

Abstract
The outer membrane of Gram-negative bacteria is essential for their survival in harsh environments and provides intrinsic resistance to many antibiotics. This membrane is remarkable; it is a highly asymmetric lipid bilayer. The inner leaflet of the outer membrane contains phospholipids whereas the fatty acyl chains attached to lipopolysaccharide (LPS) comprise the hydrophobic portion of the outer leaflet. This lipid asymmetry, and in particular the exclusion of phospholipids from the outer leaflet, is key to creating an almost impenetrable barrier to hydrophobic molecules that can otherwise pass through phospholipid bilayers. It has long been known that these lipids are not made in the outer membrane. It is now believed that conserved multi-subunit protein machines extract these lipids after their synthesis is completed at the inner membrane and transport them to the outer membrane. A longstanding question is how the cell builds and maintains this asymmetric lipid bilayer in coordination with the assembly of the other components of the cell envelope. This review describes the trans-envelope lipid transport systems that have been identified to participate in outer membrane biogenesis: LPS transport via the Lpt machine, and phospholipid transport via the Mla pathway and several recently proposed transporters.
Graphical Abstract
1. Introduction
The double layered membrane architecture of the Gram-negative cell envelope provides a robust protective barrier for the cell.1 The inner membrane is a phospholipid bilayer that encases the cytoplasm. Outside the inner membrane is the periplasmic compartment, which contains a thin cell wall composed of peptidoglycan. The outermost membrane is exposed to the environment, so it is the first line of defense for the cell. The outer membrane functions as a unique permeability barrier as we discuss below.1,2 The cell envelope of Escherichia coli is the best studied among Gram-negative bacteria and will be the main focus of this review. Although the overall architecture, composition, properties, and biogenesis of the cell envelope described in this review are conserved among Gram-negative bacteria, there is a great deal of diversity in the details.
The outer membrane is an asymmetric bilayer with an inner leaflet of glycerophospholipids and an outer leaflet mainly composed of lipopolysaccharide (LPS) molecules.3,4 Although the specific phospholipid composition varies among bacteria, in Escherichia coli, both the inner and outer membranes contain phosphatidylethanolamine, phosphatidylglycerol, and cardiolipin albeit in different ratios (Fig. 1a).5–10 It is also true that the structure of LPS varies among bacteria. In E. coli, LPS contains a Lipid A moiety composed of a di-glucosamine diphosphate with several fatty acyl chains attached to each sugar (Fig. 1b). Together these lipids comprise the hydrophobic portion of the outer leaflet of the outer membrane. The core oligosaccharide is directly attached to Lipid A and comprised of a conserved set of approximately ten sugars, some of which can be phosphorylated. The O antigen, which is ligated onto the core oligosaccharide, is a highly variable polymer that can consist of more than a hundred sugars (Fig. 1b). When properly assembled on the cell surface, the negatively charged phosphates of adjacent LPS molecules bind divalent cations (i.e. Ca2+ and Mg2+) creating strong lateral interactions and tight packing between LPS molecules. These interactions create a polyelectrolyte-like structure that is reinforced by hydrophobic interactions between acyl chains in the Lipid A component. The physicochemical properties of LPS decrease the permeability of the outer membrane to small hydrophobic compounds that would otherwise penetrate a typical phospholipid bilayer.1,11 Lipid bilayers obviously prevent diffusion of hydrophilic molecules, but small (<600 Da in E. coli) hydrophilic molecules such as nutrients can cross the outer membrane through pores in the membrane created by outer membrane integral proteins (OMPs), which adopt a β-barrel conformation.12,13 There are notable exceptions for this outer membrane architecture such as in spirochetes like Treponema pallidum. These bacteria do not produce LPS; instead, they display a large number of surface-exposed lipoproteins.14 Not surprisingly, their outer membrane is more permeable than a prototypical outer membrane. We note that many Gram-negative bacteria also have additional lipid-linked glycopolymers in the outer leaflet of their outer membranes such as the enterobacterial common antigen and capsules, which are reviewed elsewhere.15,16
Figure 1: Major lipid components of the E. coli outer membrane.

A) Structure of the three types of phospholipids found in E. coli. The whole-cell phospholipid content of E. coli is 75% phosphatidylethanolamine, 20% phosphatidylglycerol, and 5% cardiolipin.9 Although fatty acid composition can change in response to signals, the major fatty acids in phospholipids under normal growth are 16:0, 16:1, and 18:1.5 B) Structure and main components of E. coli K-12 LPS. Potential modifications of Lipid A and the corresponding modifying enzymes are color coded. PagP cleaves an acyl chain from a phospholipid and ligates it (shown in red) onto Lipid A. PagL deacylates LPS by removing the R-3-hydroxymyristate shown in green. The C1 and C4′ phosphates can be modified with L-4-aminoarabinose (pink) by ArnT and phosphoethanolamine (blue) by EptA, respectively. The core oligosaccharide is composed of glucose (Glu), heptose (Hep), galactose (Gal), and 3-deoxy-d-manno-octulosonic acid (Kdo). Heptose residues are phosphorylated (P) and modified with phosphoethanolamine (PEtN). The numbers represent glycosidic linkage positions. The structure of the highly variable O antigen is not shown.
The biogenesis of the outer membrane, including outer membrane lipoproteins and OMPs, is a highly coordinated and complex process. As we will discuss in detail below, none of the components of the outer membrane are made in this membrane. Biosynthesis of all outer membrane components occurs in the cytoplasm and/or at the inner membrane, so they must be transported across the cell envelope before they are assembled at the outer membrane. This makes their biogenesis both fascinating and challenging to study, as it can be hard to discern the role of a protein identified as necessary for outer membrane biogenesis. Is it involved in biosynthesis of a component of the outer membrane or in its transport to an assembly site at the outer membrane? Or is it a bona fide biogenesis factor that is directly involved in the assembly process? The challenge is to distinguish those players involved in synthesis, transport, and assembly, since blocking any of these processes produces outer membrane biogenesis defects.
The starting point to understand how cells build their outer membrane is to find the players (genes/proteins) required for the biogenesis of the lipids that make this bilayer - the membrane. Since there are many fine reviews on the biosynthesis of each of these outer membrane components,16–20 the aim here is to focus this review on our current understanding of the outer membrane lipid transport and assembly processes, encompassing the three known types of intramembrane lipid transport: anterograde LPS transport, and both anterograde and retrograde transport of phospholipids. Much of this review will focus on LPS transport, as it is the best understood of these systems and still surprisingly little is known about phospholipid transport. We will touch upon how, during the past 5 decades, scientists became aware of the need for an lipopolysaccharide transport (Lpt) machine and eventually discovered its components. An in-depth review about the history of the discovery of Lpt has been previously published.21 We will then describe the journey that LPS takes from the inner to the outer membrane through the Lpt system, highlighting what is known about the function of this machine and what gaps in knowledge still exist. We will then examine what is known about phospholipid transport, from the earliest models in the 60’s and 70’s to those recently proposed. In this part, the review will focus heavily on the maintenance of lipid asymmetry (Mla) system, which, in 2009, became the first phospholipid inter-membrane transport system involved in outer membrane biogenesis to be discovered. We will discuss the different evidence supporting its role in either retrograde or anterograde phospholipid transport, as well as studies proposing that other protein complexes mediate inter-membrane phospholipid transport.
2. LPS biogenesis
2.1. Brief review of LPS biosynthesis
LPS synthesis [reviewed in 18,22] starts in the cytoplasm with the precursor uridine diphosphate N-acetylglucosamine (UDP-GlcNAc). Through the action of the many Raetz pathway Lpx enzymes and KdtA, UDP-GlcNAc is first di-acylated, then, two of these molecules are condensed and the product phosphorylated to generate Lipid IVA, which is further glycosylated and acylated to produce the final product, Kdo2-Lipid A. Next, synthesis of the core oligosaccharide is accomplished by the Waa glycosyltransferase and kinase enzymes, producing lipooligosaccharide (LOS, black and brown portions of LPS in Fig. 1b), which is flipped across the inner membrane by the ATP-dependent transporter MsbA.23–27 It is now that the O antigen, a long polymer of many repeating sugar units that is independently synthesized at the inner membrane, is ligated onto LOS at the periplasmic side of the bilayer to finally produce LPS (Fig. 1b).28–31 Some bacteria such as Acinetobacter do not naturally synthesize an O antigen, so their mature glycolipid is LOS.32 In contrast, wild-type E. coli strains can synthesize a variety of O antigens, but most laboratory strains do not produce it because of a mutation in the wbbL biosynthesis gene.18,33
2.2. A model to explain the mechanism of LPS biogenesis
The problem
After its biosynthesis is completed at the inner membrane, transporting LPS across the cell envelope and assembling it at the outer membrane poses many challenges to the cell. Extracting the fatty acyl chains of LPS out of the inner membrane requires energy. Then, during its periplasmic transit to the outer membrane, the hydrophobic fatty acid chains must be shielded from the aqueous compartment. Finally, LPS must be delivered and assembled into the outer leaflet of the outer membrane. How the many (hundreds of) sugars of LPS reach the cell surface, passing through the lipid bilayer, suggested a protein-mediated process. Since, there is no ATP in the periplasm or proton-motive force at the outer membrane, LPS transport demands a different type of transporter than those described to exist to transport polysaccharides across the inner membrane.
The solution
Transport of LPS from the inner membrane to the outer membrane is mediated by the LPS transport (Lpt) machine, which is made up of seven different proteins, LptB2FGCADE (Fig. 2).34–37 According to the current model, these Lpt factors form a continuous protein bridge that spans the cell from the cytoplasm to the outer membrane and is able to extract LPS from the inner membrane, transport it across the cell envelope, and insert it directly into the outer leaflet of the outer membrane.38,39 In fact, the Lpt machine can be divided into three main parts that solve different challenges in transporting LPS: the inner membrane ABC transporter, which uses ATP hydrolysis in the cytoplasm to extract LPS from the inner membrane; the periplasmic bridge, which shields the hydrophobic portion of LPS from the aqueous periplasm; and, the outer membrane translocon, which catalyzes the final insertion of LPS into the outer leaflet of the outer membrane. Over the last five decades, key experiments from groups using very different tools have now made it possible to begin to describe the process of LPS transport and assembly at a molecular level.
Figure 2: Model for LPS transport by the Lpt system.

LPS is transported from the inner membrane (IM) to the outer membrane (OM) by the LptB2FGCADE complex in an ATP-dependent manner. Each round of ATP binding and hydrolysis by the LptB2FGC ABC transporter is thought to be used to extract one molecule of LPS from the inner membrane and place it onto the periplasmic Lpt bridge. Repeated rounds of hydrolysis extract LPS molecules that push others ahead on the bridge so that a stream of LPS travels towards the outer membrane through the Lpt bridge. LPS finally reaches the LptDE translocon and exits into the outer leaflet of the outer membrane. PG represents the peptidoglycan cell wall.
3. The study of LPS transport
Advances in electron microscopy and analytical biochemistry during the 1960s-1980s led to our understanding of the structure and composition of the Gram-negative cell envelope (reviewed in 21). Thereafter, research efforts concentrated on the identification of envelope biogenesis factors. The ability to assign lpt genes to LPS transport relied on the realization that there are hallmark phenotypes associated with LPS deficiency combined with the ability to monitor where LPS accumulates in the cell when these factors are depleted. As described below, by 2008, all members of the Lpt system were identified.21 Since then, significant progress has been made in understanding how the complex Lpt machine functions. This progress has been possible through the development of novel experimental methods and the synergistic combination of genetic, biochemical, and structural approaches.
3.1. Genetic approaches
The use of genetics to study LPS transport has been essential to identify and characterize Lpt factors because the system allows for genetic selections and phenotypic analysis. In E. coli, LPS, and therefore its transport, is essential.40 In addition, LPS provides intrinsic resistance to many antibiotics.1,41–45 Consequently, there are two key phenotypes that result from non-functional or impaired Lpt proteins: increased sensitivity to antibiotics when Lpt function is partially defective, and, in the case of complete loss of function, cell death.46 Screening for these phenotypes led to the identification and/or the characterization of the Lpt proteins.21 Since lpt genes are essential for growth in E. coli, most were characterized by regulating their transcription with inducible promoters in depletion strains. These strains have been engineered to have a specific lpt gene regulated by a known transcriptional regulator that is responsive to a chemical (i.e. inducer). When the inducer is added to the growth medium, the strain is viable because the lpt gene is transcribed; however, in the absence of inducer, transcription of the specific lpt gene stops, and the pre-existing Lpt factor is depleted as the cell divides, eventually leading to death. Observing cells depleted of Lpt factors via microscopy showed distinctive phenotypes including chaining owing to defects in daughter-cell separation and accumulation of membranous material in their periplasm.37,47 These phenotypes, together with biochemical characterizations described below, were key in the early studies describing Lpt factors, and they are still being used today to characterize lpt mutants.
The ability to determine the functional role of specific residues is required to understand the mechanism of LPS transport. This has been facilitated through the generation and phenotypic analysis of a collection of lpt mutant alleles that affect different steps of transport. If mutant alleles generated randomly or through genetic engineering confer a total or severe loss of function, they cause cell death. If mutations partially decrease function, they confer sensitivity to hydrophobic antibiotics to certain extent. This approach provides a dynamic range for measuring the functional state of the Lpt machine. Once a residue has been identified as having functional importance, its specific role can be further studied using suppressor analyses. By growing mutants with defective Lpt variants in non-permissive conditions (i.e. in the presence of hydrophobic antibiotics or conditions that lead to cell death), cells that spontaneously acquire suppressor mutations can be identified, as these mutations somehow overcome the problem caused by the defective lpt allele. Learning how suppressor mutations in lpt genes fix the disrupted function has informed us about the specific function (e.g. physical interaction with an LPS substrate or with another Lpt factor) of residues in Lpt transport.48–50 Additionally, suppressors in non-lpt genes have revealed unexpected findings about how cells cope with limiting amounts of LPS.51,52 An interesting twist of this approach that will be discussed later is how it led to the discovery that the antibiotic novobiocin interacts with LptB to increase LPS transport.53
3.2. Biochemical approaches
Studying transporters is challenging because they are highly dynamic proteins that transiently interact with a substrate that they do not modify. The Lpt system poses the additional challenge of requiring two membranes and components in every compartment of the cell (Figs. 2 and 3). Nevertheless, biochemical studies have overcome these challenges and been invaluable for characterizing Lpt factors, characterizing if and how they interact with one another and LPS, determining the role of ATP in LPS transport, and ultimately providing the full in vitro reconstitution of the Lpt machine.36,39,54–56 These accomplishments have involved a variety of methodology that we highlight below.
Figure 3: Structures of Lpt factors.

Cartoon representations of the crystal structures of the components of the Lpt machine shown in their respective cellular compartments. Components were crystalized individually or as sub-complexes as follows: The LptB2FGC complex (PDB 6MJP), LptA (PDB 2R19), and LptDE complex (PDB 4Q35). To date, a structure of the entire trans-envelope complex is not available, but it is known that the β-jellyroll domains interact in a head-to-tail fashion (see main text for details). The number of LptA molecules in each bridge is unknown, but one could be sufficient to span the periplasm.
3.2.1. Following the cellular localization of LPS
Determining the localization of LPS in cells depleted of Lpt factors was critical for the initial characterization of the Lpt system. These methods took advantage of both the ability to fractionate the inner and outer membranes of E. coli using density ultracentrifugation, and the realization that enzymes can modify LPS in a specific cellular location. Accumulation of LPS in cellular fractions of lower density than the outer membrane upon depleting Lpt factors was demonstrated.34,47 Then, the Polissi group led the work showing that this accumulation occurs at the periplasmic side of the inner membrane because the glycolipid can be aberrantly modified with colanic acid by the WaaL ligase, which normally adds the O antigen to the LPS core.46,57 This was a complementary approach to determine mislocalization of LPS in Neisseria that was developed earlier by the Tommassen group.58 Bos et al. introduced the Bordetella bronchiseptica PagL enzyme, which deacylates LPS at the outer membrane, into Neisseria as a reporter of whether LPS was assembled in the outer leaflet of the outer membrane. This classic study showed that LPS does not reach the outer leaflet of the outer membrane in the absence of LptD.58 This strategy was later extended to the outer membrane acyltransferase PagP in order to track LPS transport to the outer membrane of E. coli.59 PagP transfers an acyl chain of a phospholipid to LPS only when phospholipids mis-localize to the outer leaflet of the outer membrane, which can occur when LPS levels decrease at the outer membrane.36,60 Depletion of Lpt factors prevented PagP-dependent modification of newly synthesized LPS.35,36
3.2.2. Trapping Lpt-Lpt and Lpt-LPS interactions with site-specific photo-crosslinking
To study the mechanism of transport, one needs to be able to observe intermediate transport states of LPS within the Lpt transporter, as well as how multiple Lpt factors interact with one another. For LPS transport, a key advance was to identify sites in Lpt factors that mediate physical interactions with other Lpt factors or LPS in both live cells and the in vitro systems described in the next section. The UV photo-crosslinkable amino acid p-benzoyl phenylalanine (pBPA) can substitute a specific residue in an Lpt protein in cells through a system involving amber suppression. The pBPA-substituted Lpt protein present in live cells or purified protein samples, when exposed to UV light, will form stable covalent bonds to any interacting partners within 3–6 Å.61–63 The crosslinked partner is usually identified using immunoblotting, but the specific site of interaction with pBPA can be further characterized through mass spectrometry.64–66 This technique was first used in the outer membrane biogenesis field to identify interactions between components of the localization of lipoproteins (Lol) export system and their lipoprotein substrates.67 Photo-crosslinking has been critical to characterize the physical architecture of the Lpt transporter and transport intermediates. Indeed, if the residence time is long enough, transient interactions between LPS and pBPA substitutions in Lpt factors can be trapped through this crosslinking approach. As we will describe in sections below, this method has revealed the pathway that LPS takes along the Lpt transporter in both live cells and in vitro reconstitution assays. Unlike in Lol, the substrate LPS is not a protein, so a key to the success of this method to analyze substrate-transporter crosslinks was the availability of anti-LPS antibodies that allowed sensitive and easy detection of transport intermediates.39,54,68 Indeed, this method has been adapted to study other transport systems for non-protein substrates that can be easily detected.64,69
3.2.3. In vitro reconstitution of LPS transport
The development of in vitro reconstitution assays from purified components has been crucial for mechanistic studies of LPS transport. The first assay to be developed, which also relied on photo-crosslinking, was the reconstitution of LPS extraction from right-side-out vesicles over-producing the inner membrane Lpt complex.54 Using membrane vesicles that contained the inner membrane LptB2FGC complexes, crosslinking of LPS to pBPA-substituted LptC (or added purified pBPA-substituted LptA) could be detected, and the crosslinked products increased over time in an ATP-dependent manner.54 This assay ultimately evolved into the full reconstitution of the Lpt system.39 Proteoliposomes containing the outer membrane proteins LptDE were generated and pre-incubated with soluble LptA. These LptDEA proteoliposomes were then incubated with proteoliposomes containing LPS and the inner membrane LptB2FGC complex. As described below, by monitoring transport with photo-crosslinking in this assay and showing the need to bridge both types of proteoliposomes with LptA, Sherman et al. demonstrated both that LptB2FGCADE are both necessary and sufficient for LPS transport from the inner to the outer membrane, and that the Lpt bridge is essential for function.39 More recently, this reconstitution assay was modified to quantifiably measure LPS incorporation into the outer membrane by preloading outer-membrane-like proteoliposomes with dansylated polymyxin B nonapeptide, which binds to LPS and increases in fluorescence when it is incorporated into the membrane.70 Additional modifications to these reconstitutions and implementation of ATPase assays have been used to examine the effect of small molecules on the transporter, as well as to provide mechanistic insight into transport.53,56,66
3.3. Structural studies
Understanding the structure-function of Lpt proteins has been greatly advanced by the 3-D structures that have been produced over the years through X-ray crystallography55,68,71–76 and, more recently, cryogenic electron microscopy (cryo-EM) (Fig. 3).77,78 These structures have confirmed and been validated by findings obtained via the combined efforts of genetics and biochemistry. The great impact that solving these structures has had in advancing our understanding of the architecture and function of the Lpt system is highlighted in sections below. Notably, although structures of each Lpt protein and the inner and outer membrane complexes exist (Fig. 3), we await the challenging structure of the fully assembled trans-envelope Lpt machine.
