

Abstract
Two new cases of 16q22.3q23.3 Duplication syndrome demonstrate that phenotype can vary from severely affected to mild psychiatric concerns, even within the same family and identical duplications.
Keywords: 16q, copy number variants, Distal duplication of chromosome 16, intrafamilial variability, psychiatric predisposition
Two new cases of 16q22.3q23.3 Duplication syndrome demonstrate that phenotype can vary from severely affected to mild psychiatric concerns, even within the same family and identical duplications.

1. INTRODUCTION
Distal duplications of 16q are not well‐characterized in the literature. Previous patients include two males with midface hypoplasia and intellectual disability and one female with childhood onset schizophrenia. We describe two additional patients, and these cases demonstrate intrafamilial variability and potential sex expression differences among all cases of this duplication.
Chromosomal copy number variants (CNVs) are increasingly becoming one of the most commonly reported subsets of genetic conditions. With the evolution of genetic technology and improvement in detection rates, CNVs are currently estimated to occur in 1% of all pregnancies. 1 There has been documented incomplete penetrance and variable expression within these CNVs that have shown both inter‐ and intrafamilial variability. 2 , 3 Many CNVs are well‐characterized with disease, for example in 22q11 deletion syndrome. Information regarding the incidence, prevalence, spectrum of clinical features, and surveillance guidelines are well‐established. Other CNVs, such as duplications of 16q, are not characterized as well secondary to the limited clinical and prognostic information available. The most common derivative of 16q duplication is trisomy 16q. Trisomy 16q has previously been associated with a severe clinical picture including intrauterine growth restriction (IUGR), brain and cardiac defects, and an increased risk of both prenatal and postnatal lethality. This partial trisomy is considered a rare and phenotypically severe microduplication syndrome. 4 Furthermore, there are smaller CNVs within the q arm only that create partial trisomies of 16q. These occur even less frequently than full trisomy 16q. Currently, there are only two reported cases of distal duplications of the 16q in the Medical Genetics literature and one in the Psychiatric literature. The existing core phenotype for distal duplications of 16q is suggestive of intellectual disability and midface hypoplasia. Here, we present a set of siblings, a 15‐year‐old boy and a 17‐year‐old girl, in the hopes of reinforcing the core phenotype as well as expand the existing clinical picture for this rare microduplication syndrome.
2. CLINICAL REPORT
Patient 1 was the second pregnancy for a 21‐year‐old Nigerian mother. Prenatal care started in the second trimester and was unremarkable. He was born at 28 gestational weeks via an emergency Cesarean section for non‐reassuring fetal heart tones and premature rupture of membranes. His birth weight was 1.05kg (25th‐50th for gestational age). He remained in the neonatal intensive care unit (NICU) with complications secondary to his prematurity for the first 4 months of life. During his NICU stay, he had prominent hydrocephalus that required a ventriculoperitoneal (VP) shunt at 5 weeks of life. He required a secondary placement of his VP shunt after it became infected, but otherwise his hydrocephalus resolved. Additionally, he had his large patent ductus arteriosus (PDA) ligated. As a result of contracting necrotizing fasciitis, he had a jejunostomy tube placed and colostomy during his NICU stay. Lastly, he was noted to have a leg length discrepancy with his left leg being shorter than his right leg.
Over the course of his childhood, Patient 1 underwent a bilateral inguinal hernia repair, a tonsillectomy and a release his achilles tendon to correct a leg length discrepancy. He had scoliosis diagnosed prior to the age of 4 years and was being followed by orthopedics. Over the years, his scoliosis remained stable and has not required interventions. He has structural brain changes, including biventricular porencephalic cysts, closed‐lip schizencephaly, and polymicrogyria, for which he follows with Neurology. During childhood, he also had abnormal electroencephalograms (EEGs) that indicated absence seizures. Of recent, his EEGs are no longer abnormal and his seizures are now classified as subclinical. He does require a rescue medication for seizures, but no longer takes anti‐epileptic drugs regularly. He has been known to have diagnoses of both sleep apnea and dysautonomia, and is followed by those respective specialty clinics.
