubrinal (1.0 μM) for 1.5 hr. C Casp 3, cleaved caspase 3. (C) Apoptosis in HCT116 cells treated with Indo for 48 hr with or without Salubrinal pre-treatment as in (B) was analyzed by counting condensed and fragmented nuclei after nuclear staining with Hoechst 33258. (D)-(F) HCT116 cells transfected with control scrambled, PERK, ATF4 or CHOP siRNA were treated with 500 μM Indo. (D) Crystal violet staining of viable cells after Indo treatment for 48 hr. (E) RT-PCR analysis of DR5 expression after Indo treatment for 24 hr. (F) Analysis of apoptosis after Indo treatment for 48 hr as in (C). (G) Western blotting of pEIF2α and CHOP in HCT116 cells treated with indicated chemopreventive agents for 24 hr. Indomethacin: 500 μM; Celecoxib: 80 μM; Naproxen: 3 mM; Acetaminophen: 10 mM; Sodium Salicylate: 500 μM; Diclofenac: 600 μM; Metformin: 500 μM. (H)Apoptosis in wildtype (WT) and DR5 KO HCT116 cells treated with indicated agents as in (G) for 24 hr was analyzed as in (C). (I) Apoptosis in HCT116 cells transfected with control scrambled or CHOP siRNA and treated with indicated agents as in (G) for 24 hr was analyzed as in (C). Results in (C), (E), (F), (H) and (I) were expressed as means + SD of three independent experiments. **P < 0.01; ***P < 0.001.
We further analyzed RKO and HT29 colon cancer cells, which have different driver mutations compared to HCT116 cells [22]. Consistent with the results from HCT116 cells, SUS-induced apoptosis and caspase activation in RKO and HT29 cells was associated with the induction of DR5 and ER stress markers (Fig. S2A), and abrogated by ATF4 or CHOP knockdown and DR5 KO (Fig. S2, B–F). Indomethacin-induced apoptosis also involved the induction of DR5 and ER stress markers in RKO and HT29 cells (Fig. S2G). Together, these results suggest a broad functional role of ER stress in triggering apoptotic response to different NSAIDs in CRC cells with different genetic backgrounds.
ER stress mediates selective killing of APC-deficient colonic epithelial cells by NSAIDs
We tested if induction of ER stress is responsible for selective killing of APC-deficient intestinal epithelial cells using non-transformed NCM356 colonic epithelial cells expressing functional APC [7, 23]. While NCM356 cells are insensitive to SUS [7], knockdown of APC by siRNA triggered SUS-induced apoptosis in these cells (Fig. 3, A–C). This apoptotic response to SUS upon APC depletion was accompanied by elevated pEIF2α and induction of the ER stress effectors ATF4, CHOP and DR5 (Fig. 3, A–C). In line with the data from CRC cells, ER stress inhibition by salubrinal or knockdown of ATF4 or CHOP suppressed SUS-induced DR5 expression and apoptosis in APC-depleted NCM356 cells (Fig. 3, B–G). Importantly, pEIF2α, CHOP and DR5 induction occurred in SUS-treated control NCM356 cells without APC depletion and substantial cell death (Fig. 3, B, D and E), indicating that these events can be triggered by sulindac regardless of APC status, but lead to apoptosis only upon APC loss. These results indicate that the induction of ER stress by NSAIDs plays a key role in mediating the selective killing of colonic epithelial cells with the gatekeeper changes.
Fig. 3. ER stress mediates the killing effect of sulindac sulfide on non-transformed colonic epithelial cells with APC loss.
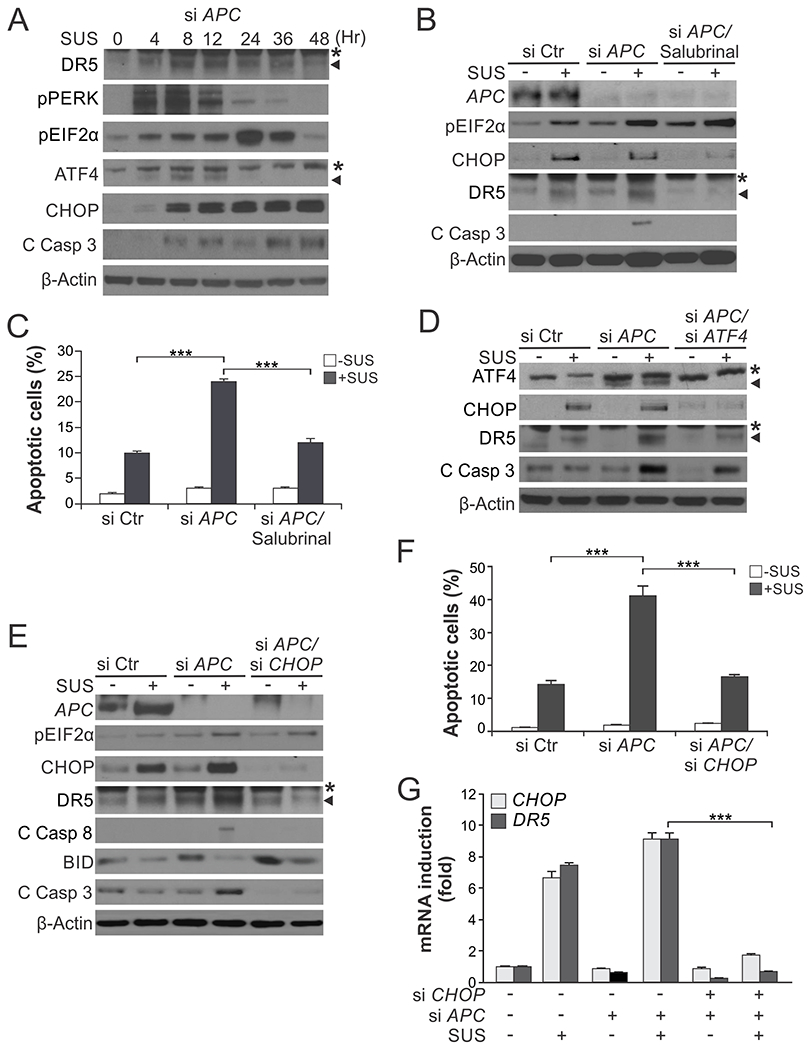
(A) Western blotting of indicated proteins in NCM356 normal colonic epithelial cells transfected with APC siRNA and then treated with 120 μM sulindac sulfide (SUS) at indicated time points. C Casp 3, cleaved caspase 3. *, non-specific bands. (B) Western blotting of indicated proteins in NCM356 cells transfected with control scrambled or APC siRNA with or without treatment with 120 μM SUS for 48 hr and pre-treatment with the ER stress inhibitor salubrinal (1.0 μM) for 1.5 hr. *, non-specific bands. (C) Apoptosis in NCM356 cells treated as in (B) was analyzed by counting condensed and fragmented nuclei after nuclear staining with Hoechst 33258. (D)-(G) NCM356 cells were transfected with indicated siRNA or combinations, and then treated with 120 μM SUS for 48 hr. (D), (E) Western blotting of indicated proteins. (F) Analysis of apoptosis as in (C). (G) RT-PCR analysis of mRNA expression of indicated genes. Results in (C), (F) and (G) were expressed as means + SD of three independent experiments. ***P < 0.001.