4. Discovering the Lpt system
The site of biosynthesis of LPS at the inner membrane as well as the transport of the newly synthesized glycolipid from the inner to the outer membrane was first demonstrated using a combination of pulse-chase and membrane separation experiments reported in two now classic papers in 1972 by Osborn et al.9,79 These studies proved that synthesis of the LPS core and O antigen occurs at the inner membrane. This information was critical to establishing that LPS transport to the outer membrane is irreversible. These studies also set the stage for the search of the LPS transporter. Before the discovery of the Lpt system, the ATP-dependence of LPS transport from the inner to the outer membrane was demonstrated in 1985 by the Osborn laboratory.80 Despite these early discoveries, it would take another two decades to identify the first Lpt factor, and most of them were identified between 2004–2008.35–37,47 Research spanning the last four decades has worked towards elucidating this mechanism of transport, starting with the studies described here that identified the factors required for this inter-membrane LPS transport.
4.1. Discovery of the LptDE outer-membrane translocon (1989–2006)
The first Lpt protein to be linked to the function of LPS in creating a permeability barrier and to outer membrane biogenesis was the outer membrane β–barrel LptD. An lptD mutant, originally named imp4213, was identified in 1989 in a genetic screen for mutants with increased membrane permeability to maltodextrins.81 Sampson, Misra, and Benson found that this mutant also exhibited sensitivity to several hydrophobic antibiotics. In fact, the imp4213 mutant allele is still widely used in small-molecule screens in order to overcome the problem of the outer-membrane permeability barrier. Interestingly, we now know that imp4213 mutants have a severe folding defect in the LptD β–barrel, a fact that led to many discoveries in BAM, the machine that folds and inserts OMPs.82–85
The functional importance of this protein remained unknown until Braun and Silhavy demonstrated its essential role in envelope biogenesis and proposed LptD (Imp) as an outer membrane biogenesis factor in E. coli.37 LptD’s specific role in LPS transport was finally demonstrated in 2004 by the Tommassen laboratory.58 Using Neisseria meningitidis, a bacterium in which LPS synthesis is not essential,86,87 Bos et al. demonstrated that a mutant lacking LptD was viable and did not have LPS assembled in their outer membrane since it could not be modified by the outer membrane deacylase PagL.58
In 2006, the Silhavy and Kahne groups discovered that the E. coli LptD co-purified in a complex with a novel outer membrane lipoprotein, RlpB (LptE).36 They also observed that depleting either LptD or LptE prevented the modification of newly synthesized LPS by the outer membrane acylase PagP and ultimately resulted in cell death. Given these phenotypes, and the localization of LptDE at the outer membrane, Wu et al. proposed that this complex assembles LPS at its final location, the outer leaflet of the outer membrane.36
4.2. Discovery of LptCAB (2004–2008)
A genetic screen searching for essential genes in E. coli led the Polissi laboratory to identify the yrbK-yhbN-yhbG locus, which would later be renamed lptCAB.34,88 Detailed phenotypic analyses of depletion strains finally demonstrated that LptCAB were necessary for LPS transport to the outer membrane.46,47 Since LptA is a periplasmic protein, it was predicted to chaperone LPS across the periplasm, while the predicted cytoplasmic ATPase LptB was thought to energize LPS extraction from the inner membrane through an unknown mechanism involving the bitopic protein LptC.34,46
4.3. At last, finding LptFG (2008)
LptB had been identified by sequence homology as a predicted ATPase belonging to the superfamily of ABC transporters,34 but its predicted transmembrane domain (TMD) partners were unknown until they were discovered using a reductionist bioinformatics approach.35 Ruiz et al. exploited the simplified genome of a Gram-negative endosymbiont that produces LPS to search for potential Lpt factors and found two candidates, YjgP and YjgQ (renamed LptF and LptG, respectively), which had been classified as putative permeases. Depletion of each of these proteins in E. coli showed the characteristic phenotypes of Lpt depletions, demonstrating their essential role in LPS transport. Given their predicted membrane topology, they were proposed to be the missing TMD partners of LptB in an ABC transporter that extracts LPS from the outer leaflet of the inner membrane.
5. Stepwise process of LPS transport
A PEZ machine
Once all Lpt factors were identified, research efforts focused on understanding the mechanism of function of the Lpt system. This body of work has led to the following model to explain how the inner membrane, periplasmic, and outer membrane Lpt components work together to transport LPS from the outer leaflet of the inner membrane to the cell surface. In this model, the LptB2FGC ABC transporter harnesses energy from ATP in the cytoplasm to power the extraction of LPS from the inner membrane (Fig. 2). A cytoplasmic LptB2 dimer constitutes the nucleotide binding domains (NBDs) of the transporter that bind and hydrolyze ATP.54,55 LptFG function as the transmembrane domains (TMDs) that directly interact with LPS to extract it from the outer leaflet of the inner membrane and place it onto the Lpt periplasmic bridge.50,77 LptC is a bitopic protein with a transmembrane (TM) helix and a periplasmic domain. Its single TM helix is located between the TM helices of LptFG, while its periplasmic domain is part of the Lpt bridge.68 The periplasmic Lpt bridge is formed by β-jellyroll domains in LptFCAD that link up to one another in a head-to-tail fashion, forming a continuous structure across the periplasm.65,68,71,72,75,76 This protein fold forms a C-shaped structure with a hydrophobic interior that shields the acyl chains of LPS from the hydrophilic periplasm. Finally, the outer-membrane translocon is made of LptDE. LptD is a large β-barrel protein in which the lipoprotein LptE resides, forming a plug-and-barrel complex.75,89 It is thought that the hydrophilic portion of LPS passes through the lumen of the LptD β-barrel guided by charge interactions with LptE and exits through a lateral gate, while the hydrophobic portion of LPS slips into the hydrophobic core of the outer membrane through an opening between the β-jellyroll and β-barrel domain of LptD.73–75,90
The Lpt machine has been likened to a PEZ dispenser where the inner-membrane ABC transporter is the spring that pushes LPS, the metaphorical candy, onto the periplasmic bridge.91 Each round of ATP-dependent transport by the LptB2FGC ABC transporter is proposed to move a newly synthesized LPS molecule onto the bridge, pushing the previously extracted molecules of LPS towards the outer membrane. As this happens repeatedly, LPS travels as a stream through the Lpt periplasmic bridge. Eventually LPS reaches the LptDE translocon, which is equivalent to the head of the PEZ dispenser, through which it exits into the outer leaflet of the outer membrane. Once there, it is stably assembled through lateral interactions with other LPS molecules and cations.
This section describes key findings and outstanding questions in each of the steps LPS takes across the envelope in the context of the PEZ model.
5.1. Mechanism of LPS extraction from the inner membrane
Most ABC transporters come in three main varieties: i) importers, which bring in substrates such as nutrients across the cell membrane, ii) exporters such as multidrug exporters that expel substrates from the cytoplasm, and iii) flippases, such as the aforementioned MsbA, which translocate lipids from one leaflet of the membrane to the other.92–94 The LptB2FGC transporter extracts a lipid from a membrane, an atypical function for an ABC transporter that is akin to the role that the LolD2CE transporter plays in extracting newly synthesized lipoproteins from the inner membrane en route to the outer membrane.95 This unusual function makes the mechanism of how this transporter couples the function of the LptB ATPase to the extraction of LPS by LptFG particularly interesting. It is therefore not surprising that structural studies have revealed unique structural features of this ABC transporter. This extraction is the most complex step in LPS transport and the one that sets the entire system into function. Therefore, it is possible that, if it exists, regulation of LPS transport is exerted by controlling the extraction function of the LptB2FGC ABC transporter.
5.1.1. LPS entry into the Lpt transporter
After being flipped by MsbA across the inner membrane,23–27 LPS must be extracted from the outer leaflet of this bilayer and placed onto the periplasmic Lpt proteins. A body of genetic, biochemical, and structural evidence has led to a model for this extraction process stating that LPS present at the outer leaflet of the inner membrane enters a cavity formed by the TMDs LptFG and their partner LptC; when this cavity collapses, LPS is squeezed out and placed onto the periplasmic Lpt bridge; after extraction, the cavity opens for another round of transport (Fig. 4 and Fig. 5). In this process, the opening and closing of the LptFGC cavity is controlled and powered by the LptB cytoplasmic ATPase. Therefore, the first step in extraction is the entry of LPS into the LptFGC cavity.
Figure 4: Conformational states of the LptB2FG(C) complex.

Cartoon representations of structures of the LptB2FG(C) transporter in different conformations thought to represent steps of the transport cycle. From left to right: crystal structure of apo LptB2FGC (PDB 6MJP) representing the resting state, the cryo-EM structure of apo LptB2FG bound to an LPS molecule (shown in orange) prior to extraction from the cavity (PDB 6MHU), and the cryo-EM structure of vanadate-trapped LptB2FG (PDB 6MHZ) representing the post-extraction state (in this structure the periplasmic β-jellyroll domains of LptFG were not resolved). Panel (a) depicts a view of these structures from the membrane, panel (b) depicts a top-down view, cut away to show the substrate-binding cavity and the movement of the TM helices.
Figure 5: Model for the mechanism of LPS extraction from the inner membrane by the LptB2FGC ABC transporter.

For simplicity, the β-jellyroll domains of LptCFG are not shown. The LptB2FGC transporter starts the cycle in an apo conformation. 1) LPS enters into the V-shaped cavity of the transporter in an ATP-independent manner. 2) The TM helix of LptC is thought to dissociate from LptFG through an unknown mechanism. This causes a partial closure of the LptFG cavity, resulting in the formation of more high-affinity contacts between LPS and LptFG. 3) ATP (yellow) binds to LptB, triggering the closure of the LptB dimer and the LptFG cavity. As the cavity closes, LPS is expelled out onto the periplasmic bridge (not shown). 4) Finally, ATP is hydrolyzed and ADP and Pi (red and black) are released, which leads to the reopening of the LptB dimer and LptF cavity and the resetting of the transporter. How and when the TM helix of LptC dissociates and re-associates with LptFG is unknown.
It is currently thought that LPS diffuses into the LptFGC cavity after its translocation across the inner membrane. Presently, there is no evidence for MsbA interacting directly with the Lpt machine. In addition, MsbA’s substrate is further modified at the periplasmic side of the inner membrane. MsbA translocates LOS (i. e. Lipid A-core), so ligation of the O antigen by the WaaL ligase occurs at the periplasmic side of the inner membrane.96–98 In many bacteria, this site is also where other chemical modifications to the Lipid A and core components can be made, such as the addition of positively charged groups to the phosphates of Lipid A that confers resistance to cationic antimicrobial peptides.99 Moreover, the in vitro reconstitution of LPS transport does not require the presence of MsbA.39 Thus, LPS is thought to enter the LptB2FGC transporter independently of other proteins.
ABC transporters start their transport cycle in the resting state or apo conformation, unbound to substrate and nucleotide. During the transport cycle, the NBDs (here LptB2) bind to ATP and hydrolyze it, while the TMDs (LptFG) interact with and translocate the substrate.94 These stages require conformational changes, but how changes in the NBDs are coupled to those in the TMDs vary among ABC transporters. There is also great deal of diversity in how ABC transporters interact with their substrates. These differences among ABC transporters are key to their respective mechanism of function. An additional difference with this Lpt transporter is that it has an additional unique component, the LptC protein. Structure-function studies driven by genetic, biochemical and structural work have recently made great strides in uncovering mechanistic details of LptB2FGC.
The majority of the structures of both LptB2FG and LptB2FGC complexes are in the apo state, with the NBD dimers and a substrate-binding V-shaped cavity in the TMDs in a relatively open conformation (Fig. 4).68,76–78,100 These structures show that LptF and LptG each have 6 TM helices that together form a central V-shaped hydrophobic cavity that opens towards the periplasm.68,76–78,100 In the absence of LptC, TM1 of one TMD (LptF or LptG) interacts with TM5 of the other TMD.68,77,78 Furthermore, the overall architecture of the LptB2FG complex suggested these LptF-LptG contact sites could potentially function as lateral gates through which LPS could enter into the LptFG cavity from the outer leaflet of the inner membrane, as the TM1(LptF) and TM5(LptG) or the TM5(LptF) and TM1(LptG) have minimal contacts with one another.68,76–78,100 However, a surprising finding pertinent to these putative gates proposal was recently made when structures of the entire LptB2FGC complex revealed that the single TM helix of LptC is inserted between the putative gate formed by TM5(LptF) and TM1(LptG).68,77,78 These structural studies raised key questions. Through which of these potential gates does LPS enter into the cavity? And, what is the function of the TM helix of LptC?
Evidence for through which gate LPS enters into the transporter has been provided by both structures and crosslinking studies. The crystal structures of LptB2FGC best resolved the periplasmic β-jellyroll domains of LptF and LptC and showed that these domains are placed above the gate formed by TM1(LptF) and TM5(LptG). This placement likely obstructs the entry of the core oligosaccharide and O antigen portions of LPS through this gate.68 In addition, site-directed photo-crosslinking revealed that LPS can be crosslinked outside of the cavity to residues in TM5(LptF) and the TM helix of LptC, as well as inside the cavity to both the TM helix of LptC and TM1(LptG). In contrast, no crosslinks were detected between LPS and the putative gate formed by TM1(LptF) and TM5(LptG). Together, these results support the model that the true “gate” that LPS enters through is the one between TM5(LptF) and TM1(LptG), where the TM helix of LptC is located. This makes us wonder how LPS can enter the transporter when LptC is located in the gate and whether the TM helix of LptC plays a role in this process.
The presence of the intervening TM helix of LptC within LptFG is a novel structural feature in ABC transporters.68,77,78 The location of this TM helix is not an artifact, as it has been reported in multiple structures and confirmed through in vivo photo-crosslinking.68,77,78 Perhaps even more surprising given this positioning and its conservation among LptC orthologs is the lack of phenotype in cells in which the TM helix of LptC is cleaved or replaced.101 The only hint to the functional importance of LptC’s TM helix comes from in vitro ATPase assays demonstrating that complexes with full-length LptC have a marked decrease in ATPase activity when compared to those without LptC or containing an LptC variant that lacks its TM helix.68,77,78 Consequently, it has been proposed that the TM helix of LptC alters ATP hydrolysis in a way that allows for more efficient coupling of ATP hydrolysis and LPS extraction, possibly by limiting futile cycles of ATP hydrolysis.68 Although the role of LptC’s helix remains mysterious, these pieces of evidence indicate that, despite being part of the entry gate, the TM helix of LptC is not required for entry of LPS into the transporter’s cavity. In agreement, cryo-EM structures have also shown that LPS is able to occupy the LptFG cavity in LptB2FG complexes lacking LptC.77,78 All evidence to date therefore suggest that LPS enters into the cavity by transiently breaking the hydrophobic interactions between the TM helices at the TM5(LptF)-TM(LptC)-TM1(LptG) gate regardless of whether the TM helix of LptC is present or not.
