

Abstract
Targeting the menin-MLL protein-protein interaction is a new therapeutic strategy for the treatment of acute leukemia carrying MLL fusion (MLL leukemia). We describe herein the structure-based optimization of a class of covalent menin inhibitors, which led to the discovery of M-808 (16) as a highly potent and efficacious covalent menin inhibitor. M-808 effectively inhibits leukemia cell growth at low nanomolar concentrations and is capable of achieving partial tumor regression in an MV4;11 xenograft tumor model in mice at a well-tolerated dose schedule. Determination of the co-crystal structure of M-808 in complex with menin provides a structural basis for their high-affinity, covalent interactions. M-808 represents a promising, covalent menin inhibitor for further optimization and evaluation toward developing a new therapy for the treatment of MLL leukemia.
Graphical Abstract
Introduction
The chromosomal translocations of the mixed lineage leukemia (MLL) gene are observed in approximately 10% of adult acute myeloid leukemia (AML) and >70% of infant acute lymphoblastic leukemias (ALL).1 The most common MLL translocation is its fusion with one of more than 70 translocation partner genes.2-3 Patients with leukemia bearing a MLL translocation (MLL leukemia) have a particularly poor prognosis and fail to respond to currently available treatments4-5, highlighting the urgent need for new treatment options for MLL leukemia.
In all the identified MLL fusion proteins, approximately 1400 amino acid residues from the wild-type MLL N-terminus are retained, which interact directly with menin protein.6 The menin-MLL protein-protein interaction has been shown to be critical for the expression of the HOXA and MEIS1 genes, which drive leukemogenesis in MLL leukemia.7-8 Therefore, targeting the protein-protein interaction between menin and MLL represents a promising therapeutic strategy for the treatment of MLL leukemia.7, 9
Analysis of a co-crystal structure of the menin-MLL complex suggests that targeting the menin-MLL protein-protein interaction with non-peptide small-molecule inhibitors (herein called menin inhibitors) is challenging but achievable6, 10. In the last few years, several classes of reversible small-molecule menin inhibitors have been reported.11 For example, inhibitors 1 (MI-503)12-13, 2 (MI-3454)14 and 3 (VTP-50469)15 (Figure 1) containing the thienopyrimidine or pyrimide, bind to menin protein with low nanomolar affinities, and demonstrate in vivo activity in an MLL leukemia model in mice. The aminomethylpiperidine class of inhibitor 4 (MIV-6)16 shows moderate inhibitory activity toward the menin-MLL interaction and leukemia cell growth. Currently, two orally bioavailable menin inhibitors such as KO-53917 and SNDX-561318, have progressed to clinical trials, although their chemical structure were not disclosed.
Figure 1.
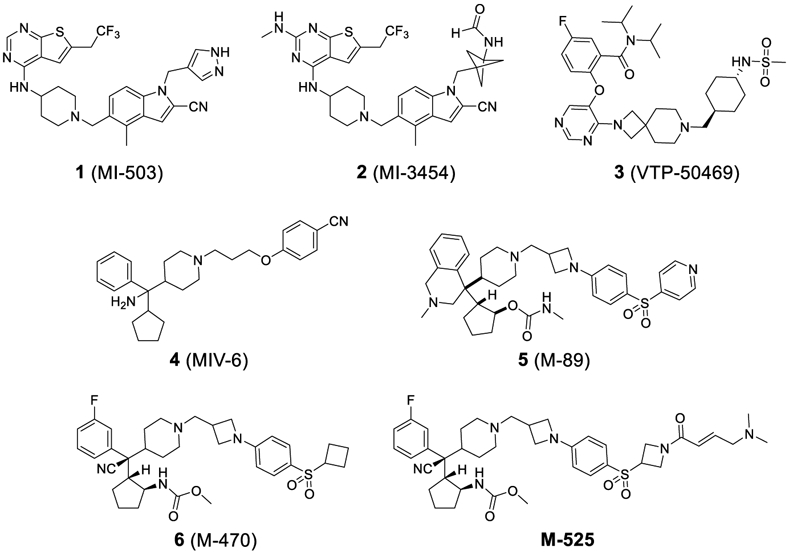
Previously reported reversible and irreversible menin inhibitors.
Recently, using MIV-6 as the initial lead compound, our laboratory has reported the discovery of compounds 5 (M-89)19 and 6 (M-470)20 as potent, reversible menin inhibitors. In addition to these reversible menin inhibitors, we also reported the discovery of M-52520 as the first-in-class irreversible, covalent menin-MLL inhibitor (Figure 1). M-525 demonstrated high potency targeting the menin-MLL interaction and achieves potent activity in inhibition of leukemia cell growth in MLL leukemia cell lines.20
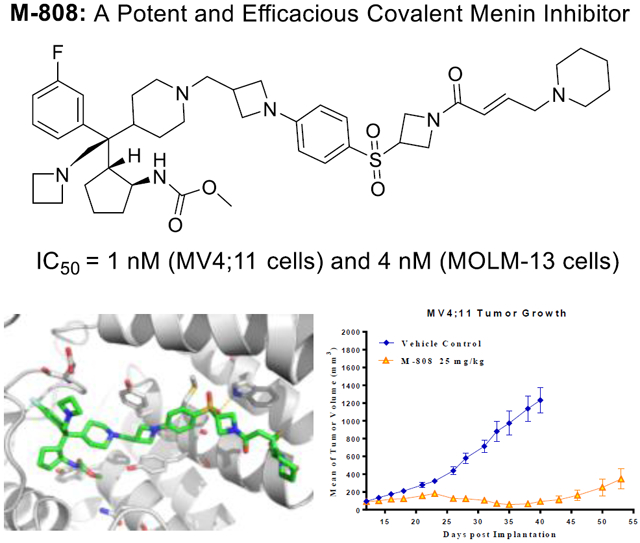
In our continuous efforts toward identification of a highly potent and efficacious menin inhibitor for advanced preclinical development and future clinical trials, we have carried out further optimization of M-525. Our efforts have yielded a set of highly potent covalent menin inhibitors with M-808 identified as the most promising compound. M-808 achieves IC50 values of 1 and 4 nM, respectively, in inhibition of cell growth in the MV4;11 and MOLM13 cell lines carrying MLL fusion and is capable of achieving partial tumor regression in the MV4;11 leukemia xenograft tumor model in mice. M-808 warrants extensive evaluation as a potential new therapy for the treatment of MLL leukemia.
Results and Discussion
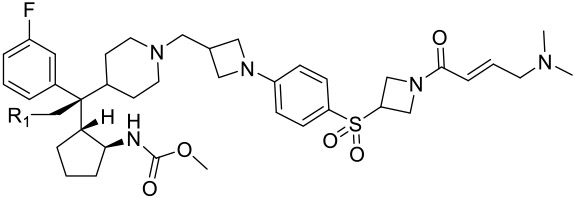
Analysis of the co-crystal structure of M-525 complexed with the menin protein shows that although the nitrile group on the quaternary carbon atom in M-525 is directed towards a solvent exposed environment, it is adjacent to two negatively charged Asp180 and Glu359 residues.20 Indeed, in our previous study, we have shown that a positively charged amino group at this site can significantly enhance the binding affinities and the cellular potencies of our designed reversible menin inhibitors19. Accordingly, we have employed a series of basic groups to replace the nitrile. For the ease of synthesis, these basic groups are connected to the quaternary carbon atom via a methylene group (Table 1).
Table 1.
Binding affinity and cell growth inhibition of menin inhibitors 7-12.
 | |||||
|---|---|---|---|---|---|
| Compd. | R1 | Binding Affinity (IC50, nM)a |
Cell growth inhibition assay (7 days) (IC50, nM)b |
||
| MV4;11 (MLL-AF4 fusion) |
MOLM13 (MLL-AF9 fusion) |
HL60 (No MLL fusion) |
|||
| M-525c | -CN | 3.3 ± 0.4 | 4 ± 3 | 19 ± 010 | 1500 ± 40 |
| 7 | 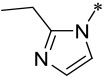 |
1.7 ± 0.3 | 2 ± 1 | 11 ± 5 | > 10,000 |
| 8 | 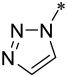 |
2 | 2 ± 1 | 14 ± 5 | 4800 ± 500 |
| 9 | 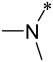 |
5 | 1.1 ± 0.1 | 2 ± 1 | 700 ± 110 |
| 10 | 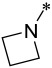 |
3 | 3 ± 2 | 8 ± 4 | 3000 ± 700 |
| 11 | 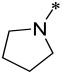 |
2 | 5 ± 2 | 15 ± 2 | 1400 ± 200 |
| 12 | 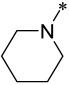 |
2 | 40 ± 14 | 157 ± 87 | 600 ± 30 |
IC50 values were determined using a FP-based competitive binding assay. The binding affinity (IC50) of all compounds is ≤ 5 nM, which exceeds the lower assay limit.
Cell viability was determined using a WST-8 assay after 7 days of treatment for each compound, with average values and SD calculated from three independent experiments.
IC50 values for M-525 were reported previously.20
Replacement of the nitrile group with a basic heteroaromatic 2-ethyl imidazole group produced compound 7 (Table 1), which has IC50 = 1.7 nM to menin in our fluorescence-polarization (FP) based, competitive binding assay21. However, since these compounds were designed as covalent menin inhibitors, the IC50 values obtained in our FP-based binding assay result from both their binding to menin and their chemical reactivity with menin. We have therefore employed the MV4;11 and MOLM-13 leukemia cell lines carrying MLL fusion to test their cellular potencies and the HL-60 leukemia cell line lacking the MLL fusion to evaluate their cellular specificity as the primary assays. Compound 7 was found to achieve IC50 values of 2 nM and 11 nM in inhibition of cell growth in the MV4;11 and MOLM-13 cell lines and to have an IC50 value of >10 μM in the HL-60 cell line. Hence, 7 demonstrates a potent cellular activity in two leukemia cell lines carrying MLL fusion proteins and a very high selectivity over a leukemia cell line lacking MLL fusion. We concluded that 7 is a potent and specific menin inhibitor.
Encouraged by the promising data for compound 7, we made additional compounds in which the nitrile group in M-525 was replaced by different basic groups. Changing the imidazole group in compound 7 with a triazole group led to 8, which has IC50 values of 2 nM and 14 nM in the MV4;11 and MOLM-13 cell lines and displays an IC50 value of 4.8 μM in the HL-60 cell line. Hence, compound 8 is also a potent and specific menin inhibitor. Replacing the triazole group in 8 with a much more basic dimethyl amine led to compound 9, which achieves IC50 values of 1.1 nM and 2 nM in the MV4;11 and MOLM-13 cell lines, respectively. Compound 9 has an IC50 value of 0.7 μM in the HL-60 cell line, thus demonstrating a cellular selectivity of >300-times for the MV4;11 and MOLM-13 cell lines over the HL-60 cell line. In direct comparison, 9 is 4-times more potent than M-525 in the MV4;11 cell line and 9.5-times more potent than M-525 in the MOLM-13 cell line, respectively. Hence, our data on compound 9 showed that a basic amino group can significantly improve the cellular activity in leukemia cells carrying MLL fusion, while retaining high cellular selectivity over leukemia cells lacking MLL fusion. Accordingly, we have made additional compounds containing a basic amine group.
Compound 10 containing an azetidine group has IC50 values of 3 nM and 8 nM, respectively, in the MV4;11 and MOLM-13 cell lines, and is thus 2-3 times less potent than 9. Compound 10 has an IC50 value of 3.0 μM in the HL-60 cell line, thus displaying a cellular selectivity of >300-times for the MV4;11 and MOLM-13 cell lines over the HL-60 cell line. Changing the 4-membered ring azetidine group in 10 to a 5-membered ring pyrrolidine yielded 11, which is 2-times less potent than 10 in both the MV4;11 and MOLM-13 cell lines. Interestingly, while 11 still retains a good cellular selectivity for MLL leukemia cells over non-MLL leukemia cells, it is less selective than 10. Changing the 4-membered ring azetidine group in compound 10 to a 6-membered ring piperidine resulted in 12, which is >10-times less potent than 10 in both the MV4;11 and MOLM-13 cell lines. However, 12 is more potent than 10 in the HL-60 cell line lacking MLL fusion and is therefore much less selective than 10.
