

Abstract
Germline mutations of TP53, which cause the cancer predisposition disorder Li-Fraumeni syndrome (LFS), can increase mitochondrial activity as well as fatty acid β-oxidation (FAO) in mice. Increased fatty acid metabolism can promote cancer malignancy, but its specific contribution to tumorigenesis in LFS remains unclear. To investigate this, we crossed LFS mice carrying the p53 R172H knockin mutation (p53172H/H, homolog of the human TP53 R175H LFS mutation) with Myoglobin knockout (MB−/−) mice known to have decreased FAO. MB−/− p53172H/H double mutant mice also showed mildly reduced FAO in thymus, a common site of T lymphoma development in LFS mice, in association with a ~40% improvement in cancer-free survival time. RNA-Seq profiling revealed that the p53 R172H mutation promotes mitochondrial metabolism and ribosome biogenesis, both of which are suppressed by the disruption of MB. The activation of ribosomal protein S6, involved in protein translation and implicated in cancer promotion, was also inhibited in the absence of MB. To further confirm the role of FAO in lymphomagenesis, mitochondrial FAO enzyme Carnitine Palmitoyltransferase 2 (CPT2) was specifically disrupted in T cells of p53172H/H mice using a Cre-loxP-mediated strategy. The heterozygous knockout of CPT2 resulted in thymus FAO haploinsufficiency and a ~30% improvement in survival time, paralleling the anti-proliferative signaling observed with MB disruption. Thus, our current study demonstrates that moderating FAO in LFS can suppress tumorigenesis and improve cancer-free survival with potential implications for cancer prevention.
Introduction
There is accumulating evidence that both the oxidation and synthesis of fatty acids contribute to tumorigenesis (1–4). Increased fatty acid β-oxidation (FAO) has been observed in diffuse large B cell lymphoma as well as chronic lymphocytic leukemia (5,6). Glioblastoma also rely on FAO for growth and reducing fatty acid availability leads to tumor senescence (7). Increased glycolysis is a well-known metabolic feature of cancer cells and dietary intake of its substrates glucose and fructose can enhance intestinal tumor growth in mouse models (8). Even in this model, the activation of glycolysis was associated with increased expression of enzymes involved in fatty acid synthesis that may support tumor growth. High fat diet and fatty acid receptor CD36 in cancer cells appear to play a role in metastasis (9). On the other hand, the consensus that fatty acid metabolism fuels cancer growth contrasts with proposals to consider high fat ketogenic diet as supplemental therapy against cancer based, in part, on its glucose and insulin lowering effects (10). Such conflicting information suggests the need to test whether modulating fat metabolism will affect tumorigenesis in vivo, for example, in mouse models of patients at high risk of developing cancer.
We previously reported that patients as well as a mouse model of the early onset cancer disorder Li-Fraumeni syndrome (LFS) caused by germline mutations of TP53 display evidence of increased oxidative metabolism (11). Mice with knockin of the p53 R172H mutation (human TP53 R175H mutation homolog) showed increased aerobic exercise endurance with a lower metabolic respiratory exchange ratio (RER) compared to wild-type, indicating increased utilization of fatty acids (11). Other studies have suggested p53 regulation of fat metabolism, and a more recent work demonstrated that another LFS mutation can activate lipolysis, an important biochemical process for fatty acid utilization (12–14). Although the general inhibition of mitochondrial respiration was observed to delay tumorigenesis in the p53 R172H mouse model (15), delineating the contribution of the specific metabolic activities affected by the p53 mutation may permit the development of more targeted strategies for cancer prevention. Furthermore, given the many differences amongst the various studies examining fatty acid metabolism in cancer, such as cancer cell type, stage of development and genetic mutations, the effect of inhibiting fatty acid metabolism on cancer development in a LFS mouse model remains unknown. As there is no chemoprevention treatment in LFS, it is imperative to identify potential therapeutic targets, such as the FAO pathway, to improve the cancer-free survival of patients affected by LFS. We therefore investigated the feasibility of inhibiting the up-regulated FAO in the LFS mouse model. The results of our current study using two different genetic approaches suggest that the inhibition of fatty acid oxidation may represent a potential strategy for delaying cancer onset in LFS.
Materials and Methods
Mice
All mice were used in accordance with, and with the approval of, an Institutional Animal Care and Use Committee (IACUC of the NHLBI). Myoglobin knockout (MB−/−) mice (provided by R.S. Balaban, created by J. Schrader) (16) were crossed with p53 R172H knockin mutant (p53172H/H) mice (NCI Frederick Mouse Repository, Frederick, MD) (17) to generate the MB−/− p53172H/H double mutant mice. CD4-Cre transgenic mice (The Jackson Laboratory) were crossed with the p53 R172H mice to generate CD4-Cre p53172H/H mice, which were in turn crossed with mice that had CPT2 alleles flanked by loxP (CPT2fl/fl) (18) to generate CD4+ T cell-specific CPT2 knockout mice. All mice were in C57BL6 background or backcrossed into C57BL6 at least 5 generations. PCR primer sequences for genotyping the mice are provided in Supplementary Table S1.
Cells
Patient samples were obtained after informed written consent as approved by the NHLBI-NIH Internal Review Board (ClinicalTrials.gov identifier NCT00406445) and primary human myoblasts prepared as previously described (11).
Mouse body composition
Measurements of lean tissue, fat, and fluid in live mice were performed on a Minispec NMR LF50 Body Composition Analyzer (Bruker).
Metabolomic screening
Gastrocnemius skeletal muscle and thymus tissues were dissected and flash frozen in liquid nitrogen. Small metabolite/limited lipid profiling and semi-quantification in tissues were performed using GC/MS and LC/MS/MS platform by Metabolon, Inc. (Durham, NC). Lipidomic profiling and semi-quantification in thymus tissue were performed by Human Metabolome Technologies America, Inc. (Boston, MA).
Lactate measurement
Lactate levels in tail blood of mice were measured using Nova Lactate Plus meter and test strip according to the manufacture’s protocol.
RNA-Seq analysis
RNA-Seq was performed on thymus tissue from 10 wk old mice with no overt evidence of tumor. The samples were sequenced on the Illumina HiSeq 2500, and the reads were aligned to mouse reference genome mm9 using Bowtie 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed with DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/).
Antibodies and immunoblotting
Antibodies used in this study were as follows: actin mouse mAb (A3853), tubulin mouse mAb (T5168) and actinin-α mouse mAb (A7811) (Sigma-Aldrich, St. Louis, MO); cyclin D1 (DCS6) rabbit mAb (2978), acetyl (ac)-lysine rabbit pAb (9441), H3K9ac rabbit mAb (9649), H3K27ac rabbit mAb (8173), H3K4me3 rabbit mAb, phospho-AMPKα (Thr172) rabbit mAb (2535), AMPKα rabbit pAb (2532), phosphor-mTOR (Ser2481) rabbit pAb (2974), phosphor-mTOR (Ser2448) rabbit pAb (2971), mTOR rabbit pAb (2983), S6 ribosomal protein rabbit mAb (2217) and phosphor-S6 ribosomal protein (Ser235/236) rabbit pAb (4856) (Cell Signaling, Danvers, MA); p53 (DO-1) mouse mAb (sc-126), myoglobin rabbit pAb (sc-25607) and hexokinase 2 goat pAb (sc-6521) (Santa Cruz, Santa Cruz, CA); histone H3 rabbit pAb (ab1791), CPT1A mouse mAb (ab128568), CPT1B rabbit pAb (ab134988) and CPT2 rabbit pAb (ab181114) (Abcam, Cambridge, UK); MAGL rabbit pAb (ABN1000) (Temecula, CA); GMPR rabbit pAb (15683–1-AP) (Proteintech, Chicago, IL); GLUT4 rabbit pAb (3945–200) (Biovision, Milpitas, CA); Cre rabbit pAb (NB100–56134) (Novus, Centennial, CO); VDAC rabbit pAb (600–401-882) (Rockland, Limerick, PA); p21 mouse mAb (OP64) (Oncogene science, Uniondale, NY).
