

Abstract
Background
Low pass sequencing has been proposed as a cost-effective alternative to genotyping arrays to identify genetic variants that influence multifactorial traits in humans. For common diseases this typically has required both large sample sizes and comprehensive variant discovery. Genotyping arrays are also routinely used to perform pharmacogenetic (PGx) experiments where sample sizes are likely to be significantly smaller, but clinically relevant effect sizes likely to be larger.
Results
To assess how low pass sequencing would compare to array based genotyping for PGx we compared a low-pass assay (in which 1x coverage or less of a target genome is sequenced) along with software for genotype imputation to standard approaches. We sequenced 79 individuals to 1x genome coverage and genotyped the same samples on the Affymetrix Axiom Biobank Precision Medicine Research Array (PMRA). We then down-sampled the sequencing data to 0.8x, 0.6x, and 0.4x coverage, and performed imputation. Both the genotype data and the sequencing data were further used to impute human leukocyte antigen (HLA) genotypes for all samples. We compared the sequencing data and the genotyping array data in terms of four metrics: overall concordance, concordance at single nucleotide polymorphisms in pharmacogenetics-related genes, concordance in imputed HLA genotypes, and imputation r2. Overall concordance between the two assays ranged from 98.2% (for 0.4x coverage sequencing) to 99.2% (for 1x coverage sequencing), with qualitatively similar numbers for the subsets of variants most important in pharmacogenetics. At common single nucleotide polymorphisms (SNPs), the mean imputation r2 from the genotyping array was 0.90, which was comparable to the imputation r2 from 0.4x coverage sequencing, while the mean imputation r2 from 1x sequencing data was 0.96.
Conclusions
These results indicate that low-pass sequencing to a depth above 0.4x coverage attains higher power for association studies when compared to the PMRA and should be considered as a competitive alternative to genotyping arrays for trait mapping in pharmacogenetics.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-021-07508-2.
Keywords: Trait mapping, Low-pass sequencing, Pharmacogenetics, Genotype imputation
Background
Research in human genetics relies on efficiently profiling the genome of large numbers of individuals. A number of approaches can be used for this, usually trading off comprehensiveness (i.e. the fraction of the genome that is measured) with cost. By far the most commonly-used approach is the genotyping array, in which a set of known polymorphisms (usually around 500,000-2,000,000) is measured. This technology is inexpensive (currently on the order of tens to hundreds of dollars), but the set of genetic variants profiled is a small number of all known variants, and the technology does not allow for the detection of new (for example rare or population-specific) genetic variants. Genotyping arrays are commonly used for pharmacogenetics (PGx) studies where typically sample numbers are more limited, but inclusion of PGx focused variants on the arrays makes them suitable tools for screening the genome for markers associated with efficacy and adverse events [3, 15, 16].
The technological alternative to genotyping technology is sequencing technology, in which specific polymorphisms are not targeted for analysis, but rather the entire genome is sampled with some average depth of coverage. As sequencing costs have dropped, low-pass sequencing (for our purposes, which we will define as sequencing in which the average coverage of the genome is equal to or lower than 1x) becomes an appealing alternative to genotyping [4, 6, 14]. As an intuition for why this approach is useful, note that a human sample sequenced at 0.4x coverage is expected to have a single sequencing read covering each of around 28 million of the 84.7 million genetic variants identified in the 1000 Genomes Project [1], while a genotyping array obtains measurements (albeit somewhat less noisy measurements) at two orders of magnitude fewer sites.
In this paper, we directly compare genotyping results from low-pass sequencing to a commonly used genotyping array, the Affymetrix Axiom Biobank Precision Medicine Research Array (PMRA). Two types of metrics are relevant for this comparison. One is simply the genome-wide coverage of the assay, which we measure using average imputation quality. The other is genotyping quality at particular genetic variants of interest. We were particularly interested in applications to PGx—the identification of genetic variants that influence drug response. In this application, genetic variants in the major histocompatibility complex (MHC) and genes involved in drug metabolism (so-called “ADME” genes, for absorption, distribution, metabolism, and excretion) are known to be particularly relevant. We thus considered these separately.