The positively charged amino “head” groups in compounds 9, 10, 11 and 12 have calculated pKa values of 9.16-9.1822, indicating a very similar basicity. Based upon our design, these head groups were also expected to be exposed to a solvent environment. However, these four compounds have significant differences in their cellular potencies in inhibition of cell growth in MV4;11 and MOLM-13 cell lines with compound 9 being the most potent and compound 12 being the least potent. To shed light on the structural basis for their different cellular potencies, we modeled their binding models with menin, based upon the co-crystal structure of M-525 in complex with menin (Figure 2), followed by a 10 ns molecular dynamic (MD) simulation for each compound. Our modeling showed that while these 4 compounds bind to menin with similar binding models, the interactions between their respective, positively charged amino “head’ group and the negatively charged Asp180 carboxylic acid differ for these four compounds. The average distance between the positively charged amino group in 9 and the nearest negatively charged oxygen atom of the carboxylic acid group of Asp180 in menin is 3.56 ± 0.38 Å, indicating a very strong charge-charge interaction. In comparison, the corresponding distances in the binding models for 10, 11 and 12 are 4.06 ± 0.60, 5.83 ± 1.38 and 10.15 ± 0.97 Å, respectively. Hence, our modeling suggested that among these compounds, compound 9 has the strongest charge-charge interactions between its positively charged amino “head’ group and the negatively charged Asp180 carboxylic acid in menin, followed by compounds 10 and 11. In contrast, compound 12 has a minimal interaction between its positively charged amino “head’ group and the negatively charged Asp180 carboxylic acid in menin. Our modeling thus provides an explanation for their different cellular potencies in MV4;11 and MOLM-13 cell lines for compounds 9-12.
Figure 2.

Predicted binding models of compounds 9-12 with menin (PDB ID: 6B41). The distance between the respective amino group of each inhibitor and the nearest oxygen atom of Asp180 in menin is shown by a black dash line.
In our previous study, we have found the cellular potency of our covalent inhibitors can be significantly enhanced by introducing the “tail” dimethyl amine group in M-525, which is able to serve as intermolecular catalytic base in the addition reaction between Michael acceptor and cysteine20, 23. Because 10 (M-734) has a very potent cellular activity in the MV4;11 and MOLM-13 cell lines and an excellent cellular selectivity over the HL-60 cell line, we have employed this compound as the template for tail modifications.
First, we examined the importance of the dimethylaminomethylene group in 10 by removal of this group, which led to 13. Compound 13 has IC50 = 23 nM in the MV4;11 cell line and IC50 = 374 nM in the MOLM-13 cell line (Table 2), which is 7- and 46-times less potent than 10, respectively. These data further confirmed the importance of the dimethylaminomethylene group in enhancing the cellular activity of our covalent menin inhibitors in leukemia cells carrying MLL fusion. We have therefore modified the dimethyl amine group with other groups containing an amine group.
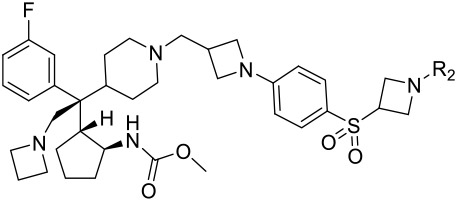
Table 2.
Binding affinity and cell growth inhibition of menin inhibitors 13-20.
 | |||||
|---|---|---|---|---|---|
| Compd. | R2 | Binding Affinity (IC50, nM) |
Cell growth inhibition assay (7 days) (IC50, μM) |
||
| MV4;11 (MLL-AF4 fusion) |
MOLM13 (MLL-AF9 fusion) |
HL60 (No MLL fusion) |
|||
| 13 | 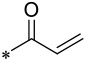 |
2 | 0.023 ± 0.009 | 0.374 ± 0.093 | 2.6 ± 0.7 |
| 14 | 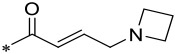 |
2 | 0.002 ± 0.001 | 0.003 ± 0.001 | 2.1 ± 0.1 |
| 15 | 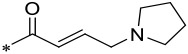 |
2 | 0.002 ± 0.0003 | 0.009 ± 0.001 | 2.1 ± 0.4 |
| 16 (M-808) | 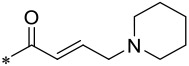 |
2.6 ± 0.5 | 0.001 ± 0.0002 | 0.004 ± 0.001 | 2.8 ± 0.6 |
| 17 | 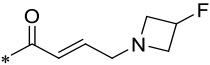 |
2 | 0.001 ± 0.0004 | 0.003 ± 0.001 | 2.0 ± 0.3 |
| 18 | 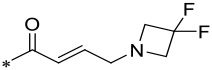 |
1 | 0.004 ± 0.001 | 0.015 ± 0.004 | 2.8 ± 0.9 |
| 19 | 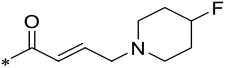 |
4 | 0.003 ± 0.0004 | 0.021 ± 0.002 | 3.6 ± 0.5 |
| 20 | 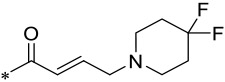 |
2 | 0.004 ± 0.001 | 0.020 ± 0.002 | 3.2 ± 0.5 |
We replaced the dimethylamino group in 10 with azetidine, which led to 14. Compound 14 achieves IC50 values of 2 nM and 3 nM in the MV4;11 and MOLM-13 cell lines, respectively. Furthermore, 14 displays an IC50 value of 2 μM in the HL-60 cell line, hence showing a cellular selectivity >700-fold for MLL leukemia cells over non-MLL leukemia cells. Encouraged with the excellent cellular potency in MLL leukemia cells and selectivity in non-MLL cells, we replaced the 4-membered ring azetidine with a 5-membered ring pyrrolidine, which yielded 15. Compound 15 is equally potent as 14 in inhibition of cell growth in the MV4;11 cell line (IC50 = 2 nM) but is 3-times less potent than 14 in the MOLM-13 cell line. 15 demonstrates a >200-fold cellular selectivity over the HL-60 cell line. Replacement of the azetidine in 15 with a 6-membered ring piperidine resulted in 16, which is essentially equally potent as 14. Compound 16 and 14 are also equally selective over the HL-60 cell line.
We next installed one F atom or two F atoms in the 3-position of azetidine in 14 to reduce the basicity of the basic nitrogen to examine the effect on the cellular potency, which yielded 17 and 18. The calculated pKa value of the basic nitrogen of the azetidine in 14 was reduced from 8.65 to 7.06 with installation of one F atom in 17. Interestingly, 17 is equally potent and selective as compared to 14. With installation of two F atoms in the 3-position of azetidine, the calculated pKa value was reduced to 5.47 in 18. Compound 18 is 4-5 times less potent than 17 in inhibition of cell growth in both the MV4;11 and MOLM-13 cell lines.
Since 16 (M-808) is highly potent and selective, we also investigated the effect of reducing the basicity of the basic nitrogen of its tail piperidine by installation of one or two F atoms in the 4-position of the piperidine ring. Installation of one F atom yielded 19, which achieves IC50 values of 3 and 21 nM in inhibition of cell growth in the MV4;11 and MOLM-13 cell lines, respectively. Hence, 19 is 3- and 5-times less potent than 16 in the MV4;11 and MOLM-13 cell lines, respectively. Installation of two F atoms at the 4-position of the piperidine ring resulted in 20, which has IC50 values of 4 and 20 nM in inhibition of cell growth in the MV4;11 and MOLM-13 cell lines, respectively, and is similarly potent as 19. Hence, installation of one or two F atoms in the 4-position of the piperidine ring in 16 resulted in modest reduction (3-5 times) in cellular potencies in inhibition of cell growth in both the MV4;11 and MOLM-13 cell lines.
To understand the mode of action of our designed covalent inhibitors, we performed mass-spectrometry analyses of the reactivity for a number of representative covalent menin inhibitors with human menin protein in vitro (Table 3). Consistent with our previous data, M-525 readily forms a covalent complex with menin protein. Within 1 h of incubation, 95% of the protein forms a covalent complex with M-525, and 100% of protein reacts with M-525 with overnight incubation. Compounds 7 and 10 also readily form a covalent complex with menin. Compound 13, which has a much reduced cellular potency in both MV4;11 and MOLM-13 cell lines as compared to M-525, also has a slower reaction kinetics with menin than M-525.
Table 3.
Mass-spectrometry analyses of the reactivity of our designed covalent menin inhibitors with human menin protein in vitro.
| Menin Inhibitor | % of menin protein forming a covalent complex with the inhibitor |
|
|---|---|---|
| 1 h | Overnight | |
| M-525 | 95 | 100 |
| 7 | 100 | 100 |
| 10 | 75 | 100 |
| 13 | 50 | 75 |
| 14 | 75 | 100 |
| 15 | 100 | 100 |
| 16 (M-808) | 75 | 100 |
| 17 | 20 | 75 |
| 18 | 0 | 0 |
| 19 | 20 | 100 |
| 20 | 0 | 0 |
Compounds 14, 15 and 16, all of which are very potent menin inhibitors, have a rapid reaction kinetics with menin. Compound 17, in which a F atom was installed on the 4-membered azetidine in 14, and 19, in which a F atom was onto the 6-membered piperidine in 16, have a much slower reaction kinetics than 14 and 16. For both 17 and 19, only 20% of menin protein forms a covalent complex with either compound with 1 h incubation time. Compound 18, in which two F atom were installed onto the 4-membered azetidine in 14, and 20, in which two F atom were installed onto the 6-membered piperidine in 16, fail to form a covalent complex with menin, even with overnight incubation time. These data strongly suggested that although 18 and 20 still potently inhibit cell growth in the MV4;11 and MOLM-13 cell lines and contain a Michael acceptor, they most likely work as reversible menin inhibitors in cells and not as covalent menin inhibitors.
To gain structural insights for their interactions with menin protein for these new covalent inhibitors, we determined a co-crystal structure of 16 (M-808) complexed with menin protein at 2.10 Å resolution (Figure 3, PDB code: 6MN9). Consistent with its design and our mass spectrometry data, M-808 forms a covalent bond between its acrylamide and the sulfur atom of Cys329 in menin, same as we have observed in the co-crystal complex for M-525 with menin, confirming its covalent binding nature with menin. While the azetidine “head” group is largely exposed to solvent, the basic nitrogen atom is 4.3 Å away from the negatively charged oxygen atom of the carboxylic acid group of Asp180 side chain, indicating a strong charge-charge interaction, consistent with our modeling results. The tail 6-membered piperidine is exposed to solvent and has no specific interactions with menin. The other interactions between M-808 and menin are essentially same as those observed in the co-crystal structure of M-525 in complex with menin. Specifically, the reverse carbamate group on the cyclopentyl ring inserts into a pocket formed by Asn282, Cys241, Met278, Tyr276 and Tyr323 in menin and its carbonyl group forms a hydrogen bond with the hydroxyl group of Tyr276, and the methyl group has hydrophobic contacts with the side chains of Met278 and Cys241. The fluorine substituted phenyl ring inserts into a large hydrophobic pocket in menin. The cyclopentyl group interacts with a hydrophobic pocket formed primarily by Phe159, Phe328 and Ala242. The 4-membered azetidine group in the middle of M-808 sandwiches between Tyr319 and Tyr323 and positions the sulfonylphenyl into a proper position and orientation for the sulfonyl group to form a strong hydrogen bond with the NH group of the indole of Trp341. This co-crystal structure provides a solid structural basis for the high-affinity, covalent binding of M-808 with menin.