For immunoblotting, tissue samples were homogenized in ice-cold RIPA buffer with protease and phosphatase inhibitors (Roche Diagnostic Corp), resolved by Tris-glycine SDS PAGE, and transferred to Immobilon-P membrane (Millipore) for standard ECL development.
Cell preparation from thymus
Thymi harvested from 10 wk old mice were cleaned off adhering tissues, rinsed in PBS buffer, and placed on a nylon mesh/filter (105 μm) in a 60 mm tissue culture dish with 3–4 ml PBS at 4 °C. The nylon mesh was then folded in half over the thymus and gently disrupted using flat edged forceps until only residual connective tissue remained. The homogenized tissue was then triturated with a pipette, filtered through a cell strainer (BD falcon 40 μm), spun down, and resuspended for subsequent use.
Fatty acid oxidation (FAO) assay using 3H-palmitate
Fatty acid oxidation was measured using 3H-palmitate as previously described (19). Briefly, 2 × 106 freshly isolated thymic lymphocytes were resuspended in 1 mL of modified KHB medium (111 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 2 mM MgSO4, 1.2 mM NaH2PO4, 2.5 mM glucose, 0.5 mM carnitine, 5 mM HEPES, pH 7.4) with or without 200 μM etomoxir. A mixture of [9,10-3H] palmitate (53.7 Ci/mmol, PerkinElmer, Boston, MA) and 10% BSA in KHB buffer was added to the cell suspension and incubated at 37 °C for 2.5 h. After incubation, the cells were spun down at 300x g and assayed for protein content while the supernatant was subjected to chloroform-methanol extraction to measure the 3H2O product in the aqueous phase. The etomoxir-sensitive 3H2O generated was used to calculate the β-oxidation rate and expressed as CPM/μg protein.
FAO assay using Seahorse XF24 Analyzer
Primary human myoblasts transduced with control empty vector or human p53 R175H mutant cDNA-lentivirus, as previously described (20), were seeded in XF24 cell culture microplates (Seahorse Bioscience) at 5 × 104 cells/well. After incubating the cells in DMEM (GlutaMax-I, Invitrogen) supplemented with 20% FBS and 2 μM uridine at 37 °C with 5% CO2 overnight, it was replaced with KHB assay medium supplemented with 200 μM palmitic acid/BSA with or without 200 μM etomoxir. Whole cell oxygen consumption was measured on an Agilent Seahorse XF24 Analyzer and normalized to protein content.
Ex vivo measurement of soleus muscle mitochondrial respiration
Whole soleus skeletal muscle was embedded into islet capture microplate and assayed for oxygen consumption in DMEM (Agilent 102353–100) supplemented with 10 mM glucose and 1 mM pyruvate using the Agilent Seahorse XF24 Analyzer according to the manufacture’s protocol.
mRNA quantification by real-time RT-PCR
RNA was isolated from cultured cells using the poly(dT) magnetic bead system (Invitrogen), reverse transcribed using Superscript II (Invitrogen), and quantified using real-time RT-PCR with SYBR green fluorescence on a LightCycler Sequence Detection System (Roche). RT-PCR primer sequences are provided in Supplementary Table S1.
Results
Myoglobin deletion reverses the increased fatty acid metabolism of p53172H/H mice
During the diurnal periods of increased activity, p53172H/H mice were observed to have a lower RER compared to wild-type mice by metabolic chamber measurements, indicating increased oxidation of fatty acids in the mitochondria (11). Given the connections between fatty acid metabolism and cancer, we wondered whether inhibiting FAO activity could prevent tumorigenesis in this LFS mouse model. As a simple but global approach to reversing this fatty acid metabolic phenotype, we crossed p53172H/H mice with Myoglobin knockout (MB−/−) mice which are known to have a higher respiratory exchange ratio (RER) indicative of reduced FAO (21,22). Because fatty acid oxidation has a greater stoichiometric requirement for oxygen compared with carbohydrates, the absence of oxygen reservoir myoglobin would be expected to result in substrate switching from fatty acids to carbohydrates. Myoglobin has been reported to be expressed at elevated levels in various cancers at their earliest stages and may promote neoplastic growth (23), so the deletion of myoglobin afforded an opportunity to test whether it affects the tumor susceptibility of p53172H/H mice.
The absence of myoglobin was first confirmed in the skeletal muscle of MB−/− p53172H/H double mutant mice by immunoblotting which also revealed increased levels of glucose transporter 4 (GLUT4) and hexokinase 2 (HK2) (Fig. 1A). This suggested a compensatory increase in glycolysis due to reduced oxygen availability. Furthermore, the double mutant MB−/− p53172H/H compared with p53172H/H mice had a higher body fat composition, consistent with decreased fatty acid metabolism (Fig. 1B). Metabolomic profiling of skeletal muscle also suggested lower FAO and increased glucose utilization as reflected by lower ketone body metabolite 1,2-propanediol and higher lactate and citrate steady-state levels, respectively (Fig. 1C). In addition, multiple metabolites involved in one-carbon metabolism, known to be affected by mitochondrial function and to play a role in tumorigenesis (24,25), were altered in MB−/− p53172H/H skeletal muscle (Fig. 1C). As in skeletal muscle, lactate levels were elevated in MB−/− p53172H/H mouse blood (Fig. 1D). Supporting diminished intracellular oxygen availability in the absence of myoglobin, the pale colored MB−/− p53172H/H soleus muscle, a slow fiber known to be enriched in mitochondria with a preference for fatty acid substrates {Baskin, 2015 #3383, showed decreased ex vivo oxygen consumption rate suggesting altered FAO (Fig. 1E; Supplementary Fig. S1). These observations were consistent with the metabolomic data and prompted us to investigate further whether modulating the aberrant fatty acid oxidative metabolism in MB−/− p53172H/H mice can affect cancer development.
Figure 1.