NSAID-induced apoptosis is immunogenic cell death (ICD)
ER stress-mediated apoptosis can trigger a robust immune response and is considered as immunogenic cell death (ICD) [17]. We determined the immunogenic consequences of NSAID-induced apoptosis by analyzing the ICD hallmarks of attracting and engaging phagocytic dendritic cells (DCs) [24]. Immunostaining and flow cytometry detected markedly enhanced cell-surface translocation of calreticulin (CRT), a pro-phagocytic signal [17], in HCT116 cells upon SUS and Indo treatment (Fig. 4A). The CRT translocation was abrogated by salubrinal, CHOP or PERK knockdown, and by blocking apoptosis with BID KO or caspase 8 knockdown (Fig. 4, A–C). Similar observation was made in RKO cells undergoing SUS-induced apoptosis (Fig. S3A). We then directly tested if dying CRC cells can be engulfed by DCs. SUS-treated HCT116 cells and DCs differentiated from healthy donor’s human monocytes were labelled with red and green fluorescence, respectively, and co-incubated. DC phagocytosis was analyzed by detecting fused cells by fluorescence microscopy and quantified by flow cytometry. We observed a significant increase in DC phagocytosis of dying HCT116 cells, which was attenuated by ATF4, CHOP knockdown or BID KO (Fig. 4D, S3B and S3C). Furthermore, increased CRT cell-surface translocation and DC phagocytosis was observed in SUS-treated NCM356 cells with APC knockdown, but not in those without APC depletion (Fig. 4, E and F, and S3D). Together, these results indicate that NSAID-induced ER stress and apoptosis in CRC cells and APC-deficient colonic epithelial cells favors DC phagocytosis, which is the first step for an adaptative antitumor immune response [12, 13].
Fig. 4. NSAID-induced apoptosis is immunogenic cell death.
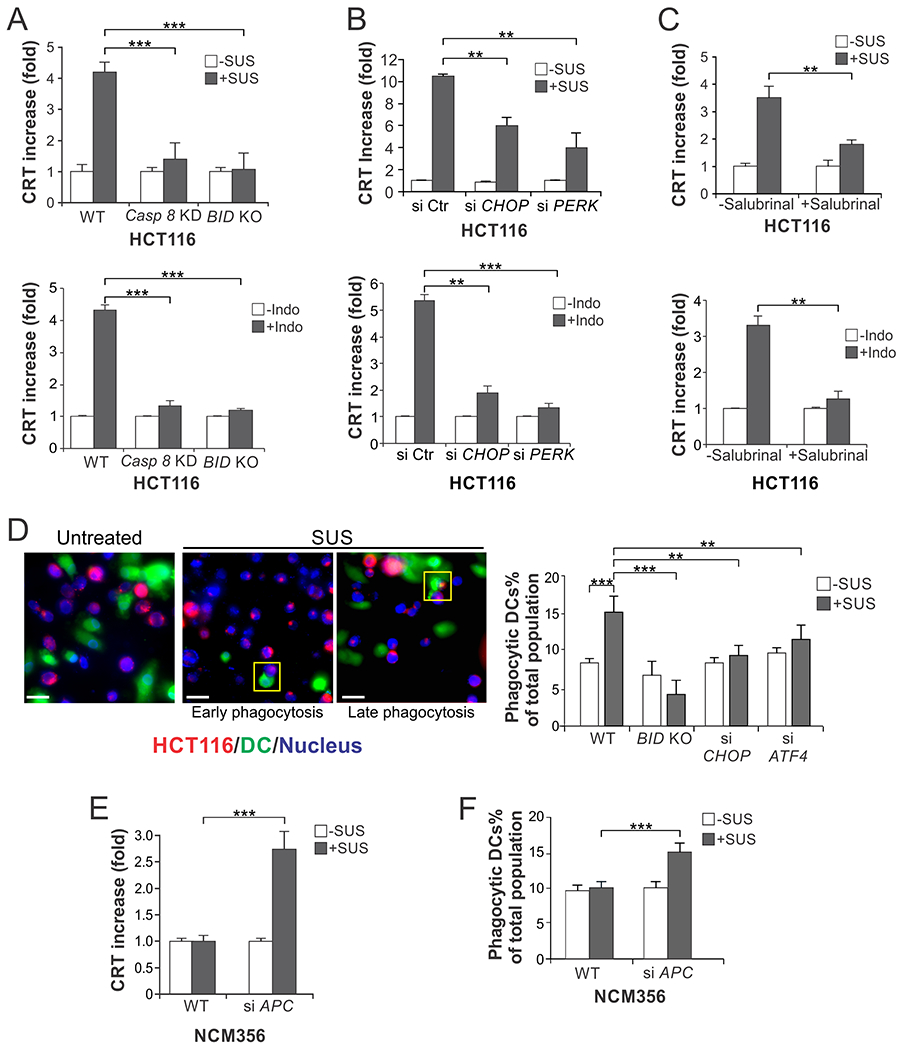
(A)-(C) HCT116 cells with inhibition of apoptosis or ER stress were treated with 120 µM sulindac sulfide (SUS) or 500 µM indomethacin (Indo) for 12 hr. CRT translocation was examined by immunostaining followed by flow cytometry. (A) Cells with stable shRNA knockdown of caspase 8 (Casp 8 KD) or BID KO. (B) Cells with transfection of control scrambled, CHOP, or PERK siRNA. (C) Cells with pretreatment with Salubrinal (1.0 μM) for 1.5 hr. (D) HCT116 cells with or without SUS treatment as in (A) and dendritic cells differentiated from healthy donors’ human monocytes were labelled with red (Far Red) and green (CFSE) fluorescence, respectively, and co-incubated at 1:1 ratio. DC phagocytosis was analyzed by detecting fused cells by fluorescence microscopy and quantified by flow cytometry. Left, representative pictures of fluorescence microscopy showing early and late DC phagocytosis in indicated boxes; scale bars: 10 µm. Right, quantification of fused HCT 116 and phagocytic DC cells by flow cytometry. (E), (F) NCM356 cells transfected with control scrambled or APC siRNA and treated with 120 μM sulindac sulfide for 12 hr were analyzed for (E) CRT translocation as in (A), and for (F) DC phagocytosis as in (D) by flow cytometry. Results were expressed as means + SD of three independent experiments. **P <0.01; ***P < 0.001
Inhibition of ER stress suppresses NSAID-mediated chemoprevention and tumor infiltration of lymphocytes in APCMin/+mice