Figure 3.

Co-crystal structure of compound 16 (M-808) (cyan carbons) complexed with menin at 2.1 Å resolution (PDB code: 6MN9). Hydrogen bonds depicted as black dashed lines.
In our previous study20, we have shown that a single dose of M-525 is capable of achieving strong suppression of the expression of MEIS1 and HOXA9 genes in the MV4;11 xenograft tumor tissue at 50 mg/kg intravenous dosing. Based upon the potent cellular activity in both the MV4;11 and MOLM-13 cell lines, we have selected compounds M-525, 9, 10, 14-17 for their pharmacodynamics (PD) in mice bearing the MV4;11 xenograft tumors (Figure 4). Our PD data showed that 10, 15 and 16 are most effective in suppression of the expression of MEIS1 and HOXA9 genes in the MV4;11 tumors among these 7 menin inhibitors tested. Compounds 10, 15 and 16 at 10 mg/kg are effective in reducing the expression of MEIS1 gene by >2-fold at 24 hr time-point. Compounds 10, 15 and 16 have a significant but modest effect in suppressing the expression of HOXA9 gene.
Figure 4.

Pharmacodynamic effect of Menin inhibitors on expression of HOXA9 and MEIS1 genes in MV4;11 xenograft tumors. Mice bearing MV4;11 tumors were treated with a single intravenous of each menin inhibitor at 10 mg/kg and tumors were collected at 24 hr time point for qRT-PCR analysis. ** (p < 0.01).
Based upon the PD data, we selected 10, 15 and 16 (M-808) to evaluate their tolerability in mice. It was found that 10, 15 and 16 were well tolerated in SCID mice with intravenous administration of 10 mg/kg, every other day dosing (three times a week) for one week. However, 10 and 15 induced animal weight loss (>10%) at 25 mg/kg every other day dosing (three times a week) for one week. In comparison, 16 was found to be well tolerated at 25 mg/kg every other day dosing (three times a week) for one week. Based upon the tolerability data, we decided to evaluate 16 further for its pharmacodynamic effect and for its efficacy in vivo.
Mice bearing MV4;11 tumors were treated with a single intravenous dose of M-808 at 25 mg/kg. Mice were sacrificed at 6, 24 and 48 h time-points and tumors were collected for qRT-PCR analysis of HOXA9 and MEIS1 gene expression (Figure 5). The PD data showed that a single dose of M-808 at 25 mg/kg effectively suppresses the expression of MEIS1 and HOXA9 genes in the tumor tissue at all the three time-points. While M-808 is equally effective in suppressing HOXA9 gene expression at 6, 24 and 48 h, it becomes more effective in reducing the expression of MEIS1 gene at later time points. The long-lasting PD effect of M-808 on MV4;11 tumors provided a basis for us to evaluate the in vivo efficacy of M-808 at a dosing schedule less frequent than daily administration.
Figure 5.

Pharmacodynamic effect of 16 (M-808) on expression of HOXA9 and MEIS1 genes in MV4;11 xenograft tumors. Mice bearing MV4;11 tumors were treated with a single intravenous of M-808 at 25 mg/kg and tumors were collected at 6, 24 and 48 hr time points for qRT-PCR analysis. ** (p < 0.01).
M-808 was tested for its in vivo antitumor efficacy in the MV4;11 xenograft model in SCID mice. Mice were treated with M-808 at 10 or 25 mg/kg every other day, 3 times per week for a total of 11 doses. While M-808 at 10 mg/kg has no effect on tumor growth over the vehicle control, M-808 at 25 mg/kg is very efficacious. M-808 at 25 mg/kg achieves a maximum tumor growth inhibition (TGI) of 97% during treatment (day 35) (Figure 6) and reduces the average tumor volume from 92 mm3 at the beginning of the treatment to 59 mm3 at day 35. Mice treated with M-808 at both doses have no significant weight change and show no signs of drug-related toxicity during and after the treatment. Hence, our efficacy data show that M-808 is very efficacious in vivo at a well-tolerated dose-schedule.
Figure 6.

In vivo antitumor efficacy of compounds 16 (M-808) in the MV4;11 xenograft model in SCID mice. Compound was administered IV at the indicated dose schedules. (A) Tumor growth inhibition. (B) Body weight of mice.
Chemistry
The synthetic routes to these compounds are shown below. The intermediates 21, 27 and compound M-525 were prepared according to published methods20. The syntheses of compounds 7-20 are shown in Scheme 1. Intermediate 21 was reduced successively by DIBAL-H and NaBH4 to give the amine 22. The free amine of compound 22 was modified to yield 23a-f, which were further deprotected with trifluoroacetic acid to form 24a-f. Compounds 24a-f were reacted with dimethyl dicarbonate, then the benzyl protecting group was removed to afford the deprotected piperidine intermediates 26a-f, which were reacted with the separately prepared20 tail 27, to form compounds 28a-f. Removal of the Boc protecting group of 28a-f produced the corresponding amine intermediates, which reacted with diverse α,β-unsaturated carboxylic acids or acryloyl chloride afforded the final products 7-20.
Scheme 1:
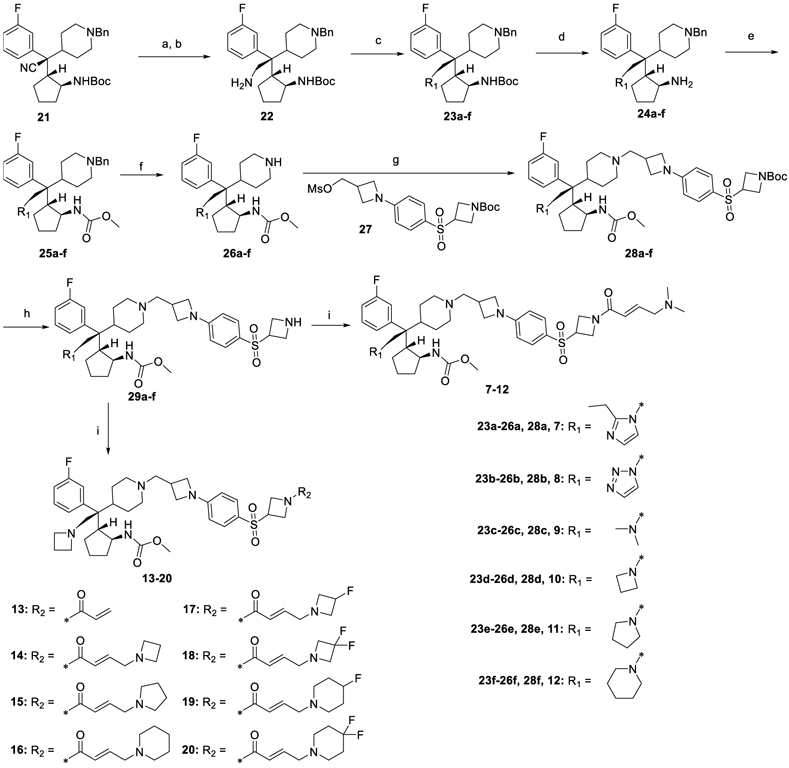
Syntheses of compounds 7-20.
Reagents and conditions: (a) DIBAL-H, toluene, 0 °C to rt; (b) NaBH4, MeOH, 0 °C to rt; (c) propionaldehyde, glyoxal, ammonium acetate, MeOH, 60 °C (23a); p-toluenesulfonyl hydrazide, glyoxal, AcOH, MeOH, 60 °C (23b); HCHO, NaBH(AcO)3, DCM, rt (23c); 1,3-dibromopropane, K2CO3, MeCN, 80 °C (23d); 1,4-dibromobutane, K2CO3, MeCN, 80 °C (23e); 1,5-dibromopentane, K2CO3, MeCN, 80 °C (23f); (d) TFA, DCM, 0 °C to rt; (e) dimethyl dicarbonate, DIPEA, DCM, 0 °C to rt; (f) 10% Pd/C, H2, MeOH, rt; (g) K2CO3, KI, MeCN, 80 °C; (h) TFA, DCM, 0 °C to rt; (i) α,β-unsaturated carboxylic acids, HATU, DIPEA, DCM, 0 °C to rt (7-12, 14-20); acryloyl chloride, DIPEA, DCM, 0 °C to rt (13).
Conclusion
In the present study, we have described the optimization of a class of covalent menin inhibitors based upon our previously published covalent inhibitor M-525. Our efforts have yielded M-808 as a highly potent and efficacious covalent menin inhibitor. Mass spectrometric analysis and co-crystal structure of M-808 complexed with menin clearly show that M-808 readily forms a covalent bond with Cys329 in menin. M-808 achieves IC50 values of 1-4 nM in inhibition of cell growth in MV4;11 and MOLM13 leukemia cell lines carrying MLL fusion proteins and displays an excellent selectivity over the HL-60 cell line lacking MLL fusion. M-808 effectively suppresses expression of MEIS1 and HOXA9 genes in the MV4;11 xenograft tumor tissues and is capable of achieving partial tumor regression in mice bearing the MV4;11 xenograft tumor at a well-tolerated dose-schedule. Taken together, our study shows that M-808 is a promising covalent menin inhibitor for further evaluations and optimization toward developing a new therapy for the treatment MLL leukemia.
EXPERIMENTAL SECTION
General Methods for Chemistry.
Unless otherwise noted, commercial solvents and reagents were used without further purification with the exception of THF which was freshly distilled from sodium wire. Proton nuclear magnetic resonance (1H NMR) was recorded on a Bruker Advance 400 MHz spectrometer. 1H NMR spectra were reported in parts per million (ppm) downfield from tetramethylsilane (TMS). In the spectral data reported, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. MS analyses were carried out with a Waters UPLC–mass spectrometer. The final compounds were all purified by C18 reverse phase preparative HPLC column with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in MeCN) as eluents. The purity of all the final compounds was confirmed to be > 95% by UPLC analysis (10% to 100% MeCN in H2O containing 0.1% TFA in 10 min).
Methyl ((1S,2R)-2-((S)-1-(1-((1-(4-((1-((E)-4-(dimethylamino)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-2-(2-ethyl-1H-imidazol-1-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (7).
Compound 7 was synthesized using the method described for 10 from the intermediate 23a. 1H NMR (400 MHz, MeOD) δ 7.70 (d, J = 8.8 Hz, 2H), 7.57-7.52 (m, 1H), 7.50-7.44 (m, 2H), 7.33 (s, 1H), 7.20 (t, J = 8.4 Hz, 1H), 6.97 (s, 1H), 6.77-6.70 (m, 1H), 6.52 (d, J = 8.8 Hz, 2H), 6.47 (d, J = 15.2 Hz, 1H), 4.84 (d, J = 16.0 Hz, 1H), 4.59 (d, J = 15.6 Hz, 1H), 4.51 (d, J = 6.0 Hz, 2H), 4.29-4.15 (m, 5H), 3.99-3.93 (m, 3H), 3.78-3.73 (m, 2H), 3.71-3.69 (m, 1H), 3.65 (s, 3H), 3.50-3.42 (m, 3H), 3.28-3.21 (m, 1H), 3.14-3.07 (m, 2H), 3.05-2.99 (m, 1H), 2.89 (s, 6H), 2.87-2.84 (m, 1H), 2.71-2.64 (m, 2H), 2.31 (d, J = 13.6 Hz, 1H), 2.02 (d, J = 15.6 Hz, 1H), 1.96-1.90 (m, 1H), 1.66-1.61 (m, 1H), 1.58-1.52 (m, 1H), 1.44-1.35 (m, 5H), 1.24-1.16 (m, 2H), 1.07-0.96 (m, 1H); ESI-MS calculated for C44H60FN7O5S [M + H]+ = 818.44, found: 818.61.