Patient 1 has had a history of global developmental delay. With correction for prematurity, he did not stand until 4 and a half years, walk until 7 years, and currently at 15 years of age has no identifiable words and communicates primarily by using gestures. With regards to behavior, he has diagnoses of both autism spectrum disorders and attention‐deficit/hyperactivity disorder (ADHD). He is currently in 8th grade in special education classes and has moderate intellectual disability.
At age 15, on physical examination (Figure 1), his demeanor is very calm and cooperative. He has facial asymmetry with turicephaly. His facial dysmorphisms include midface hypoplasia and prognathism. His extremities show bilateral 5th finger clinodactyly and outward deviated toes on his left foot. He wears ankle‐foot orthosis (AFOs) and is able to walk at a slow pace.
FIGURE 1.
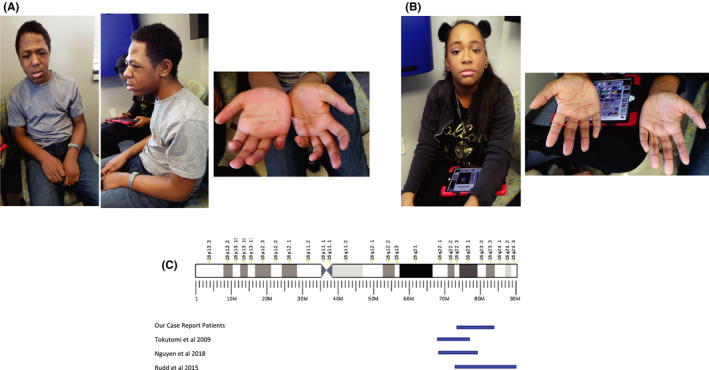
A, Photographs of Patient 1 showing dysmorphic facial features and 5th finger clinodactyly. B, Photographs of Patient 2 showing her to be non‐dysmorphic but with 5th finger clinodactyly present in both hands. C, Schematic of the duplications in Patient 1 and 2 referenced with the duplications mentioned in the literature to compare size and location
Patient 2 was the first pregnancy of a 19‐year‐old Nigerian mother and the full sister of Patient 1. Prenatal care began in the second trimester and later ultrasounds were notable for oligohydramnios. She was born at 33 weeks vaginally due to premature rupture of membranes. Her birth weight was 1.96 kg (50th percentile for gestational age). She spent two weeks in the NICU for difficulties with feeding and growth, but otherwise had an unremarkable neonatal course.
When corrected for prematurity, Patient 2 had appropriate motor and speech development. She walked at 15.5 months and had words at 15.5 months. She had a single febrile seizure at 18 months. Her subsequent EEGs were normal and she had no other signs of epilepsy. She was diagnosed with ADHD at 4 years old and was well controlled on medication. She is in typical classes and getting average to above average grades. She has an educational plan in place to allow for extra time on tests based on her diagnosis of ADHD, but has normal intellect.
Patient 2 first presented to psychiatry with the initial concern for anxiety in her early teens. This was expressed in the form of trichotillomania to the point of noticeable hair loss. Her psychiatrist initiated a regimen of talk therapy sessions and medication that decreased the frequency and severity of her trichotillomania. She is not a part of team or school activities, but does express engaging socially with friends and does not have a diagnosis of autism spectrum disorders.
On physical examination at age 17 (Figure 1), she was cooperative and engaged. She responded appropriately to questions directed at her. She was non‐dysmorphic aside from bilateral 5th finger clinodactyly.
Chromosomal Microarray Study was performed on peripheral blood samples taken from both patients and their mother, through informed consent via genetic counseling. SNP microarray analysis with the Affymetrix 6.0 platform was performed in a clinical lab. The CMA revealed a duplication of 8.85 Mb at 16q22.3q23.3 spanning from position 72,243,843 to 81,099,024 (NCBI36/hg18). (Figure 1).
Karyotyping confirmed the imbalances via high resolution karyotyping in both Patient 1 and Patient 2. This revealed both 46,XY,der(5)(p15.3ins16q22.3q23.3) and 46,XX,der(5)(p15.3ins16q22.3q23.3) karyotypes, respectively, that had a noted derivative 5th chromosome. This was consistent with the maternal karyotype which was balanced and revealed an insertion of distal 16q into 5p consistent with 46,XX,ins(5;16)(p15.3;q22.3q23.3).