We then used the APCMin/+ mouse model to investigate the role of ER stress and ICD in NSAID-mediated chemoprevention. To initiate intervention at an early timepoint, four-week-old APCMin/+ mice were treated with diet containing the prodrug sulindac (200 ppm) alone, or in combination with salubrinal (IP; 1 mg/kg/day) for 2 weeks. After 4 weeks when polyps became visible, the same treatment was repeated for 1 week, followed by sacrifice of mice at week 11 and analysis of polyp numbers as described [7, 25] (Fig. 5A). As expected, sulindac treatment markedly reduced the numbers of small intestinal and colonic polyps in APCMin/+ mice. Salubrinal, which had no effect by itself, significantly inhibited the activity of sulindac by ~50% in both small intestines and colon (Fig. 5, B and C). To determine if this change was due to inhibition of ER stress and apoptosis, we analyzed mice on sulindac treatment for 1 week (Fig. 5A) [7, 25]. Consistent with polyp phenotypes, salubrinal treatment abrogated sulindac-induced CHOP expression (Fig. 5D), as well as caspase 3 activation in the small intestines and colon of APCMin/+ mice (Fig. 5, E and F). These results indicate that ER stress plays a critical role in mediating the chemopreventive and apoptotic effects of sulindac in APCMin/+ mice.
Fig. 5. Inhibition of ER stress suppresses the chemopreventive and apoptotic effects of sulindac in APCMin/+ mice.
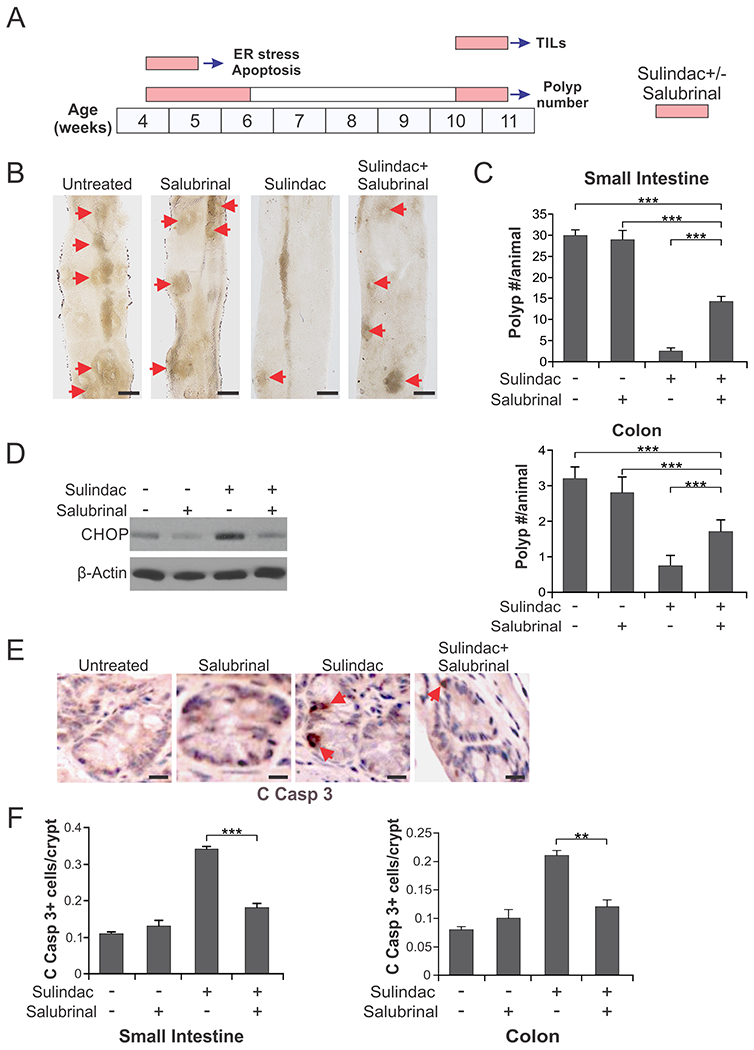
(A) Schemes of sulindac (dietary; 200 ppm) and salubrinal (IP; 1 mg/kg/day) treatments for analyzing ER stress and apoptosis, polyp numbers, and TILs in APCMin/+mice. (B), (C) Four-week-old APCMin/+mice were treated with sulindac +/− salubrinal as in (A) for analyzing polyp numbers. (B) Representative images of the small intestine of control and treated mice, with arrows indicating microscopic lesions (polyps/adenomas). Scale bars: 2 mm, n=4. (C) Mean numbers + SD of adenomas (>0.5mm in diameter) in the small intestine (upper panel) and colon (lower panel). (D)-(F) Four-week-old APCMin/+mice treated with sulindac +/− salubrinal as in (A) for analyzing ER stress and apoptosis. (D) Western blotting of CHOP in the small intestinal mucosa isolated from the treated mice. (E) Representative pictures of active caspase 3 (C Casp 3) immunostaining of small intestinal sections, with arrows indicating example cells with positive staining. Scale bars: 25 µm. (F) Quantification of active caspase signals in the sections of small intestine (left panel) and colon of the treated mice (right panel). In (C) and (F), means + SD are shown, n=3. **P <0.01; ***P < 0.001
To determine if the chemopreventive activity of sulindac is mediated by an immune response, we analyzed influx of tumor infiltrating lymphocytes (TILs), which was shown to be associated with ER stress [26, 27]. CD3 and CD8 immunostaining was performed on intestinal tissues isolated from 10-week-old APCMin/+ mice treated with sulindac +/− salubrinal for 1 week (Fig. 5A). We found sulindac treatment markedly induced CD3+ and CD8+ T lymphocyte infiltration in the small intestinal and colonic polyps of APCMin/+ mice. ER stress inhibition by salubrinal abrogated this effect (Fig. 6, A–C and S4A). Interestingly, blocking sulindac-induced apoptosis by BID KO suppressed both apoptosis and TILs to a slightly greater extent (Fig. 6, A–C and S4A). Immunostaining of other immune cell markers revealed ER stress- and BID-dependent enrichment of CD69 (active leucocytes including T cells), CD11c (DCs), and MHCII (antigen presenting cells including DCs) positive cells in the small intestinal polyps of sulindac-treated APCMin/+ mice (Fig. 6, D–G; Fig. S4, B and C), suggesting activation of T cells and DCs by sulindac treatment. These results demonstrate that sulindac-induced ER stress and cell death is required to activate an immune response, which may play a critical role in tumor suppression by sulindac in APCMin/+ mice.
