

Abstract
Patients with soft tissue sarcomas should be assessed for neurotrophic tropomyosin receptor kinase (NTRK) gene fusions as neoadjuvant treatment with larotrectinib may prevent amputation.
Keywords: larotrectinib, neoadjuvant, NTRK gene fusions, soft tissue sarcoma
Patients with soft tissue sarcomas should be assessed for neurotrophic tropomyosin receptor kinase (NTRK) gene fusions as neoadjuvant treatment with larotrectinib may prevent amputation.

1. INTRODUCTION
Neurotrophic tropomyosin receptor kinase gene‐fusions are rare but can occur in diverse cancers. The NTRK inhibitor larotrectinib is newly approved by the FDA. This paper reports the first case of a man with a NTRK‐rearranged soft tissue sarcoma treated with larotrectinib in the neoadjuvant setting, in Belgium, to avoid limb amputation.
Soft tissue sarcomas (STS) are very rare tumors with over 80 histological entities and even more molecular subsets. Surgery is the cornerstone of treatment and the standard surgical procedure is a wide excision to achieve negative margins.
About forty percent of STS occur in the extremities of the limbs, and less than ten percent of cases need amputation. 1 Neoadjuvant chemotherapy can facilitate surgery by inducing sufficient shrinkage of the tumor to allow limb‐sparing surgery. 2
Neurotrophic tropomyosin receptor kinase (NTRK) gene fusions result in the constitutive activation of TRK receptors. The fusion proteins can lead to oncogenesis by activating the PI3K/AKT, RAS/MAPK/ERK and PLC‐gamma signaling pathways. These pathways lead to cell differentiation, proliferation, and survival. 3 , 4 , 5 NTRK gene fusions are found in about one percent of all solid tumors and can be a target for NTRK‐inhibitors. 6
Larotrectinib is an oral, highly selective, pan‐TRK inhibitor. 7
Here we describe the first case of a man with a NTRK‐rearranged STS treated with larotrectinib in the neoadjuvant setting in Belgium to avoid limb amputation.
2. CASE
A 30‐year‐old man presented to the outpatient clinic in February 2019 with a bleeding mass on his distal left leg. He had scars on both sides of the ankle due to multiple previous childhood surgeries between 1992 and 2002 for a tumor classified as pseudosarcomatous dysplasia with no sign of malignancy.
The mass was painless and nonpulsatile but bled upon removal of the bandages and continued to bleed without the application of a compression bandage. The patient described the lesion as fast‐growing.
A complete oncologic workup of the lesion was performed revealing a voluminous, fleshy, heterogenous tumor measuring approximately 11 × 14 × 8 centimeters (cm). The tumor invaded the Kager space, destroying the posterior cortical of the tibia, and surrounded the left Achilles tendon (Figure 1A,B).
FIGURE 1.