Methyl ((1S,2R)-2-((S)-1-(1-((1-(4-((1-((E)-4-(dimethylamino)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)-2-(1H-1,2,3-triazol-1-yl)ethyl)cyclopentyl)carbamate (8).
Compound 8 was synthesized using the method described for 10 from the intermediate 23b. 1H NMR (400 MHz, MeOD) δ 8.01 (s, 1H), 7.79 (s, 1H), 7.70 (d, J = 8.8 Hz, 2H), 7.47-7.37 (m, 3H), 7.09 (t, J = 8.0 Hz, 1H), 6.78-6.70 (m, 1H), 6.54 (d, J = 8.8 Hz, 2H), 6.47 (d, J = 15.2 Hz, 1H), 5.28 (s, 2H), 4.52 (d, J = 10.4 Hz, 2H), 4.29-4.23 (m, 2H), 4.20-4.17 (m, 4H), 3.93 (d, J = 6.4 Hz, 2H), 3.76-3.74 (m, 2H), 3.55-3.47 (m, 3H), 3.42 (s, 3H), 3.26-3.20 (m, 1H), 3.07-2.94 (m, 2H), 2.90 (s, 6H), 2.85-2.78 (m, 1H), 2.52 (t, J = 11.2 Hz, 1H), 2.23 (d, J = 14.0 Hz, 1H), 1.95-1.92 (m, 1H), 1.86-1.79 (m, 1H), 1.60-1.51 (m, 2H), 1.49-1.42 (m, 1H), 1.38-1.15 (m, 4H), 0.78-0.67 (m, 1H); ESI-MS calculated for C41H55FN8O5S [M + H]+ = 791.40, found: 791.51.
Methyl ((1S,2R)-2-((S)-2-(dimethylamino)-1-(1-((1-(4-((1-((E)-4-(dimethylamino)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (9).
Compound 9 was synthesized using the method described for 10 from the intermediate 23c. 1H NMR (400 MHz, MeOD) δ 7.71 (d, J = 8.8 Hz, 2H), 7.52-7.46 (m, 1H), 7.42-7.39 (m, 2H), 7.17-7.13 (m, 1H), 6.78-6.70 (m, 1H), 6.54 (d, J = 8.8 Hz, 2H), 6.47 (d, J = 15.2 Hz, 1H), 4.52 (d, J = 6.4 Hz, 2H), 4.29-4.16 (m, 5H), 3.95-3.86 (m, 5H), 3.82-3.78 (m, 2H), 3.68-3.60 (m, 2H), 3.50-3.45 (m, 4H), 3.31 (s, 3H), 3.14-2.94 (m, 6H), 2.90 (m, 6H), 2.87-2.81 (m, 1 H), 2.71-2.63 (m, 1H), 2.50-2.40 (m, 1H), 2.22-2.12 (m, 2H), 1.98-1.96 (m, 1H), 1.76-1.64 (m, 3H), 1.59-1.50 (m, 3H), 1.44-1.37 (m, 1H); ESI-MS calculated for C41H59FN6O5S [M + H]+ = 767.43, found: 767.54.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-((1-(4-((1-((E)-4-(dimethylamino)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (10).
The trifluoroacetic acid salt of 29d (300 mg, 0.38 mmoL) was dissolved in dry DCM (10 mL) and MeCN (1 mL). Then, DIPEA (0.2 mL, 1.15 mmol), (E)-4-(dimethylamino)but-2-enoic acid hydrochloride (76 mg, 0.46 mmol) and HATU (175 mg, 0.46 mmol) were added at 0 °C. After stirring for 30 min at rt, the reaction mixture was concentrated under vacuum. The residue was purified by reverse phase preparative HPLC to give the title compound as a trifluoroacetate salt (200 mg, 67%). 1H NMR (400 MHz, MeOD) δ 7.70 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.13 (m, 2H), 7.05 (d, J = 7.6 Hz, 1H), 6.77-6.70 (m, 1H), 6.53 (d, J = 8.8 Hz, 2H), 6.48 (d, J = 15.2 Hz, 1H), 4.56-4.50 (m, 4H), 4.39-4.32 (m, 2H), 4.29-4.22 (m, 1H), 4.19-4.12 (m, 5H), 3.95-3.93 (m, 2H), 3.81-3.74 (m, 3H), 3.56-3.47 (m, 3H), 3.40 (d, J = 7.8 Hz, 2H), 3.25-3.19 (m, 1H), 3.04-2.95 (m, 2H), 2.90 (s, 6H), 2.81-2.74 (m, 1H), 2.57-2.50 (m, 1H), 2.48-2.41 (m, 1H), 2.07 (d, J = 14.8 Hz, 2H), 2.01-1.97 (m, 2H), 1.92-1.86 (m, 1H), 1.81-1.75 (m, 1H), 1.71-1.61 (m, 3H), 1.50-1.41 (m, 1H), 1.14-1.04 (m, 1H); ESI-MS calculated for C42H59FN6O5S [M + H]+ = 779.43, found: 779.51.
Methyl ((1S,2R)-2-((S)-1-(1-((1-(4-((1-((E)-4-(dimethylamino)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)-2-(pyrrolidin-1-yl)ethyl)cyclopentyl)carbamate (11).
Compound 11 was synthesized using the method described for 10 from the intermediate 23e. 1H NMR (400 MHz, MeOD) δ 7.70 (d, J = 8.8 Hz, 2H), 7.50-7.44 (m, 1H), 7.32 (d, J = 8.8 Hz, 2H), 7.16-7.11 (m, 1H), 6.77-6.70 (m, 1H), 6.52 (d, J = 9.2 Hz, 2H), 6.47 (d, J = 15.2 Hz, 1H), 4.51 (d, J = 6.4 Hz, 2H), 4.29-4.22 (m, 1H), 4.21-4.15 (m, 4H), 4.12-4.06 (m, 1H), 3.95-3.93 (m, 2H), 3.85 (d, J = 15.6 Hz, 1H), 3.78-3.71 (m, 5H), 3.57 (d, J = 11.6 Hz, 2H), 3.48-3.33 (m, 5H), 3.31 (s, 3H), 3.28-3.22 (m, 1H), 3.07-3.01 (m, 1H), 2.98-2.92 (m, 1H), 2.89 (s, 6H), 2.85-2.79 (m, 1H); ESI-MS calculated for C43H61FN6O5S [M + H]+ = 793.44, found: 793.52.
Methyl ((1S,2R)-2-((S)-1-(1-((1-(4-((1-((E)-4-(dimethylamino)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)-2-(piperidin-1-yl)ethyl)cyclopentyl)carbamate (12).
Compound 12 was synthesized using the method described for 10 from the intermediate 23f. 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 8.8 Hz, 2H), 7.47-7.41 (m, 1H), 7.36 (d, J = 8.8 Hz, 2H), 7.14-7.10 (m, 1H), 6.77-6.70 (m, 1H), 6.51 (d, J = 8.8 Hz, 2H), 6.46 (d, J = 15.6 Hz, 1H), 4.50 (d, J = 6.4 Hz, 2H), 4.28-4.14 (m, 5H), 4.04-4.00 (m, 1H), 3.94-3.92 (m, 2H), 3.79-3.74 (m, 3H), 3.69-3.51 (m, 5H), 3.40-3.32 (m, 5H), 3.29-3.22 (m, 4H), 3.02 (t, J = 12.0 Hz, 1H), 2.95-2.81 (m, 2H), 2.89 (s, 6H), 2.15-2.03 (m, 5H), 1.91-1.71 (m, 8H), 1.59-1.46 (m, 3H); ESI-MS calculated for C44H63FN6O5S [M + H]+ = 807.46, found: 807.50.
Methyl ((1S,2R)-2-((S)-1-(1-((1-(4-((1-acryloylazetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-2-(azetidin-1-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (13).
Compound 29d (41 mg, 0.061 mmol) was dissolved in dry DCM (5 mL) and DIPEA (16 mg, 0.122 mmol) and acryloyl chloride (7 mg, 0.074 mmol) were added at 0 °C. After stirring for 2 h at rt, the reaction mixture was concentrated under vacuum. The residue was purified by reverse phase preparative HPLC to give the title product (30 mg, 59%) as the trifluoroacetate salt. 1H NMR (400 MHz, MeOD) δ 7.70 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.12 (m, 2H), 7.06 (d, J = 7.6 Hz, 1H), 6.52 (d, J = 8.8 Hz, 2H), 6.33-6.20 (m, 2H), 5.77-5.74 (m, 1H), 4.51-4.44 (m, 4H), 4.39-4.32 (m, 2H), 4.26-4.11 (m, 6H), 3.80-3.72 (m, 3H), 3.56-3.48 (m, 3H), 3.41-3.39 (m, 2H), 3.25-3.20 (m, 1H), 3.04-2.92 (m, 2H), 2.81-2.74 (m, 1H), 2.57-2.41 (m, 2H), 2.08-1.97 (m, 4H), 1.89-1.77 (m, 2H), 1.71-1.63 (m, 3H), 1.51-1.42 (m, 1H), 1.15-1.08 (m, 1H); ESI-MS calculated for C39H52FN5O5S [M + H]+ = 722.37, found: 722.16.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-((1-(4-((1-((E)-4-(azetidin-1-yl)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (14).
Azetidine (4.3 mg, 0.074 mmol) was added at room temperature to a solution of (E)-4-bromobut-2-enoic acid (12 mg, 0.074 mmol) and DIPEA (19 mg, 0.150 mmol) in DMF (1 mL). After stirring for 1 h at 60 °C, compound 29d (25 mg, 0.037 mmol) and HATU (28 mg, 0.074 mmol) were added at 0 °C. After stirring for 30 min at rt, the reaction mixture was concentrated under vacuum. The residue was purified by reverse phase preparative HPLC to give the title compound as a trifluoroacetate salt (15 mg, 44%). 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.12 (m, 2H), 7.05 (d, J = 7.2 Hz, 1H), 6.65-6.58 (m, 1H), 6.53 (d, J = 8.8 Hz, 2H), 6.39 (d, J = 15.6 Hz, 1H), 4.59-4.49 (m, 4H), 4.37-7.31 (m, 2H), 4.28-4.21 (m, 3H), 4.18-4.15 (m, 4H), 4.13-4.09 (m, 2H), 4.01-3.99 (m, 2H), 3.81-3.74 (m, 3H), 3.57-3.44 (m, 3H), 3.40 (d, J = 6.8 Hz, 2H), 3.31 (s, 3H), 3.26-3.19 (m, 1H), 3.04-2.90 (m, 3H), 2.80-2.74 (m, 1H), 2.60-2.42 (m, 4H), 2.08-1.97 (m, 4H), 1.92-1.85 (m, 1H), 1.82-1.74 (m, 1H), 1.71-1.58 (m, 3H), 1.51-1.41 (m, 1H), 1.18-1.05 (m, 1H); ESI-MS calculated for C43H59FN6O5S [M + H]+ = 791.43, found: 791.44.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(3-fluorophenyl)-1-(1-((1-(4-((1-((E)-4-(pyrrolidin-1-yl)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)ethyl)cyclopentyl)carbamate (15).