Family History was pertinent as Patient 1 and 2’s mother, based on karyotyping, had a known 5p insertion with a karyotype 46,XX,ins(5;16)(p15.3;q22.3q23.3). Mother is now a G4P0403 whose only significant history is 4 preterm deliveries. Mother had no intellectual disability or psychiatric concerns. After Patient 1 and Patient 2, mother experienced one loss due to non‐viability of the fetus at delivery secondary to prematurity and unspecified anomalies. Patient 1 and 2 have a full younger brother with the reciprocal 16q deletion syndrome. Their full brother reflects the 16q deletion phenotype with moderate intellectual disability (ID), autism, behavioral abnormalities, genitourinary abnormalities, and gastroesophageal reflux. The maternal family history is positive for a loss in the first week of life for an uncle of unknown causes. There are three maternal uncles all reportedly healthy, and one maternal aunt who is healthy. All maternal first cousins are alive and well. The paternal family is positive for a father with an unspecified psychiatric diagnosis that required institutionalization.
3. DISCUSSION
The current literature on distal duplications of 16q suggests a core phenotype of intellectual disability and midface hypoplasia in conjunction with a high degree of variability between patients. 5 Our sibling pair add to the existing literature both to confirm the core phenotype and suggest further variability within this microduplication syndrome (Table 1).
TABLE 1.
Adapted from 5
| Tokutomi (2009) | Nguyen (2018) | Rudd (2015) | Patient 1 (This Report) | Patient 2 (This Report | |
|---|---|---|---|---|---|
| Duplication Size | 6.1 Mb | 8.3 Mb | 17.6 Mb | 8.85 Mb | 8.85 Mb |
| Cytoband | Chr16q22.1q23.1 | Chr16q22.1q23.1 | Chr16q22.3q24.3 | Chr16q22.3q23.3 | Chr16q22.3q23.3 |
| Sex | M | M | F | M | F |
| Height | ‐3 SD | ‐2 SD | Not reported | 0 SD | +1 SD |
| Weight | ‐2 SD | ‐2.5 SD | Not reported | ‐1 SD | ‐1 SD |
| OFC | ‐0.6 SD | <−2 SD | Not reported | ‐2.6 SD | +2 SD |
| ID | + | + | ‐ | + | ‐ |
| Seizures | + | ‐ | ‐ | + | ‐ |
| Dysmorphic Features (including midface hypoplasia) | + | + | ‐ |
+ |
‐ |
| Congenital anomalies | + | + | ‐ | + | ‐ |
| Neurologic Features | + | + | + | + | ‐ |
| Psychiatric Features | ‐ | ‐ | + | ‐ | + |
| Other | 5th finger clinodactyly, flat foot, wide gait, cryptorchidism, mild anemia, vesicouretic reflux | Vision loss, hypothyroidism | None | 5th finger clinodactyly, left toes with outward deviation | 5th finger clinodactyly |
Comparison of clinical features of the five known patients carrying duplications of the16q22.3q23.1 region, including the patient in the Psychiatric literature, and the two patients presented in this paper.
The three previously reported patients with distal duplication of 16q include a 13‐year‐old Vietnamese boy with an 8.3 Mb duplication, a 10‐year‐old Japanese boy with a 6.1 Mb duplication, and a girl with a 17.6 Mb duplication. 5 , 6 , 7 Both male cases reported thus far were adopted boys of East Asian decent with little to no birth and family history. Both duplications were 16q22.1q23.1. Between these two cases, variability of features was appreciated, but a core phenotype was suggested to be postnatal growth restriction, intellectual disability, and midface hypoplasia. Furthermore, these two cases had reported differences in regards to the presence or absence of autistic features, other dysmorphic features, and epilepsy. 5 , 7
The third patient with a distal duplication of 16q was reported in the Psychiatric literature. This duplication was at 16q22.3q24.3. This duplication is larger and extends more distally than our patients and the males reported in 5 and. 7 Her reported phenotype was limited to Childhood Onset Schizophrenia (COS). 6 Otherwise, the patient had typical birth and development until teenage years. She was diagnosed with COS of the disorganized type at 12 years old, and was given an additional diagnosis of intellectual disability at 18 years old, secondary to the COS. The patient's chromosomal microarray revealed three chromosomal aberrations: a paternally inherited 2.2 Mb 3p12.2p12.1 deletion, de novo 17.6 Mb duplication of 16q22.3q24.1, and de novo 43 Mb duplication of Xq23q24. Based on existing candidate genes, 6 hypothesizes that the distal duplication of 16q is the cause of her COS.