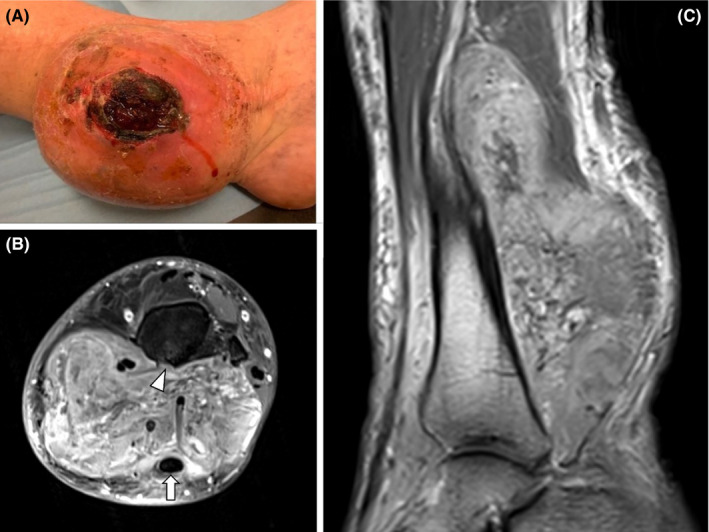
Lesion at diagnosis assessed by (A) photograph and contrast‐enhanced MRI (B) fat‐suppressed axial T1‐weighted images and (C) sagittal T1‐weighted images. MRI demonstrated a well‐defined enhancing mass of the posterior compartment of the leg surrounding the Achilles tendon (arrow) and eroding the posterior tibial cortex (arrowhead)
We performed a needle biopsy. The pathology revealed a CD34 and S100 co‐expressing spindle cell tumor, morphologically resembling lipofibromatosis‐like neural tumor variant and staining for pan‐TRK. We then performed a surgical biopsy to obtain sufficient material to carry out next generation sequencing. It confirmed a NTRK‐rearranged lipofibromatosis‐like neural tumor with a new fusion partner SPECC1L. 8
It was decided to treat the lesion with neoadjuvant lacotrectinib in an attempt to save the limb as the patient was strongly against amputation. While the authorization for larotrectinib use was under consideration, the tumor proliferated, and the skin ruptured at the biopsy site. Bleeding remained constant, there was a putrid smell related to the necrosis, and the mass on the inner part of the distal tibia interfered with walking ability. We decided to debulk the extrophic part of the lesion in May 2019 (Figure 1C). The pathology once again showed a NTRK‐rearranged so‐called lipofibromatosis‐like neural tumor that was now progressing to undifferentiated sarcoma.
In June 2019, the tumor measured 17.4 × 11 × 6.8 cm on MRI and the patient started larotrectinib (Figure 2A). Treatment was well‐tolerated. The only adverse event reported was grade 1 fatigue. After 2 weeks of treatment, the bleeding had stopped, and we observed a decrease in the size of the lesion after 6 weeks (Figure 2B).
FIGURE 2.

Photographs of the left leg (A) before and (B) after 3 mo of larotrectinib therapy
In February 2020, after the patient had undergone 8 months of treatment and following discussion with the sarcoma multidisciplinary team, we decided to perform limb salvage surgery to remove the tumor. Given that several neurovascular structures were enclosed, R1 resection was planned followed by radiotherapy (Figure 3).
FIGURE 3.

Lesion before surgery assessed by (A) photograph and contrast‐enhanced MRI (B) fat‐suppressed axial T1‐weighted images and (C) sagittal T1‐weighted images. MRI showed a significant decrease in tumor volume with large nonenhanced areas suggestive of tissue necrosis (arrows)
The main goal of surgery was to remove the tumor while preserving the mobility, stability, and sensitivity of the foot. The lesion was removed en bloc using a double medial and lateral approach. The surgery proved to be difficult as the lesion had induced local fibrosis and was partly involuted. We first isolated the posterior tibial pedicle proximal to the lesion and dissected it out of the lesion. The artery was obliterated by the tumor, but we were able to salvage the nerve. The Achilles, tibialis posterior, flexor digitorum longus, and flexor hallucis longus tendons were preserved. The periosteum of the tibia was removed with the lesion. The superficial sensitive nerves (intermediate dorsal cutaneous nerve and lateral branch of the peroneal nerve) were preserved. The lesion was removed whole with the skin fistula existing on the inner part of the calf where the tumor had previously grown out of the limb. The specimen was sent for pathological analysis.
The pathologist found a grade 3 NTRK‐related spindle cell sarcoma measuring 12.5 × 5.0 × 2.5 cm. The distal and medial margins were microscopically positive (pre‐planned‐R1). The tumor was classified as ypT3N0M0. 9 Comparison with the previous sample showed more severe nuclear atypia with more spindle cells.
Adjuvant radiotherapy consisting of 65 Gray in 30 fractions was carried out between April 6 and May 19, 2020.
Three months after the completion of radiotherapy, the patient was disease‐free, able to walk adequately on the affected limb, and back to living normal life.
At the time of writing, there is no evidence of disease and the patient remains in active follow‐up.