Compound 15 was synthesized using the method described for 10 from the intermediate 29d and (E)-4-(pyrrolidin-1-yl)but-2-enoic acid hydrochloride. 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 8.8 Hz, 2H), 7.48-7.43 (m, 1H), 7.15-7.12 (m, 2H), 7.06 (d, J = 7.2 Hz, 1H), 6.78-6.71 (m, 1H), 6.52 (d, J = 8.8 Hz, 2H), 6.45 (d, J = 15.2 Hz, 1H), 4.52-4.50 (m, 4H), 4.37-4.28 (m, 2H), 4.27-4.22 (m, 1H), 4.20-4.11 (m, 5H), 4.00 (d, J = 6.0 Hz, 2H), 3.80-3.73 (m, 3H), 3.66-3.60 (m, 2H), 3.56-3.48 (m, 3H), 3.40 (d, J = 7.2 Hz, 2H), 3.31 (s, 3H), 3.26-3.20 (m, 1H), 3.14-3.09 (m, 2H), 3.04-2.92 (m, 2H), 2.81-2.74 (m, 1H), 2.56-2.49 (m, 1H), 2.46-2.40 (m, 1H), 2.19-2.13 (m, 2H), 2.08-1.91 (m, 7H), 1.80-1.73 (m, 1H), 1.70-1.59 (m, 3H), 1.52-1.43 (m, 1H), 1.20-1.09 (m, 1H); ESI-MS calculated for C44H61FN6O5S [M + H]+ = 805.44, found: 805.57.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(3-fluorophenyl)-1-(1-((1-(4-((1-((E)-4-(piperidin-1-yl)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)ethyl)cyclopentyl)carbamate (16).
Compound 16 was synthesized using the method described for 10 from the intermediate 29d and (E)-4-(piperidin-1-yl)but-2-enoic acid hydrochloride. 1H NMR (400 MHz, MeOD) δ 7.70 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.13 (m, 2H), 7.05 (d, J = 6.8 Hz, 1H), 6.78-6.71 (m, 1H), 6.53 (d, J = 8.8 Hz, 2H), 6.47 (d, J = 15.2 Hz, 1H), 4.53-4.49 (m, 4H), 4.39-4.32 (m, 2H), 4.28-4.12 (m, 6H), 3.91 (d, J = 6.4 Hz, 2H), 3.80-3.73 (m, 3H), 3.56-3.46 (m, 5H), 3.40 (d, J = 6.8 Hz, 2H), 3.31 (s, 3H), 3.26-3.20 (m, 1H), 3.04-2.92 (m, 4H), 2.81-2.74 (m, 1H), 2.57-2.45 (m, 2H), 2.09-1.95 (m, 6H), 1.87-1.84 (m, 2H), 1.79-1.73 (m, 3H), 1.69-1.56 (m, 3H), 1.53-1.41 (m, 2H), 1.15-1.05 (m, 1H); 13C NMR (100 MHz, MeOD) δ 165.8, 165.2, 162.7, 162.3, 162.0, 161.6, 159.8, 156.0, 139.8, 132.9, 131.3, 128.3, 125.6, 123.9, 122.2, 119.3, 117.2, 116.9, 116.4, 115.8, 115.5, 111.6, 62.0, 60.8, 60.3, 58.0, 56.1, 54.8, 54.3, 54.0, 53.0, 52.2, 51.9, 51.0, 50.1, 41.3, 33.7, 26.9, 26.7, 26.4, 26.0, 24.3, 22.5, 21.2, 17.0; ESI-MS calculated for C45H63FN6O5S [M + H]+ = 819.46, found: 819.06.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-((1-(4-((1-((E)-4-(3-fluoroazetidin-1-yl)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (17).
Compound 17 was synthesized using the method described for 14 from the intermediate 29d and 3-fluoroazetidine hydrochloride. 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.12 (m, 2H), 7.05 (d, J = 7.6 Hz, 1H), 6.67-6.60 (m, 1H), 6.52 (d, J = 8.8 Hz, 2H), 6.42 (d, J = 15.6 Hz, 1H), 5.51-5.32 (m, 1H), 4.62-4.55 (m, 2H), 4.53-4.47 (m, 4H), 4.42-4.28 (m, 4H), 4.26-4.22 (m, 1H), 4.19-4.15 (m, 4H), 4.09 (d, J = 6.4 Hz, 2H), 3.80-3.73 (m, 3H), 3.56-3.48 (m, 3H), 3.40 (d, J = 6.8 Hz, 2H), 3.31 (s, 3H), 3.30-3.19 (m, 2H), 3.04-2.90 (m, 2H), 2.80-2.74 (m, 1H), 2.57-2.41 (m, 2H), 2.08-1.97 (m, 4H), 1.92-1.86 (m, 1H), 1.81-1.75 (m, 1H), 1.70-1.59 (m, 3H), 1.51-1.41 (m, 1H), 1.17-1.06 (m, 1H); ESI-MS calculated for C43H58F2N6O5S [M + H]+ = 809.42, found: 809.50.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-((1-(4-((1-((E)-4-(3,3-difluoroazetidin-1-yl)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (18).
Compound 18 was synthesized using the method described for 14 from the intermediate 29d and 3,3-difluoroazetidine hydrochloride. 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 9.2 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.12 (m, 2H), 7.05 (d, J = 7.6 Hz, 1H), 6.70-6.63 (m,, 1H), 6.53 (d, J = 8.8 Hz, 2H), 6.36 (d, J = 15.6 Hz, 1H), 4.55-4.49 (m, 8H), 4.38-4.32 (m, 2H), 4.28-4.21 (m, 1H), 4.18-4.11 (m, 5H), 3.98 (d, J = 6.4 Hz, 2H), 3.80-3.73 (m, 3H ), 3.56-3.47 (m, 3H), 3.40 (d, J = 7.2 Hz, 2H), 3.31 (s, 3H), 3.26-3.19 (m, 1H), 3.04-2.92 (m, 2H), 2.80-2.74 (m, 1H), 2.54-2.42 (m, 2H), 2.08-1.97 (m, 4H), 1.92-1.86 (m, 1H), 1.80-1.75 (m, 1H), 1.71-1.58 (m, 3H), 1.51-1.45 (m, 1H), 1.15-1.05 (m, 1H); ESI-MS calculated for C43H58F3N6O5S [M + H]+ = 827.41, found: 827.49.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(3-fluorophenyl)-1-(1-((1-(4-((1-((E)-4-(4-fluoropiperidin-1-yl)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)ethyl)cyclopentyl)carbamate (19).
Compound 19 was synthesized using the method described for 14 from the intermediate 29d and 4-fluoropiperidine hydrochloride. 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.12 (m, 2H), 7.06 (d, J = 7.6 Hz, 1H), 6.79-6.72 (m, 1H), 6.53 (d, J = 8.8 Hz, 2H), 6.48 (d, J = 15.2 Hz, 1H), 5.07-5.00 (m, 1H), 4.53-4.47 (m, 4H), 4.37-4.32 (m, 2H), 4.27-4.11 (m, 6H), 3.97 (d, J = 6.8 Hz, 2H), 3.80-3.74 (m, 3H), 3.56-3.39 (m, 7H), 3.31 (s, 3H), 3.27-3.22 (m, 2H), 3.04-2.92 (m, 2H), 2.83-2.74 (m, 1H), 2.55-2.44 (m, 2H), 2.24-1.89 (m, 9H), 1.82-1.63 (m, 5H), 1.51-1.41 (m, 1H), 1.16-1.11 (m, 1H); ESI-MS calculated for C45H62F2N6O5S [M + H]+ = 837.45, found: 837.60.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-((1-(4-((1-((E)-4-(4,4-difluoropiperidin-1-yl)but-2-enoyl)azetidin-3-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (20).
Compound 20 was synthesized using the method described for 14 from the intermediate 29d and 4,4-difluoropiperidine hydrochloride. 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.16-7.12 (m, 2H), 7.05 (d, J = 8.4 Hz, 1H), 6.79-6.72 (m, 1H), 6.52 (d, J = 8.8 Hz, 2H), 6.48 (d, J = 15.6 Hz, 1H), 4.52-4.51 (m, 4H), 4.40-4.32 (m, 2H), 4.28-4.22 (m, 1H), 4.20-4.10 (m, 5H), 4.02-4.00 (m, 2H), 3.80-3.73 (m, 3H), 3.56-3.39 (m, 8H), 3.31 (s, 3H), 3.25-3.19 (m, 2H), 3.04-2.92 (m, 2H), 2.81-2.74 (m, 1H), 2.56-2.45 (m, 2H), 2.41-2.32 (m, 4H), 2.08-1.97 (m, 4H), 1.91-1.75 (m, 2H), 1.71-1.58 (m, 3H), 1.51-1.41 (m, 1H), 1.13-1.02 (m, 1H); ESI-MS calculated for C45H61F3N6O5S [M + H]+ = 855.44, found: 855.55.
tert-Butyl ((1S,2R)-2-((S)-2-amino-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)ethyl)-cyclopentyl)carbamate (22).
Intermediate 2120 (3g, 6.1 mmol) was added to a dry round bottomed flask then covered with a kimwipe and placed in a desiccator that was maintained under vacuum for 1-2 days. Then the flask was removed from the desiccator and quickly capped with a septum and the system was vacuumed under an N2 atmosphere. The anhydrous toluene (30 ml) was added to the flask, which was then cooled to 0 °C in an ice-bath. Diisobutylaluminiumhydride (25% in toluene, 16.4 mL, 24.4 mmol) was injected slowly into the reaction mixture with a syringe at 0 °C with stirring. Then the ice-bath was removed and the reaction was monitored using UPLC-Mass. After the intermediate 21 was consumed, in about 4 h, NaOH solution (1M, 20 mL) was added slowly into the reaction mixture at 0 °C to quench the reaction. After stirring for 5 min, the ice-bath was removed and additional saturated brine (20 mL) was added. Then EtOAc (~50 mL) was added, the solid was filtered with celite, and was washed with EtOAc. The solution was extracted with EtOAc and DCM twice respectively. The combined organic solvent was dried with Na2SO4, filtered, and concentrated in a rotatory evaporator. Then the residue was redissolved in MeOH (100 mL), NaBH4 (461 mg, 12.2 mmol) was added slowly at 0 °C and the reaction mixture was stirred at rt for 2 days. Then the reaction mixture was concentrated, and diluted with H2O. The solution was extracted with EtOAc and DCM twice respectively. The combined organic solvent was dried with Na2SO4, filter, and concentrated in a rotatory evaporator to give crude title product 22 (2.8 g, 93%) without further purification. 1H NMR (400 MHz, MeOD) δ 7.41-7.35 (m, 1H), 7.33-7.23 (m, 6H), 7.18 (d, J = 11.6 Hz, 1H), 6.99-6.95 (m, 1H), 4.07-4.02 (m, 1H), 3.52-3.44 (m, 2H), 3.24 (d, J = 14.4 Hz, 1H), 3.09 (d, J = 14.4 Hz, 1H), 2.98 (d, J = 11.2 Hz, 1H), 2.91 (d, J = 10.8 Hz, 1H), 2.35-2.29 (m, 1H), 2.12-2.04 (m, 2H), 2.01-1.94 (m, 2H), 1.77-1.69 (m, 1H), 1.61-1.58 (m, 1H), 1.54-1.47 (m, 2H), 1.44 (s, 9H), 1.41-1.29 (m, 3H), 1.22-1.14 (m, 2H); ESI-MS calculated for C30H42FN3O2 [M + H]+ = 496.33, found: 496.48.
tert-Butyl ((1S,2R)-2-((S)-1-(1-benzylpiperidin-4-yl)-2-(2-ethyl-1H-imidazol-1-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (23a).