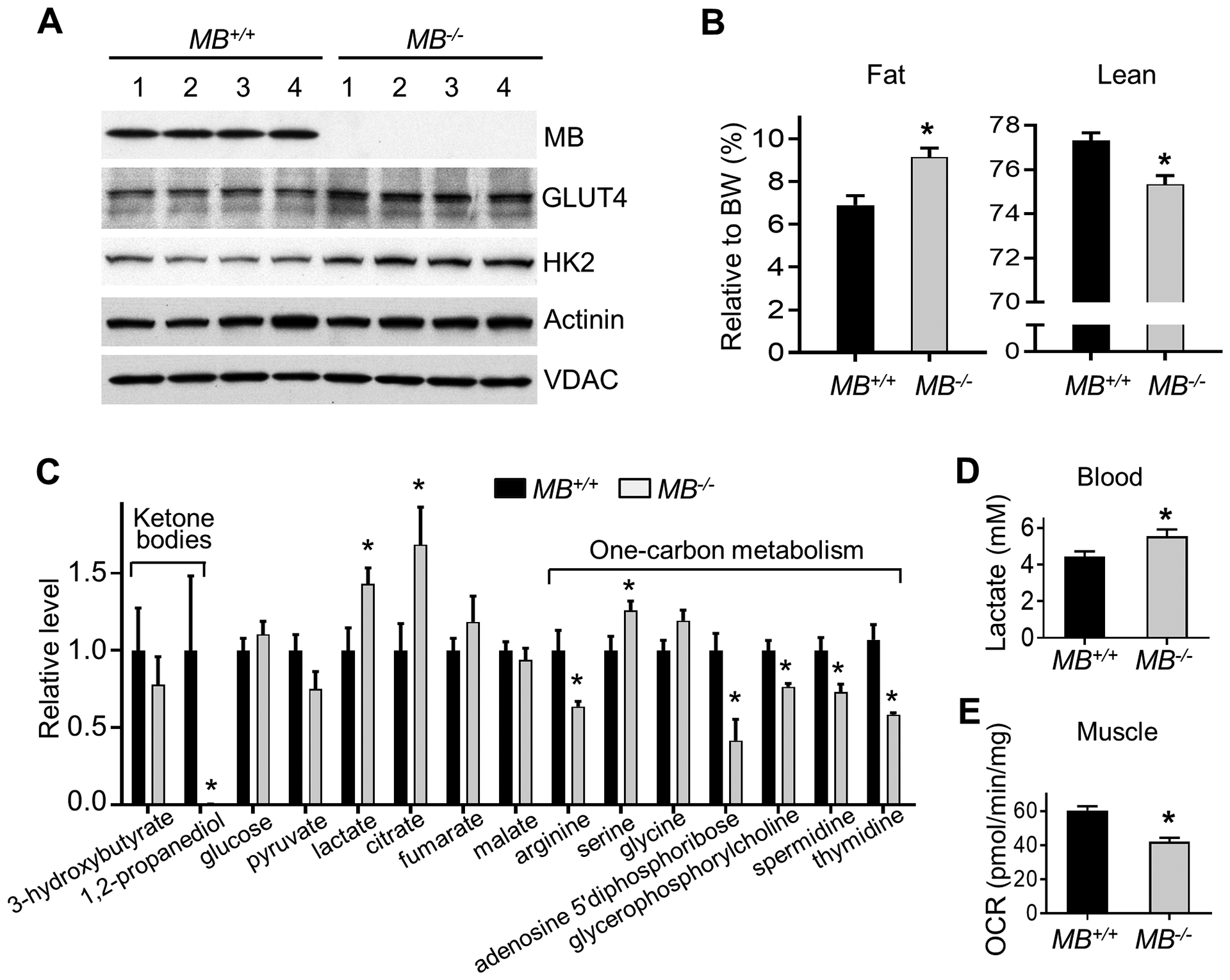
MB ablation alters metabolism in p53172H/H mice. Male mice of the indicated genotypes without evidence of tumor were used for the following experiments. A, Immunoblot of indicated proteins in skeletal muscle of 10 wk old mice. Actinin and VDAC serve as protein loading controls. B, Measurement of body composition relative to body weight (BW) in 10 wk old mice (n = 21). C, Relative levels of some metabolites related to mitochondrial activity identified by metabolomic profiling of skeletal muscle (10 wk old mice, n = 7–8). D, Lactate levels in tail blood of ~12 wk old mice (n = 19). E, Ex vivo oxygen consumption rate (OCR) of whole soleus muscle from aged-matched mice (n = 6). Statistical difference by 2-tailed unpaired t test. Values are mean ± SEM. *P < 0.05.
MB deletion inhibits FAO in thymus and delays thymic lymphoma development in p53172H/H mice
Remarkably, the disruption of MB increased the median survival time of p53172H/H mice from 131 d to 188 d (~40%) (Fig. 2A). The tumor spectrum of the double mutant MB−/− p53172H/H mice was not significantly changed, and most animals still died with thymic lymphoma, suggesting mainly a delay in tumorigenesis caused by the metabolic alteration (Fig. 2B). We performed gene expression analysis of p53+/+, p53172H/H and double mutant MB−/− p53172H/H thymi to examine globally how mutant p53 affects the tissue where the tumor develops and whether disrupting MB can rescue any of the changes. RNA-Seq profiling and KEGG pathway analysis prior to the development of overt tumor revealed increased mitochondrial activity and ribosome biogenesis in p53172H/H versus p53+/+samples while these increases were down-regulated by MB deletion (Table 1). Notably, the up-regulated one-carbon metabolism observed in p53172H/H thymus was also decreased by MB disruption, consistent with the metabolomic results (Fig. 1C; Table 1). Furthermore, the thymic lymphocytes dissociated from p53172H/H thymus showed increased mitochondrial FAO activity relative to wild-type, which could be reversed in the MB deficient state (Fig. 2C). Deleting MB in the p53 wild-type state also reduced FAO, confirming the specific role of MB in facilitating FAO independently of p53 mutation {Hendgen-Cotta, 2017 #3281}.
Figure 2.
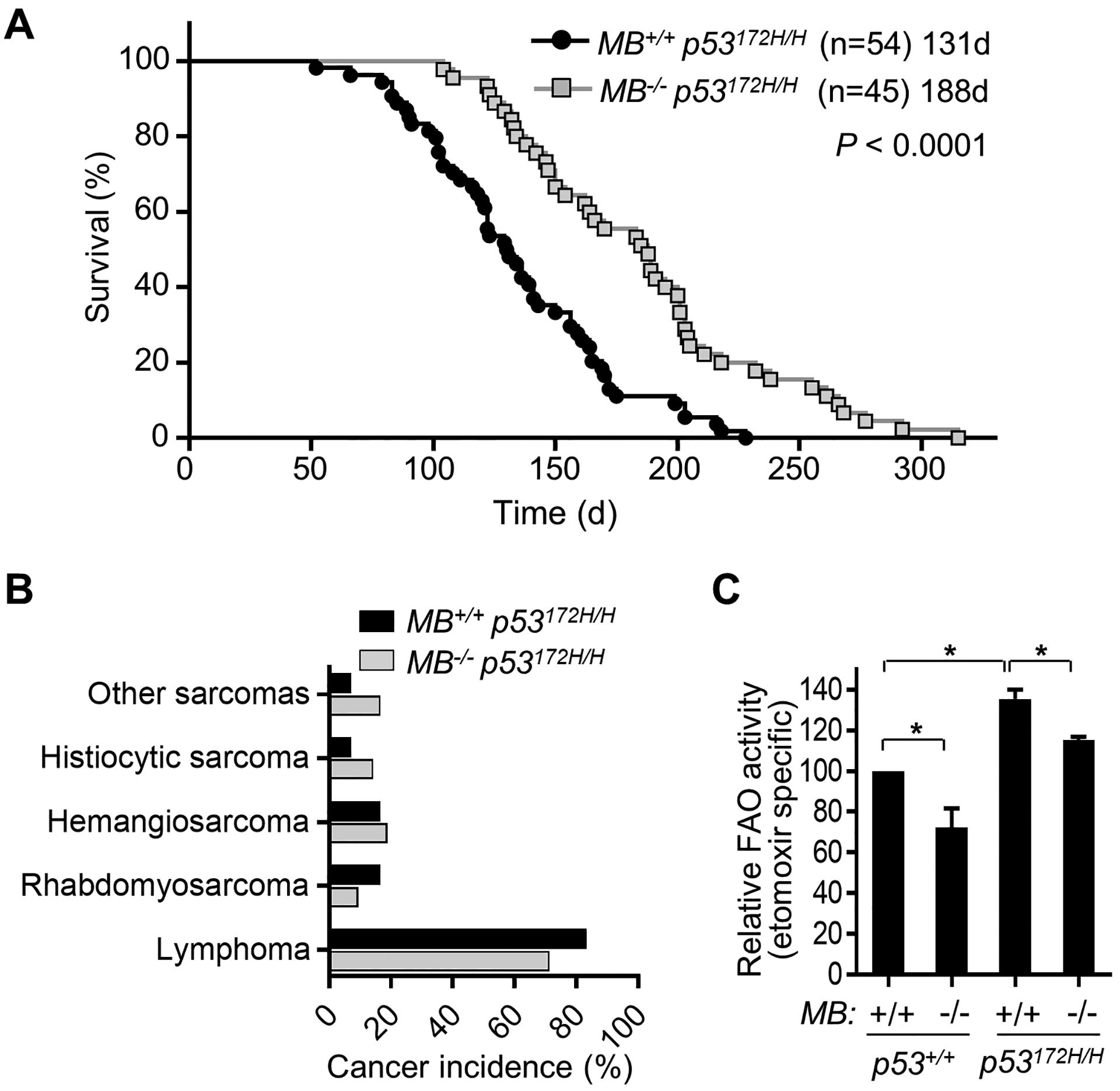
MB ablation decreases fatty acid oxidation and increases the survival time of p53172H/H mice. A, Kaplan-Meier survival plot of p53172H/H mice with or without MB deletion. Statistical difference by log-rank (Mantel-Cox) test. B, Spectra of cancer types diagnosed by necropsy and histopathology. C, The effect of MB deletion on fatty acid oxidation was measured in thymic lymphocytes using 3H-palmitate as substrate (n = 3). *P < 0.05.