Fig. 6. ER stress inhibition and BID KO suppresses NSAID-mediated immune cell infiltration.
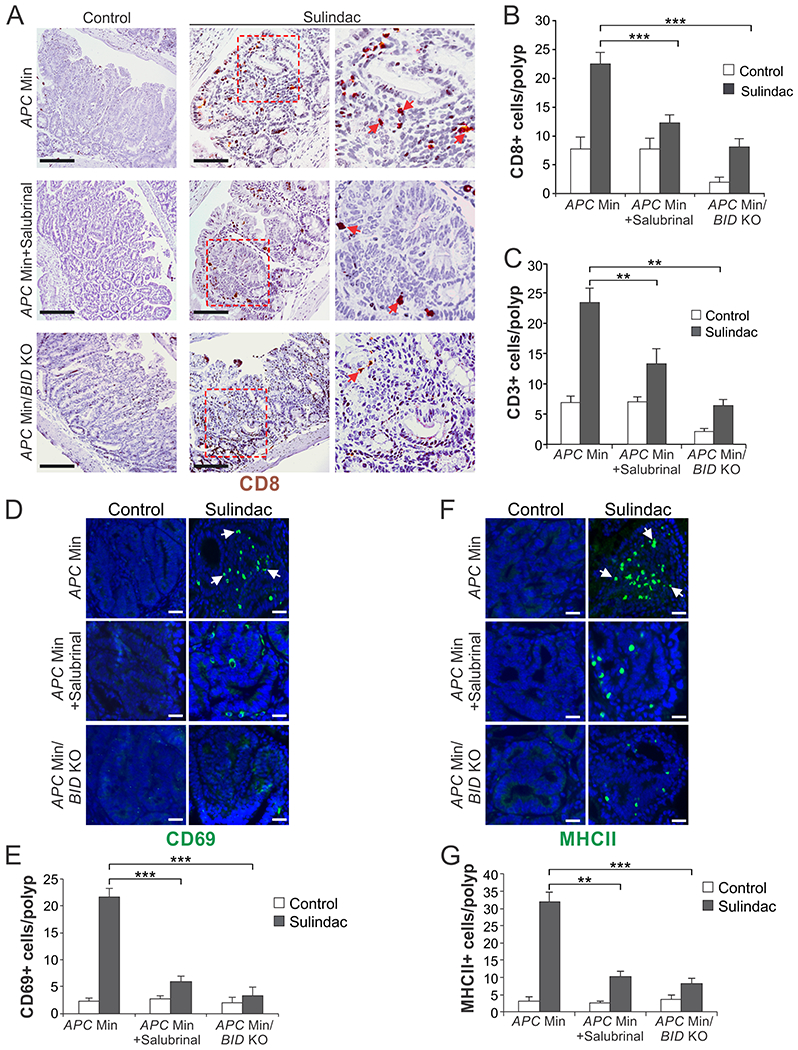
Ten-week-old APCMin/+mice with WT or KO of BID were treated with sulindac +/− salubrinal as in Fig. 5A. Sections of small intestine were analyzed by immunostaining for indicated immune cell markers. (A) Representative pictures and (B) quantification of CD8 staining. (C) Quantification of CD3 staining. (D) Representative pictures and (E) quantification of CD69 staining. (F) Representative pictures and (G) quantification of MHCII staining. In (A), (D) and (F), arrows indicate example cells with positive staining. Scale bars: 25 µm. In (B), (C), (E) and (G), means + SD are shown, n=5. **P <0.01; ***P < 0.001.
Induction of ER stress and lymphocyte infiltration in NSAID-treated human adenomas
We previously found that advanced human adenomas from NSAID-taking patients have increased apoptosis and caspase activation [7]. Coinciding with increased caspase 8 activation (Fig. 7A), a substantial increase in pEIF2α in the adenomas of NSAID-taking patients was observed compared to untreated patients (Fig. 7B). Using a counting algorithm based on positive pixels, we detected higher numbers of CD8+ T cells and slightly increased CD3+ T cells in the adenomas from NSAID-taking patients (Fig. 7C and S5). However, the differences in TILs lack statistical significance (P= 0.067 for CD8) and were not as strong as those seen in APCMin/+ mice, which may be due to limited sample size and long-term NSAID use by these patients [7]. Nonetheless, our results from human adenomas support the findings from cells and animal models and suggest that the antitumor effect of NSAIDs is mediated by antitumor immunity and restoration of immunosurveillance.
Fig. 7. Induction of apoptosis, ER stress, and lymphocyte infiltration in advanced adenomas from NSAID-treated patients.

(A), (B) Advanced adenomas from 9 NSAID-treated patients and 7 untreated patients were analyzed by immunostaining for (A) active caspase 8 and (B) pEIF2α (S51). Left panels: representative staining pictures with arrows indicating example positive signals. Scale bars: 25 µm. Right panels: quantification of positive signals. Means + SD are shown. *** P < 0.001. (C) Advanced adenomas from 16 NSAID-treated and 14 untreated patients were analyzed for CD8 by immunostaining. Left panels: representative staining pictures. Scale bars: 200 µm. Right panel: quantification of positive signals. Means + SEM are shown. (D) Proposed mechanism of NSAID-induced immunogenic cell death in APC-deficient normal epithelial cells.