Propionaldehyde (0.75 mL, 10.09 mmol), glyoxal (0.35 mL, 3.03 mmol, 40%) and ammonium acetate (227 mg, 3.03 mmol) were added to a solution of 22 (0.5 g, 1.01 mmol) in MeOH (6 mL). The reaction mixture was stirred at 60 °C overnight under an N2 atmosphere. Then the solution was concentrated and purified by reverse phase HPLC to afford the title product as the trifluoroacetate salt (323 mg, 46%). 1H NMR (400 MHz, MeOD) δ 7.56-7.50 (m, 1H), 7.47-7.42 (m, 7H), 7.31 (s, 1H), 7.19 (t, J = 8.4 Hz, 1H), 6.96 (s, 1H), 4.80 (d, J = 15.6 Hz, 1H), 4.58 (d, J = 15.6 Hz, 1H), 4.22 (s, 2H), 3.92-3.87 (m, 1H), 3.57 (d, J = 12.0 Hz, 1H), 3.42 (d, J = 11.6 Hz, 1H), 3.10-3.02 (m, 3H), 2.96 (t, J = 12.0 Hz, 1H), 2.70-2.61 (m, 2H), 2.27 (d, J = 14.8 Hz, 1H), 2.00 (d, J = 14.8 Hz, 1H), 1.92-1.90 (m, 1H), 1.58-1.52 (m, 2H), 1.46 (s, 9H), 1.40 (t, J = 7.6 Hz, 3H), 1.35-1.26 (m, 3H), 1.04-0.90 (m, 2H); ESI-MS calculated for C35H47FN4O2 [M + H]+ = 575.37, found: 575.49.
tert-Butyl ((1S,2R)-2-((S)-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)-2-(1H-1,2,3-triazol-1-yl)ethyl)cyclopentyl)carbamate (23b).
p-Toluenesulfonylhydrazide (207 mg, 1.11 mmol), glyoxal (0.29 mL, 2.52 mmol, 40% wt in H2O) and AcOH (12 mg, 0.2 mmol) were added to a solution of intermediate 22 (0.5 g, 1.01 mmol) in MeOH (10 mL). The reaction mixture was stirred overnight at 60 °C under an N2 atmosphere. Then, the solution was concentrated and purified by reverse phase HPLC to afford the title product as the trifluoroacetate salt (520 mg, 78%). 1H NMR (400 MHz, MeOD) δ 8.04 (s, 1H), 7.77 (s, 1H), 7.48-7.36 (m, 8H), 7.07 (t, J = 7.6 Hz, 1H), 5.27-5.11 (m, 2H), 4.21 (s, 2H), 4.20-4.10 (m, 1H), 3.44-3.38 (m, 2H), 3.04-2.94 (m, 2H), 2.80-2.74 (m, 1H), 2.55 (t, J = 11.6 Hz, 1H), 2.21 (d, J = 13.6 Hz, 1H), 1.90-1.76 (m, 2H), 1.55-1.45 (m, 2H), 1.40-1.35 (m, 1H), 1.31 (s, 9H), 1.28-1.05 (m, 3H), 0.72-0.61 (m, 1H); ESI-MS calculated for C32H42FN5O2 [M + H]+ = 548.33, found: 548.47.
tert-Butyl ((1S,2R)-2-((S)-1-(1-benzylpiperidin-4-yl)-2-(dimethylamino)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (23c).
Formaldehyde (0.12 mL, 1.61 mmol, 37% wt. in H2O) and sodium triacetoxyborohydride (342 mg, 1.61 mmol) were added to a solution of intermediate 22 (0.16 g, 0.32 mmol) in DCM (10 mL). The reaction mixture was stirred at rt overnight. Then the solution was concentrated and purified by reverse phase HPLC to afford the title product as the trifluoroacetate salt (150mg, 73%). 1H NMR (400 MHz, MeOD) δ 7.49-7.43 (m, 6H), 7.37 (d, J = 8.8 Hz, 2H), 7.14-1.10 (m, 1H), 4.24 (s, 2H), 3.97-3.93 (m, 1H), 3.83-3.77 (m, 2H), 3.52-3.47 (m, 2H), 3.07-2.90 (m, 8H), 2.75-2.65 (m, 1H), 2.34-2.20 (m, 1H), 2.14-2.03 (m, 2H), 2.00-1.93 (m, 1H), 1.68-1.46 (m, 7H), 1.29 (s, 9H); ESI-MS calculated for C32H46FN3O2 [M + H]+ = 524.36, found: 524.51.
tert-Butyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (23d).
1,3-Dibromopropane (0.74 ml, 7.26 mmol), K2CO3 (2.51 g, 18 mmol) and KI (100 mg, 0.6 mmol) were added to a solution of the intermediate 22 (3 g, 6.05 mmol) in MeCN (150 mL). The mixture was stirred at 80 °C for 1~2 days then it was filtered with celite to remove solid K2CO3. The filtrate was concentrated and dissolved in H2O, extracted with EtOAc and DCM twice respectively, and dried over Na2SO4, and the solvent was evaporated under vacuum. The residue was purified by the chromatography column to afford the title product (3g, 93%). 1H NMR (400 MHz, MeOD) δ 7.47-7.40 (m, 6H), 7.16-7.03 (m, 3H), 4.52-4.46 (m, 2H), 4.38-4.31 (m, 1H), 4.19-4.10 (m, 2H), 4.19 (s, 2H), 3.70-3.66 (m, 1H), 3.44-3.40 (m, 3H), 3.01-2.90 (m, 2H), 2.79-2.73 (m, 1H), 2.56-2.46 (m, 1H), 2.42-2.36 (m, 1H), 2.05-1.93 (m, 4H), 1.82-1.73 (m, 2H), 1.68-1.57 (m, 3H), 1.37-1.29 (m, 1H), 1.22 (s, 9H), 1.06-0.98 (m, 1H). 1H NMR (400 MHz, MeOD) δ ; ESI-MS calculated for C33H46FN3O2 [M + H]+ = 536.36, found: 536.44.
tert-Butyl ((1S,2R)-2-((S)-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)-2-(pyrrolidin-1-yl)ethyl)cyclopentyl)carbamate (23e).
23e was synthesized using the method described for 23d from the intermediate 22 and 1,4-dibromobutane. 1H NMR (400 MHz, MeOD) δ 7.49-7.41 (m, 6H), 7.33-7.30 (m, 2H), 7.14-7.09 (m, 1H), 4.19 (s, 2H), 4.14-4.10 (m, 1H), 3.81-3.64 (m, 4H), 3.49-3.33 (m, 4H), 3.02-2.90 (m, 2H), 2.87-2.80 (m, 1H), 2.21-2.14 (m, 2H), 2.05-1.93 (m, 7H), 1.80-1.68 (m, 4H), 1.38-1.27 (m 2H), 1.19 (s, 9H). ESI-MS calculated for C34H48FN3O2 [M + H]+ = 550.37, found: 550.53.
tert-Butyl ((1S,2R)-2-((S)-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)-2-(piperidin-1-yl)ethyl)cyclopentyl)carbamate (23f).
23f was synthesized using the method described for 23d from the intermediate 22 and 1,5-dibromopentane. 1H NMR (400 MHz, MeOD) δ 7.48-7.34 (m, 8H), 7.11-7.07 (m, 1H), 4.18 (s, 2H), 4.06-4.03 (m, 2H), 3.74-3.72 (m, 1H), 3.61-3.55 (m, 3H), 3.43-3.41 (m, 2H), 3.29-3.22 (m, 2H), 3.01-2.89 (m, 3H), 2.07-1.86 (m, 9H), 1.80-1.70 (m, 5H), 1.59-1.50 (m, 1H), 1.34-1.23 (m, 2H), 1.18 (s, 9H). ESI-MS calculated for C35H50FN3O2 [M + H]+ = 564.39, found: 564.56.
(1S,2R)-2-((S)-2-(Azetidin-1-yl)-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentan-1-amine (24d).
Compound 23d (2.55 g, 4.76 mmol) was dissolved in DCM (5 mL), then trifluoroacetic acid (10 mL) was added slowly at 0 °C. After stirring for 2 h at rt, the reaction mixture was concentrated under vacuum, and redissolved in DCM (100 mL). Amberlyst® a21 (3g) was added and stirred for 30 min to neutralize the remaining trifluoroacetic acid. Then the resin was filtered, and the organic solvent was evaporated to give the crude title product (1.8 g, 87%) without further purification. ESI-MS calculated for C28H38FN3 [M + H]+ = 436.30, found: 436.32.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)-ethyl)cyclopentyl)carbamate (25d).
Compound 24d (2.07 g, 4.75 mmol) was dissolved in dry DCM (50 mL). Then, DIPEA (3.31 mL, 19 mmol) and dimethyl dicarbonate (764 mg, 5.7 mmol) were added at 0 °C. After stirring for 2 h at rt, the reaction mixture was concentrated under vacuum. The residue was purified by reverse phase HPLC to give the title product (2.5 g, 87%) as a trifluoroacetate salt. 1H NMR (400 MHz, MeOD) δ 7.48-7.40 (m, 6H), 7.14-7.10 (m, 2H), 7.02 (d, J = 7.6 Hz, 1H), 4.52-4.47 (m, 2H), 4.38-4.31 (m, 2H), 4.21 (s, 2H), 4.11 (d, J = 15. 6 Hz, 1H), 3.76 (d, J = 15.6 Hz, 1H), 3.46-3.41 (m, 3H), 3.29 (s, 3H), 3.02-2.90 (m, 2H), 2.77-2.71 (m, 1H), 2.55-2.48 (m, 1H), 2.46-2.40 (m, 1H), 2.05-2.02 (m, 2H), 1.99-1.95 (m, 2H), 1.88-1.82 (m, 1H), 1.77-1.73 (m, 1H), 1.69-1.61 (m, 3H), 1.43-1.34 (m, 1H), 1.07-0.97 (m, 1H); ESI-MS calculated for C30H40FN3O2 [M + H]+ = 494.31, found: 494.45.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(3-fluorophenyl)-1-(piperidin-4-yl)ethyl)cyclopentyl)carbamate (26d).
10% Pd/C (280 mg, 10% wt.) was added to a solution of the trifluoroacetate salt 25d (1.6 g, 2.63 mmol) in MeOH (50 mL) under an N2 atmosphere. Then, the flask was degassed three times with stirring. Then the mixture was stirred for 2 h at room temperature under a normal pressure H2 atmosphere. After the Pd/C catalyst was filtered off, the solvent was removed by rotary evaporation to give the title product (0.95 g, 89%). 1H NMR (400 MHz, MeOD) δ 7.48-7.43 (m, 1H), 7.16-7.06 (m, 3H), 4.51-4.45 (m, 2H), 4.38-4.27 (m, 2H), 4.10 (d, J = 15.6 Hz, 1H), 3.77 (d, J = 15.2 Hz, 1H), 3.55-3.52 (m, 1H), 3.40-3.33 (m, 2H), 3.31 (s, 3H), 3.01-2.89 (m, 2H), 2.78-2.72 (m, 1H), 2.58-2.48 (m, 1H), 2.46-2.39 (m, 1H), 2.05-1.93 (m, 5H), 1.78-1.70 (m, 1H), 1.68-1.54 (m, 3H), 1.39-1.30 (m, 1H), 1.08-1.02 (m, 1H); ESI-MS calculated for C23H34FN3O2 [M + H]+ = 404.26, found: 404.42.
tert-Butyl 3-((4-(3-((4-((S)-2-(azetidin-1-yl)-1-(3-fluorophenyl)-1-((1R,2S)-2-((methoxy-carbonyl)amino)cyclopentyl)ethyl)piperidin-1-yl)methyl)azetidin-1-yl)phenyl)sulfonyl)-azetidine-1-carboxylate (28d).