Some ABC transporters require ATP binding to interact with their substrates.102,103 However, entry of LPS into LptB2FGC is ATP-independent. Using photo-crosslinking experiments, Owens et al. showed that without ATP, LPS can accumulate within the LptFGC cavity of the transporter.68 These data, in combination with the fact that LPS-bound structures lack nucleotide, indicate that ATP is not required for LPS entering into the cavity.68,77,78 One outstanding question about LPS entry into the transporter is whether entry of LPS into LptB2FGC is selective or not. It is possible that other lipids such as phospholipids flow through the transporter’s cavity but are not recognized as substrates, so they do not engage in transport; according to this model, LPS would have to be recognized as substrate in the cavity, likely through (some of) the interactions described in the next section. Alternatively, LPS molecules could specifically be recognized outside of the cavity, which would trigger selective entry. At present, these models remain untested.
5.1.2. Interactions between LPS and the LptFGC cavity
Specific interactions between substrates and their transporters are critical for proper transport. This is challenging for the Lpt system since LPS structure can vary between different species with respect to the type and number of sugars and fatty acid chains, and even between strains of one same species with respect to the structure of the core and O antigens. Interestingly, E. coli strains producing a minimal “LPS” structure (an intermediate of the Raetz pathway) can survive in the laboratory.104 This intermediate, called lipid IVA, is only composed of a phosphorylated glucosamine disaccharide with four fatty acid chains. Consequently, and as we describe below, the Lpt system must at least recognize features in this conserved minimal structure.
The first insight into how the TMDs LptFG interact with LPS came from studies in Burkholderia cenocepacia.105 The negatively charged phosphates on Lipid A can be modified in some bacteria to make them either neutral or positively charged, which confers resistance to cationic antimicrobial peptides.99 In B. cenocepacia, these Lipid A phosphates are constitutively modified with the positively charged 4-amino-4-deoxy-L-arabinose (L-Ara4N). Unlike in other organisms such as E. coli, this modification is essential for viability of B. cenocepacia.106 Hamad et al., found that lethality caused by the loss of this modification can be suppressed by changing a residue in TM1(LptG) with the lptG(D31H) allele. Bertani et al. later realized that this LptG residue covaries in organisms depending on the charge at the C1 and C4′ positions on their respective Lipid A structures.50 Structure-function analyses in E. coli guided by this observation and structural information led Bertani et al. to identify a cluster of functionally important residues in LptG’s TM1 that face the interior of the cavity. Substitutions at these residues result in phenotypes that are typical of Lpt defects, including increased sensitivity to hydrophobic compounds. Furthermore, this sensitivity can be suppressed by constitutively modifying the Lipid A phosphates with positively charged L-Ara4N and phosphoethanolamine. These results led to the proposal that the cluster of residues (K34, D37, Q38, K40, and K41) in the TM1 of LptG in E. coli establish critical contacts with the phosphates on Lipid A, providing the first evidence of LPS directly interacting with the LptFG cavity. It was also suggested that when these contacts cannot occur, changing the charge of Lipid A improves LPS transport because the modified substrate establishes different contacts with the transporter’s cavity.
Recent cryo-EM structures of LptB2FG and LptB2FGC complexes containing LPS have revealed that the hydrophobic acyl-chains of Lipid A contact the hydrophobic interior wall of the V-shaped cavity, while the phosphates at positions C1 and C4′ in Lipid A interact with charged and hydrophilic residues on the periplasmic rim of the transporter’s cavity (Fig. 4).77,78 Notably, the phosphate at the C1 position interacts with a charged pocket in TM1 of LptG containing the aforementioned residues (K34, D37, Q38, K40, and K41) that were identified by Bertani et al.50,77,78 Comparing the structures of LptB2FGC and LptB2FG complexes bound to LPS also reveals a shift in the contacts made between LPS and LptFG.77,78 As one might expect, when the TM helix of LptC is not between LptFG, the cavity is smaller (Fig. 4 and Fig. 5). This LptFG cavity makes more contacts with LPS than the larger LptFGC cavity. Additionally, both structures of the LptB2FGC-LPS complex have relatively poor resolution of LPS. The comparative difficulty of imaging LPS in this state suggests that LPS does not make optimal contacts with the transporter when the TM helix of LptC is present. Since the presence of the TM helix of LptC displaces TM1–3 of LptG, keeping the aforementioned charged pocket away from LPS, it was suggested by Li et al. that the formation of tight contacts between the TM1 of LptG with LPS might displace the TM helix of LptC out of the cavity.77 However, we currently do not know what causes the TM helix of LptC to move out from the LptFG cavity, when this step happens in the transport cycle, or how this helix re-associates with LptFG for a subsequent round of transport.
Together, these studies establish several interactions between both hydrophobic and hydrophilic portions of Lipid A and the transporter’s cavity. Some of these interactions specifically recognize LPS as substrate and can still occur in the minimal life-supporting LPS structure mentioned above. Whether other parts of the LPS molecule are recognized by this ABC transporter remains unclear. Biochemical data suggest that the LPS core affects transport since the efficiency of LPS transport in the in vitro full reconstitution system is affected by the structure of the core oligosaccharide.70 However, it remains unknown whether this effect is due to differences in substrate recognition by the transporter or differences in the physical properties of the molecules that alter their tendency to aggregate or diffuse into the transporter, which might indirectly affect the transport assay.
5.1.3. Collapse and re-opening of the cavity: expelling LPS and resetting the extractor
The extraction of LPS from the inner membrane requires energy derived from ATP in the cytoplasm (Fig. 5). With LPS inside the LptFGC cavity, the cytoplasmic NBDs (LptB2) need to bind and hydrolyze ATP, and coordinate this ATP cycle with the collapse and opening of the V-shaped cavity. Cavity collapse squeezes out LPS and moves it to the periplasmic Lpt bridge, while re-opening the cavity to the LPS-free state resets the transport cycle so that a new LPS molecule can enter the LptB2FGC extractor. These movements by the TMDs LptFG are driven by the ATP-dependent opening and closure cycle that the LptB dimer undergoes. In ABC transporters, the dimeric ATPase is in an open state when not bound to ATP.94 When the dimer binds to ATP, the NBDs attain a closed conformation that sandwiches two ATP molecules at the dimer interface. Each half of the two ATP-binding sites is asymmetrically provided by a monomer so that each site is mainly composed of the Walker A motif from one monomer and the signature motif from the other monomer. ATP hydrolysis then leads to the opening of the closed NBD dimer. Direct physical connections between LptB2 and LptFG, together with allosteric networks, are critical in coupling the ATP-driven opening and closure of the LptB dimer to the LptFGC transport cycle. To understand how the ATP hydrolysis cycle is coupled to the closing and opening of the LptFGC cavity, we need to know i) how and when in the transport cycle LptB is binding and hydrolyzing ATP, ii) how LptB is physically linked to LptFG, and iii) the residues that are important for coordinating NBD-TMD coupling along great distances in the transporter.
The NBDs are relatively well conserved across ABC transporters and share a set of motifs that are important for binding and hydrolyzing ATP, as well as for interacting with their TMD partners.94 By comparing sequence and structural homology to other NBDs, these motifs were identified in the first X-ray structure of LptB.55 Some of these motifs were altered to demonstrate the requirement for ATP hydrolysis in LPS transport through phenotypic analyses of mutants producing catalytically defective LptB variants.55 ABC transporters also share a general conserved structure, including the way in which their NBDs physically link to their TMDs. The first structures of the LptB dimer clearly showed a groove region that was later determined to accommodate coupling helices of LptFG.55 These are short helical segments of TMDs that serve as the primary points of NBD-TMD contact in ABC transporters.66,76 In LptFG, the coupling helices are located in the cytoplasm connecting their respective TM2 and TM3.66,76 They were first identified by Simpson et al. using bioinformatics, genetics, and biochemistry,66 and later confirmed by structures of the LptB2FG complex.76,100 Direct physical interaction between the coupling helices of LptFG and the structural groove of LptB in cells was demonstrated via in vivo site-directed photo-crosslinking and mass spectrometry; their functional connection was revealed through suppression analysis, since defects in LPS transport caused by substituting conserved glutamates located in the coupling helices of LptFG are suppressed by a specific change in the LptB groove region.66 These studies also revealed that although they are structurally similar, the coupling helices of LptFG play distinct roles in the transport cycle, as changing equivalent residues in each coupling helix does not confer the same phenotypes.66 Indeed, as we describe below, recent studies have proposed that the conserved glutamate in the coupling helix of LptF, but not the structurally equivalent residue in that of LptG, is critical in mediating the closure of the LptFG cavity.107
Further insight into the NBD-TMD coupling that is mediated by the interactions between the LptFG coupling helices and the groove region in LptB surprisingly came from the antibiotic novobiocin.53 This antibiotic targets DNA gyrase and is often used in assessing the effect of Lpt defects on outer-membrane permeability because it is hydrophobic 41,66,108,109 Serendipitously, novobiocin was found to suppress defects in LPS transport caused by specific mutations that we now know affect NBD-TMD coupling.53,56 Structural and biochemical studies have demonstrated that, in addition to binding to its canonical target, DNA gyrase, novobiocin also binds to the groove region of LptB, where it forms contacts with residues that were previously identified as being important for LptB function.53,56,66,107 As demonstrated by an in vitro reconstitution LPS release assay, through this binding, novobiocin increases LPS transport, which leads to suppression of coupling defects.53 The mechanism for how novobiocin increases LPS transport remains to be elucidated, but these findings open the door for the development of small molecules that might interfere with LPS transport. It also suggests that the activity of the transporter could be subject to regulation.
Although aforementioned work clearly demonstrated that the function of the LptB ATPase is needed for LPS transport,55 the role of the nucleotide in transport was undefined until recently. As described above, entry of LPS into the transporter does not require ATP. This raises the question of whether the collapse of the LptFG cavity that leads to the translocation of LPS onto the periplasmic bridge is dependent on ATP binding or hydrolysis. This question was first addressed by Simpson et al. when investigating a novel essential domain in LptB.56 The C-terminal domain (CTD) of LptB (residues 230–241 in E. coli) is conserved but unique among LptB orthologs. Its essential function depends on its ability to contact two motifs that are critical in ABC transporters, the Walker A and the switch helix.56 Suppression analysis of defects conferred by altering the CTD indicated that this domain is functionally connected to the ATP-binding sites, specifically the Walker A the signature helix that constitute the two halves of each site. Further analyses uncovered a striking genetic interaction between alleles altering the CTD and the signature helix. A change (R144H substitution) in the signature helix of LptB is conditionally lethal and one in the CTD (F239A) of LptB is lethal under all conditions; however, a mutant producing an LptB variant that has both of these lethal changes exhibits wild-type-like LPS transport. This surprising co-suppression indicates that these lethal changes have opposite effects on the same function of LptB so that they can compensate each other when combined. Combining these genetic results with biochemical characterization of ATP binding and ATP hydrolysis kinetics of reconstituted purified LptBFGC complexes led to the proposal that the R144H and F239A changes affect the opening and closing of the LptB dimer in opposite ways: while the R144H change causes ATP-binding defects that result in the dimer aberrantly favoring the open-conformation state, the F239A change prevents the opening of the closed LptB dimer, as it causes a defect in the ATP hydrolysis step that is required for its reopening. The data suggested that these defects reach a functional equilibrium so that the LptB(R144H/F239A) dimer can once again transition between the open and closed states, but hydrolyzing less ATP than wild-type LptB. Since LPS transport can be achieved without full restoration of ATP hydrolysis, it was proposed that ATP binding, and not hydrolysis, triggers the collapse of the LptFG cavity to transport LPS. Hydrolysis was proposed to be used by LptB to reset the transporter. Recent structural studies have further supported this model. A cryo-EM structure of the LptB2FGC transporter bound to the non-hydrolyzable ATP analog β-γ-imidoadenosine 5′-triphosphate shows the LptFG cavity in the collapsed state.78 This structural conformation is also nearly identical to the vanadate-trapped structure representing the ATP hydrolysis transition state (Fig. 4). Although this latter structure could not resolve the nucleotide, both studies show that binding to the nucleotide causes the closure of the LptFG cavity.77,78
More recently, genetics have provided additional mechanistic insight into the closure of the LptFG cavity that LptB triggers when binding ATP. Changes to the essential groove-region (residue E86 of LptB), like the R144H change, can reduce the ability of the LptB dimer to bind ATP and attain the closed-dimer conformation that triggers cavity collapse.107 Both of these defects in LptB can be suppressed by changing either the structure of LPS via ΔlpxM, or TM helices in LptG. LpxM is the last enzyme in the Raetz pathway and is responsible for adding the 6th acyl chain onto LPS. Thus, a deletion of lpxM results in cells that produce only penta-acylated LPS. The suppressing changes in LptG are located in TM2 and TM3 at the bottom of the substrate-binding cavity of the transporter. Thus, either reducing the hydrophobicity of Lipid A or altering the Lipid A-binding cavity suppresses defects in the ability of the LptFG cavity to close. These results are in agreement with the expectation that as the cavity closes to squeeze out LPS, hydrophobic interactions between Lipid A and residues lining the LptFG cavity are replaced by cavity-to-cavity hydrophobic interactions. The suppressors must be facilitating this transition by changing either the substrate or the substrate-binding cavity. These genetic studies by Lundstedt et al. provide evidence that the substrate LPS and its interactions with the LptFG cavity can directly affect the activity of the LptB ATPase. Furthermore, they support a model for the bidirectional coupling of LptB and LptFG where their movements are linked through a rigid body mechanism of transport that has been proposed for other ABC transporters and by structural studies on Lpt.77,78,110,111 In other words, the closure of the LptB dimer drives the closure of the LptFG cavity, and vice versa. Notably, the same suppressors that alter LPS structure and LptG can also fix defects conferred by substitutions at the conserved glutamate of the coupling of LptF, but not the equivalent substitution in that of LptG. Consequently, this evidence led Lundstedt et al. to suggest that the coupling helix of LptF must also be important in the cavity collapse step.107
Together, this large body of evidence leaves us with a model where LPS present at the outer leaflet of the inner membrane enters into the LptFGC cavity through the gate formed by TM1(LptG)-TM(LptC)-TM5(LptF) in an ATP-independent manner (Fig. 5). This entry likely involves the transient loss of hydrophobic contacts between these gate helices. The TM helix of LptC is likely to then leave the cavity through an unknown mechanism. This partially reduces the size of the cavity, which causes LPS to be pushed up in the cavity, where it can form more contacts with LptF and LptG. Next, LptB2 binds ATP, simultaneously closing the NBDs and TMDs. The collapse of the LptFG cavity causes the expulsion of LPS and its placement onto the periplasmic bridge. ATP is then hydrolyzed, and ADP and Pi are released, causing the reopening of the transporter.
5.1.4. Out of the cavity and onto the periplasmic bridge
Once LPS is squeezed out of the LptFG cavity, it somehow makes its way onto the β-jellyroll of LptC.54 Both LptF and LptG also have periplasmic β-jellyroll domains,76,100 either of which could plausibly link up to LptC and facilitate passing of LPS from the cavity. The crystal structures of LptB2FGC in the apo form for both Vibrio cholerae and Enterobacter cloacae obtained by Owens et al. have the β-jellyroll domains of LptFGC fully resolved.68 Both structures show the β-jellyroll portion of LptF connected to that of LptC, suggesting that after extraction, LPS travels to the β-jellyroll of LptF and then to that of LptC. There is significant evidence demonstrating that these structures are not artifacts, nor do they fail to report an alternative bridge formed between LptG and LptC. The first data suggesting that the β-jellyroll of LptF is part of the Lpt bridge was provided by Benedet et al., who found that lethality caused by an lptC deletion can be suppressed by changing LptF’s β-jellyroll residue R212.49 The authors suggested that LptF normally connects to LptC, and that changing R212 allows the β-jellyroll of LptF to now link to that of LptA, overcoming the need for LptC. Interestingly, the aforementioned structures by Owens et al. showed that LptF residue R212 is near the end of the β-jellyroll that normally associates with the β-jellyroll of LptC.68 In addition, in vivo crosslinking studies by Owens et al. showed contacts between the edges of the β-jellyroll of LptF and LptC, confirming that LptF and LptC do in fact connect to one another in cells. Similar crosslinks were not detected between LptG and LptC.68 Importantly, LPS crosslinks to a residue in the concave portion of the β-jellyroll in LptF that is along the LptF-LptC interface, but no crosslinks between LPS and the β-jellyroll of LptG can be found. All these data suggest that LPS travels from the β-jellyrolls of LptF to that of LptC. This model was further supported by using an LptF variant with two cysteine substitutions strategically positioned in its β-jellyroll domain.68 These substituted cysteines are close enough to form a disulfide bond, which would close the entry into the bottom of the β-jellyroll of LptF. Individually, each cysteine substitution confers no defects, but together they are lethal and prevent LPS transport. Furthermore, although we lack direct biochemical evidence of the formation of the disulfide bond between the two substituted cysteines, it has been shown that transport can be restarted in vitro by the addition of reducing agents, which are thought to break the putative disulfide bond and allow LPS to move through the β-jellyroll of LptF once more. Thus, the data we have to date strongly supports a model where LPS is only able to travel through the β-jellyroll of LptF to reach the periplasmic bridge. However, we must still recognize that the lack of evidence is not evidence for the lack of interactions between LPS and/or LptC and the β-jellyroll of LptG. It is therefore still unclear what the functional role of the β-jellyroll of LptG is.
We should also note that, unlike in other ABC transporters, where substrates diffuse away from their transporter, LPS is loaded onto a protein bridge that is physically connected to the TMDs LptFG. How is it that newly extracted LPS does not slide back into the cavity of LptFG when it opens for the next round of transport? Owens et al. proposed that the bottom of the β-jellyroll of LptF has a valve-like mechanism that opens and closes to prevent LPS from sliding back into the cavity of the transporter once it reaches the β-jellyroll of LptF.68 The aforementioned putative disulfide crosslink suggests that the closure of LptF is physically possible and does prevent LPS transport. The bottom loops of the β-jellyroll domain of LptF also are in a more open or more closed state in the two different crystal structures, which implies that this movement is possible. An alternative but not exclusive model for preventing back-flow is that direction of transport after extraction is simply affinity driven. Partly supporting this idea is evidence showing that LPS cannot move from LptA to LptC in vitro.72 It is also currently not known if an LPS molecule that has been newly extracted from the LptFG cavity stays within the β-jellyroll of LptF or somehow travels onto LptC.