Discussion
The antitumor mechanisms of NSAIDs have been extensively studied but remained obscure [28]. We show NSAID-induced ER stress is responsible for selective killing of emerging tumor cells with APC deficiency and subsequent oncogene activation by activating and enhancing death receptor signaling to trigger synthetic lethality (Fig. 7D). Activation of c-Myc and other oncogenes due to APC deficiency primes pre-malignant cells to NSAID-induced apoptosis via death receptor signaling [7, 29], which explains the selective killing effect of NSAID. ER stress is a physiological response to misfolded proteins or variety of other stimuli including oncogenes [30]. Severe or unresolved ER stress promotes apoptosis via pEIF2α and CHOP-mediated DR5 induction [31, 32]. Recent studies from other groups also suggest that ER stress-mediated apoptosis is critical for the chemopreventive effects of NSAIDs and other anticancer agents [33, 34]. However, the direct molecular targets of NSAIDs and upstream signaling events leading to ER stress are still unclear, which may be related to the inhibition of cyclooxygenases by NSAIDs [7]. The elongated ER morphology (Fig. 1C), distinct from swollen ER induced by other stimuli [19, 20], may provide a clue for understanding the mechanism of NSAID-induced ER stress.
We used sulindac sulfide at 120 μM or high concentrations to induce apoptosis in colon cancer cells. These concentrations, though higher than those measured in blood [18], is justifiable for several reasons. It was shown that sulindac is converted to the active sulfide form by aerobic gut flora on the surface of the colon and rectum [35, 36]. Systemic sulindac sulfide concentrations were significantly reduced in patients with colectomies [37]. Therefore, gut may be physiologically exposed to sulindac sulfide at higher concentrations than those measured in blood. This unusual mode of metabolism can create locally high concentrations of sulindac sulfide, which may topically suppress tumor growth [38]. It may also explain why sulindac is most effective against colorectal tumors in mice and humans. Furthermore, many previous studies used sulindac sulfide at 100 μM or high concentrations to induce apoptosis in colon cancer cells [8, 9, 39–41]. Our previous work showed the results of such in vitro studies were informative and consistent with those from animal model studies [7, 25].
Accumulating evidence suggests that effective cancer prevention requires not only a direct effect on pre-malignant cells, but also indirect effects such as antitumor immune response [11]. Recent epidemiological studies indicate that NSAIDs contribute to immunoediting by restoring immunosurveillance and reversing the immune evasive mechanisms that pre-malignant lesions utilize [14]. Such an immunogenic effect may be essential for complete and durable elimination of emerging tumor cells. However, the underlying mechanisms of restoring immunosurveillance remain to be elucidated. We found that inhibiting ER stress or blocking cell death abrogated NSAID-induced CRT cell-surface translocation, DC phagocytosis, and influx of TILs to adenomas of APCMin/+ mice, suggesting ER stress-mediated apoptosis as a trigger of immune response (Fig. 7D). Consistent with our findings, other chemopreventive agents such as metformin induce cell death to modulate the number and function of TILs and therefore provide a protective immune response [42]. Induction of ER stress was correlated with the influx of TILs in breast tumors [26]. ER stress-mediated apoptosis was shown to promote release of so called “Damage-Associated Molecular Pattern” (DAMP) molecules, such as CRT, ATP, and high mobility group box 1 (HMGB1), which can act as immunogenic signals to restore antitumor immunity [43], and lead to improved antitumor responses [15, 17]. The ICD induced by NSAIDs is similar to type II ICD described in previous studies, which specifically targets the ER to trigger apoptosis and ensuing immune response [44]. The exact mechanism of NSAID-induced ICD needs to be further examined using in vivo systems such as vaccination and immuno-deficient mouse models.
Analysis of immune responses may provide markers useful for predicting chemoprevention efficacy. Epidemiological studies suggest that regular use of aspirin is associated with reduced risk of CRCs with low-level TILs, but may not be beneficial to those with high levels of TILs without prior NSAID use [45]. Our data showed a trend of increased CD8+ T cells in adenomas from NSAID-treated patients compared to untreated patients (Fig. 7C). However, our results should be cautiously explained as they are moderate and lack statistical significance. The adenoma patients we analyzed had variations in NSAID use frequency (1-7 tablets/week), types of NSAIDs used (aspirin or related drugs, ibuprofen or related drugs, and COX-2 inhibitors). It is unlikely that they took sulindac, which was focused in this study. The lack of statistical significance is likely due to limited sample size (16 vs. 14) and/or long-term NSAID use by the patients (ranging from 5 to up to 15-20 years). Our animal studies suggest that a short-term NSAID exposure is optimal for analyzing the immune response associated with ER stress and apoptosis (Fig. 5 and 6) [25]. Consistent with this notion, a recent, short-term study showed that pre-operative treatment of CRC patients with NSAIDs such as indomethacin and celecoxib led to a robust immune response as shown by increased levels of TILs and reduced immunosuppressive regulatory T cells (Tregs) in the stroma and tumor tissues [46].
Establishing and understanding the immunogenic effects of NSAIDs provides a rationale for combining different types of agents for developing more effective and less toxic cancer prevention strategies [47]. Chemo/immuno-based combinations may represent a promising approach for CRC prevention. Chemopreventive agents such as NSAIDs are often efficacious by eliminating emerging tumor cells [25], but limited by short-term responses and potential toxicity associated with long-term drug use. For this reason, NSAIDs have not been approved for cancer prevention in general population. In contrast, immunoprevention by approaches such as vaccination is less efficacious, but much more durable and less toxic. The promise of such combinations has been illustrated in recent studies for combining CAR-T therapy with ICD-inducing DC vaccines or other inducers [48].
In summary, our results indicate that ER stress-dependent induction of ICD plays a critical role in chemoprevention by NSAIDs by establishing lasting antitumor immunity. Further studies of NSAID-induced ER stress and associated immune response may hold the key for developing improved and safer cancer prevention strategies.
Materials and methods
Cell culture and drug treatment
Human CRC cell lines, including HCT116, RKO and HT29 (purchased from American Type Culture Collection) and derivatives, were cultured in McCoy’s 5A (Gibco; ThermoFisher) modified media supplemented with 10% defined FBS (Hyclone), 100 U/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). NCM356 non-transformed colonic epithelial cells (INCELL) and derivatives were cultured in M3 Media (INCELL). DR5 KO HCT116 and RKO cells were described previously [49].
Cell lines were authenticated by genotyping and analysis of protein expression by Western blotting, and routinely checked for Mycoplasma contamination by PCR. All cell lines were maintained at 37°C and an atmosphere of 5% CO2.
For drug treatment, cells were plated in 12-well plates at 20-30% density 24 hr prior to treatment. Chemical agents, including sulindac sulfide (Merck), indomethacin (Sigma), celecoxib (Sigma), acetaminophen (Sigma), metformin (Cayman Chemical), diclofenac (sodium salt; Cayman Chemical), sodium salicylate (Sigma), and salubrinal (Apex Bio), were prepared as DMSO stock solutions, except for Naproxen sodium (Apex Bio), which was dissolved in dH2O. Stock solution of each agent was diluted to appropriate concentrations in cell culture medium, and then added to cells.