Compound 27 (548 mg, 1.19 mmol), K2CO3 (274 mg, 1.98 mmol) and KI (16 mg, 0.099 mmol) were added to a solution of the intermediate 26d (400 mg, 0.991 mmol) in MeCN (5 mL). The mixture was stirred at 80 °C overnight. Then, the mixture was extracted with DCM, washed with brine, dried over Na2SO4, and the solvent was evaporated under vacuum. The residue was purified by reverse phase preparative HPLC to give the trifluoroacetate salt of 28d (650 mg, 74%). 1H NMR (400 MHz, MeOD) δ 7.68 (d, J = 8.8 Hz, 2H), 7.49-7.43 (m, 1H), 7.17-7.07 (m, 3H), 6.52 (d, J = 8.8 Hz, 2H), 4.51-4.46 (m, 2H), 4.39-4.28 (m, 2H), 4.18-4.07 (m, 8H), 3.81-3.74 (m, 3H), 3.55-3.51 (m, 3H), 3.41 (d, J = 6.8 Hz, 2H), 3.33(s, 3H), 3.26-3.20 (m 1H), 3.07-2.94 (m, 2H), 2.81-2.75 (m, 1H), 2.57-2.49 (m, 1H), 2.47-2.39 (m, 1H), 2.10-1.95 (m, 5H), 1.78-1.74 (m, 1H), 1.70-1.57 (m, 3H), 1.52-1.48 (m, 1H), 1.42 (s, 9H), 1.23-1.18 (m, 1H); ESI-MS calculated for C41H58FN5O6S [M + H]+ = 768.41, found: 768.50.
Methyl ((1S,2R)-2-((S)-2-(azetidin-1-yl)-1-(1-((1-(4-(azetidin-3-ylsulfonyl)phenyl)-azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (29d).
The trifluoroacetic acid salt of 28d (650 mg, 0.737 mmol) was dissolved in DCM (5 mL) and trifluoroacetic acid (5 mL) was added at 0 °C. After stirring for 1 h at rt, the reaction mixture was concentrated under vacuum to give the trifluoroacetate salt of 29d (500 mg, 87%). 1H NMR (400 MHz, MeOD) δ 7.69 (d, J = 8.8 Hz, 2H), 7.48-7.43 (m, 1H), 7.15-7.11 (m, 2H), 7.07 (d, J = 7.2 Hz, 1H), 6.52 (d, J = 9.2 Hz, 2H), 4.52-4.47 (m, 2H), 4.41-4.26 (m, 7H), 4.19-4.11 (m, 3H), 3.80-3.74 (m, 3H), 3.56-3.51 (m, 3H), 3.41 (d, J = 7.2 Hz, 2H), 3.32 (s, 3H), 3.27-3.20 (m, 1H), 3.05-2.93 (m, 2H), 2.81-2.74 (m, 1H), 2.56-2.49 (m, 1H), 2.47-2.39 (m, 1H), 2.08-2.05 (m, 2H), 2.01-1.95 (m, 3H), 1.80-1.73 (m, 1H), 1.70-1.59 (m, 3H), 1.53-1.44 (m, 1H), 1.21-1.11 (m, 1H); ESI-MS calculated for C36H50FN5O4S [M + H]+ = 668.36, found: 668.53.
Fluorescence Polarization (FP)-Based Binding Assay.
Compounds binding activity was measured using a FP assay described previously.21 Briefly, 5 μL of compounds with varying concentrations dissolved in DMSO was added to 195 μL of mixture of menin and the fluorescein-labeled tracer (compound 37 in our previous paper21) in the assay buffer (phosphate buffered saline, 100 μg/mL bovine γ-globulin, with 0.01% triton-x 100), and were incubated for 1 h at room temperture. Final concentrations of menin protein and the fluorescein-labeled tracer were 4 nM and 2 nM, respectively. FP values were measured using the Infinite M-1000 plate reader (Tecan, Morrisville, NC). The IC50 values were determined by nonlinear regression fitting of the sigmoidal dose-dependent FP decreases as a function of total compound concentrations using Graphpad Prism 5.0 software.
Cell Growth Inhibition.
MV4;11 (catalog no. CRL-9591) and HL-60 (catalog no. CRL-9591) cell lines were purchased from the American Type Culture Collection (ATCC), and MOLM-13 (catalog no. ACC 554) cell line was purchased from the DSMZ German cell bank. Cells were cultured in either Iscove's Modified Dulbecco's Medium or RPMI 1640 medium (ATCC) supplemented with 10% fetal bovine serum and 100 U/L penicillin-streptomycin and incubated at 37 °C under 5% CO2, according to the cell supplier’s instructions.
For cell growth experiments, compounds were diluted in the corresponding medium and then serially diluted 3-fold in a 96-well tissue culture plate to a final volume of 100 μL/well. Two to three thousand cells/well in 100 μL were added to each well containing compound to a final volume of 200 μL/well. Cells were incubated for 7 days at 37 °C under 5% CO2. Cell growth was evaluated utilizing a lactate dehydrogenase-based WST-8 assay (Dojindo Molecular Technologies, MD). An adequate volume of WST-8 reagent was added to each well, incubated for at least 1 h in the cell culture incubator, and read at 450 nm in a Tecan Infinite M-1000 multimode microplate reader (Tecan, Morrisville, NC). The readings were normalized to the untreated cells and fitted using a nonlinear regression analysis with the Graphpad Prism 5.0 software to obtain the IC50 value for each compound.
Computational modeling.
The ligands of interest covalently bind to Menin protein, thus the binding modes were investigated using covalent docking mode available in GOLD (version 5.2)24-25. Co-crystal structure of M-525/Menin (PDB code: 6B41) was used and “protonate 3D” module in MOE26 was applied to assign the physiological protonation states considering pH = 7.0. Ligand 3D structures were drawn and the carbon-carbon double bonds in the Michael acceptor of the ligands were replaced by sulfur-carbon single bonds to mimic the status after covalently binding. This sulfur was also used to assign the covalently binding atom in GOLD for docking simulation. 15 runs were performed for each ligand and the top-ranked poses by ChemPLP scoring function was selected.
Considering the high flexibility of our ligands, a 10 ns MD simulation was performed for each of the predicted binding modes using AMBER18.27 The covalent bonds of ligands-Cys329 require additional setup. RESP charge fitting28 needs to be firstly given to the special ligand-cysteine fragment. Both backbone amine and carbonyl of the cysteine are capped by methyl group.29 The charge profile are computed in HF/6-31G* using Gaussian0930 and the atomic partial charge are assigned in antechamber package available in AMBER18. The partial charges on the backbone atoms (e.g. Ca, N, H, C, O) in Cys329 were reserved, in order to be consistent with natural amino acids, but charges on the side chain were assigned based on the different ligand binding. Required force field parameters such as bond, angles, torsions associated with the C-S covalent bond were added using the available parameters in small molecular force field GAFF2. The complex system was then solvated using TIP3P solvation model31 and neutralized by adding counter ions, followed by a 12000 steps minimization (first 3000 steps in steepest descent and followed by conjugate gradient). A 0.2 ns NVT simulation with a 5 kcal/mol/Å2 force constraint was given to the protein-ligand system (not solvent) to gradually raised the temperature to physiological condition (310K); another constrained 0.2 ns NPT simulation with P = 1 atm was then performed. The whole system was then relaxed by gradually reducing the force constant to 0 in a 2 ns NPT simulation. A 10 ns unconstraint simulation was performed on the fully relaxed system. Over the simulation, the bond vibration associated with hydrogens are fixed using the SHAKE algorithm32, and the long-range non-bonding interactions were treated by applying PME method33 with a cutoff of 10Å. Figures were prepared using the PyMOL program (www.pymol.org).
X-ray Crystallography.
Menin (residues 2-610 containing a deletion from 460-519) was purified as described previously.20 Prior to crystallization, menin (25 mg/mL in 25 mM Tris 8.0, 150 mM NaCl and 5mM DTT) were incubated with M-808 in a protein to compound ratio of 1:1.2. Menin:M-808 was immediately allowed to crystallize at 4 °C.
All crystals were grown in drops containing 1 μL of complex and 1 μL of well solution (1.96 M NaCl, 89 mM Bis-Tris PH 6.8, 0.178 M MgCl2 and 10.7 mM Pr Acetate). The crystals were cryoprotected by progressively soaking crystals in well solution containing increasingly higher amounts of sodium formate (1 M - 5 M in steps of 1M). Diffraction data were collected on a Mar300 detector at the Advanced Photon Source LS-CAT 21-ID-F beamlines at Argonne National Laboratory. All data were processed with HKL200034. The structures of the menin inhibitors were solved by molecular replacement (Molrep35) using the apo menin structure (PDB ID 3U84) as the search model. Each structure went through iterative rounds of electron density fitting and structural refinement using Coot36 and Buster37, respectively. The coordinates and restraint files for the ligands were created from smiles in Grade38 with the mogul+qm option. The initial Fo-Fc electron density map showed the presence of each compound covalently bound to C329 (Figure S1). The following regions were disordered in the structures: 582-610 in menin:M-808. Data collection and structural refinement statistics are provided in Table S1 in SI.
Mass-spectroscopic analysis of human menin protein incubated with menin inhibitors.
Samples of menin (25 mg/mL in 25 mM T ris 8.0, 150 mM NaCl and 5mM DTT) were incubated with compounds in a protein to compound molar ratio of 1:1.2 for 1 h or overnight at 4 °C. Following incubation, the sample was diluted to 1 mg/mL with H2O. 0.1 mL of each sample was applied to a reverse phase HPLC column (Phenomenex Aeris widepore C4 column 3.6 μM, 50 × 2.10 mm) at a flow rate of 0.5 mL/min in H2O with 0.2% (v/v) HCOOH. Protein was eluted using a gradient of 5-100% MeCN with 0.2% (v/v) HCOOH over 4 min. The LC-MS experiment (Agilent Q-TOF 6545) was carried out under the following conditions: fragmentor voltage, 300 V; skimmer voltage, 75 V; nozzle voltage, 100 V; sheath gas temperature, 350 °C; drying gas temperature, 325 °C. MassHunter Qualitative Analysis Software (Agilent) was used to analyze the data. Intact protein masses were obtained using the maximum entropy deconvolution algorithm.
Animal Experiments.
All animal experiments were performed under the guidelines of the University of Michigan Committee for Use and Care of Animals and using an approved animal protocol (PI, Shaomeng Wang).
Tolerability studies in mice.
The compounds were tested at indicated doses, with three times a week (days 1, 3 and 5) for one week. Three female SCID mice were used per group. Mice were weighed and checked for clinical signs of toxicity. Greater than 10% weight loss was considered toxic.
In Vivo pharmacodynamics and efficacy studies in an MV4;11 human AML xenograft model in mice.
MV4;11 cells were grown in suspension and collected in the log phase. A cell sample was mixed 1:1 with Trypan Blue (GIBCO™, Invitrogen Corp.) and counted on a hemocytometer to determine the number of live/dead cells. Cells were washed twice with 1 × PBS (GIBCO™, Invitrogen Corp.) and resuspended in an ice cold mixture of 1:1 PBS and Matrigel (BD Biosciences, Invitrogen Corp.) for a final Matrigel protein concentration of 5 mg/mL. MV4;11 cells were inoculated into female C.B-17 SCID mice at 5 × 106 cells in 0.1 mL with Matrigel. Cells were injected by subcutaneous route into the flank region of each mouse. The size of tumors growing in the mice was measured in two dimensions using calipers. Tumor volume (mm3) = (A×B2)/2 where A and B are the tumor length and width (mm), respectively.
For pharmacodynamics studies, mice bearing MV4;11 tumors were injected with a single dose of either vehicle or compound at 10 mg/kg or 25 mg/kg administered IV. Tumors were harvested at 6 h, 24 h or 48 h after the injection, with 3 mice/tumors for each time point. Tumor tissue was immediately frozen in liquid N2, ground into fine powder, placed on dry ice and stored at −80 °C for RT-PCR analysis.
For efficacy studies, before treatment, tumors were allowed to grow to 60-140 mm3 in volume. Mice with tumors within the acceptable size range were randomized into treatment groups of 7 mice per group. Experimental compound M-808 was given IV, in a solution of 10% PEG 400:3% Cremophor EL:87% PBS. The control group was given vehicle only. During treatment, tumor volume and body weight was measured two or three times per week. After the treatment was stopped, tumor volume and body weight was measured at least once per week.
Real-Time PCR.