5.2. Traversing the periplasm
The Lpt bridge mediates transit of LPS from the inner to the outer membrane while protecting the hydrophobic portion of Lipid A from the aqueous periplasm. All bridge proteins, LptFCAD, have β-jellyroll domains with a C-shaped fold that contains a hydrophobic interior that is believed to conceal the fatty acyl chains of Lipid A (Fig. 3).71,72,75,76 These domains have been shown to interact with LPS through photo-crosslinking.54,68,112 In addition, in vitro experiments have shown that LptA co-elutes with “LPS” regardless of the presence of O antigen or core oligosaccharide, indicating that the Lipid A portion of LPS is sufficient for the interaction.113
There were initially two models for how Lpt could facilitate LPS movement across the periplasm: i) LptA is a soluble chaperone that binds LPS and freely diffuses between LptFGC at the inner membrane and LptDE at the outer membrane to transport LPS, analogous to how LolA functions in the Lol system to transport lipoproteins to the outer membrane.114 Or ii) LptA is physically connected to both LptC and LptDE, forming a continuous bridge across the periplasm. However, strong evidence accumulated early on against LptA functioning in the same way as LolA. Using a method previously developed for LolA,115 Tefsen et al. showed in 2004, before LptA was even identified, that newly synthesized lipoproteins were released from spheroplasts when periplasmic contents (which contain LolA) were added back to the spheroplasts.59 In contrast, the authors could not observe release of newly synthesized LPS into the soluble fraction. Importantly, they could demonstrate that LPS could still be transported to remnants of the outer membrane that were physically connected to the spheroplasts. This was the first evidence that LPS transport did not involve a soluble periplasmic chaperone like LolA. Following this observation, Chng et al. used sucrose gradient fractionation and co-purification to show that all Lpt proteins co-fractionate, suggesting for the first time that they form a trans-envelope protein bridge connecting the inner and outer membranes.38
In 2012, two landmark papers provided strong evidence for the bridge architecture and function. Using in vivo site-specific photo-crosslinking, Freinkman et al. showed head-to-tail connections between the edges of the β-jellyrolls of bridge proteins.65 The C-terminus of LptC connects to the N-terminus of LptA, whose C-terminus connects to the N-terminus of LptD (Fig. 5). This study also showed that LptA can oligomerize with itself in vivo.65 Although in vitro LptA can also oligomerize,71,116 it is still unknown if one or more LptA proteins form the bridge in the periplasm. Nevertheless, we refer the reader to the crystal structure of an LptA oligomer, as it illustrates how β-jellyroll domains could form a bridge.46 Okuda et al. further demonstrated that LPS is able to crosslink to the inside, but not outside, of the β-jellyrolls of LptA and LptC. Notably, this work was also the first to reconstitute the inner membrane portion of the Lpt complex in vitro, show that continuous rounds of ATP hydrolysis are required for LPS movement onto LptA, and propose the idea that LPS travels as a stream through the Lpt bridge - the PEZ model.54 Ultimately, the full in vitro reconstitution was accomplished by Sherman et al. in 2018. This tour de force reconstituted LPS transport from inner-membrane-like liposomes containing purified LptB2FGC complexes to outer-membrane-like liposomes containing purified LptDE complexes. This transport required LptA to physically bridge both types of liposomes, demonstrating once and for all that the Lpt machine forms and functions as a continuous bridge. This body of work provides support of the PEZ model for transport in which each new LPS molecule entering the transporter at the inner membrane pushes those already in the bridge towards the outer membrane.39 Whether the β-jellyrolls are passive structures or contribute somehow to the directional movement of LPS is unclear.
5.3. LPS insertion into the outer leaflet of the outer membrane
After LPS reaches the last β-jellyroll of the periplasmic bridge, the N-terminal domain of LptD, it must be inserted into the outer leaflet of the outer membrane by the LptDE translocon (Figs. 2 and 3). There is no indication that LPS ever exists in the inner leaflet of this membrane. LptD and LptE form a plug-and-barrel structure where the LptE lipoprotein resides in the lumen of the C-terminal β-barrel portion of LptD.73,75,117 How does this complex accomplish the task of properly inserting LPS exclusively into the outer leaflet, all while navigating both hydrophilic and hydrophobic portions of the molecule and in the absence of an obvious energy source? Before we discuss its mechanism of function, it is important to understand the structure of this complex.
Preceding the crystal structures of LptDE, genetic and biochemical studies showed not only that LptDE form a plug-and-barrel structure, but also that LptE plays a key role in the biogenesis of LptD.89,117,118 In fact, studying the biogenesis of the LptDE complex has significantly contributed to our understanding of folding and insertion of β-barrel proteins in the outer membrane. The first lpt mutant allele to be discovered (lptD4213) encodes a defective substrate of the BAM complex, which assembles OMPs.81,83–85 LptD assembly, and especially that of LptD4213, is very slow, which has enabled the analysis of folding intermediates.82,119 Interested readers should refer to studies cited here.48,82,118–124
The first x-ray structures of the LptDE translocon offered important clues as to how LPS might be translocated from the periplasmic β-jellyroll of LptD to the outer leaflet of the outer membrane.73,75 These structures show that LptD is a 26-strand β-barrel with a hydrophilic interior that is closed off from the environment by extracellular loops that fold into the lumen. As expected, the lumen of the LptD β-barrel also accommodates LptE, which serves as a plug (Fig. 3). Where the β-jellyroll of LptD meets its β-barrel, there is a hole that is predicted to be in the membrane. This hole is lined on either side with hydrophobic residues that have been shown to be functionally important.75,90 The current model is that the Lipid A portion of LPS moves from the β-jellyroll of LptD through the hole directly into the outer membrane. According to this model, the hydrophilic portion of LPS would then need assistance to traverse the hydrophobic core of the membrane. Structures of the LptDE complex have suggested a path. The hydrophilic portion of LPS is predicted to first enter the periplasmic side of the LptD barrel when the hydrophobic portion of the molecule transitions from the β-jellyroll of LptD into the membrane. The hydrophilic portion of LPS is then proposed to exit from the LptD barrel through a lateral gate formed between β strands 1 and 26, the seam of the barrel. Typically, β-barrels are closed at the seam formed between the first and last β strands by a series of stable hydrogen bonds.125 However, in LptD, the first β-strand is distorted by two conserved prolines located in β strands 1 and 2, which results in β-strand strands 1 and 26 only forming three main-chain hydrogen bonds with each other.75 Molecular dynamics simulations propose that this is in fact the weakest part of LptD’s β-barrel and that substituting those prolines with alanines stabilizes it.73,126 Indeed, these prolines in β strands 1 and 2 are critical for function in cells.126 There are also four aromatic residues along this putative lateral gate that have been proposed to interact with the hydrophilic core of LPS as it passes through the lumen. Interestingly, below this gate are two luminal loops that are either in open or closed states depending on the LptD structure. Since deletion of these loops is lethal, as is introducing cysteines within each loop that can putatively form disulfide bonds, it has been suggested that they must open to allow the hydrophilic portion of LPS to exit the LptD lumen.90 More recently, a molecular dynamics study has supported this model for LPS translocation across the outer membrane and proposed that the presence of LPS may trigger the opening of both the loops and the β-barrel gate.127
What is LptE’s role in this transport process? LptE has been shown to bind to LPS in vitro.74,89 Surface plasmon resonance and electron microscopy showed that not only LptE binds to LPS but also prevents its aggregation.74,128 Based on these results and LptE’s location in the lumen of the LptD barrel, it was proposed that LptE might facilitate transport by interacting with LPS and breaking the strong lateral interactions that LPS molecules form with one another, facilitating its insertion into the outer membrane. However, more work is needed to fully understand how LptE facilitates LPS transport.
Interestingly, recent evidence suggests that the LptDE translocon may control the activity of the Lpt system.128 In a fully reconstituted in vitro system, Xie et al. observed that if the outer-membrane-like liposomes containing LptDE were preloaded with saturating amounts of LPS, it prevented additional transport of LPS from the inner-membrane-like liposomes containing the LptB2FGC transporter. Curiously, it also inhibited the ATPase activity of these LptB2FGC complexes. The simplest explanation for these observations is that the outer membrane components can signal back to the inner membrane components to control LPS transport and avoid overloading the outer membrane with LPS. The outer leaflet of the outer membrane is not very fluid because of the close packing of LPS molecules and the high density of acyl chains in LPS.1 In comparison, phospholipid bilayers are highly fluid and allow high rate of lateral diffusion of lipids and proteins. Lpt machines might then have to be shut off once they have filled a particular area of the membrane with LPS molecules. The mechanism for this suggested regulation of LPS transport across the cell envelope and how it might affect the stability of Lpt bridges is not understood. It is also unclear how LPS transport may be coordinated with the biogenesis of other envelope components to ensure proper assembly of the outer membrane and growth of the entire cell envelope. In section 9.2.2., we describe some recent developments that are shedding some light into the coordination of phospholipid and LPS synthesis, but whether they are connected to their transport is unknown.
We note that cells likely regulate LPS transport at several earlier points in addition to regulating the process at the point of LPS assembly at the outer membrane. The most obvious way to regulate the flux of LPS is by controlling LPS biosynthesis. The first committed step in that pathway involves the enzyme LpxC, and we discuss this regulation briefly at the end of the review. Another obvious point of regulation is by controlling the assembly and/or disassembly of the Lpt machine. We currently know the sites of interaction between Lpt proteins, and that the inner- and outer-membrane sub-complexes can be stably purified from cells. However, we do not understand how cells assemble Lpt proteins to form the trans-envelope machine. We can envision that cells might assemble this machine in such a way as to prevent sending LPS molecules through an incomplete bridge to nowhere. There is evidence that interactions between the β-jellyroll domains of LptA and LptD do not occur unless the LptDE translocon is properly assembled.117 Whether the remaining contacts between β-jellyroll domains in the periplasmic bridge are also subject to regulation driven by quality control of the assembly of the inner-membrane sub-complex is unknown. We also do not know if the assembly of the power engine of transport, the LptB2FGC ABC transporter, is orderly regulated to control when LPS transport can occur, but point out the unusual operon structure that controls expression of lptCAB could allow for regulation here as well. Specifically, ATPases of ABC transporters are usually encoded in operons with their transmembrane domain partners, but LptB is encoded with LptC and LptA, not LptFG; in addition, the first 31 base pairs of the lptA gene overlap with lptC, which might increase the efficiency in assembling their respective factors into a complex. Lastly, there is nothing known about the stability of these bridges in a cell (or even their existence) and it is possible that when LPS transport stops, cells disassemble the machine.
So far, we have reviewed the main findings that have led to remarkable progress on understanding LPS transport. This is not only critical for our understanding of bacterial physiology, but also in the fight to combat the increasing threat of antibiotic resistance in Gram-negative bacteria. The development of new antibiotics has been greatly inhibited by the impermeability of the outer membrane conferred by its asymmetric structure. Not surprisingly, efforts are being made to target the Lpt system with inhibitors that would break this barrier. For further reading on this topic, we refer readers to a review by Lehman and Grabowicz.2
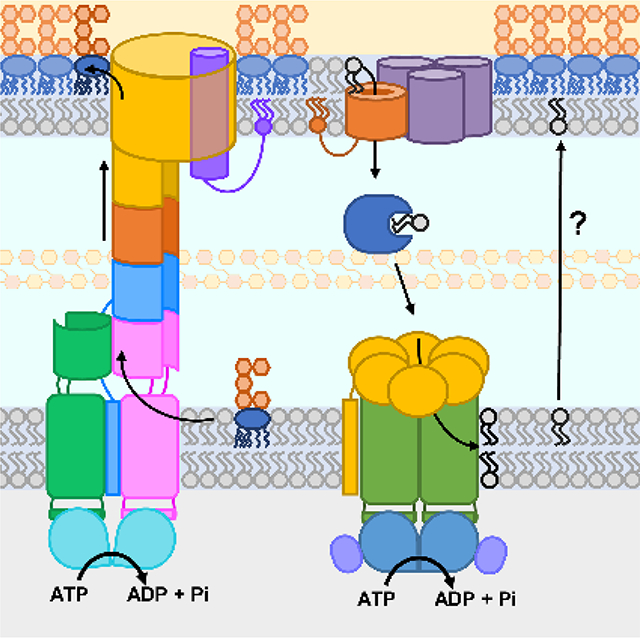
6. Phospholipid transport between the two membranes is bidirectional
In order to build the outer membrane, newly synthesized phospholipids must be assembled into the outer membrane. As we discussed, LPS is delivered to the outer leaflet of the outer membrane directly via a protein bridge. But how phospholipids are asymmetrically positioned in the inner leaflet of the outer membrane remains a mystery despite almost half a century of work. Two basic models can be considered: either the cell assembles a phospholipid bilayer and then converts the outer membrane to an asymmetric membrane with the insertion of LPS selectively into the outer leaflet and the concomitant removal of phospholipids, or alternatively the cell directly makes an asymmetric bilayer by specifically delivering phospholipids to the inner leaflet of the outer membrane somehow coordinated with LPS assembly.
In 1977, Jones and Osborn demonstrated that phospholipids added outside the cell could be trafficked to the inner membrane and then equilibrated with the outer membrane after being modified at the inner membrane.129 This simple experiment demonstrated that mechanisms existed for bidirectional transport of phospholipids in cells. The biosynthesis of phospholipids is completed at the inner membrane; therefore, anterograde transport from inner to outer membrane is needed to fill the outer membrane. To maintain the asymmetric LPS/phospholipid structure of the outer membrane, retrograde transport of phospholipids in cells could be required to specifically remove phospholipids that somehow mis-localize to the outer leaflet of the outer membrane or to remove phospholipids from the inner leaflet in order to balance the growth of the inner leaflet with that of the outer leaflet. Perhaps the simplest explanation, and one first considered by Osborn herself, is that diffusive bidirectional transport of phospholipids could occur through zones of adhesion between the inner and outer membranes. Because in this model, the outer leaflet of the inner membrane and the inner leaflet of the outer membrane are continuous, this mechanism would allow direct coordination of growth of the inner and outer membranes.
Phospholipid transport in Gram-negative bacteria has been challenging to study because it is difficult to identify and characterize the factors involved. As we discuss in sections below, it has also been difficult to determine the directionality of phospholipid transport even when components have been identified. We still lack reliable biochemical approaches and a broader collection of mutants to allow these biochemical methods to be validated so the field can move forward as described in the Lpt system. Nevertheless, recent advances in the study of phospholipid transport is generating considerable excitement and debate. Here we describe the current understanding of phospholipid transport, focusing on the Mla (maintenance of lipid asymmetry) system, as it is the best characterized phospholipid transporter to date.
7. Maintaining the lipid asymmetry of the outer membrane
There is a characteristic increase in sensitivity of cells to antibiotics and detergents when LPS transport is impaired. These outer membrane defects arise because the levels of phospholipids in the outer leaflet of the outer membrane increase. Having phospholipids rather than LPS in the outer leaflet is detrimental to the cell. It is thought that phospholipids and LPS do not pack well when they are in the same leaflet, leading to their segregation into domains.43,44 This segregation creates phospholipid bilayer patches in the outer membrane, which are more permeable to antibiotics and detergents.1,45
The cell has various mechanisms to remove phospholipids from the outer leaflet of the outer membrane. As mentioned earlier, the outer membrane enzyme PagP is capable of transferring a palmitoyl acyl chain from a phospholipid to the hexa-acylated disaccharide of LPS, Lipid A.130 Both of these PagP substrates are present in the outer leaflet if a phospholipid bilayer forms in the outer membrane. Under these conditions, PagP activity increases, as detected by the increased ratio of hepta-acylated to hexa-acylated LPS. The cell can also remove phospholipids from the outer leaflet of the outer membrane by breaking down phospholipids with the phospholipase PldA. This enzyme is capable of sequentially removing acyl chains of phospholipids.131,132 The resultant fatty acids released are proposed to be recycled to the cytoplasm and serve as signals that increase LPS synthesis, thus PldA serves as a sensor of outer membrane lipid asymmetry.133
As mentioned earlier, Osborn and collaborators showed that phospholipids transport between the inner and outer membranes is bidirectional.129 The enzymes mentioned above clearly remove, but do not move phospholipids. However, three decades after Osborn’s studies, Malinverni and Silhavy finally discovered components of a phospholipid transport system.134 They proposed this transporter mediates retrograde transport from the outer to the inner membrane and named it Mla to reflect its function in the maintenance of lipid asymmetry at the outer membrane. The system is composed of six Mla proteins (MlaABCDEF) and the outer membrane β-barrel protein OmpC (Fig. 6).134,135 Although there is controversy surrounding the directionality of Mla (see section 9.2.1.), the most reasonable model for how Mla functions is that the outer membrane lipoprotein MlaA associates with an OmpC trimer, allowing binding of phospholipids in the outer leaflet of the outer membrane and transfers these phospholipids to the periplasmic chaperone MlaC. MlaC then shuttles these phospholipids to the MlaFEDB ABC transporter, which delivers them to the inner membrane. In this section, we describe the most salient work on the Mla system starting with its discovery and describing its function following phospholipids through the proposed retrograde path (Fig. 6).
Figure 6: Model for retrograde phospholipid transport by the Mla system.