3. DISCUSSION
This case describes the first patient in Belgium to receive larotrectinib in the neoadjuvant setting for a NTRK‐rearranged soft tissue sarcoma. The patient achieved a partial response, offering the possibility of curative limb‐sparing surgery.
The cornerstone of soft tissue sarcoma treatment is complete surgical resection with histologically negative margins by an experienced surgeon. As forty percent of soft tissue sarcomas are localized in the limbs, this approach leads, however, to significant patient morbidity. 1 The use of adjuvant treatments is facilitating limb‐sparing curative strategies. In one study, a limb‐sparing curative strategy with adjuvant radiotherapy on the pre‐planned margin showed similar results to amputation with better quality of life. 10 For large (> 5 cm) or deep tumor with high‐grade histology, neoadjuvant chemotherapy with anthracyclines plus ifosfamide remains standard. 11
Evaluation of this case revealed a NTRK‐rearranged lipofibromatosis‐like neural tumor. NTRK gene fusions are very rare in soft tissue sarcomas. One group found them in only eight out of 1272 patients with soft tissue sarcoma. 12 In our patient, NTRK was rearranged with a new fusion partner, SPECC1L, which, to our knowledge, has not been previously reported in the literature. This gene is located at 22q11.23 and is involved in the migration of neural crest cells during embryology via regulation of Cytospin‐A. 8 As a result of the NTRK‐fusion gene, our patient was able to be treated with larotrectinib with a successful outcome and less side‐effects than would be expected with standard anthracycline‐based treatment.
Larotrectinib has been approved by the US Food and Drug administration since November 2018 for adult and pediatric patients with solid tumors harboring a NTRK gene fusion. 13 Three initial trials (LOXO‐TRK‐14001 [NCT02122913]; SCOUT [NCT02637687], NAVIGATE [NCT02576431]) assessed the efficacy of larotrectinib in adults and children with various tumor types harboring NTRK fusions for an overall response rate of approximately seventy‐five percent. 2 , 13 Although generally well‐tolerated, adverse events reported with larotrectinib include fatigue, gastrointestinal disorders (nausea, vomiting, diarrhea, and constipation), dizziness, cough and increased alanine and aspartate transaminases. Long‐term administration is feasible. 13
Larotrectinib has also been assessed in soft tissue sarcoma. In the pediatric phase I trial of larotrectinib, five children with TRK‐fusion soft tissue sarcoma all responded to treatment: two had a partial response, two had a pathologic complete response, and one had a nearly complete pathologic response (>98% treatment effect). 14 One adult patient with metastatic uterine sarcoma harboring NTRK fusion also had a complete response after 16 months of larotrectinib. 15 Another 8‐year‐old girl with recurrent NTRK fusion positive soft tissue sarcoma had a complete response after 6 months of larotrectinib. 16 A separate phase I/II study in pediatric patients confirmed an objective response rate of 93% and a good safety profile. 17
In 2020, an updated pooled analysis of the three initial larotrectinib studies showed an overall median duration of response, progression‐free survival and overall survival of 35.2, 28.3, and 44.4 months, respectively, in 260 treated patients. In the 153 patients evaluable for efficacy, the overall response rate was 79% with 16% complete responses. 18 This analysis also confirmed the drug's favorable safety profile.
Despite encouraging study results, resistance can occur with acquired mutations in the NTRK gene. Second generation TRK inhibitors are under clinical development, and preclinical trials are proving efficacy in inhibiting the mutant NTRK fusion‐gene. 12
Our patient case, and other reports, highlight the importance of searching for NTRK gene fusions even if they are rare. Larotrectinib is a well‐tolerated target therapy and can save limbs. Further research is required to validate its use in the neoadjuvant setting as an alternative to chemotherapy.
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
CP: wrote the entire paper. TS: wrote the clinical part of the paper. CG: contributed data in molecular biology. TK: wrote the radiologic description of the figures. FM: collected the data and wrote the entire paper.
ETHICS STATEMENT, FUNDING AND DATA AVAILABILITY STATEMENT
We have not received any funding or grant for this case and the patient consents to the use of his anonymous data.