Immunofluorescence imaging
HCT116 cells seeded on sterile glass chamber slides were treated with sulindac sulfide for 24 hr. After treatment, cells were fixed with 2% paraformaldehyde in PBS for 1 hr, permeabilized with 0.1% Triton X-100 for 15 min, washed 3 times with PBS, and blocked with 5% normal goat serum for 45 min at room temperature. Immunostaining was performed by overnight incubation at 4 °C with anti-CHOP and anti-BiP antibodies (Table S1). After 3 washes with PBS, slides were incubated with proper secondary antibodies (Table S1) for 1 hr at room temperature. Upon nuclear counter staining with Hoechst 33258 for 30-45 sec, cell images were captured with a Nikon A1 confocal microscope.
Transmission electron microscopy (TEM) and immunogold TEM
Control HCT116 cells, and those treated with sulindac sulfide were analyzed for ER morphology by TEM as previously described [50]. For immunogold TEM, cells were fixed in cold 2% paraformaldehyde in 0.01M PBS (pH 7.4), rinsed in PBS, dehydrated through a graded series of ethanol, and infiltrated with and embedded in LR White resin (Electron Microscopy Sciences). Semi-thin (300 nm) sections were obtained on a Leica Ultracut 7, stained with 0.5% Toluidine Blue in 1% sodium borate, and examined under the light microscope to determine specific areas. Ultrathin sections (65 nm) were picked up on 100 mesh nickel grids, labeled with 1:100 dilution of rabbit anti-BiP (Abcam) overnight at 4°C, and then labeled with a 6 nm goat anti-rabbit colloidal gold conjugated secondary at a dilution of 1:10 at room temperature for 1 hr. After rinsing in PBS and dH2O, the sections were counterstained with 2% uranyl acetate and examined on JEOL 1011 transmission electron microscope with a side mount AMT 2k digital camera (Advanced Microscopy Techniques).
Analysis of apoptosis and cell viability
Apoptosis was analyzed by nuclear staining with Hoechst 33258 (Invitrogen) followed by counting cells with condensed and fragmented nuclei as described [7]. Each sample was analyzed in duplicate with at least 300 cells counted. Results were obtained from at least three independent experiments. Viable cells in 12-well plates were visualized by crystal violet (Sigma) staining as described [50].
siRNA transfection
Cells with 20-30% confluency seeded in 12-well plates were used for siRNA transfection, which was performed using siRNAs listed in Table S2 and Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Drug treatment was initiated at ~24 hr after transfection.
Western blotting
Western blotting was performed using antibodies listed in Table S1 as previously described [51].
Real-time reverse-transcriptase (RT) PCR
Total RNA was isolated using the Mini RNA Isolation II kit (ZYMO Research) according to the manufacturer’s protocol. One-μg of total RNA was used to generate cDNA by using the SuperScript III reverse transcriptase (Invitrogen). Real-time PCR was performed with primers listed in Table S3 and previously described conditions [52].
Analysis of CRT cell-surface translocation
After sulindac treatment for 12 hr, cells were stained with a rabbit anti-CRT antibody (Table S1), followed by Alexa Fluor488 anti-rabbit (1:1000; Table S1) as described [53]. Isotype-matched IgG antibodies were used as control. Cell-surface CRT was analyzed by flow cytometry with ACCURI C6 (BD Biosciences) with gating of propidium iodide (PI)-negative cells.
Analysis of phagocytosis by DCs
Phagocytosis by DCs was analyzed as described with modifications [17]. HCT116 and NCM356 cells with or without siRNA transfection were stained with 5 μM of Cell Trace Far Red dye (Invitrogen) or 2 nM CellVue Claret Far Red (Sigma) and seeded in 48-well plates at 1×105 cells/well (for flow cytometry) or in 4-well chamber slides (Nunc) (for fluorescence microscopy). After 24 hr, cells were treated with sulindac sulfide for 12 hr. DCs were prepared as previously described with modifications [54]. Briefly, peripheral mononuclear cells (PBMCs) were prepared from healthy human buffy coats (Central Blood Bank, Pittsburgh, PA or BioIVT) by Ficoll-Hypaque density gradient centrifugation. CD14+ cells were isolated from PBMCs using CD14 MACS (Miltenyi) following the manufacturer’s instructions. CD14+ cells were cultured at a density of 1×106/ml in AIM V medium (Gibco; ThermoFisher) supplemented with 2% FBS, 1000 IU/ml IL-4, and 1000 IU/ml GM-CSF for 6 days. The purity and maturity of DCs was checked by flow cytometry analysis of CD14, CD86, HLA-DR and CD83. DCs were stained with 2.5 μM of carboxyfluorescein succinimidyl ester (CFSE; Invitrogen), incubated with an excess of hIgG (20 μg/106 cells; Sigma) for 30 min at room temperature, and added to sulindac-treated HCT116 or NCM356 cells in 1:1 ratio, followed by co-incubation for 2 hr. Phagocytosis was analyzed by detecting fused cells with red/green double staining by flow cytometry (ACCURI C6) and confirmed by fluorescence microscopy on a Nikon Ti inverted fluorescence microscope, with a Z stack of 0.2 μm, and deconvoluted and analyzed using NIS-Elements Advanced Research software.
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Mice were housed in micro-isolator cages in a room illuminated from 07:00 to 19:00 hours (12:12-hr light-dark cycle) and fed AIN-93G diets (Dyets) with access to water and chow ad libitum. C57/BL6J BID+ (BID KO) mice [55] were crossed with APCMin/+mice (Jackson Laboratory) to generate BID KO APCMin/+ mice as described in [7]. Sulindac treatment was performed by switching from regular AIN-93G diet to that containing 200 ppm sulindac (Sigma). Salubrinal treatment was done by daily intraperitoneal injection of salubrinal at 1 mg/kg, which was freshly prepared by diluting a stock solution (10 mg/ml) in 50/50 Poly (ethylene glycol)/saline solution. After treatment, mice were sacrificed and tissues from small intestine and colon were harvested as previously described [7]. Adenoma numbers were counted under a dissecting microscope as described [7]. Mucosal scrapings from small intestine were analyzed for CHOP by western blotting, and paraffin-embedded Swiss rolls were stained by immunohistochemistry (IHC) for active caspase 3 as described [7]. Sections of small intestines and colon were also analyzed by IHC for CD3, CD8, CD69, CD11c and MHCII. A total of 10-15 polyps were analyzed for each mouse to calculate the average T cell infiltration (TIL) per polyp. All antibodies are listed in Table S1. Sample size estimation was based on previous experience [7]. All treated animals were analyzed without randomization and blinding to the group allocation.