Patient 1 and 2 demonstrate the basis of intrafamilial variability as they have identical duplications and vastly different phenotypes. Although their degrees of prematurity differ, it does not fully explain the phenotypic differences. Chromosomal microduplications have been described with variable expressivity in the literature time and time again. 2 Even with the few known cases of distal duplication of 16q, we can already appreciate variable expressivity.
Patient 1 confers the core phenotype associated with 16q duplication. Even with the confounding variable of prematurity, the similarities to the previously reported cases include absence and subclinical epilepsy, developmental delay, and intellectual disability. He had skeletal birth defects including a leg length discrepancy and developed scoliosis. This could be related to the duplication, as the Japanese patient had flat feet as well as an abnormal gait that was not further characterized. 7 His PDA and hydrocephalus are likely the result of extreme prematurity, but would not explain his schizencephaly and dysautonomia. Lastly, the Japanese patient had brain atrophy and abnormalities that could be consistent with Patient 1’s brain changes. 7
Patient 2 disputes the existing core phenotype of distal duplications of 16q and highlights the variable expressivity that can be appreciated in this syndrome. Outside of the diagnosis of ADHD, Patient 2 had an unremarkable childhood until psychiatric concerns emerged in her early teen years. She had no dysmorphic features on examination aside from 5th finger clinodactyly and pertinently had no midface hypoplasia. She is in mainstream classes and has typical intelligence. Although Patient 2 has the same duplication coordinates as her brother, who is severely affected, Patient 2 has no features of the core duplication.
When considering Patient 2’s phenotype in the context of the patient in Rudd et al 2015, 6 they have similar past medical history including normal birth, developmental milestone attainment, and normal academic achievement until onset of symptoms. In their early teenage years, both females progressed to a psychiatric phenotype. Patient 2 appears milder as she has no intellectual disability or loss of skills while the patient in Rudd et al 2015 6 was given an additional diagnosis of intellectual disability in the spectrum of her COS. It is striking that the only reported female patients defy the previous core phenotype of midface hypoplasia and intellectual disability and display a primarily psychiatric phenotype. This could be suggestive of a potential sex difference in phenotypic expression for this microduplication syndrome. Furthermore, it more clearly demarcates a potential critical region for neuropsychiatric conditions first proposed in Rudd et al 2015. 6 Rudd et al 2015 described 20 candidate genes within the larger distal duplication of 16q that could be related to the COS seen in their patient. The overlapping regions between Patient 2 and the patient in Rudd et al 2015 6 is 16q22.3q 23.3, in turn, narrows the differential candidate genes for psychiatric illness predisposition. There are seven candidate genes that are mutually shared by these two patients within this microduplication including ATP2C2, JPH3, CDH15, FA2H, ZNRF1, CNTNAP4, and DYNLRB2. One of these genes may be more likely the source of the psychiatric predisposition as it has been observed in two patients with diagnosed psychiatric conditions and 16q duplication. If there is a psychiatric phenotype‐only presentation of this duplication, it is very likely to be severely underdiagnosed as a chromosomal microarray is not standard of care for most psychiatric illnesses.
Furthermore, it is noted that both Patients 1 and 2 were the result of an unbalanced insertion resulting from a translocation of chromosome 16q material into chromosome 5 causing a derivative chromosome on the maternal karyotype. The previous patient reported in 5 had a chromosomal insertion into chromosome 13. Of the previous patients in the literature and our two patients, there are two assumed de novo duplications and three as a result of insertion. This could highlight an area of genetic instability that makes 16q vulnerable to recombination, specifically this region being more likely to be inserted into other chromosomal regions.