Total RNA was isolated from tumor samples from pharmacodynamic studies, using the RNEASY kit (QIAGEN) according to the manufacturer’s protocol. The cDNA was generated using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR amplifications of HOXA9, MEIS1, and GAPDH genes were carried out with primers specific for each gene, using TaqMan gene expression assays (Applied Biosystems). Relative quantification of each gene transcript was calculated by a comparative cycle threshold (Ct) method. The results were presented as relative expression to vehicle treatment after normalizing to an internal loading control GAPDH. The catalog numbers for primers of each genes are: Hs00365956_m1 (HOXA9), Hs00180020_m1 (MEIS1), and Hs02786624_g1 (GAPDH)
Supplementary Material
ACKNOWLEDGMENT
We thank G.W. A. Milne for his critical reading and editing of the manuscript.
Funding Sources: This work is supported in part by the National Institutes of Health/National Cancer Institute (R01 CA208267 to SW), the Prostate Cancer Foundation (to SW) and the Rogel Cancer Center Core Grant from the National Cancer Institutes, NIH (P30 CA046592). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
ABBREVIATIONS
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- DCM
dichloromethane
- DIBAL-H
Diisobutylaluminium hydride
- DIPEA
N,N-Diisopropylethylamine
- ESI-MS
electrospray ionization mass spectrometry
- FP
Fluorescence polarization
- MD
molecular dynamic
- MEN1
multiple endocrine neoplasia 1
- MLL
mixed lineage leukemia protein
- MTD
maximum tolerated dose
- PBS
phosphate buffered saline
- PD
pharmacodynamics
- RT
room temperature
- SCID
severe combined immunodeficient
- TFA
trifluoroacetic acid
- TGI
tumor growth inhibition
- THF
tetrahydrofuran
- UPLC
Ultra performance liquid chromatography
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Modeled structures for compounds 9-12 in complex with menin protein in PDB format. Crystallography data collection and refinement statistics. Purity for the all final compounds. A molecular string file for all the final target compounds (CSV).
Accession Codes. The coordinated for M-808 complexed with menin has been deposited into the Protein Data Bank with the PDB ID code: 6MN9. Authors will release the atomic coordinates upon article publication.
Notes: The authors declare the following competing financial interest(s): The University of Michigan has filed a number of patent applications on this class of menin inhibitors, which have been licensed to Medsyn Biopharma, LLC. S. Wang, S. Xu, A. Aguilar, L. Huang, T. Xu, K. Zheng, D. McEachern, and J. Stuckey are co-inventors on one or more of these patents, and receive royalties on these patents from the University of Michigan. S. Wang is a co-founder of Medsyn. The University of Michigan and S. Wang own stock in Medsyn.
REFERENCES
- 1.Krivtsov AV; Armstrong SA, MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7 (11), 823–833. [DOI] [PubMed] [Google Scholar]
- 2.Guest EM; Hirsch BA; Kolb EA; Alonzo TA; Gerbing R; Aplenc R; Pollard J; Sung L; Meshinchi S; Gamis AS; Raimondi SC, Prognostic significance of 11q23/MLL fusion partners in children with acute myeloid leukemia (AML) - results from the children's oncology group (COG) trial AAML0531. Blood 2016, 128 (22), 1211–1211. [Google Scholar]
- 3.Meyer C; Kowarz E; Hofmann J; Renneville A; Zuna J; Trka J; Ben Abdelali R; Macintyre E; De Braekeleer E; De Braekeleer M; Delabesse E; de Oliveira MP; Cave H; Clappier E; van Dongen JJM; Balgobind BV; van den Heuvel-Eibrink MM; Beverloo HB; Panzer-Grumayer R; Teigler-Schlegel A; Harbott J; Kjeldsen E; Schnittger S; Koehl U; Gruhn B; Heidenreich O; Chan LC; Yip SF; Krzywinski M; Eckert C; Moricke A; Schrappe M; Alonso CN; Schafer BW; Krauter J; Lee DA; zur Stadt U; Te Kronnie G; Sutton R; Izraeli S; Trakhtenbrot L; Lo Nigro L; Tsaur G; Fechina L; Szczepanski T; Strehl S; Ilencikova D; Molkentin M; Burmeister T; Dingermann T; Klingebiel T; Marschalek R, New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23 (8), 1490–1499. [DOI] [PubMed] [Google Scholar]
- 4.Popovic R; Zeleznik-Le NJ, MLL: how complex does it get? J. Cell. Biochem 2005, 95 (2), 234–242. [DOI] [PubMed] [Google Scholar]
- 5.Slany RK, When epigenetics kills: MLL fusion proteins in leukemia. Hematol. Oncol 2005, 23 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- 6.Grembecka J; Belcher AM; Hartley T; Cierpicki T, Molecular basis of the mixed lineage leukemia-menin interaction: implication for targeting mixed lineage leukemias. J. Biol. Chem 2010, 285 (52), 40690–40698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokoyama A; Somervaille TCP; Smith KS; Rozenblatt-Rosen O; Meyerson M; Cleary ML, The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123 (2), 207–218. [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama A; Cleary ML, Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14 (1), 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cierpicki T; Grembecka J, Challenges and opportunities in targeting the menin-MLL interaction. Future Med. Chem 2014, 6 (4), 447–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi A; Murai MJ; He S; Lund G; Hartley T; Purohit T; Reddy G; Chruszcz M; Grembecka J; Cierpicki T, Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120 (23), 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grembecka J; He S; Shi A; Purohit T; Muntean AG; Sorenson RJ; Showalter HD; Murai MJ; Belcher AM; Hartley T; Hess JL; Cierpicki T, Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol 2012, 8 (3), 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borkin D; Pollock J; Kempinska K; Purohit T; Li X; Wen B; Zhao T; Miao H; Shukla S; He M; Sun D; Cierpicki T; Grembecka J, Property focused structure-based optimization of small molecule inhibitors of the protein–protein interaction between menin and mixed lineage leukemia (MLL). J. Med. Chem 2016, 59 (3), 892–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borkin D; He S; Miao H; Kempinska K; Pollock J; Chase J; Purohit T; Malik B; Zhao T; Wang J; Wen B; Zong H; Jones M; Danet-Desnoyers G; Guzman Monica L.; Talpaz M; Bixby Dale L.; Sun D; Hess Jay L.; Muntean Andrew G.; Maillard I; Cierpicki T; Grembecka J, Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 2015, 27 (4), 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klossowski S; Miao H; Kempinska K; Wu T; Purohit T; Kim E; Linhares BM; Chen D; Jih G; Perkey E; Huang H; He M; Wen B; Wang Y; Yu K; Lee SC-W; Danet-Desnoyers G; Trotman W; Kandarpa M; Cotton A; Abdel-Wahab O; Lei H; Dou Y; Guzman M; Peterson L; Gruber T; Choi S; Sun D; Ren P; Li L-S; Liu Y; Burrows F; Maillard I; Cierpicki T; Grembecka J, Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Invest 2020, 130 (2), 981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krivtsov AV; Evans K; Gadrey JY; Eschle BK; Hatton C; Uckelmann HJ; Ross KN; Perner F; Olsen SN; Pritchard T; McDermott L; Jones CD; Jing D; Braytee A; Chacon D; Earley E; McKeever BM; Claremon D; Gifford AJ; Lee HJ; Teicher BA; Pimanda JE; Beck D; Perry JA; Smith MA; McGeehan GM; Lock RB; Armstrong SA, A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell 2019, 36 (6), 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He S; Senter TJ; Pollock J; Han C; Upadhyay SK; Purohit T; Gogliotti RD; Lindsley CW; Cierpicki T; Stauffer SR; Grembecka J, High-affinity small-molecule inhibitors of the menin-mixed lineage leukemia (MLL) interaction closely mimic a natural protein-protein interaction. J. Med. Chem 2014, 57 (4), 1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.ClinicalTrials.gov identifier for KO-539: NCT04067336.
- 18.ClinicalTrials.gov identifier for SNDX-5613: NCT04065399.
- 19.Aguilar A; Zheng K; Xu T; Xu S; Huang L; Fernandez-Salas E; Liu L; Bernard D; Harvey KP; Foster C; McEachern D; Stuckey J; Chinnaswamy K; Delproposto J; Kampf JW; Wang S, Structure-based discovery of M-89 as a highly potent inhibitor of the menin-mixed lineage leukemia (menin-MLL) protein–protein interaction. J. Med. Chem 2019, 62 (13), 6015–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu S; Aguilar A; Xu T; Zheng K; Huang L; Stuckey J; Chinnaswamy K; Bernard D; Fernández - Salas E; Liu L; Wang M; McEachern D; Przybranowski S; Foster C; Wang S, Design of the first - in - class, highly potent irreversible inhibitor targeting the menin - MLL protein – protein interaction. Angew. Chem. Int. Ed 2018, 57 (6), 1601–1605. [DOI] [PubMed] [Google Scholar]
- 21.Zhou H; Liu L; Huang J; Bernard D; Karatas H; Navarro A; Lei M; Wang S, Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein-protein interaction. J. Med. Chem 2013, 56 (3), 1113–1123. [DOI] [PubMed] [Google Scholar]
- 22.ChemDraw Professional 18, Waltham, MA, USA: PerkinElmer Inc., 2018. [Google Scholar]
- 23.Wissner A; Overbeek E; Reich MF; Floyd MB; Johnson BD; Mamuya N; Rosfjord EC; Discafani C; Davis R; Shi X; Rabindran SK; Gruber BC; Ye F; Hallett WA; Nilakantan R; Shen R; Wang Y-F; Greenberger LM; Tsou H-R, Synthesis and structure–activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J. Med. Chem 2003, 46 (1), 49–63. [DOI] [PubMed] [Google Scholar]
- 24.Jones G; Willett P; Glen RC; Leach AR; Taylor R, Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267 (3), 727–748. [DOI] [PubMed] [Google Scholar]
- 25.Verdonk ML; Cole JC; Hartshorn MJ; Murray CW; Taylor RD, Improved protein–ligand docking using GOLD. Proteins 2003, 52 (4), 609–623. [DOI] [PubMed] [Google Scholar]
- 26.Molecular operating environment (MOE), 2013.08, Montreal, QC, Canada: Chemical Computing Group, 2018. [Google Scholar]
- 27.Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TEI; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; Salomon-Ferrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA AMBER 2018, University of California, San Francisco, CA, USA, 2018. [Google Scholar]
- 28.Bayly CI; Cieplak P; Cornell W; Kollman PA, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem 1993, 97 (40), 10269–10280. [Google Scholar]
- 29.Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA, A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc 1995, 117 (19), 5179–5197. [Google Scholar]
- 30.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian, Wallingford, CT, USA, 2016. [Google Scholar]
- 31.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79 (2), 926–935. [Google Scholar]
- 32.Ryckaert J-P; Ciccotti G; Berendsen HJC, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23 (3), 327–341. [Google Scholar]
- 33.Darden T; York D; Pedersen L, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98 (12), 10089–10092. [Google Scholar]
- 34.Otwinowski Z; Minor W, [20] Processing of X-ray diffraction data collected in oscillation mode. In Meth. Enzymol, Carter CW, Ed. Academic Press: San Diego, USA, 1997; Vol. 276, pp 307–326. [DOI] [PubMed] [Google Scholar]
- 35.Vagin A; Teplyakov A, MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr 1997, 30 (6), 1022–1025. [Google Scholar]
- 36.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Cryst. D 2004, 60 (12 Part 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- 37.Roversi P; Sharff A; Smart OS; Vonrhein C; Womack TO BUSTER version 2.11.2 Cambridge, United Kingdom: Global Phasing Ltd., 2011. [Google Scholar]
- 38.Smart OS; Womack TO; Sharff A; Flensburg C; Keller P; Paciorek W; Vonrhein C; Bricogne G GRADE, 1.2.13, Cambridge, United Kingdom: Global Phasing Ltd., 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.