Analysis of human colonic adenomas
Human colonic adenomas were acquired from the Pitt Biospecimen Core at University of Pittsburgh. Acquisition of the tissue samples was approved by the Institutional Review Board. Informed consent was received from all participating patients. All patients had advanced adenomas by virtue of having polyps ≥ 1 cm in size. They previously filled an NSAID Use Query Form, which included questions on frequency (from 1-3 times/month to 7 times/week), duration (from <5 years to more than 20 years), and type of NSAIDs (aspirin or aspirin-containing products, ibuprofen or ibuprofen-containing products, and COX-2 inhibitors). Subjects taking NSAIDs reported use ranging from 1-3 tablets per week to >7 tablets per week over the preceding year. The specific NSAIDs in use were not recorded. Frozen specimens from 9 NSAID-treated patients and 7 untreated patients were analyzed by immunofluorescence for active caspase 8 and phospho-eIF2α (peIF2α; S51) using antibodies listed in Table S1 as previously described [7]. Results were quantified by counting positive signals in representative fields in each slide. Paraffin blocks from 16 NSAID-treated patients and 14 untreated patients were analyzed by IHC for CD3 and CD8 at the Histology Lab of the Department of Pathology, University of Pittsburgh School Medicine. Results were quantified by scanning each slide and using the positive pixel algorithm.
Statistical Analysis
Statistical analyses were performed by using GraphPad Prism VI software. Multiple comparisons of the responses were analyzed by one-way ANOVA and Dunnett’s post hoc test, whereas those between two groups were made by an unpaired t test. All statistical tests were two-tailed. Differences were considered significant if P < 0.05.
Supplementary Material
Acknowledgements
The authors thank Dr. Michael T. Lotze and our lab members for discussion and critical reading and Ms. Dorothy Coe for technical assistance. This work is supported by U.S. National Institute of Health grants (R01CA172136, R01CA203028, R01CA217141, R01CA236271, and R01CA247231 to LZ; U19AI068021 and R01CA215481 to JY; U01CA152753 to RES.). This project used the UPMC Hillman Cancer Center shared facilities that were supported in part by award P30CA047904.
Abbreviations:
- APC
adenomatous polyposis coli
- ATF
activating transcription factor
- BID
BH3-interacting domain death agonist
- CD3
cluster of differentiation 3
- CD8
cluster of differentiation 8
- CFSE
carboxyfluorescein succinimidyl ester
- CHOP
CCAAT-enhancer-binding protein homologous protein
- COX
cyclooxygenase
- CRC
colorectal cancer
- CRT
calreticulun
- CTLA-4
cytotoxic T-lymphocyte-associated antigen 4
- DAMP
damage-associated molecular pattern
- DCs
dendritic cells
- DR
death receptor
- eIF2α
eukaryotic translation initiation factor 2α
- ER
endoplasmic reticulum
- HMGB1
high mobility group box 1
- ICD
immunogenic cell death
- IHC
immunohistochemistry
- Indo
indomethacin
- KO
knockout
- NSAIDs
non-steroidal anti-inflammatory drugs
- PBMC
peripheral blood mononuclear cell
- PD-1
program death 1
- PDI
protein disulfide isomerase
- PD-L1
programmed death ligand 1
- PERK
protein kinase R-like endoplasmic reticulum kinase
- PEG
polyethylene glycol
- PI
propidium iodide
- siRNA
small-interfering RNA
- RT PCR
reverse-transcriptase PCR
- SUS
sulindac sulfide
- TEM
transmission electron microscopy
- TILs
tumor-infiltrating lymphocytes
- Tregs
regulatory T cells
- WT
wildtype
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Drew DA, Cao Y, Chan AT. Aspirin and colorectal cancer: the promise of precision chemoprevention. Nat Rev Cancer 2016;16:173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keller JJ, Giardiello FM. Chemoprevention strategies using NSAIDs and COX-2 inhibitors. Cancer Biol Ther 2003;2:S140–9. [PubMed] [Google Scholar]
- 4.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10:789–99. [DOI] [PubMed] [Google Scholar]
- 5.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2008;457:608–11. [DOI] [PubMed] [Google Scholar]
- 6.Qiu W, Carson-Walter EB, Kuan SF, Zhang L, Yu J. PUMA suppresses intestinal tumorigenesis in mice. Cancer Res 2009;69:4999–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leibowitz B, Qiu W, Buchanan ME, Zou F, Vernon P, Moyer MP, et al. BID mediates selective killing of APC-deficient cells in intestinal tumor suppression by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A 2014;111:16520–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science 2000;290:989–92. [DOI] [PubMed] [Google Scholar]
- 9.Kohli M, Yu J, Seaman C, Bardelli A, Kinzler KW, Vogelstein B, et al. SMAC/Diablo-dependent apoptosis induced by nonsteroidal antiinflammatory drugs (NSAIDs) in colon cancer cells. Proc Natl Acad Sci U S A 2004;101:16897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bank A, Wang P, Du C, Yu J, Zhang L. SMAC mimetics sensitize nonsteroidal anti-inflammatory drug-induced apoptosis by promoting caspase-3-mediated cytochrome c release. Cancer Res 2008;68:276–84. [DOI] [PubMed] [Google Scholar]
- 11.Fletcher R, Wang YJ, Schoen RE, Finn OJ, Yu J, Zhang L. Colorectal cancer prevention: Immune modulation taking the stage. Biochimica et biophysica acta Reviews on cancer 2018;1869:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011;331:1565–70. [DOI] [PubMed] [Google Scholar]
- 13.Finn OJ. Cancer immunology. N Engl J Med 2008;358:2704–15. [DOI] [PubMed] [Google Scholar]
- 14.Marzbani E, Inatsuka C, Lu H, Disis ML. The invisible arm of immunity in common cancer chemoprevention agents. Cancer Prev Res (Phila) 2013;6:764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nature reviews Immunology 2017;17:97–111. [DOI] [PubMed] [Google Scholar]
- 16.Wang YJ, Fletcher R, Yu J, Zhang L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes & Diseases 2018;5:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A, et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med 2016;22:624–31. [DOI] [PubMed] [Google Scholar]
- 18.Berg AK, Mandrekar SJ, Ziegler KL, Carlson EC, Szabo E, Ames MM, et al. Population pharmacokinetic model for cancer chemoprevention with sulindac in healthy subjects. Journal of clinical pharmacology 2013;53:403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kratochvilova K, Moran L, Padourova S, Stejskal S, Tesarova L, Simara P, et al. The role of the endoplasmic reticulum stress in stemness, pluripotency and development. European journal of cell biology 2016;95:115–23. [DOI] [PubMed] [Google Scholar]