4. CONCLUSION
With now a total of five reported cases of distal duplication of 16q, Patients 1 and 2 highlight the presence of inter‐ and intrafamilial variability as well as additional information regarding the potential spectrum of clinical features. There remains the question of a potential sex difference in phenotypic expression of the distal duplication of 16q given the clinical features of the two known female patients. Additionally, the observation that three out of five patients with a distal duplication of 16q arose from insertions could highlight an area vulnerable to chromosomal rearrangement. The duplication is rarely reported in literature and likely underdiagnosed, especially in patients that may only have a psychiatric phenotype. Reports of more patients with distal duplication of 16q will be helpful in further delineating the clinical spectrum and the possibility of differences in sex expression.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR’S CONTRIBUTIONS
Each author's contribution on this manuscript is as follows: Kathryn Gunther collected the clinical data, literature research, and writing. Laura S Farach provided clinical guidance and assisted in manuscript editing. Kate Mowrey assisted in manuscript editing and collecting clinical data.
ETHICS STATEMENT
Written informed consent was obtained from the patient for the publication of this case report. IRB approval not required from this study.
ACKNOWLEDGEMENTS
The authors thank the Laboratory Corporation of America® for the access to raw data and cytogenetic technology access for the purposes of publication in this familial case. Published with written consent of the patient.
Gunther K, Mowrey K, Farach LS. Two new reported cases of 16q22.3q23.3 duplication syndrome highlight intrafamilial variability and potential sex expression differences within a rare duplication syndrome. Clin Case Rep. 2021;9:1629–1633. 10.1002/ccr3.3862
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author, Kathryn Gunther, at Kathryn.a.gunther@uth.tmc.edu. The data are not publicly available as they are private medical information.
REFERENCES
- 1. Martin CL, Kirkpatrick BE, Ledbetter DH. Copy Number Variants, Aneuploidies, and Human Disease. Clin Perinatol. 2015;42(2):227‐242. 10.1016/j.clp.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Banka S, Fitzgibbon GJ, Gaunt L, Rankin WJ, Clayton‐Smith J. A novel 800 kb microduplication of chromosome 16q22.1 resulting in learning disability and epilepsy may explain phenotypic variability in a family with 15q13 microdeletion. Am J Med Genet A. 2011;155(6):1453‐1457. 10.1002/ajmg.a.34034 [DOI] [PubMed] [Google Scholar]
- 3. Bateman MS, Collinson MN, Bunyan DJ, et al. Incomplete penetrance, variable expressivity, or dosage insensitivity in four families with directly transmitted unbalanced chromosome abnormalities. Am J Med Genet A. 2017;176(2):319‐329. 10.1002/ajmg.a.38564 [DOI] [PubMed] [Google Scholar]
- 4. Chen C‐P, Lin S‐P, Lin C‐C, et al. Perinatal findings and molecular cytogenetic analysis ofde novo partial trisomy 16q (16q22.1?qter) and partial monosomy 20q (20q13.3?qter). Prenat Diagn. 2005;25(2):112‐118. 10.1002/pd.1083 [DOI] [PubMed] [Google Scholar]
- 5. Nguyen HH, Pham VA, Barcia G, et al. Distal duplication of chromosome 16q22.1q23.1 in a Vietnamese patient with midface hypoplasia and intellectual disability. Am J Med Genet A. 2018;176(9):1981‐1984. 10.1002/ajmg.a.40375 [DOI] [PubMed] [Google Scholar]
- 6. Rudd D, Axelsen M, Epping EA, Andreasen N, Wassink T. Childhood‐onset schizophrenia case with 2.2 Mb deletion at chromosome 3p12.2‐p12.1 and two large chromosomal abnormalities at 16q22.3‐q24.3 and Xq23‐q28. Clin Case Rep. 2015;3(4):201‐207. 10.1002/ccr3.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tokutomi T, Wada T, Nakagawa E, Saitoh S, Sasaki M. A de novo direct duplication of 16q22.1 → q23.1 in a boy with midface hypoplasia and mental retardation. Am J Med Genet A. 2009;149A(11):2560‐2563. 10.1002/ajmg.a.33049 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author, Kathryn Gunther, at Kathryn.a.gunther@uth.tmc.edu. The data are not publicly available as they are private medical information.