Table 1.
Effect of p53172H/H and MB−/− genotypes on thymus by RNA-Seq analysis
| KEGG pathway Term | p53172H/H | MB−/− p53172H/H | ||
|---|---|---|---|---|
| p53+/+ | MB+/+ p53172H/H | |||
| Up-regulated | Down-regulated | |||
| Count | P | Count | P | |
| Carbon metabolism | 42 | 1.30E-05 | 16 | 9.70E-03 |
| Purine metabolism | 55 | 1.10E-04 | 21 | 1.50E-02 |
| Ribosome biogenesis | 33 | 1.60E-10 | 12 | 2.10E-02 |
| Calcium signaling | 50 | 2.70E-03 | 20 | 3.00E-02 |
| Calcium signaling | 12 | 1.60E-04 | 5 | 3.40E-02 |
Myoglobin ablation down-regulates the metabolic pathways induced by p53172H/H genotype. RNA-Seq was performed using thymus of 10 wk old male mice of the indicated genotypes, and KEGG pathway analysis was performed with DAVID Bioinformatics Resources 6.8 (n = 3–6). The pathways significantly induced in p53172H/H vs. p53+/+ state while being reversed by MB−/− state in the p53172H/H background are shown.
Aberrant lipid metabolism and cell signaling in p53172H/H thymus are rescued by MB deletion
In order to obtain further insights into how MB deficiency affects lipid metabolism and prevents tumorigenesis, we performed metabolomic profiling of thymic tissues from p53+/+, p53172H/H, and MB−/− p53172H/H mice. Out of the 377 identified metabolites, only 14 were significantly changed in MB−/− p53172H/H mice compared with p53172H/H mice when ranked by statistical significance (P < 0.05) (Fig. 3A). The majority of these metabolites (11 out of 14) were increased by MB deletion in p53172H/H thymus and some stood out as being involved in lipid metabolism. The second most significantly ranked compound was 2-oleoylglycerol, a known substrate of monoacylglycerol lipase (MAGL) that has been implicated in promoting tumorigenesis (Fig. 3B) (26). p53172H/H thymus also showed significant reductions in 5’-GMP which is converted to IMP via GMP reductase (GMPR), potentially activating lipolysis and mitochondrial activity (Fig. 3B) (27). Consistent with the changes in 2-oleoylglycerol and 5’-GMP levels, the increased expression of MAGL and GMPR in p53172H/H thymus was partially or completely reversed in the MB−/− p53172H/H genotype state (Fig. 3C). The FAO rate limiting enzyme carnitine palmitoyltransferase 1A (CPT1A) showed an expression pattern by p53 and MB genotypes, which was similar to that of MAGL and GMPR, consistent with its known induction by the product of FAO acetyl-CoA (Fig. 3C) (28). Furthermore, the levels of myoglobin were increased in p53172H/H thymus, perhaps reflecting the increased oxygen requirement for FAO. These data suggested that the p53 R172H mutation may increase lipolysis and fatty acid metabolism in part through the concerted activities of multiple mediators such as MAGL, GMPR and CPT1, and that these changes can be dampened by MB deletion.
Figure 3.
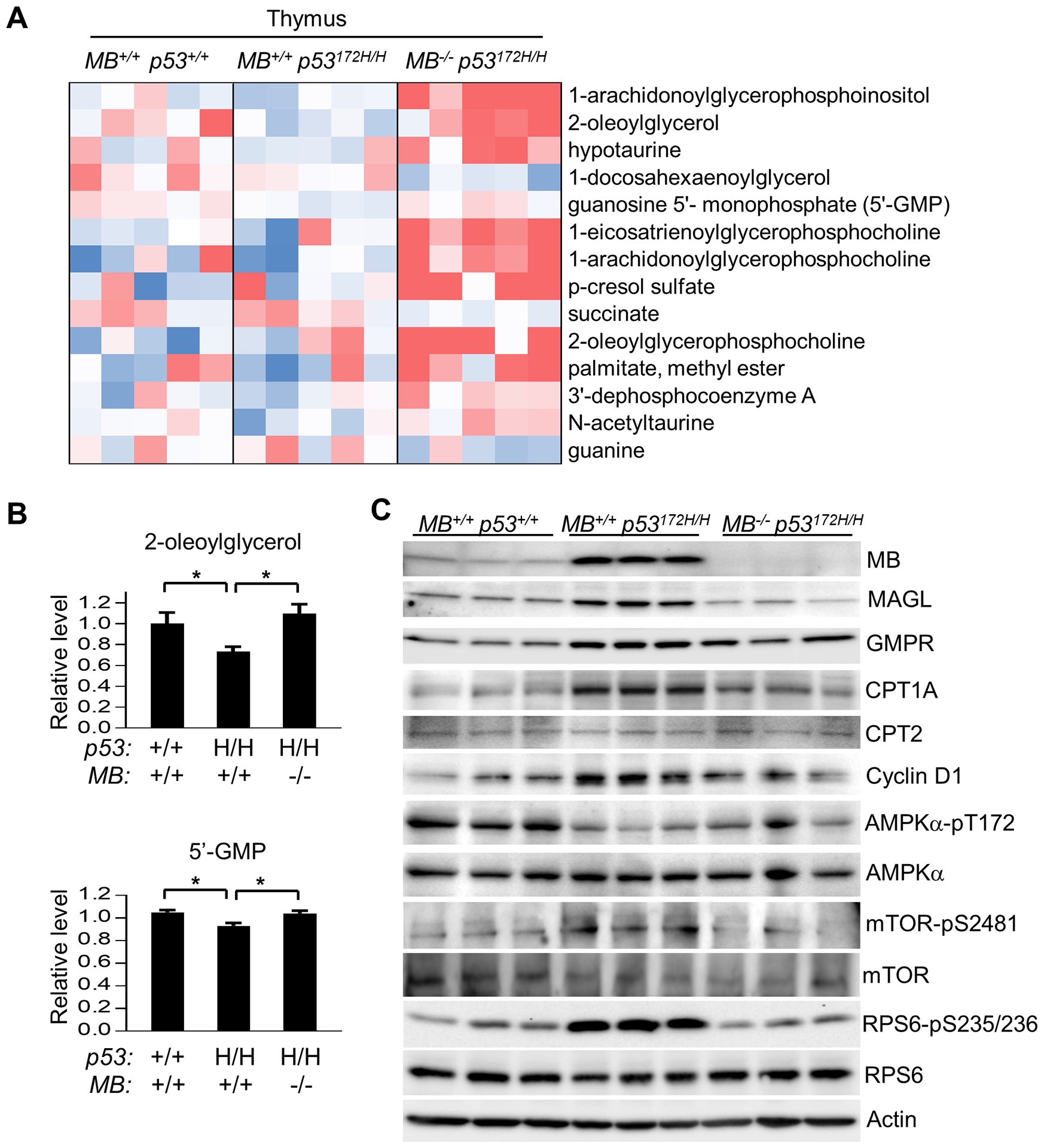
Aberrant lipid metabolism and cell signaling in p53172H/H thymus are corrected by MB ablation. Thymi of male mice (10 wk old) were used for the following experiments. A, Heat map of metabolites identified by metabolomic profiling that were significantly different between MB+/+ p53172H/H and MB−/− p53172H/H mice (P < 0.05, n = 5). Metabolite ion abundance was normalized to the median and then log2 transformed. Color is based on the normalized ion abundance in the range of -1.0 to 0.6 (blue, lowest abundance; red, highest abundance). B, Effect of MB deletion on the reduced levels of 5’-GMP and 2-oleoylglycerol in p53172H/H mice (n = 5). C, Immunoblots of thymic tissues from mice of indicated genotypes. *P < 0.05.