- 20.Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods in enzymology 2011;490:71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005;307:935–9. [DOI] [PubMed] [Google Scholar]
- 22.Tong J, Tan S, Zou F, Yu J, Zhang L. FBW7 mutations mediate resistance of colorectal cancer to targeted therapies by blocking Mcl-1 degradation. Oncogene 2017;36:787–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Ren X, Alt E, Bai X, Huang S, Xu Z, et al. Chemoprevention of colorectal cancer by targeting APC-deficient cells for apoptosis. Nature 2010;464:1058–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuler PJ, Harasymczuk M, Visus C, Deleo A, Trivedi S, Lei Y, et al. Phase I dendritic cell p53 peptide vaccine for head and neck cancer. Clin Cancer Res 2014;20:2433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu W, Wang X, Leibowitz B, Liu H, Barker N, Okada H, et al. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis. Proc Natl Acad Sci U S A 2010;107:20027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JY, Heo SH, Song IH, Park IA, Kim YA, Gong G, et al. Activation of the PERK-eIF2alpha Pathway Is Associated with Tumor-infiltrating Lymphocytes in HER2-Positive Breast Cancer. Anticancer Res 2016;36:2705–11. [PubMed] [Google Scholar]
- 27.Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buque A, Huang X, et al. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol 2015;33:86–92. [DOI] [PubMed] [Google Scholar]
- 28.Gurpinar E, Grizzle WE, Piazza GA. NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res 2014;20:1104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Engels IH, Knee DA, Nasoff M, Deveraux QL, Quon KC. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 2004;5:501–12. [DOI] [PubMed] [Google Scholar]
- 30.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011;13:184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 2004;279:45495–502. [DOI] [PubMed] [Google Scholar]
- 32.Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, et al. Cell death. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014;345:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Lee SH, Min KW, McEntee MF, Jeong JB, Li Q, et al. The involvement of endoplasmic reticulum stress in the suppression of colorectal tumorigenesis by tolfenamic acid. Cancer Prev Res (Phila) 2013;6:1337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, et al. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ 2004;11:1009–16. [DOI] [PubMed] [Google Scholar]
- 35.Strong HA, Renwick AG, George CF, Liu YF, Hill MJ. The reduction of sulphinpyrazone and sulindac by intestinal bacteria. Xenobiotica 1987;17:685–96. [DOI] [PubMed] [Google Scholar]
- 36.Lee SC, Renwick AG. Sulphoxide reduction by rat intestinal flora and by Escherichia coli in vitro. Biochem Pharmacol 1995;49:1567–76. [DOI] [PubMed] [Google Scholar]
- 37.Strong HA, Warner NJ, Renwick AG, George CF. Sulindac metabolism: the importance of an intact colon. Clin Pharmacol Ther 1985;38:387–93. [DOI] [PubMed] [Google Scholar]
- 38.Seed MP, Brown JR, Freemantle CN, Papworth JL, Colville-Nash PR, Willis D, et al. The inhibition of colon-26 adenocarcinoma development and angiogenesis by topical diclofenac in 2.5% hyaluronan. Cancer Res 1997;57:1625–9. [PubMed] [Google Scholar]
- 39.Chan TA, Morin PJ, Vogelstein B, Kinzler KW. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc Natl Acad Sci, USA 1998;95:681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y, He Q, Hillman MJ, Rong R, Sheikh MS. Sulindac sulfide-induced apoptosis involves death receptor 5 and the caspase 8-dependent pathway in human colon and prostate cancer cells. Cancer Res 2001;61:6918–24. [PubMed] [Google Scholar]
- 41.He Q, Montalbano J, Corcoran C, Jin W, Huang Y, Sheikh MS. Effect of Bax deficiency on death receptor 5 and mitochondrial pathways during endoplasmic reticulum calcium pool depletion-induced apoptosis. Oncogene 2003;22:2674–9. [DOI] [PubMed] [Google Scholar]
- 42.Pereira FV, Melo ACL, Low JS, de Castro IA, Braga TT, Almeida DC, et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018;9:25808–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 2009;28:578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rufo N, Garg AD, Agostinis P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends in cancer 2017;3:643–58. [DOI] [PubMed] [Google Scholar]
- 45.Cao Y, Nishihara R, Qian ZR, Song M, Mima K, Inamura K, et al. Regular Aspirin Use Associates With Lower Risk of Colorectal Cancers With Low Numbers of Tumor-Infiltrating Lymphocytes. Gastroenterology 2016;151:879–92 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lonnroth C, Andersson M, Arvidsson A, Nordgren S, Brevinge H, Lagerstedt K, et al. Preoperative treatment with a non-steroidal anti-inflammatory drug (NSAID) increases tumor tissue infiltration of seemingly activated immune cells in colorectal cancer. Cancer immunity 2008;8:5. [PMC free article] [PubMed] [Google Scholar]
- 47.Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol 2015;6:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019;365:162–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan X, Tong J, Wang YJ, Fletcher R, Schoen RE, Yu J, et al. BET inhibitors potentiate chemotherapy and killing of SPOP-mutant colon cancer cells via induction of DR5. Cancer Res 2019;79:1191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc Natl Acad Sci U S A 2018;115:3930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knickelbein K, Tong J, Chen D, Wang YJ, Misale S, Bardelli A, et al. Restoring PUMA induction overcomes KRAS-mediated resistance to anti-EGFR antibodies in colorectal cancer. Oncogene 2018;37:4599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang P, Yu J, Zhang L. The nuclear function of p53 is required for PUMA-mediated apoptosis induced by DNA damage. Proc Natl Acad Sci U S A 2007;104:4054–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010;29:482–91. [DOI] [PubMed] [Google Scholar]
- 54.Lopez-Albaitero A, Mailliard R, Hackman T, Andrade Filho PA, Wang X, Gooding W, et al. Maturation pathways of dendritic cells determine TAP1 and TAP2 levels and cross-presenting function. Journal of immunotherapy 2009;32:465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999;400:886–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.