Phospholipids that are mislocalized to the outer leaflet of the outer membrane first interact with the MlaA-OmpC complex and transverse the bilayer through a central pore in MlaA. Next, the phospholipid molecule is transferred to the periplasmic chaperone MlaC. MlaC moves across the periplasm and docks onto the IM MlaFEDB complex at the inner membrane. The phospholipid molecule is moved through this complex back into the inner membrane in an ATP-dependent fashion. It is unknown whether the phospholipid is placed in the inner or outer leaflet of the inner membrane. To avoid cluttering, the transmembrane domain of only one of the six MlaD monomers is depicted.
7.1. Discovery of the Mla system
The Mla system was first identified by the homology of a subset of Mla proteins (MlaF, MlaE, and MlaD) to conserved transporters that play a role in uptake of lipids in Actinobacteria and inter-membrane retrograde phospholipid transport in chloroplasts.136–138 Malinverni and Silhavy set out to find the Gram-negative homologs of these proteins and clarify their function.134 They found homologs encoded at the mlaFEDCB locus, which in some bacteria often also includes the mlaA gene.134 In E. coli, mlaA is not part of the mlaFEDCB locus, but when any of the mla genes were deleted, cells became hypersensitive to detergents, suggesting that Mla’s normal role in maintaining outer membrane integrity is to prevent phospholipids from accumulating in the outer leaflet of the outer membrane. Indeed, their findings showing that sensitivity of an mla null could be suppressed by overexpressing the outer membrane lipase PldA, and that mla deletions caused a PagP-dependent increase in hepta-acylated LPS implied that mla mutants accumulate phospholipids at the cell surface. Together, these results functionally linked Mla to maintaining the lipid asymmetry of the outer membrane.
Chng and coworkers later showed that the outer membrane lipoprotein MlaA co-purifies with the trimeric OmpC and OmpF beta-barrels;135 however, only the interaction with OmpC was found to be important for Mla function. Recent in vivo photo-crosslinking and molecular dynamics simulations further support the relevance of the OmpC-MlaA interaction in cells.139 In support of retrograde transport, deleting ompC also results in accumulation of phospholipids at the outer leaflet of the outer membrane. Epistasis analysis of the accumulation of phospholipids caused by ompC and mlaA alleles combined with their physical interactions led to the proposal that OmpC functions in the Mla pathway.135
7.2. Entry of phospholipids into MlaA at the outer membrane
In the model described above and in Fig. 6, the phospholipids that accumulate in the outer leaflet of the outer membrane are the substrates of the Mla transporter. Therefore, to understand this transport system, it is critical to elucidate how the outer membrane lipoprotein MlaA (and its partner OmpC) can specifically transport outer leaflet phospholipids. A major contribution in addressing this question came from x-ray crystallography studies of MlaA-OmpF complexes by the van den Berg group. Abellón-Ruiz et al. revealed that MlaA assembles into a ring-shaped α-helical structure that contains a central pore (Fig. 7).140 The relative positioning of MlaA and OmpF in the structure would place the MlaA ring flat on the inner leaflet of the outer membrane, leaving its pore accessible only to phospholipids in the outer leaflet of the bilayer. The structures of OmpF and OmpC are very similar. Even though OmpC is the functional partner in vivo, it is reasonable to assume that the structures of the two protein complexes would be similar. Consistent with this, both OmpF and OmpC interact with MlaA in cells. The inside of MlaA is relatively hydrophilic and was shown to bind to the headgroup of phospholipids via molecular dynamics.140 Cysteines have also been introduced into MlaA in an attempt to lock and prevent the opening of its pore through the formation of a disulfide bond. Although direct biochemical evidence of the formation of the putative disulfide bond was not provided, this approach abolished MlaA function. Importantly, loss of MlaA function required the presence of both cysteines and could be reversed in the presence of a reducing agent, supporting the hypothesis that this pore facilitates phospholipid translocation. More recently, Yeow et al. used in vivo structural probing to show that the central pore of MlaA is solvent-exposed (presumably to the periplasm).139 Based on in vivo structure-function analysis, these authors proposed that a group of aspartates within the pore and an adjacent hairpin loop might move to open and close the pore in order to facilitate transport.
Figure 7: Structures of Mla factors.

Cartoon representations of the structures of Mla proteins shown in their respective cellular compartments. Structures were obtained from individual components or sub-complexes as follows: The MlaD 6E2F2B2 complex (PBD 6IC4), MlaD bound to a PL molecule (PBD 5UWA), MlaA complexed with the OmpF trimer (PDB 5NUQ). MlaA can associate with OmpF and OmpC (not shown), but only OmpC has been shown to be functionally relevant.
Abellón-Ruiz et al. also provided structural insight into a defective gain-of-function Mla variant that Sutterlin et al. had previously proposed to mediate the aberrant translocation of phospholipids from the inner leaflet to the outer leaflet of the outer mebrane.52,140 In a suppressor selection using an lpt mutant with reduced LPS transport, Sutterlin et al. serendipitously found a dominant gain-of-function mla allele referred to as mla*.52 Mla* has a two-residue deletion in the first helix of Mla and, in otherwise wild-type cells, causes mislocalization of phospholipids to the cell surface even when other mla genes are deleted. The crystal structure of MlaA revealed that the two-residue deletion in Mla* is predicted to open up the central pore, leaving it accessible to inner leaflet phospholipids.140 This structural defect would then explain how MlaA* can seemingly work in reverse and cause the accumulation of phospholipids in the outer leaflet of the outer membrane even in the absence of other Mla proteins.
Although multiple approaches have offered significant support for MlaA being able to shuttle phospholipids and have highlighted the function of its central structural pore, we still do not understand how phospholipids are driven through MlaA, the conformational changes that MlaA might undergo during this process, and how OmpC functions with MlaA. Moreover, MlaA has thus far not been shown to directly bind phospholipids.
7.3. Transfer of phospholipids from the outer membrane to the periplasm
MlaA is proposed to transfer phospholipids to MlaC, which shuttles them across the periplasm to the IM (Fig. 6). In agreement, MlaA was shown to interact in cells with MlaC via in vivo photo-crosslinking.141 However, the two proteins do not co-purify, nor does MlaC co-purify with the MlaFEDB complex, likely due to the transient nature of these interactions. MlaC is a soluble periplasmic protein akin to LolA, the periplasmic chaperone for lipoprotein transport.114,142 MlaC binds with high affinity to phospholipids, as it readily purifies already bound to phospholipids.141,143 Indeed, its crystal structure revealed that MlaC has a hydrophobic pocket that was bound to a phospholipid molecule.144 In this structure, the phospholipid headgroup is solvent-exposed, while its acyl chains are sheltered in the interior of the pocket. Interestingly, the aforementioned sites on MlaC that crosslink to MlaA are adjacent to this phospholipid-binding pocket,141,144 but it is not currently known what facilitates the transfer of phospholipids from MlaA to MlaC. Given MlaC’s high affinity for phospholipids and the lack of an obvious energy source in the periplasm and outer membrane, the current model suggests that transfer is affinity driven.141 Understanding the mechanism will require the biochemical in vitro reconstitution of phospholipid transfer between MlaA and MlaC, and the use of mutant proteins. Developing this reconstitution is challenging, as it requires distinguishing the location of phospholipids within liposomes.
7.4. Transferring phospholipids from periplasmic MlaC to the IM
After receiving a phospholipid molecule from MlaA at the outer membrane, MlaC transports it through the periplasm to MlaFEDB at the inner membrane (Figs 5 and 6). The MlaFEDB proteins co-purify as a complex and constitute an ABC transporter.144–146 MlaF and MlaE homodimers function as the NBDs and TMDs, respectively. The cytoplasmic accessory protein MlaB, which interacts with the MlaF ATPase, is important for both stabilizing the MlaFEDB complex and its ATPase activity.146 However, how and why MlaB is required for proper function of the transporter remains to be elucidated. MlaD is anchored to the inner membrane by a single TM helix, and its periplasmic region contains a mammalian cell entry (MCE) domain. In didermic bacteria, MCE domains have been implicated in lipid uptake, while in eukaryotic organelles, they have been proposed to transport phospholipids in retrograde fashion.147 Several studies have shown that the MCE periplasmic domain of MlaD readily co-purifies with phospholipids and forms a homo-hexameric ring-like structure with a hydrophobic pore.141,143,144 In vivo photo-crosslinking studies also showed that the periplasmic side of the MlaD ring interacts with MlaC.141 These MlaC-contact sites lie along MlaD dimer interfaces near the opening to the central hydrophobic pore of the hexameric ring structure.
The structure of the MlaF2E2D6B2 complex shows that the MlaD hexamer rests on top of the periplasmic side of the MlaE dimer, the TMDs of the ABC transporter.144 This structure, combined with the findings described above, provide a model for how the transfer of phospholipid cargo from MlaC to MlaFEDB and ultimately the inner membrane might occur. In this model, MlaC docks onto MlaD and delivers a phospholipid molecule to the MlaD hexameric ring. The MlaFEDB ABC transporter could provide the energy required to promote this transfer of the phospholipid molecule from the high-affinity binding MlaC protein to MlaD. Phospholipids could then travel through the central hydrophobic pore of the MlaD ring and, ultimately, be delivered to either the outer leaflet of the inner membrane or somehow translocated to the inner leaflet possibly through the action of the Mla ABC transporter.
Proposing the above model for the transfer of phospholipids from MlaC to the inner membrane and that Mla functions in retrograde transport seems at odds with biochemical experiments that surprisingly showed that MlaD spontaneously transfers phospholipids to MlaC in vitro.141,143 This transfer order appears to suggest that Mla functions in anterograde transport of phospholipids from the inner to the outer membrane instead of retrograde transport. In section 9.2.1., we will discuss the recent controversy that those and other results have sparked about Mla directionality.148 However, we want to stress here that the aforementioned genetic work that first led Malinverni and Silhavy to propose that Mla functions in retrograde transport in E. coli is incompatible with the anterograde transport model.134 The loss of Mla leads to an accumulation of phospholipids in the outer leaflet of the outer membrane, a defect that can be suppressed by the action of the outer membrane lipase PldA. In contrast, as pointed out by Chng and collaborators, the fact that MlaD spontaneously transfers phospholipids to MlaC in vitro is still compatible with retrograde phospholipid transport.141 The strong binding of MlaC to phospholipids might be required in cells to drive the transfer of phospholipids from MlaA to MlaC at the outer membrane. The energy derived from ATP provided by the MlaFEDB ABC transporter is then likely to be required in releasing the tightly bound phospholipid from periplasmic MlaC to MlaD. This proposed mechanism resembles how substrates are transferred from periplasmic high-affinity substrate-binding proteins to the TMDs of some ABC importers.149 According to this explanation, the reported ATP-independent transfer of phospholipids from MlaD to MlaC that spontaneously occurs in vitro would not be physiologically relevant. We believe that Mla catalyzes retrograde phospholipid transport (Fig. 6), however, we also recognize that much work is still needed to understand how Mla functions. We expect this will require developing the in vitro reconstitution of this transport system and the synergistic combination of genetic and biochemical studies - allowing one to compare in vivo and in vitro results - which proved to be critical to study the Lpt system. [The work proposing that Mla mediates anterograde transport is discussed in section 9.2.1., which focuses on anterograde phospholipid transport].
8. Tol-Pal: maintaining balanced outer membrane biogenesis through retrograde phospholipid transport
As we discussed in section 7, the Mla system has been proposed to fix problems in lipid asymmetry caused by the accumulation of phospholipids in the outer leaflet of the outer membrane. This situation can arise when LPS synthesis or transport are defective. It can also occur when there are not enough divalent cations to bridge proximal phosphates on adjacent LPS molecules at the cell surface, as this situation leads to the release of LPS molecules into the environment.52,150 Ultimately, it is a break in the balance of the growth or homeostasis of the two leaflets of the outer membrane that creates a lipid asymmetry.
How cells coordinate growth and assembly of all envelope layers and compartments is not understood. For the outer membrane, this coordination must ensure not only that both leaflets of the outer membrane ultimately grow while maintaining the asymmetric lipid composition in each leaflet, but also that there be mechanisms to allow for insertion of large numbers of OMPs during growth that span both leaflets of the outer membrane. The huge amount of membrane proteins added to the outer membrane by BAM suggests that the cell may therefore require other systems that remove bulk phospholipid quickly during growth. Indeed, cells coordinate synthesis of phospholipids and LPS by balancing the distribution of a shared precursor.151–154 As discussed in section 9, little is known about inner-to-outer membrane anterograde phospholipid transport, so it is unclear how it might be coordinated with LPS transport. In addition, Shrivastava and Chng have recently proposed that cells normally transport excess phospholipids to the outer membrane to ensure that there is always enough material to build both leaflets.155 This model would explain how there are enough phospholipid molecules to fill the outer leaflet of the outer membrane when levels of LPS decrease, but it would also demand the need for retrograde phospholipid transport systems when LPS and OMP biogenesis are fully functional. Shrivastava and Chng have proposed that the Tol-Pal system, which is distinct from Mla, mediates this type of phospholipid retrograde transport in order to maintain outer membrane lipid homeostasis.
Tol-Pal is a five-protein trans-envelope complex that has long been implicated in cell division and maintenance of outer membrane integrity.156–159 For decades, we have known that loss of Tol-Pal leads to sensitivity to antibiotics and detergents. More recently, Chng and coworkers have shown that E. coli tol-pal deletion strains accumulate phospholipids in the outer membrane, which likely results in the observed increase in phospholipids at the outer leaflet.155 The accumulation of phospholipids at the outer membrane has also been observed in Salmonella strains lacking a functional Tol-Pal complex.160Additionally, by following turnover of pulse-labeled outer membrane phospholipids in vivo, Shrivastava et al. showed that E. coli tol-pal deletion strains are defective in retrograde phospholipid transport, a defect that can be partially suppressed by overexpressing the Mla system.155 The authors specifically proposed that Tol-Pal is involved in transporting back to the inner membrane the aforementioned excess of phospholipids that cells normally send out to the outer membrane during growth. According to their model, in wild-type cells, the Tol-Pal system mediates the bulk of retrograde phospholipid transport, while Mla transports the small population of phospholipids that are mislocalized to the outer leaflet of the outer membrane.155,161 This model would implicate Tol-Pal in the retrograde transport described by Osborn decades ago.129 However, the mechanism by which the Tol-Pal complex affects phospholipid transport remains elusive, and an important outstanding question is whether its proposed role is direct or indirect. In fact, as reviewed by Szczepaniak et al., recent work has led to the proposal that Tol-Pal functions to coordinate the remodeling of the peptidoglycan cell wall and to stabilize contacts between the cell wall and the outer membrane during cell division.162 These more recent Tol-Pal findings could suggest it affects retrograde transport indirectly. Or, alternatively, Tol-Pal could function in retrograde phospholipid transport as suggested by the Chng group but, in a more general way, to integrate multiple biogenesis pathways at the outer membrane that may require the cell create space in one or both leaflets.
9. Anterograde phospholipid transport
Anterograde transport of phospholipids is essential to build the outer membrane, since these molecules are synthesized at the inner membrane. Unlike the great progress made in understanding the biogenesis of other outer membrane components, there is astonishingly little understood about the transport of phospholipids from the inner to the outer membrane. It is unclear why identifying the factors involved has been so difficult, but an obvious explanation is that several systems are capable of mediating anterograde phospholipid transport and their functional redundancy has made their discovery and characterization difficult. It is also possible that anterograde phospholipid transport is not directly mediated by a proteinaceous transporter. Two types of transport mechanisms have been proposed to possibly mediate inner-to-outer membrane anterograde phospholipid: by passive diffusion through hemi-fusion contacts sites between the inner and outer membranes, or by protein-dependent systems involving either protein bridges or soluble chaperones (Fig. 8). Vesicle-mediated transport has been disfavored given the size of the periplasm and the fact that the size of vesicles would be expected to be too large to pass through the relatively small “pores” in the lattice-like peptidoglycan cell wall in the periplasm.163,164 In this section, we discuss evidence supporting each of the remaining two models.
Figure 8: Potential mechanisms of anterograde phospholipid transport.

From left to right: i) Zones of hemi-fusion between the inner and outer membranes, termed Bayer junctions, would allow free bidirectional diffusion of phospholipids between the two membranes. ii) A chaperone-mediated mechanism would rely on a soluble protein that moves phospholipids through the periplasm. This chaperone would likely be loaded by an inner membrane component and eventually dock to an outer membrane protein that would incorporate the phospholipid molecule into the inner leaflet of the outer membrane iii) A protein bridge or tunnel between inner- and outer-membrane components would allow phospholipids to transverse the periplasm. The inner membrane component would load phospholipids on to the bridge, while the outer membrane portion would incorporate the PLs into the inner leaflet of the outer membrane. The outer membrane components in the protein-based models are depicted as lipoproteins that are embedded in the inner leaflet similarly to MlaA, but there is no evidence suggesting this type of factor.
9.1. Bidirectional diffusion of phospholipids through membrane contact sites
Phospholipid transport between the inner and outer membranes was first proposed to be mediated through physical hemi-fusions or adhesion sites between these two membranes (Fig. 8). In 1968, Manfred Bayer examined thin-sections of plasmolyzed E. coli cells with electron microscopy and reported areas of fusion or adhesion between the inner and outer membranes that became known as Bayer junctions or Bayer bridges.165 A decade later, as we mentioned earlier, Jones and Osborn demonstrated that Salmonella could import exogenous labelled phospholipids to the inner membrane, where they could be modified by specific enzymes.129 Since these phospholipids could rapidly equilibrate between the inner and outer membranes, Jones and Osborn proposed that phospholipid transport was likely to occur by diffusion through zones of adhesion between the inner and outer membrane. However, the existence of zones of adhesion was challenged by Kellenberger, who deemed Bayer junctions as artifacts of chemical fixation of electron-microscopy samples.166 Although Bayer later rebutted Kellenberger’s claim, the existence of Bayer junctions remained controversial.167
The idea that zones of adhesion exist and mediate diffusional flow of phospholipids between the inner and outer membranes has been recently revived by the Silhavy and Huang groups in their studies of the aforementioned mlaA* mutant.52 Recall that MlaA* is a dominant-defective protein that translocates phospholipids into the outer leaflet of the outer membrane. By following membrane dynamics in wild-type and mlaA* through live-cell microscopy, Sutterlin et al. observed energy-independent lipid flow between both membranes. The authors proposed that zones of hemi-fusion are responsible for the diffusive transport of phospholipids between the inner and outer membranes, although they could not visualize them owing to the technical challenges involved. If these zones of adhesion between the inner and outer membrane exist, there is no information as to how they might form and prevent the diffusion of lipoproteins and newly synthesized glycolipids like LPS between the two membranes. It is also unclear whether the observations by Sutterlin et al. could be related to the aforementioned retrograde transport involving the Tol-Pal system.52,155 Evidence for these hemi-fusion zones will likely depend on their visualization, which will be challenging and require the development of new microscopy methods.