The delayed tumorigenesis caused by MB deficiency paralleled previous observations whereby mild disruption of mitochondrial respiration was sufficient to improve cancer free survival in p53172H/H mice, potentially by inducing anti-proliferative cell signaling caused by bioenergetic stress (15). Mutants of p53 can lose their capacity to repress cyclin D1 expression while acquiring gain-of-function to prevent the activating phosphorylation of AMPKα at Thr-172 by tumor suppressor LKB1 (29–31). On the other hand, inhibiting FAO can activate AMPKα by increasing Thr-172 phosphorylation (32). Indeed, the increased cyclin D1 and decreased phospho-AMPKα levels observed in p53172H/H thymus were reversed by MB deletion in association with decreased FAO (Fig. 2C and 3C). Mutant p53 inhibition of AMPKα with subsequent activation of mTOR (Ser-2481 autophosphorylation) and its effector ribosomal protein S6 (RPS6 Ser-235/236 phosphorylation) were also reversed by MB deletion in p53172H/H thymus (Fig 3C) (33). Importantly, RPS6 activation plays a critical role in cancer initiation as well as tumor growth and is known to promote the up-regulation of the ribosome biogenesis transcriptional program which was identified independently by RNA-Seq analysis (Table 1) (34–36). Together, these screening studies suggest that the aberrant fatty acid metabolism caused by mutated p53 can be moderated indirectly by deleting MB, and that this metabolic inhibition activates AMPKα which in turn inhibits the mTOR pathway activated by mutant p53 with resultant tumor suppression.
LFS mutant p53 can promote FAO in cultured human muscle cells
To examine whether the in vivo observation of increased FAO in the LFS mouse model could apply to humans and to gain additional insights into mechanism, human myoblasts were transduced with lentivirus expressing human p53 R175H protein, homolog of the mouse p53 R172H mutation. Compared with control, expression of p53 R175H markedly promoted mitochondrial oxygen consumption using palmitate as FAO substrate in human myoblasts (Fig. 4A). Consistent with the observation in mice (Fig. 3C), mutant p53 increased the protein levels of some important enzymes involved in lipid metabolism while decreasing the levels of p21, the prototypical wild-type p53 target gene (Fig. 4B). Furthermore, the mRNA levels of FAO related enzymes CPT1B (muscle-specific) and GMPR were induced by mutant p53 while that of p21 was suppressed as expected (Fig. 4C). Thus, a human LFS mutant p53 can cell autonomously promote the expression of more than one gene involved in FAO and fatty acid metabolism as they are complex processes requiring the concerted actions of multiple enzymes.
Figure 4.
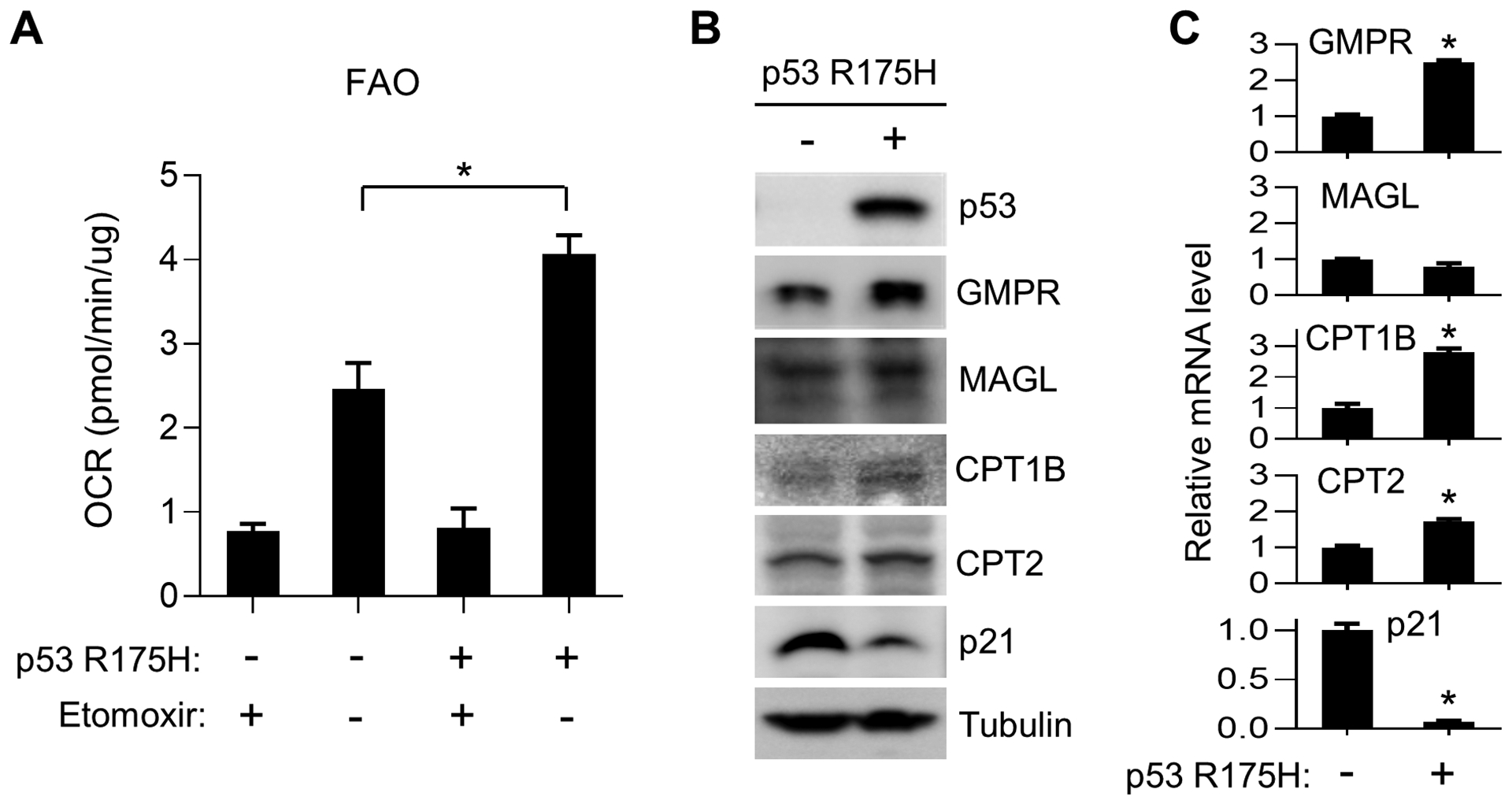
Expression of human R175H mutant p53 (mouse p53 R172H homolog) in human myoblasts promotes FAO. Myoblasts were transduced with empty vector control (-) or human p53 R175H cDNA lentivirus and used for the following assays. A, Palmitate-induced oxygen consumption rate (OCR) was measured by Seahorse with or without FAO inhibitor etomoxir (n =5). B, Immunoblots of enzymes involved in FAO. Note that CPT1B is specific to skeletal muscle tissue. C, Relative mRNA levels of the respective genes involved in FAO quantified by RT-PCR. p21 expression serves as a control for the dominant negative activity of the mutant p53 (n = 6). *P < 0.05.