ACKNOWLEDGMENTS
The authors would like to thank Aileen Eiszele of A&L Medical Communications for writing assistance and manuscript preparation.
Percy C, Schubert T, Galant C, Kirchgesner T, Mazzeo F. Larotrectinib in a NTRK‐rearranged soft tissue sarcoma in the neoadjuvant setting: A case report. Clin Case Rep. 2021;9:1694–1698. 10.1002/ccr3.3878
REFERENCES
- 1. von Mehren M, Lor Randall R, Benjamin RS, et al. Soft tissue sarcoma, version 2.2018. NCCN clinical practice guidelines in oncology. J Natl Comp Canc Netw. 2018;16(5):526‐563. [DOI] [PubMed] [Google Scholar]
- 2. Drilon A, Laetsch T, Kummar S, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378(8):731‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amatu A, Sartore‐Bianchi A, Bencardino K, et al. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019;30(Supplement_8):viii5‐viii15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amatu A, Sartore‐Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016;1(2):e000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gatalica Z, Xiu J, Swensen J, et al. Molecular characterization of cancers with NTRK gene fusions. Mod Pathol. 2019;32(1):147‐153. [DOI] [PubMed] [Google Scholar]
- 6. Doebele R, Davis LE, Vaishnavi A, et al. An oncogenic NTRK fusion in a soft tissue sarcoma patient with response to the tropomyosin‐related kinase (TRK) inhibitor LOXO‐101. Cancer Discov. 2015;5(10):1049‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saleh K, Khalifeh‐Saleh N, Kourie HR. TRK inhibitors: toward an era of agnostic targeted therapies in oncology. Pharmacogenomics. 2019;20(13):927‐927. [DOI] [PubMed] [Google Scholar]
- 8. Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 gene fusion define a novel subset of locally aggressive lipofibromatosis‐like neural tumor. Am J Surg Pathol. 2016;40(10):1407‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brierley JD, Gospodarowicz MK, Wittekind C, eds. TNM classification of malignant tumours, 8th edn. Oxford, UK: Wiley Blackwell; 2017. [Google Scholar]
- 10. Gundle KR, Karchinski L, Gupta S, et al. Analysis of margin classification systems for assessing the risk of local recurrence after soft tissue sarcoma resection. J Clin Oncol. 2018;36(7):704‐709. [DOI] [PubMed] [Google Scholar]
- 11. Casali PG, Abecassis N, Aro HT, et al. Soft tissue and visceral sarcomas: ESMO‐EURACAN Clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2018;29(Suppl 4):iv51‐iv67. [DOI] [PubMed] [Google Scholar]
- 12. Drilon A, Nagasubramanian R, Blake JF, et al. A next‐generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion‐positive solid tumors. Cancer Discov. 2017;7(9):963‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. U.S. Food & Drug Administration . FDA approves larotrectinib for solid tumors with NTRK gene fusions (14/12/2018). https://www.fda.gov/drugs/fda‐approves‐larotrectinib‐solid‐tumors‐ntrk‐gene‐fusions. Accessed September 19, 2020.
- 14. DuBois SG, Laetsch TW, Federman N, et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer. 2018;124(21):4241‐4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rabban JT, Devine WP, Sangoi AR, et al. NTRK fusion cervical sarcoma: a report of three cases, emphasising morphological and immunohistochemical distinction from other uterine sarcomas, including adenosarcoma. Histopathology. 2020;77(1):100‐111. [DOI] [PubMed] [Google Scholar]
- 16. Kato S, Fujimura J, Nozaki Y, et al. A case of pediaric soft tissue sarcoma with LMNA‐NTRK1 gene fusion treated with larotrectinib under single patient expanded access system. Gan To Kagaku Ryoho. 2019;46(10):1595‐1597. [PubMed] [Google Scholar]
- 17. Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open‐label, phase 1/2 study. Lancel. Oncol. 2018;19(4):705‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion‐positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020;21(4):531‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]