9.2. Protein-mediated models for anterograde phospholipid transport
In the model described above, bidirectional phospholipid transport would be mediated by a continuous lipid bilayer connecting the outer leaflet of the inner membrane to the inner leaflet of the outer membrane. Although proteins might be involved in the biogenesis of these structures and their ability to prevent diffusion of other molecules, the act of transport would not be mediated by proteins. In contrast, the alternative model posits that phospholipids could travel from the inner to the outer membrane through protein-mediated transport. There are two types of protein-based transport systems that could shuttle phospholipids between the two membranes: one type involves a periplasmic chaperone intermediate, while the other type is mediated by a bridge-like protein structure like in the Lpt system.
9.2.1. Soluble-chaperone model for anterograde phospholipid transport
Section 7 described Mla as a system that transports phospholipids in retrograde fashion from the outer leaflet of the outer membrane to the inner membrane. The genetic data obtained in E. coli strongly supports this model and argues against a role in anterograde phospholipid transport, since its loss leads to the accumulation of phospholipids at the cell surface and can be suppressed by increasing the levels of the PldA outer membrane phospholipase.134,141 More recently, additional support for Mla functioning in retrograde transport has been independently provided by the Kahne and Trent groups in Acinetobacter baumannii, a Gram-negative bacterium that naturally produces LOS (i. e. LPS lacking O antigen) but can survive without it.32,168,169 Although viable, A. baumannii cells lacking LOS have severe growth defects. These defects are mitigated by the loss of the Mla system and the PldA phospholipase.168,169 The simplest explanation for these findings is that cells lacking LOS require phospholipids to build the outer leaflet of their outer membrane, and removing Mla and PldA improves their growth because they normally remove phospholipids from the outer leaflet of the outer membrane through retrograde transport or degradation, respectively.
Nevertheless, controversy about the directionality of Mla-dependent phospholipid transport has recently stemmed from independent studies led by the Miller and Knowles groups.143,145 The opposing views on the directionality of Mla have been thoroughly reviewed elsewhere.148 In summary, as mentioned in section 7.4., biochemical studies have demonstrated that purified MlaD loaded with phospholipids can transfer them to the periplasmic protein MlaC in vitro, but not vice-versa.141,143 The inner membrane MlaFEDB ABC transporter was also shown to transfer phospholipids to MlaC independently of ATP hydrolysis.143 In addition, Kamischke et al. showed, using membrane fractionation and pulse-chase experiments in A. baumannii, that mla deletions result in an overall decrease in phospholipids in the outer membrane.145 Based on these results, it has been proposed that Mla facilitates anterograde transport. However, there are caveats to consider from these biochemical experiments. Disrupting Mla function causes cells to release outer membrane vesicles,170,171 but these vesicles were not accounted for in the pulse-chase experiments measuring phospholipids in the inner and outer membrane fractions in A. baumannii.145 An additional complication of this experiment is that altering membrane lipid composition can affect how membranes will fractionate in density gradients. In addition, in the in vitro experiments, transfer of phospholipids from MlaFEDB and MlaD to MlaC was shown to occur in the absence of ATP.141,143 As described earlier, this transfer is likely to not be physiologically relevant, but the result of MlaC’s high affinity for phospholipids, which is needed for periplasmic MlaC to be able to pull phospholipids from MlaA at the outer membrane; phospholipid transfer from MlaC to MlaD is likely dependent on ATP hydrolysis catalyzed by the MlaDEFB ABC transporter.141 Importantly, there is still no genetic evidence supporting a role for Mla in anterograde transport. Instead, the aforementioned genetic data from both E. coli and A. baumannii strongly argues against anterograde directionality but supports the retrograde model.134,169 Thus, this controversy is based on the disagreement between genetic and biochemical work. It is critical that future studies combine in vivo and in vitro work that complements each other and that employs both a collection of mutants affecting different steps in the pathway and challenging in vitro reconstitution experiments. This strategy has proven fruitful in elucidating mechanistic details of LPS transport by the Lpt system.
Given these conflicting views, and the lack of any other candidate pathway, we currently lack solid evidence for the existence of any system involving a soluble chaperone that facilitates anterograde phospholipid transport.
9.2.2. Protein-bridge model for anterograde (or bidirectional?) phospholipid transport
The last model for phospholipid transport to the outer membrane is one facilitated by a protein bridge or tunnel forming a continuous inner-to-outer membrane connection. This type of transport could potentially be anterograde or bidirectional, and driven by an energy-demanding extraction at the inner membrane or passive diffusion, respectively. Although there is no direct evidence for this type of transporter, several potential candidates have emerged.
Recently, several proteins containing MCE domains have been proposed to function as trans-membrane periplasmic tunnels that could transport phospholipids between the two membranes. Originally discovered in mycobacteria, MCE domains have been implicated in the maintenance of the cell envelope and lipid transport in two-membrane bacteria and organelles.172 E. coli encodes three proteins with MCE domains: MlaD (from the Mla system), PqiB, and YebT (renamed LetB for lipophilic envelope-spanning tunnel B).147,173 As we described for MlaD, these proteins co-purify with phospholipids,144,146,174,175 and their loss confers varying degrees of sensitivity to detergents, indicating that they could play a role in phospholipid transport.147 As we have described MlaD in previous sections, we describe here recent studies on PqiB and LetB.
The model for PqiB and LetB functioning as tunnel-like transporters is mainly founded on structural studies. PqiB is encoded in an operon with pqiA and pqiC, which code for an integral inner membrane protein and an outer membrane lipoprotein, respectively. Based on this operon structure, these proteins have been proposed to form a complex spanning the envelope, but we lack experimental evidence of this interaction.173 Similarly, letB is in an operon with letA, which encodes an integral inner membrane protein predicted to interact with LetB because of this genetic arrangement. Both PqiB and LetB possess several MCE domains that, like in MlaD, were shown to form ring-like structures with central hydrophobic pores through which phospholipids might travel.144 Ekiert et al. showed that PqiB has an N-terminal TM helix that anchors the protein to the inner membrane, and a periplasmic region composed of three stacked ring-like MCE domains and a long C-terminus that forms a syringe-like structure. These latter domains are predicted to span the periplasm.144 LetB also has an N-terminal TM helix, but its periplasmic region is composed of seven stacked MCE rings that make a continuous tube that likely spans the periplasm.144,174,175 These massive structures have been proposed to serve as tubes through which phospholipids can travel. In support of this model, the length of LetB’s tube is functionally relevant, since deleting MCE domains results in functional defects.175 Moreover, the fact that substrate(s) labeled with 32P can be photo-crosslinked to the interior of the LetB channel has led to the proposal that LetB could transport phospholipids between the two membranes. However, the identity of the phosphate-containing substrate(s) remains unknown. Although the structure is certainly suggestive of this function and purified LetB has been shown to bind phospholipids,144 it is important to note that: i) the recent crosslinking experiments were done with a non-functional LetB variant lacking its TM helix; and ii) the crosslinking step was done in lysed cells.175 Although the physiological relevance of this crosslinking is questionable, one of the LetB structures shows density in the tunnel that could correspond to phospholipids according to modeling.174 In addition, the recent high-resolution structures of LetB from Isom et al. and Liu et al. show rings in different conformations, suggesting that they can undergo conformational changes between constricted and open states.174,175 Based on these observations, it has been proposed that lipids could potentially be moved through the channel by being squeezed through these conformational changes.175
Despite their ability to bind phospholipids and their impressive tube-like structures suggesting a role in phospholipid transport across the cell envelope,144,174,175 the phenotypes so far reported by the loss of these proteins are modest. In E. coli, deleting either pqiAB or letAB on their own only results in mild sensitivity to select detergents.147 The triple mlaD pqiAB letAB deletion mutant showed increased sensitivity to detergents and more phospholipids at the outer leaflet of the outer membrane than the mlaD single mutant, despite the single and double pqiAB or letAB mutants behaving like wild type.147,173 The authors concluded that PqiAB and LetAB have distinct but somewhat overlapping functions with MlaD; this explanation could suggest that LetB and PqiB can at least transport phospholipids in the same direction as Mla.147 Moreover, even if Mla mediated anterograde transport, the mild defects of the pqiAB and letAB mutants, and the fact that the mlaD pqiAB letAB double and triple deletion mutants grow without severe defects indicate that these proteins are not the only or the primary transporters of phospholipids to the outer membrane. Regardless of their specific role in maintaining the outer membrane, pqiAB and letAB play, unlike mla genes, relatively minor roles as indicated by the mild phenotypes resulting from their deletion. Additional work is clearly needed to elucidate the functional role of PqiB and LetB in the cell.
The only other protein that has been implicated in anterograde transport of phospholipids is YejM and its homolog PbgA in Salmonella and Shigella. However, recent work calls into question this proposed role and has provided strong evidence for a new function in envelope biogenesis. YejM/PbgA is an essential tetrameric inner membrane protein that contains an essential N-terminal TM portion, and a non-essential linker and periplasmic domain.176,177 Mutations to the C-terminal periplasmic domain of YejM/PbgA result in an increased sensitivity to hydrophobic antibiotics and temperature sensitivity, suggesting it plays important role in membrane biogenesis.176,178,179 In Salmonella, the C-terminal periplasmic domain of YejM/PbgA was proposed to be essential for the transport of cardiolipin to the outer membrane, but only when cardiolipin levels increase in response to the activation of the PhoPQ two-component system, indicating that it is not the primary CL transporter.177 Shigella mutants lacking the C-terminal periplasmic domain of YejM/PbgA show no transport of cardiolipin to the outer membrane irrespective of PhoPQ activation, but curiously maintain wild-type levels of cardiolipin in the inner membrane.180 Cardiolipin was also shown to form contacts with YejM/PbgA, but these contacts are in both the linker region and TM portion of the protein.181 Although this set of evidence links YejM/PbgA and cardiolipin, we still lack direct evidence for YejM/PbgA being a transporter. Importantly, the essential function of YejM/PbgA can also not be attributed to cardiolipin transport. The periplasmic domain, which is implicated in cardiolipin transport, is not essential, nor is the presence of cardiolipin itself.177,182 Furthermore, yejM E. coli mutants lacking the periplasmic C-terminal domain exhibit outer membrane permeability defects independent of the presence of cardiolipin.183
Recently, the essential function of YejM/PbgA has been attributed to a novel role in regulating LPS biosynthesis. A link between YejM/PbgA and LPS synthesis was first established by Cian et al. in Salmonella. These authors found that outer-membrane permeability and virulence defects caused by the loss of the periplasmic domain of YejM/PbgA could be suppressed by changes in genes involved in regulating LPS synthesis: lapB, ftsH, and lpxC.184 Three recent independent studies by Guest et al., Fivenson and Bernhardt, and Clairfeuille et al. have provided further insight into this regulation. They demonstrated that the essential and primary function of YejM/PbgA in E. coli is to regulate LPS levels by repressing the function of LapB (also known as YciM), which normally activates the FtsH protease to degrade LpxC.185–187 LpxC catalyzes the first committed step in LPS synthesis,188 and levels of LPS synthesis are controlled through degradation of LpxC by the LapB-FtsH protease complex.151,152,189 YejM/PbgA downregulates the LapB-FtsH-dependent degradation of LpxC through an unknown mechanism involving direct physical contact with LapB.185–187 Structural studies by Clairfeuille et al. uncovered a functionally-relevant lipid A-binding motif in YejM/PbgA located along the periplasmic leaflet of the inner membrane.187 Furthermore, the authors demonstrated that accumulation of LPS at the outer leaflet of the inner membrane decreases LpxC levels, while its accumulation at the inner leaflet of the outer membrane increases LpxC levels. As a result, they proposed that the levels of LPS in the outer leaflet of the inner membrane regulate LPS synthesis by binding or unbinding the YejM/PbgA-LapB complex, which controls the degradation of LpxC by FtsH. Future studies should focus on elucidating the mechanistic basis of this regulation.
10. Conclusions
Over the last 50 years, monumental progress has been made towards understanding the processes involved in the biogenesis of the outer membrane. Multi-protein machines have been discovered to transport outer membrane components across the cell envelope so that they can be assembled into an asymmetric lipid bilayer, and we are just beginning to elucidate details about their mechanism of function. Studying transport of lipids between the inner and outer membranes has proven challenging. LPS transport is the best understood process, although, as we have pointed out in this review, mechanistic details of the Lpt system and how its function may be regulated remain to be elucidated. Significant progress has also been made to understand how transport of phospholipids maintains the asymmetry of the outer membrane via the Mla system, but we critically need the development of a full reconstitution of the Mla system to study its function and resolve the controversy of transport directionality. Unfortunately, the mechanism for anterograde transport remains mysterious, with no clear identified mechanism of bulk transport to the outer membrane, although exciting candidates have been recently identified. Much work is needed to understand their function, but their identification as potential lipid transporters has been an important step. We also still know very little about how cells regulate the different outer membrane lipid transport systems so that, by coordinating their activities, the asymmetric structure of the outer membrane can be established and maintained. We believe that the next frontier in outer membrane research is bound to be focused on addressing these fundamental questions and that progress will undoubtedly rely on understanding the mechanistic details of how the different lipid transporters function. Drawing from the great progress made in understanding the Lpt machine, we anticipate that the synergistic combination of genetic, biochemical, and structural studies will be key in these future endeavors to elucidate how Gram-negative bacteria build their complex cell envelope.
Acknowledgements
This work was supported by the National Institutes of Health awards R01-AI081059 and R01-AI153358 to D.K. and R01-GM100951 to N.R.
Biographies
Emily Lundstedt received her B.S. in Medical Microbiology in 2011 from the University of New Hampshire in 2011. She is currently a Ph.D. student in the laboratory of Natividad Ruiz at The Ohio State University. Her research interest is studying the mechanism of LPS transport by the Lpt machine.
Daniel Kahne is the Higgins Professor of Chemistry and Chemical Biology and of Molecular and Cellular Biology at Harvard University, Massachusetts, USA, as well as Professor of Biological Chemistry and Molecular Pharmacology at Harvard Medical School. He has longstanding interests in understanding the biogenesis of the cell envelope of Gram-negative bacteria, in particular peptidoglycan biosynthesis and outer membrane assembly.
Natividad Ruiz obtained her B.A. from the University of Kansas and her Ph.D. from Washington University in St. Louis, where she worked with Professor Michael Caparon. She then joined Professor Thomas J. Silhavy’s laboratory at Princeton University to become a postdoc and later a research scientist. In 2010, she joined The Ohio State University, where she is a Professor of Microbiology. Her laboratory investigates the biogenesis of the cell envelope in Gram-negative bacteria, with an emphasis on glycolipid transport.