T cell-specific deletion of CPT2 reduces FAO in thymus and improves the survival of p53172H/H mice
We next set out to examine whether FAO plays a cell autonomous role in the thymic T cell lymphoma development of p53172H/H mice. We targeted CPT2 which converts acyl-carnitine, produced by CPT1A on the outer mitochondrial membrane, back to acyl-CoA on the inner membrane for entry into the β-oxidation cycle in the matrix. To specifically knockout CPT2 in T cells, we took advantage of a previously described strategy for conditionally disrupting CPT2 using a cell type-specific promoter driven Cre-loxP strategy (18). CPT2 was deleted in T cells by crossing CD4 promoter driven-Cre transgenic mice with p53172H/H mice carrying floxed CPT2 alleles, resulting in heterozygous (CPT+/fl) or homozygous (CPT2fl/fl) CPT2 knockout states. Although the incidence and types of cancer were not significantly different at this sample size as with MB deletion, the LFS mouse model with heterozygous knockout of CPT2 (CD4-Cre CPT2+/fl p53172H/H) showed a ~30% improvement in survival time but, unexpectedly, not in the CPT2 homozygous knockout state (Fig. 5A, B). Immunoblotting for CPT2 protein and measurement of FAO activity in the thymus showed that they were reduced in a CPT2 allele dose-dependent manner; 3H-palmitate oxidation was decreased by ~20% and ~50% in the CPT2+/fl and CPT2fl/fl genotype states, respectively (Fig 5C, D). Thus, the delay in tumorigenesis was not proportional to the degree of FAO inhibition, suggesting that the beneficial effect derived by mildly inhibiting FAO through CPT2 haploinsufficiency may be masked by the more severe consequences of complete CPT2 absence (Fig. 5A, D).
Figure 5.
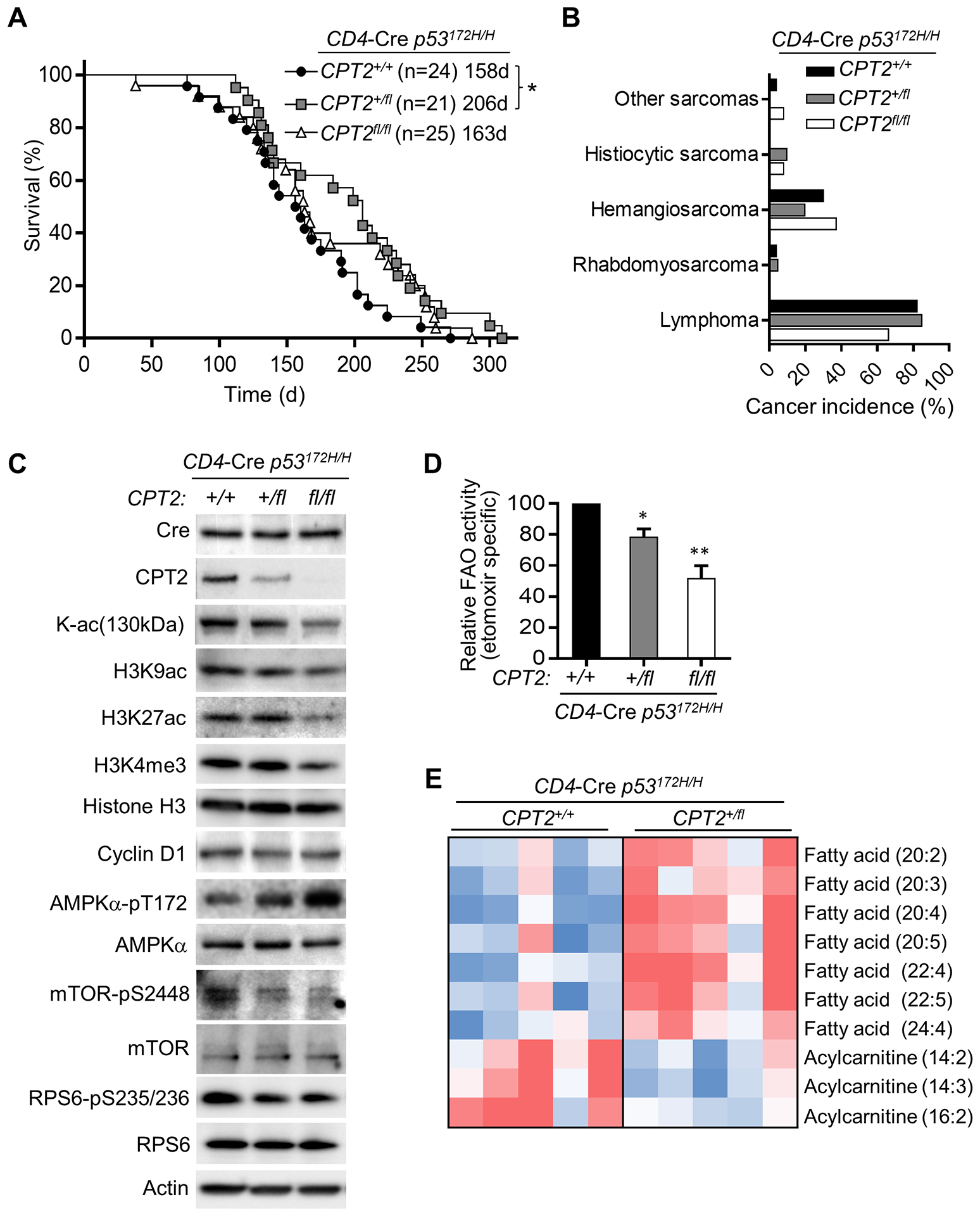
Haploinsufficiency of CPT2 in T cells decreases FAO and mTOR activity while increasing the survival time of p53172H/H mice. A, Kaplan-Meier survival plot of CD4-Cre+ p53172H/H mice with the indicated CPT2 genotypes. Statistical testing by log-rank (Mantel-Cox) test shows a significant difference between CPT2+/+ and CPT2+/fl genotypes. B, Spectra of cancer types diagnosed by necropsy and histopathology. C, Immunoblots of thymic lymphocytes from 10-wk old male mice of the indicated genotypes. D, In vitro fatty acid oxidation assay of thymic lymphocytes using 3H-palmitate as substrate (n = 3–4). Statistical difference by 1-way ANOVA. E, Heat map of metabolites identified by lipidomic profiling. Lipid species which were significantly different (P < 0.05) between the two genotypes are shown. Note the accumulation of long-chain free fatty acids in CPT2 deficiency. Metabolite ion abundance was normalized to the median and then log2 transformed. Color is based on the normalized ion abundance in the range of -1.0 to 0.6 (blue, lowest abundance; red, highest abundance) (n = 5). *P < 0.05, **P < 0.001.