References
- (1).Nikaido H Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lehman KM; Grabowicz M Countering Gram-negative antibiotic resistance: Recent progress in disrupting the outer membrane with novel therapeutics. Antibiotics (Basel) 2019, 8, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Muhlradt PF; Golecki JR Asymmetrical distribution and artifactual reorientation of lipopolysaccharide in the outer membrane bilayer of Salmonella typhimurium. Eur. J. Biochem. 1975, 51, 343–352. [DOI] [PubMed] [Google Scholar]
- (4).Kamio Y; Nikaido H Outer membrane of Salmonella typhimurium: accessibility of phospholipid head groups to phospholipase c and cyanogen bromide activated dextran in the external medium. Biochemistry 1976, 15, 2561–2570. [DOI] [PubMed] [Google Scholar]
- (5).Lopez-Lara IM; Geiger O Bacterial lipid diversity. Biochim. Biophys. Acta. Mol. Cell. Biol. Lipids. 2017, 1862, 1287–1299. [DOI] [PubMed] [Google Scholar]
- (6).Yasuhiro K; Yuzuru A; Shoshichi N Composition and turnover of the phospholipids in Escherichia coli. Biochim. Biophys. Acta, Lipids Lipid Metab. 1967, 144, 382–390. [PubMed] [Google Scholar]
- (7).Yamagami A; Yoshioka T; Kanemasa Y Differences in phospholipid composition between wild strains and streptomycin resistant mutants of certain enteric bacteria. Jpn. J. Microbiol. 1970, 14, 174–176. [DOI] [PubMed] [Google Scholar]
- (8).Furse S; Scott DJ Three-dimensional distribution of phospholipids in gram negative bacteria. Biochemistry 2016, 55, 4742–4747. [DOI] [PubMed] [Google Scholar]
- (9).Osborn MJ; Gander JE; Parisi E; Carson J Mechanism of assembly of the outer membrane of Salmonella typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J. Biol. Chem. 1972, 247, 3962–3972. [PubMed] [Google Scholar]
- (10).Ishinaga M; Kanamoto R; Kito M Distribution of phospholipid molecular species in outer and cytoplasmic membrane of Escherichia coli. J. Biochem. 1979, 86, 161–165.383707 [Google Scholar]
- (11).Zgurskaya HI; Rybenkov VV Permeability barriers of Gram-negative pathogens. Ann. N. Y. Acad. Sci. 2020, 1459, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vergalli J; Bodrenko IV; Masi M; Moynie L; Acosta-Gutierrez S; Naismith JH; Davin-Regli A; Ceccarelli M; van den Berg B; Winterhalter M et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Microbiol. 2020, 18, 164–176. [DOI] [PubMed] [Google Scholar]
- (13).Masi M; Winterhalter M; Pagès J-M In Bacterial cell walls and membranes; Springer International Publishing: Cham, 2019; 79–123 [Google Scholar]
- (14).Radolf JD; Kumar S The Treponema pallidum outer membrane. Curr. Top. Microbiol. Immunol. 2018, 415, 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Holst O The structures of core regions from enterobacterial lipopolysaccharides - an update. FEMS Microbiol. Lett. 2007, 271, 3–11. [DOI] [PubMed] [Google Scholar]
- (16).Willis LM; Whitfield C Structure, biosynthesis, and function of bacterial capsular polysaccharides synthesized by ABC transporter-dependent pathways. Carbohydr. Res. 2013, 378, 35–44. [DOI] [PubMed] [Google Scholar]
- (17).Kuhn HM; Meier-Dieter U; Mayer H ECA, the enterobacterial common antigen. FEMS Microbiol. Rev. 1988, 4, 195–222. [DOI] [PubMed] [Google Scholar]
- (18).Bertani B; Ruiz N Function and biogenesis of lipopolysaccharides. EcoSal Plus 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang YM; Rock CO Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [DOI] [PubMed] [Google Scholar]
- (20).Raetz CR; Whitfield C Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ruiz N; Kahne D; Silhavy TJ Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat. Rev. Microbiol. 2009, 7, 677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Whitfield C; Trent MS Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem. 2014, 83, 99–128. [DOI] [PubMed] [Google Scholar]
- (23).Polissi A; Georgopoulos C Mutational analysis and properties of the msbA gene of Escherichia coli, coding for an essential ABC family transporter. Mol. Microbiol. 1996, 20, 1221–1233. [DOI] [PubMed] [Google Scholar]
- (24).Zhou Z; White KA; Polissi A; Georgopoulos C; Raetz CR Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 1998, 273, 12466–12475. [DOI] [PubMed] [Google Scholar]
- (25).Doerrler WT; Gibbons HS; Raetz CR MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem. 2004, 279, 45102–45109. [DOI] [PubMed] [Google Scholar]
- (26).Mi W; Li Y; Yoon SH; Ernst RK; Walz T; Liao M Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 2017, 549, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Karow M; Georgopoulos C The essential Escherichia coli msbA gene, a multicopy suppressor of null mutations in the htrB gene, is related to the universally conserved family of ATP-dependent translocators. Mol. Microbiol. 1993, 7, 69–79. [DOI] [PubMed] [Google Scholar]
- (28).Samuel G; Reeves P Biosynthesis of O-antigens: genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res. 2003, 338, 2503–2519. [DOI] [PubMed] [Google Scholar]
- (29).Liston SD; Mann E; Whitfield C Glycolipid substrates for ABC transporters required for the assembly of bacterial cell-envelope and cell-surface glycoconjugates. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862, 1394–1403. [DOI] [PubMed] [Google Scholar]
- (30).Kalynych S; Morona R; Cygler M Progress in understanding the assembly process of bacterial O-antigen. FEMS Microbiol. Rev. 2014, 38, 1048–1065. [DOI] [PubMed] [Google Scholar]
- (31).Whitfield C; Williams DM; Kelly SD Lipopolysaccharide O-antigens-bacterial glycans made to measure. J. Biol. Chem. 2020, 295, 10593–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Moffatt JH; Harper M; Harrison P; Hale JD; Vinogradov E; Seemann T; Henry R; Crane B; St Michael F; Cox AD et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob. Agents Chemother. 2010, 54, 4971–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liu D; Reeves PR Escherichia coli K12 regains its O antigen. Microbiology 1994, 140, 49–57. [DOI] [PubMed] [Google Scholar]
- (34).Sperandeo P; Pozzi C; Deho G; Polissi A Non-essential KDO biosynthesis and new essential cell envelope biogenesis genes in the Escherichia coli yrbG-yhbG locus. Res. Microbiol. 2006, 157, 547–558. [DOI] [PubMed] [Google Scholar]
- (35).Ruiz N; Gronenberg LS; Kahne D; Silhavy TJ Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5537–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wu T; McCandlish AC; Gronenberg LS; Chng SS; Silhavy TJ; Kahne D Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11754–11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Braun M; Silhavy TJ Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol. Microbiol. 2002, 45, 1289–1302. [DOI] [PubMed] [Google Scholar]
- (38).Chng SS; Gronenberg LS; Kahne D Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry 2010, 49, 4565–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sherman DJ; Xie R; Taylor RJ; George AH; Okuda S; Foster PJ; Needleman DJ; Kahne D Lipopolysaccharide is transported to the cell surface by a membrane-to-membrane protein bridge. Science 2018, 359, 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhang G; Meredith TC; Kahne D On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ruiz N; Wu T; Kahne D; Silhavy TJ Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem. Biol. 2006, 1, 385–395. [DOI] [PubMed] [Google Scholar]
- (42).Ruiz N; Kahne D; Silhavy TJ Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 2006, 4, 57–66. [DOI] [PubMed] [Google Scholar]
- (43).Lasch P; Schultz CP; Naumann D The influence of poly-(L-lysine) and porin on the domain structure of mixed vesicles composed of lipopolysaccharide and phospholipid: an infrared spectroscopic study. Biophys. J. 1998, 75, 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Takeuchi Y; Nikaido H Persistence of segregated phospholipid domains in phospholipid--lipopolysaccharide mixed bilayers: studies with spin-labeled phospholipids. Biochemistry 1981, 20, 523–529. [DOI] [PubMed] [Google Scholar]
- (45).Vaara M Antibiotic-supersusceptible mutants of Escherichia coli and Salmonella typhimurium. Antimicrob. Agents Chemother. 1993, 37, 2255–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sperandeo P; Lau FK; Carpentieri A; De Castro C; Molinaro A; Deho G; Silhavy TJ; Polissi A Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J. Bacteriol. 2008, 190, 4460–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sperandeo P; Cescutti R; Villa R; Di Benedetto C; Candia D; Deho G; Polissi A Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J. Bacteriol. 2007, 189, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chimalakonda G; Ruiz N; Chng SS; Garner RA; Kahne D; Silhavy TJ Lipoprotein LptE is required for the assembly of LptD by the beta-barrel assembly machine in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 2492–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Benedet M; Falchi FA; Puccio S; Di Benedetto C; Peano C; Polissi A; Deho G The lack of the essential LptC protein in the trans-envelope lipopolysaccharide transport machine Is circumvented by suppressor mutations in LptF, an inner membrane component of the Escherichia coli transporter. PLoS One 2016, 11, e0161354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bertani BR; Taylor RJ; Nagy E; Kahne D; Ruiz N A cluster of residues in the lipopolysaccharide exporter that selects substrate variants for transport to the outer membrane. Mol. Microbiol. 2018, 109, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yao Z; Davis RM; Kishony R; Kahne D; Ruiz N Regulation of cell size in response to nutrient availability by fatty acid biosynthesis in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E2561–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Sutterlin HA; Shi H; May KL; Miguel A; Khare S; Huang KC; Silhavy TJ Disruption of lipid homeostasis in the Gram-negative cell envelope activates a novel cell death pathway. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).May JM; Owens TW; Mandler MD; Simpson BW; Lazarus MB; Sherman DJ; Davis RM; Okuda S; Massefski W; Ruiz N et al. The antibiotic novobiocin binds and activates the ATPase that powers lipopolysaccharide transport. J. Am. Chem. Soc. 2017, 139, 17221–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Okuda S; Freinkman E; Kahne D Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science 2012, 338, 1214–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Sherman DJ; Lazarus MB; Murphy L; Liu C; Walker S; Ruiz N; Kahne D Decoupling catalytic activity from biological function of the ATPase that powers lipopolysaccharide transport. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 4982–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Simpson BW; Pahil KS; Owens TW; Lundstedt EA; Davis RM; Kahne D; Ruiz N Combining mutations that inhibit two distinct steps of the ATP hydrolysis cycle restores wild-type function in the lipopolysaccharide transporter and Shows that ATP binding triggers transport. mBio 2019, 10, e01931–01919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Meredith TC; Mamat U; Kaczynski Z; Lindner B; Holst O; Woodard RW Modification of lipopolysaccharide with colanic acid (M-antigen) repeats in Escherichia coli. J. Biol. Chem. 2007, 282, 7790–7798. [DOI] [PubMed] [Google Scholar]
- (58).Bos MP; Tefsen B; Geurtsen J; Tommassen J Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9417–9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Tefsen B; Geurtsen J; Beckers F; Tommassen J; de Cock H Lipopolysaccharide transport to the bacterial outer membrane in spheroplasts. J. Biol. Chem. 2005, 280, 4504–4509. [DOI] [PubMed] [Google Scholar]
- (60).Jia W; Zoeiby AE; Petruzziello TN; Jayabalasingham B; Seyedirashti S; Bishop RE Lipid trafficking controls endotoxin acylation in outer membranes of Escherichia coli. J. Biol. Chem. 2004, 279, 44966–44975. [DOI] [PubMed] [Google Scholar]
- (61).Chin JW; Martin AB; King DS; Wang L; Schultz PG Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11020–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Liu CC; Schultz PG Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- (63).Dormán G; Prestwich GD Benzophenone photophores in biochemistry. Biochemistry 1994, 33, 5661–5673. [DOI] [PubMed] [Google Scholar]
- (64).Nickerson NN; Mainprize IL; Hampton L; Jones ML; Naismith JH; Whitfield C Trapped translocation intermediates establish the route for export of capsular polysaccharides across Escherichia coli outer membranes. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 8203–8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Freinkman E; Okuda S; Ruiz N; Kahne D Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry 2012, 51, 4800–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Simpson BW; Owens TW; Orabella MJ; Davis RM; May JM; Trauger SA; Kahne D; Ruiz N Identification of residues in the lipopolysaccharide ABC transporter that coordinate ATPase activity with extractor function. mBio 2016, 7, e01729–01716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Okuda S; Tokuda H Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5877–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Owens TW; Taylor RJ; Pahil KS; Bertani BR; Ruiz N; Kruse AC; Kahne D Structural basis of unidirectional export of lipopolysaccharide to the cell surface. Nature 2019, 567, 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Rubino FA; Mollo A; Kumar S; Butler EK; Ruiz N; Walker S; Kahne DE Detection of transport intermediates in the peptidoglycan flippase MurJ identifies residues essential for conformational cycling. J. Am. Chem. Soc. 2020, 142, 5482–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Xie R; Taylor RJ; Kahne D Outer membrane translocon communicates with inner membrane ATPase to stop lipopolysaccharide transport. J. Am. Chem. Soc. 2018, 140, 12691–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Suits MD; Sperandeo P; Deho G; Polissi A; Jia Z Novel structure of the conserved gram-negative lipopolysaccharide transport protein A and mutagenesis analysis. J. Mol. Biol. 2008, 380, 476–488. [DOI] [PubMed] [Google Scholar]
- (72).Tran AX; Dong C; Whitfield C Structure and functional analysis of LptC, a conserved membrane protein involved in the lipopolysaccharide export pathway in Escherichia coli. J. Biol. Chem. 2010, 285, 33529–33539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Dong H; Xiang Q; Gu Y; Wang Z; Paterson NG; Stansfeld PJ; He C; Zhang Y; Wang W; Dong C Structural basis for outer membrane lipopolysaccharide insertion. Nature 2014, 511, 52–56. [DOI] [PubMed] [Google Scholar]
- (74).Malojcic G; Andres D; Grabowicz M; George AH; Ruiz N; Silhavy TJ; Kahne D LptE binds to and alters the physical state of LPS to catalyze its assembly at the cell surface. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9467–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Qiao S; Luo Q; Zhao Y; Zhang XC; Huang Y Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 2014, 511, 108–111. [DOI] [PubMed] [Google Scholar]
- (76).Luo Q; Yang X; Yu S; Shi H; Wang K; Xiao L; Zhu G; Sun C; Li T; Li D et al. Structural basis for lipopolysaccharide extraction by ABC transporter LptB2FG. Nat. Struct. Mol. Biol. 2017, 24, 469–474. [DOI] [PubMed] [Google Scholar]
- (77).Li Y; Orlando BJ; Liao M Structural basis of lipopolysaccharide extraction by the LptB2FGC complex. Nature 2019, 567, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Tang X; Chang S; Luo Q; Zhang Z; Qiao W; Xu C; Zhang C; Niu Y; Yang W; Wang T et al. Cryo-EM structures of lipopolysaccharide transporter LptB2FGC in lipopolysaccharide or AMP-PNP-bound states reveal its transport mechanism. Nat. Commun. 2019, 10, 4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Osborn MJ; Gander JE; Parisi E Mechanism of assembly of the outer membrane of Salmonella typhimurium. Site of synthesis of lipopolysaccharide. J. Biol. Chem. 1972, 247, 3973–3986. [PubMed] [Google Scholar]
- (80).Marino PA; Phan KA; Osborn MJ Energy dependence of lipopolysaccharide translocation in Salmonella typhimurium. J. Biol. Chem. 1985, 260, 14965–14970. [PubMed] [Google Scholar]
- (81).Sampson BA; Misra R; Benson SA Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 1989, 122, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Lee J; Xue M; Wzorek JS; Wu T; Grabowicz M; Gronenberg LS; Sutterlin HA; Davis RM; Ruiz N; Silhavy TJ et al. Characterization of a stalled complex on the beta-barrel assembly machine. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 8717–8722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Eggert US; Ruiz N; Falcone BV; Branstrom AA; Goldman RC; Silhavy TJ; Kahne D Genetic basis for activity differences between vancomycin and glycolipid derivatives of vancomycin. Science 2001, 294, 361–364. [DOI] [PubMed] [Google Scholar]
- (84).Wu T; Malinverni J; Ruiz N; Kim S; Silhavy TJ; Kahne D Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 2005, 121, 235–245. [DOI] [PubMed] [Google Scholar]
- (85).Ruiz N; Falcone B; Kahne D; Silhavy TJ Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 2005, 121, 307–317. [DOI] [PubMed] [Google Scholar]
- (86).Steeghs L; den Hartog R; den Boer A; Zomer B; Roholl P; van der Ley P Meningitis bacterium is viable without endotoxin. Nature 1998, 392, 449–450. [DOI] [PubMed] [Google Scholar]
- (87).Albiger B; Johansson L; Jonsson AB Lipooligosaccharide-deficient Neisseria meningitidis shows altered pilus-associated characteristics. Infect. Immun. 2003, 71, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Serina S; Nozza F; Nicastro G; Faggioni F; Mottl H; Deho G; Polissi A Scanning the Escherichia coli chromosome by random transposon mutagenesis and multiple phenotypic screening. Res. Microbiol. 2004, 155, 692–701. [DOI] [PubMed] [Google Scholar]
- (89).Chng SS; Ruiz N; Chimalakonda G; Silhavy TJ; Kahne D Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5363–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Gu Y; Stansfeld PJ; Zeng Y; Dong H; Wang W; Dong C Lipopolysaccharide is inserted into the outer membrane through an intramembrane hole, a lumen gate, and the lateral opening of LptD. Structure 2015, 23, 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Okuda S; Sherman DJ; Silhavy TJ; Ruiz N; Kahne D Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat. Rev. Microbiol. 2016, 14, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Hollenstein K; Dawson RJ; Locher KP Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17, 412–418. [DOI] [PubMed] [Google Scholar]
- (93).Thomas C; Tampe R Multifaceted structures and mechanisms of ABC transport systems in health and disease. Curr. Opin. Struct. Biol. 2018, 51, 116–128. [DOI] [PubMed] [Google Scholar]
- (94).Locher KP Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [DOI] [PubMed] [Google Scholar]
- (95).Zuckert WR Secretion of bacterial lipoproteins: through the cytoplasmic membrane, the periplasm and beyond. Biochim. Biophys. Acta, Mol. Cell Res. 2014, 1843, 1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Han W; Wu B; Li L; Zhao G; Woodward R; Pettit N; Cai L; Thon V; Wang PG Defining function of lipopolysaccharide O-antigen ligase WaaL using chemoenzymatically synthesized substrates. J. Biol. Chem. 2012, 287, 5357–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Abeyrathne PD; Lam JS WaaL of Pseudomonas aeruginosa utilizes ATP in in vitro ligation of O antigen onto lipid A-core. Mol. Microbiol. 2007, 65, 1345–1359. [DOI] [PubMed] [Google Scholar]