Immunoblotting revealed further differences between the CPT2+/fl and CPT2fl/fl genotypes. LFS mouse thymus with homozygous but not heterozygous knockout of CPT2 showed reductions in lysine acetylation and H3 histone acetylation and methylation, complementing the previous observation of decreased histone acetylation upon loss of CPT1A with resultant depletion of acetyl CoA levels (Fig. 5C) (37). These results also suggested that severe inhibition of FAO in homozygous CPT2fl/fl thymus can interfere with normal cellular process causing us to focus on the heterozygous CPT2+/fl state to understand its tumor suppressive effect under more physiologic conditions. The decrease in cyclin D1 caused by CPT2 deficiency was marginal, but the activation of AMPKα by Thr-172 phosphorylation, a sensor of bioenergetic stress, correlated well with CPT2 allele copy number loss (Fig. 5C). These observations were consistent with the general consensus that fatty acids cannot be replaced entirely by glucose in vivo, both as a mitochondrial oxidative substrate and for non-bioenergetic activities (2). As observed in the MB−/− p53172/H/H double mutant mice, CPT2 deficiency also suppressed the mTOR pathway in p53172/H/H mice, presumably due to AMPKα activation (Fig. 5C) (38). Thus, a mild reduction in FAO can activate AMPKα and delay tumorigenesis in LFS mice without causing significant changes in the common epigenetic modifications, such as histone acetylation and methylation, as observed in the CPT2fl/fl homozygous knockout state (Fig. 5C).
To confirm whether the mild inhibition of FAO caused by CPT2 haploinsufficiency measured in T cells in vitro is sufficient to alter lipid metabolism in vivo, we performed lipidomic profiling of CPT2 wild-type and heterozygous knockout (CD4-Cre CPT2+/fl p53172H/H) thymus tissue. As expected with reduction of long-chain fatty acid β-oxidation in the mitochondria, there was accumulation of long-chain fatty acids (LCFAs) in thymus of heterozygous CPT2+/fl LFS mice (Fig. 5E). Furthermore, the steady state levels of three acyl-carnitines (14:2, 14:3, 16:2) were significantly decreased while there was a pattern of increased longer chain acyl-carnitines in the CD4-Cre CPT2+/fl p53172H/H thymus, indicating decreased conversion of long-chain acyl-carnitines to acyl-CoA due to CPT2 deficiency (Fig. 5E; Supplementary Fig. S2). In summary, these results suggest that the partial inhibition of FAO activates AMPKα with subsequent down-regulation of the mutant p53 activated mTOR pathway, which may contribute to delaying tumorigenesis (Fig. 6).
Figure 6.
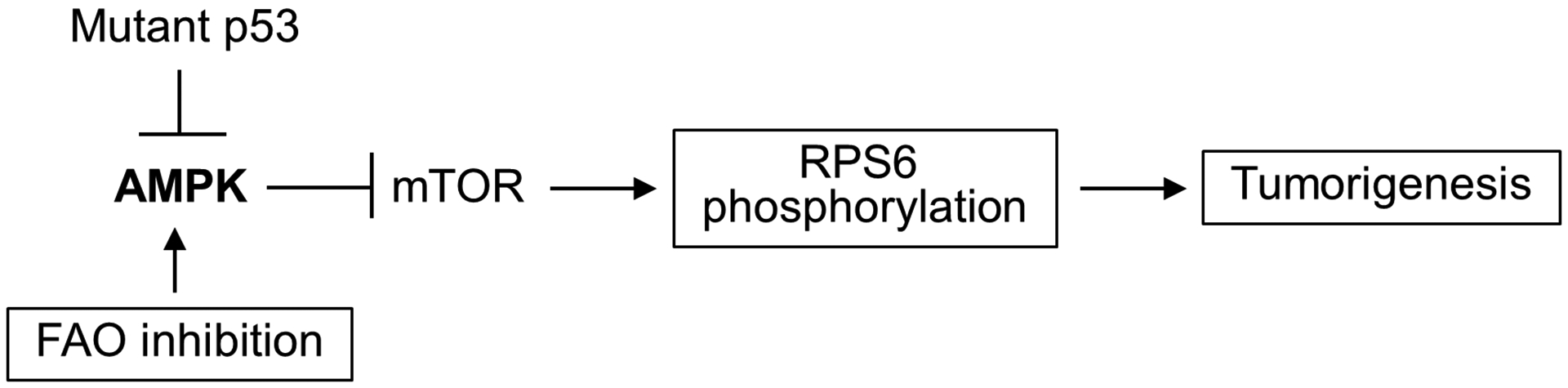
Potential mechanism by which inhibiting fatty acid oxidation exerts tumor suppressive effects. Decreasing FAO metabolism counteracts the inhibition of AMPK by mutant p53, thereby down-regulating the mTOR-RPS6 pathway that is important for cancer development.
Discussion
In the present study, we have used two different genetic modalities to reduce fatty acid oxidation in a mouse model of Li-Fraumeni syndrome for which no cancer preventive treatments are currently available. We demonstrate that partial inhibition of FAO globally or specifically in CD4+ T cells that are cancer-prone can significantly increase the cancer-free survival time of LFS mice. It should be noted that the mild inhibition of FAO appears to be beneficial in delaying tumorigenesis while more severe inhibition results in the loss of this survival benefit. Although the health consequences of inhibiting FAO over a lifetime are unclear, the benefits of partially inhibiting FAO in this LFS mouse model appear to prevail and suggest its consideration as a potential strategy for cancer chemoprevention.
Because FAO impacts many different cellular processes including the generation of acetyl-CoA necessary for histone acetylation (37), the loss of such an essential metabolic function in the homozygous CPT2 knockout mice may compromise multiple cellular activities directly or indirectly related to cancer such as tumor suppressive immune function, confounding the interpretation of their survival time. Complete loss of CPT2 could also result in abnormal T cell development as suggested by the significantly decreased thymic lymphocyte cell count in homozygous CPT2fl/fl but not heterozygous CPT2+/fl or MB−/− genotype states (Supplementary Fig. S3A) (39). In addition to the synthesis of fatty acids, its catabolism by lipolysis and FAO is also appreciated to promote cancer cell survival as the FAO inhibitor etomoxir can promote apoptosis in leukemia cells (1,26,40). In vitro treatment of p53172H/H thymic lymphocytes with etomoxir did not promote cell death, making it more likely that the cancer prevention mechanism by FAO reduction in the LFS mouse model is through alterations in cell signaling such as AMPK mediated inhibition of the mTOR pathway (Supplementary Fig. S3B) (33). In this light, the pharmacologic inhibition of FAO using clinically approved medications such as trimetazidine or ranolazine could be considered for chemoprevention in LFS.
A preponderance of clinical studies suggests that low fat diet decreases the risk of cancer as well as cardiovascular diseases. A large randomized clinical trial recently showed that low-fat diet significantly decreases the risk of mortality from breast cancer, commonly observed in patients with LFS (41). Given the extremely high incidence of breast cancer in women with LFS, being on a low-fat diet may confer relatively greater benefit for cancer prevention in this specific group compared with the general population. Based on the observation that LFS patients and a mouse model of this condition have increased oxidative phosphorylation and fatty acid utilization, we have now targeted both mitochondrial respiration and FAO in a LFS mouse model and observe decreased tumorigenesis and improved cancer-free survival (15). It remains to be seen whether inhibiting mitochondrial respiration, such as by metformin, and FAO together can have a synergistic effect on cancer prevention in LFS. Our current study reveals an alternative strategy for cancer prevention by targeting fatty acid metabolism in LFS patients who, for example, cannot tolerate metformin for mitochondrial inhibition or wish to consider dietary methods for decreasing the lifetime risk of cancer.
Supplementary Material
Significance:
FAO is increased in a mouse model of LFS and its genetic inhibition modulates cellular signaling pathways involved in cancer development, suggesting a strategy for cancer prevention.
Acknowledgements
We wish to thank Robert S. Balaban/Jurgen Schrader and Toren Finkel for providing us with the MB−/− and CPT2fl/fl mice, respectively. We also thank Yuesheng Li (NHLBI DNA Sequencing and Genomics Core) and Martin P. Playford for their assistance and advice. This work was supported by the NHLBI-NIH Division of Intramural Research (HL005101-15) (to PMH).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Currie E, Schulze A, Zechner R, Walther TC, Farese RV, Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013;18:153–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 2013;13:227–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seton-Rogers S Tumour metabolism: Building up and breaking down fatty acids. Nat Rev Cancer 2016;16:677. [DOI] [PubMed] [Google Scholar]
- 4.Goncalves MD, Hopkins BD, Cantley LC. Dietary Fat and Sugar in Promoting Cancer Development and Progression. Annual Review of Cancer Biology 2019;3:255–73 [Google Scholar]
- 5.Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012;22:547–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, et al. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood 2014;123:2663–72 [DOI] [PubMed] [Google Scholar]
- 7.Duman C, Yaqubi K, Hoffmann A, Acikgoz AA, Korshunov A, Bendszus M, et al. Acyl-CoA-Binding Protein Drives Glioblastoma Tumorigenesis by Sustaining Fatty Acid Oxidation. Cell Metab 2019;30:274–89 e5 [DOI] [PubMed] [Google Scholar]
- 8.Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019;363:1345–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41–5 [DOI] [PubMed] [Google Scholar]
- 10.Klement RJ. The emerging role of ketogenic diets in cancer treatment. Curr Opin Clin Nutr Metab Care 2019;22:129–34 [DOI] [PubMed] [Google Scholar]
- 11.Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N Engl J Med 2013;368:1027–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell 2011;44:491–501 [DOI] [PubMed] [Google Scholar]
- 13.Parrales A, Iwakuma T. p53 as a Regulator of Lipid Metabolism in Cancer. Int J Mol Sci 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang JG, Lago CU, Lee JE, Park JH, Donnelly MP, Starost MF, et al. A Mouse Homolog of a Human TP53 Germline Mutation Reveals a Lipolytic Activity of p53. Cell reports 2020;30:783–92 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang PY, Li J, Walcott FL, Kang JG, Starost MF, Talagala SL, et al. Inhibiting mitochondrial respiration prevents cancer in a mouse model of Li-Fraumeni syndrome. J Clin Invest 2017;127:132–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Godecke A, Flogel U, Zanger K, Ding Z, Hirchenhain J, Decking UK, et al. Disruption of myoglobin in mice induces multiple compensatory mechanisms. Proc Natl Acad Sci U S A 1999;96:10495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004;119:847–60 [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Ellis JM, Wolfgang MJ. Adipose fatty acid oxidation is required for thermogenesis and potentiates oxidative stress-induced inflammation. Cell reports 2015;10:266–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thapa D, Wu K, Stoner MW, Xie B, Zhang M, Manning JR, et al. The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial beta-oxidation enzyme HADHA. J Biol Chem 2018;293:17676–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Wang PY, Long NA, Zhuang J, Springer DA, Zou J, et al. p53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc Natl Acad Sci U S A 2019;116:19626–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flogel U, Laussmann T, Godecke A, Abanador N, Schafers M, Fingas CD, et al. Lack of myoglobin causes a switch in cardiac substrate selection. Circ Res 2005;96:e68–75 [DOI] [PubMed] [Google Scholar]
- 22.Merx MW, Godecke A, Flogel U, Schrader J. Oxygen supply and nitric oxide scavenging by myoglobin contribute to exercise endurance and cardiac function. FASEB J 2005;19:1015–7 [DOI] [PubMed] [Google Scholar]
- 23.Flonta SE, Arena S, Pisacane A, Michieli P, Bardelli A. Expression and functional regulation of myoglobin in epithelial cancers. Am J Pathol 2009;175:201–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 2013;13:572–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010;140:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salvatore D, Bartha T, Larsen PR. The guanosine monophosphate reductase gene is conserved in rats and its expression increases rapidly in brown adipose tissue during cold exposure. J Biol Chem 1998;273:31092–6 [DOI] [PubMed] [Google Scholar]
- 28.McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, et al. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell reports 2016;17:1463–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rocha S, Martin AM, Meek DW, Perkins ND. p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Mol Cell Biol 2003;23:4713–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dudgeon C, Chan C, Kang W, Sun Y, Emerson R, Robins H, et al. The evolution of thymic lymphomas in p53 knockout mice. Genes Dev 2014;28:2613–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol Cell 2014;54:960–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Estan MC, Calvino E, Calvo S, Guillen-Guio B, Boyano-Adanez Mdel C, de Blas E, et al. Apoptotic efficacy of etomoxir in human acute myeloid leukemia cells. Cooperation with arsenic trioxide and glycolytic inhibitors, and regulation by oxidative stress and protein kinase activities. PLoS One 2014;9:e115250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez A, Hall MN, Lin SC, Hardie DG. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab 2020;31:472–92 [DOI] [PubMed] [Google Scholar]
- 34.Khalaileh A, Dreazen A, Khatib A, Apel R, Swisa A, Kidess-Bassir N, et al. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res 2013;73:1811–20 [DOI] [PubMed] [Google Scholar]
- 35.Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau-Aveilla C, Dreazen A, et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014;33:474–83 [DOI] [PubMed] [Google Scholar]
- 36.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017;168:960–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong BW, Wang X, Zecchin A, Thienpont B, Cornelissen I, Kalucka J, et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 2017;542:49–54 [DOI] [PubMed] [Google Scholar]
- 38.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577–90 [DOI] [PubMed] [Google Scholar]
- 39.O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014;41:75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 2010;120:142–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chlebowski RT, Anderson GL, Manson JE, Prentice RL, Aragaki AK, Snetselaar L, et al. Low-Fat Dietary Pattern and Cancer Mortality in the Women’s Health Initiative (WHI) Randomized Controlled Trial. JNCI Cancer Spectr 2018;2:pky065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.