- (98).Ruan X; Loyola DE; Marolda CL; Perez-Donoso JM; Valvano MA The WaaL O-antigen lipopolysaccharide ligase has features in common with metal ion-independent inverting glycosyltransferases. Glycobiology 2012, 22, 288–299. [DOI] [PubMed] [Google Scholar]
- (99).Simpson BW; Trent MS Pushing the envelope: LPS modifications and their consequences. Nat. Rev. Microbiol. 2019, 17, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Dong H; Zhang Z; Tang X; Paterson NG; Dong C Structural and functional insights into the lipopolysaccharide ABC transporter LptB2FG. Nat. Commun. 2017, 8, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Villa R; Martorana AM; Okuda S; Gourlay LJ; Nardini M; Sperandeo P; Deho G; Bolognesi M; Kahne D; Polissi A The Escherichia coli Lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains. J. Bacteriol. 2013, 195, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Chen J Molecular mechanism of the Escherichia coli maltose transporter. Curr. Opin. Struct. Biol. 2013, 23, 492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Vigonsky E; Ovcharenko E; Lewinson O Two molybdate/tungstate ABC transporters that interact very differently with their substrate binding proteins. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5440–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Meredith TC; Aggarwal P; Mamat U; Lindner B; Woodard RW Redefining the requisite lipopolysaccharide structure in Escherichia coli. ACS Chem. Biol. 2006, 1, 33–42. [DOI] [PubMed] [Google Scholar]
- (105).Hamad MA; Di Lorenzo F; Molinaro A; Valvano MA Aminoarabinose is essential for lipopolysaccharide export and intrinsic antimicrobial peptide resistance in Burkholderia cenocepacia. Mol. Microbiol. 2012, 85, 962–974. [DOI] [PubMed] [Google Scholar]
- (106).Ortega XP; Cardona ST; Brown AR; Loutet SA; Flannagan RS; Campopiano DJ; Govan JR; Valvano MA A putative gene cluster for aminoarabinose biosynthesis is essential for Burkholderia cenocepacia viability. J. Bacteriol. 2007, 189, 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Lundstedt EA; Simpson BW; Ruiz N LptB-LptF coupling mediates the closure of the substrate-binding cavity in the LptB2 FGC transporter through a rigid-body mechanism to extract LPS. Mol. Microbiol. 2020, 114, 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Gellert M; O’Dea MH; Itoh T; Tomizawa J Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc. Natl. Acad. Sci. U. S. A. 1976, 73, 4474–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Bisacchi GS; Manchester JI A new-class antibacterial-almost. Lessons in drug discovery and development: A critical analysis of more than 50 years of effort toward ATPase inhibitors of DNA gyrase and topoisomerase IV. ACS Infect Dis 2015, 1, 4–41. [DOI] [PubMed] [Google Scholar]
- (110).Khare D; Oldham ML; Orelle C; Davidson AL; Chen J Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell 2009, 33, 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Oldham ML; Davidson AL; Chen J Structural insights into ABC transporter mechanism. Curr. Opin. Struct. Biol. 2008, 18, 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Li X; Gu Y; Dong H; Wang W; Dong C Trapped lipopolysaccharide and LptD intermediates reveal lipopolysaccharide translocation steps across the Escherichia coli outer membrane. Sci. Rep. 2015, 5, 11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Tran AX; Trent MS; Whitfield C The LptA protein of Escherichia coli is a periplasmic lipid A-binding protein involved in the lipopolysaccharide export pathway. J. Biol. Chem. 2008, 283, 20342–20349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Grabowicz M Lipoproteins and their trafficking to the outer membrane. EcoSal Plus 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Matsuyama S; Tajima T; Tokuda H A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J. 1995, 14, 3365–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Merten JA; Schultz KM; Klug CS Concentration-dependent oligomerization and oligomeric arrangement of LptA. Protein Sci. 2012, 21, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Freinkman E; Chng SS; Kahne D The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 2486–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Ruiz N; Chng SS; Hiniker A; Kahne D; Silhavy TJ Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Chng SS; Xue M; Garner RA; Kadokura H; Boyd D; Beckwith J; Kahne D Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science 2012, 337, 1665–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Lee J; Sutterlin HA; Wzorek JS; Mandler MD; Hagan CL; Grabowicz M; Tomasek D; May MD; Hart EM; Silhavy TJ et al. Substrate binding to BamD triggers a conformational change in BamA to control membrane insertion. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 2359–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Lee J; Tomasek D; Santos TM; May MD; Meuskens I; Kahne D Formation of a beta-barrel membrane protein is catalyzed by the interior surface of the assembly machine protein BamA. eLife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Soltes GR; Martin NR; Park E; Sutterlin HA; Silhavy TJ Distinctive roles for periplasmic proteases in the maintenance of essential outer membrane protein assembly. J. Bacteriol. 2017, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Schwalm J; Mahoney TF; Soltes GR; Silhavy TJ Role for Skp in LptD assembly in Escherichia coli. J. Bacteriol. 2013, 195, 3734–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Vertommen D; Ruiz N; Leverrier P; Silhavy TJ; Collet JF Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 2009, 9, 2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Konovalova A; Kahne DE; Silhavy TJ Outer membrane biogenesis. Annu. Rev. Microbiol. 2017, 71, 539–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Botos I; Majdalani N; Mayclin SJ; McCarthy JG; Lundquist K; Wojtowicz D; Barnard TJ; Gumbart JC; Buchanan SK Structural and functional characterization of the LPS transporter LptDE from Gram-negative pathogens. Structure 2016, 24, 965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Lundquist KP; Gumbart JC Presence of substrate aids lateral gate separation in LptD. Biochim. Biophys. Acta, Biomembr. 2020, 1862, 183025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Xie R; Taylor RJ; Kahne D Outer membrane translocon communicates with inner membrane ATPase to stop lipopolysaccharide transport. J. Am. Chem. Soc. 2018, 140, 12691–12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Jones NC; Osborn MJ Translocation of phospholipids between the outer and inner membranes of Salmonella typhimurium. J. Biol. Chem. 1977, 252, 7405–7412. [PubMed] [Google Scholar]
- (130).Bishop RE; Gibbons HS; Guina T; Trent MS; Miller SI; Raetz CR Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. EMBO J. 2000, 19, 5071–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Snijder HJ; Ubarretxena-Belandia I; Blaauw M; Kalk KH; Verheij HM; Egmond MR; Dekker N; Dijkstra BW Structural evidence for dimerization-regulated activation of an integral membrane phospholipase. Nature 1999, 401, 717–721. [DOI] [PubMed] [Google Scholar]
- (132).Dekker N Outer-membrane phospholipase A: known structure, unknown biological function. Mol. Microbiol. 2000, 35, 711–717. [DOI] [PubMed] [Google Scholar]
- (133).May KL; Silhavy TJ The Escherichia coli phospholipase PldA regulates outer membrane homeostasis via lipid signaling. mBio 2018, 9, e00379–00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Malinverni JC; Silhavy TJ An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8009–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Chong ZS; Woo WF; Chng SS Osmoporin OmpC forms a complex with MlaA to maintain outer membrane lipid asymmetry in Escherichia coli. Mol. Microbiol. 2015, 98, 1133–1146. [DOI] [PubMed] [Google Scholar]
- (136).Benning C A role for lipid trafficking in chloroplast biogenesis. Prog. Lipid Res. 2008, 47, 381–389. [DOI] [PubMed] [Google Scholar]
- (137).Mohn WW; van der Geize R; Stewart GR; Okamoto S; Liu J; Dijkhuizen L; Eltis LD The actinobacterial mce4 locus encodes a steroid transporter. J. Biol. Chem. 2008, 283, 35368–35374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Pandey AK; Sassetti CM Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Yeow J; Tan KW; Holdbrook DA; Chong ZS; Marzinek JK; Bond PJ; Chng SS The architecture of the OmpC-MlaA complex sheds light on the maintenance of outer membrane lipid asymmetry in Escherichia coli. J. Biol. Chem. 2018, 293, 11325–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Abellon-Ruiz J; Kaptan SS; Basle A; Claudi B; Bumann D; Kleinekathofer U; van den Berg B Structural basis for maintenance of bacterial outer membrane lipid asymmetry. Nat. Microbiol. 2017, 2, 1616–1623. [DOI] [PubMed] [Google Scholar]
- (141).Ercan B; Low W-Y; Liu X; Chng S-S Characterization of interactions and phospholipid transfer between substrate binding proteins of the OmpC-Mla system. Biochemistry 2019, 58, 114–119. [DOI] [PubMed] [Google Scholar]
- (142).Tajima T; Yokota N; Matsuyama S; Tokuda H Genetic analyses of the in vivo function of LolA, a periplasmic chaperone involved in the outer membrane localization of Escherichia coli lipoproteins. FEBS Lett. 1998, 439, 51–54. [DOI] [PubMed] [Google Scholar]
- (143).Hughes GW; Hall SCL; Laxton CS; Sridhar P; Mahadi AH; Hatton C; Piggot TJ; Wotherspoon PJ; Leney AC; Ward DG et al. Evidence for phospholipid export from the bacterial inner membrane by the Mla ABC transport system. Nat. Microbiol. 2019, 4, 1692–1705. [DOI] [PubMed] [Google Scholar]
- (144).Ekiert DC; Bhabha G; Isom GL; Greenan G; Ovchinnikov S; Henderson IR; Cox JS; Vale RD Architectures of lipid transport systems for the bacterial outer membrane. Cell 2017, 169, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Kamischke C; Fan J; Bergeron J; Kulasekara HD; Dalebroux ZD; Burrell A; Kollman JM; Miller SI The Acinetobacter baumannii Mla system and glycerophospholipid transport to the outer membrane. eLife 2019, 8, e40171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Thong S; Ercan B; Torta F; Fong ZY; Wong HYA; Wenk MR; Chng S-S Defining key roles for auxiliary proteins in an ABC transporter that maintains bacterial outer membrane lipid asymmetry. eLife 2016, 5, e19042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Isom GL; Davies NJ; Chong Z-S; Bryant JA; Jamshad M; Sharif M; Cunningham AF; Knowles TJ; Chng S-S; Cole JA et al. MCE domain proteins: conserved inner membrane lipid-binding proteins required for outer membrane homeostasis. Sci. Rep. 2017, 7, 8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Powers MJ; Trent MS Intermembrane transport: Glycerophospholipid homeostasis of the Gram-negative cell envelope. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 17147–17155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Lewinson O; Livnat-Levanon N Mechanism of action of ABC importers: Conservation, divergence, and physiological adaptations. J. Mol. Biol. 2017, 429, 606–619. [DOI] [PubMed] [Google Scholar]
- (150).Leive L Release of lipopolysaccharide by EDTA treatment of E. coli. Biochem. Biophys. Res. Commun. 1965, 21, 290–296. [DOI] [PubMed] [Google Scholar]
- (151).Klein G; Kobylak N; Lindner B; Stupak A; Raina S Assembly of lipopolysaccharide in Escherichia coli requires the essential LapB heat shock protein. J. Biol. Chem. 2014, 289, 14829–14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Mahalakshmi S; Sunayana MR; SaiSree L; Reddy M yciM is an essential gene required for regulation of lipopolysaccharide synthesis in Escherichia coli. Mol. Microbiol. 2014, 91, 145–157. [DOI] [PubMed] [Google Scholar]
- (153).Nicolaes V; El Hajjaji H; Davis RM; Van der Henst C; Depuydt M; Leverrier P; Aertsen A; Haufroid V; Ollagnier de Choudens S; De Bolle X et al. Insights into the function of YciM, a heat shock membrane protein required to maintain envelope integrity in Escherichia coli. J. Bacteriol. 2014, 196, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Bittner LM; Arends J; Narberhaus F When, how and why? Regulated proteolysis by the essential FtsH protease in Escherichia coli. Biol. Chem. 2017, 398, 625–635. [DOI] [PubMed] [Google Scholar]
- (155).Shrivastava R; Jiang XE; Chng S-S Outer membrane lipid homeostasis via retrograde phospholipid transport in Escherichia coli. Mol. Microbiol. 2017, 106, 395–408. [DOI] [PubMed] [Google Scholar]
- (156).Llamas MA; Ramos JL; Rodriguez-Herva JJ Mutations in each of the tol genes of Pseudomonas putida reveal that they are critical for maintenance of outer membrane stability. J. Bacteriol. 2000, 182, 4764–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Bernadac A; Gavioli M; Lazzaroni JC; Raina S; Lloubes R Escherichia coli tol-pal mutants form outer membrane vesicles. J. Bacteriol. 1998, 180, 4872–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Gerding MA; Ogata Y; Pecora ND; Niki H; de Boer PA The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol. Microbiol. 2007, 63, 10081025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Yakhnina AA; Bernhardt TG The Tol-Pal system is required for peptidoglycancleaving enzymes to complete bacterial cell division. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 6777–6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Masilamani R; Cian MB; Dalebroux ZD Salmonella Tol-Pal reduces outer membrane glycerophospholipid levels for envelope homeostasis and survival during bacteremia. Infect. Immun. 2018, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Shrivastava R; Chng SS Lipid trafficking across the Gram-negative cell envelope. J. Biol. Chem. 2019, 294, 14175–14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Szczepaniak J; Press C; Kleanthous C The multifarious roles of Tol-Pal in Gram-negative bacteria. FEMS Microbiol. Rev. 2020, 44, 490–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Kulp A; Kuehn MJ Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Demchick P; Koch AL The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J. Bacteriol. 1996, 178, 768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Bayer ME Areas of adhesion between wall and membrane of Escherichia coli. J. Gen. Microbiol. 1968, 53, 395–404. [DOI] [PubMed] [Google Scholar]
- (166).Kellenberger E The ‘Bayer bridges’ confronted with results from improved electron microscopy methods. Mol. Microbiol. 1990, 4, 697–705. [DOI] [PubMed] [Google Scholar]
- (167).Bayer ME Zones of membrane adhesion in the cryofixed envelope of Escherichia coli. J. Struct. Biol. 1991, 107, 268–280. [DOI] [PubMed] [Google Scholar]
- (168).Nagy E; Losick R; Kahne D Robust suppression of lipopolysaccharide deficiency in Acinetobacter baumannii by growth in minimal medium. J. Bacteriol. 2019, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Powers MJ; Trent MS Phospholipid retention in the absence of asymmetry strengthens the outer membrane permeability barrier to last-resort antibiotics. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E8518–E8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Roier S; Zingl FG; Cakar F; Durakovic S; Kohl P; Eichmann TO; Klug L; Gadermaier B; Weinzerl K; Prassl R et al. A novel mechanism for the biogenesis of outer membrane vesicles in Gram-negative bacteria. Nat. Commun. 2016, 7, 10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Baarda BI; Zielke RA; Le Van A; Jerse AE; Sikora AE Neisseria gonorrhoeae MlaA influences gonococcal virulence and membrane vesicle production. PLoS Pathog. 2019, 15, e1007385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Arruda S; Bomfim G; Knights R; Huima-Byron T; Riley LW Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science 1993, 261, 1454. [DOI] [PubMed] [Google Scholar]
- (173).Nakayama T; Zhang-Akiyama QM pqiABC and yebST, putative mce operons of Escherichia coli, encode transport pathways and contribute to membrane integrity. J. Bacteriol. 2017, 199, e00606–00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Liu C; Ma J; Wang J; Wang H; Zhang L Cryo-EM Structure of a bacterial lipid transporter YebT. J. Mol. Biol. 2020, 432, 1008–1019. [DOI] [PubMed] [Google Scholar]
- (175).Isom GL; Coudray N; MacRae MR; McManus CT; Ekiert DC; Bhabha G LetB structure reveals a tunnel for lipid transport across the bacterial envelope. Cell 2020, 181, 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).De Lay NR; Cronan JE Genetic interaction between the Escherichia coli AcpT phosphopantetheinyl transferase and the YejM inner membrane protein. Genetics 2008, 178, 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Dalebroux Zachary D.; Edrozo Mauna B.; Pfuetzner Richard A.; Ressl S; Kulasekara Bridget R.; Blanc M-P; Miller Samuel I. Delivery of cardiolipins to the Salmonella outer membrane Is necessary for survival within host tissues and virulence. Cell Host Microbe 2015, 17, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Hirvas L; Nurminen M; Helander IM; Vuorio R; Vaara M The lipid A biosynthesis deficiency of the Escherichia coli antibiotic-supersensitive mutant LH530 is suppressed by a novel locus, ORF195. Microbiology 1997, 143 73–81. [DOI] [PubMed] [Google Scholar]
- (179).Nurminen M; Hirvas L; Vaara M The outer membrane of lipid A-deficient Escherichia coli mutant LH530 has reduced levels of OmpF and leaks periplasmic enzymes. Microbiology 1997, 143, 1533–1537. [DOI] [PubMed] [Google Scholar]
- (180).Rossi RM; Yum L; Agaisse H; Payne SM Cardiolipin synthesis and outer membrane localization are required for Shigella flexneri virulence. mBio 2017, 8, e01199–01117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Fan J; Petersen EM; Hinds TR; Zheng N; Miller SI Structure of an inner membrane protein required for PhoPQ-regulated increases in outer membrane cardiolipin. mBio 2020, 11, e03277–03219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Tan BK; Bogdanov M; Zhao J; Dowhan W; Raetz CR; Guan Z Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16504–16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Qiu N; Misra R Overcoming iron deficiency of an Escherichia coli tonB mutant by increasing outer membrane permeability. J. Bacteriol. 2019, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Cian MB; Giordano NP; Masilamani R; Minor KE; Dalebroux ZD Salmonella enterica serovar Typhimurium uses PbgA/YejM to regulate lipopolysaccharide assembly during bacteremia. Infect. Immun. 2019, 88, e00758–00719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Guest RL; Same Guerra D; Wissler M; Grimm J; Silhavy TJ YejM modulates activity of the YciM/FtsH protease complex to prevent lethal accumulation of lipopolysaccharide. mBio 2020, 11, e00598–00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Fivenson EM; Bernhardt TG An essential membrane protein modulates the proteolysis of LpxC to control lipopolysaccharide synthesis in Escherichia coli. mBio 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Clairfeuille T; Buchholz KR; Li Q; Verschueren E; Liu P; Sangaraju D; Park S; Noland CL; Storek KM; Nickerson NN et al. Structure of the essential inner membrane lipopolysaccharide-PbgA complex. Nature 2020, 584, 479–483. [DOI] [PubMed] [Google Scholar]
- (188).Anderson MS; Bull HG; Galloway SM; Kelly TM; Mohan S; Radika K; Raetz CR UDP-N-acetylglucosamine acyltransferase of Escherichia coli. The first step of endotoxin biosynthesis is thermodynamically unfavorable. J. Biol. Chem. 1993, 268, 19858–19865. [PubMed] [Google Scholar]
- (189).Ogura T; Inoue K; Tatsuta T; Suzaki T; Karata K; Young K; Su LH; Fierke CA; Jackman JE; Raetz CR et al. Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli. Mol. Microbiol. 1999, 31, 833–844. [DOI] [PubMed] [Google Scholar]