Results
We selected 79 individuals to be both genotyped and sequenced. These individuals derive from a pool of volunteers based out of Cambridge, UK for which prior consent was obtained. Each individual was genotyped on the Affymetrix Axiom Biobank PMRA, and sequenced by Gencove, Inc. to an average of 1x coverage using the Illumina HiSeq 4000 platform with paired-end 150 base pair reads. Sequencing reads were then sampled at random to obtain an average of 0.8x, 0.6x, and 0.4x coverage of the genome (Methods).
We then performed genotyped imputation of genetic variants in the 1000 Genomes Phase 3 release. This imputation was performed using minimac2 (for the genotyping array data) or Gencove’s loimpute software v0.18 (for the low-pass sequencing data, see Methods for details). Both the unimputed PMRA data and the imputed low-pass sequencing data were then used to impute HLA genotypes using HIBAG [18].
The relevant metrics to use when comparing the two technologies depend on the downstream use cases. Specifically, if an investigator is interested in identifying genetic variants associated with a trait but has no a priori knowledge of where in the genome such variants are likely to be located, then the relevant metric is the average correlation between imputed genotype calls and true genotypes. On the other hand, if the investigator knows that specific variants are most likely to be relevant to the trait of interest, then the relevant metric is the concordance between the technologies at those specific sites. Since in PGx applications there are some specific genes and variants of interest, we computed metrics in both of these classes.
Overall genotype concordance
We first examined the overall concordance between the genotyping arrays and imputed sequences at different depths. To do this, we removed genotypes imputed with low confidence (with less than 90% posterior probability on a single genotype), and assessed the concordance between the two platforms, averaging across individuals, using metrics from the draft guidance of the United States Food and Drug Administration [19]. These metrics measure concordance for variants present and absent in a reference genome—a “positive percent agreement” (PPA) for variants that are different from the reference and a “negative percent agreement” (NPA) for variants that match a reference genome. For our purposes we considered the genotypes from the PMRA as “truth”; in this case the PPA ranged from 98.2% for 0.4x coverage sequencing to 99.2% for 1x coverage sequencing, while the NPA ranged from 99.8% for 0.4x coverage to 99.9% for 1x coverage (Table 1).
Table 1.
Genotype concordance between genotyping and sequencing platforms
| Comparison | PPA (%) | NPA (%) | No Calls (Average) |
|---|---|---|---|
| Accuracy, .4x vs PMRA | 98.22% | 99.82% | 2535 |
| Accuracy, .6x vs PMRA | 98.76% | 99.85% | 1848 |
| Accuracy, .8x vs PMRA | 99.01% | 99.86% | 1508 |
| Accuracy, 1x vs PMRA | 99.19% | 99.88% | 1251 |
In all cases the genotyping array was treated as ‘Truth’. Positive % Agreement (PPA)– The percent of non-reference calls in the Truth dataset detected by Test, ignoring no calls in Test. (True Positives / True Positives + False Negatives). Negative % Agreement (NPA) – The percent of reference calls in the Truth dataset detected by Test, ignoring no calls in Test. (True Negatives / True Negatives + False Positives). No Calls– Count of No Calls in test that were variant in Truth. No calls are averaged across all 79 individuals. The total number of overlapping variants between the PMRA and the imputed sequence data is ~ 423 k
Genotype concordance at ADME genes
We then specifically compared the concordance between the genotypes at variants in ADME genes as defined by Hoverlson et al. [7]. There were 216 such variants that were directly genotyped on the PMRA. We thus computed the same concordance metrics specifically at these 216 variants. For these analyses we excluded low-confidence genotype calls from the low-pass sequencing data; the percentage of excluded calls range from 1.6% of genotype calls in the 0.4x data down to 0.8% of genotype calls in the 1x data.
Concordance results are presented in Fig. 1a. At common variants (where the minor allele is present in more than five copies in the sample, corresponding to a minor allele frequency over 3%), PPA ranged from 98.5% (for 0.4x coverage) up to 99.4% (for 1x coverage). The lowest concordance metric was the PPA at rarer variants, which ranged from 82.1% (for 0.4x coverage) to 95.2% (for 1x coverage).
Fig. 1.
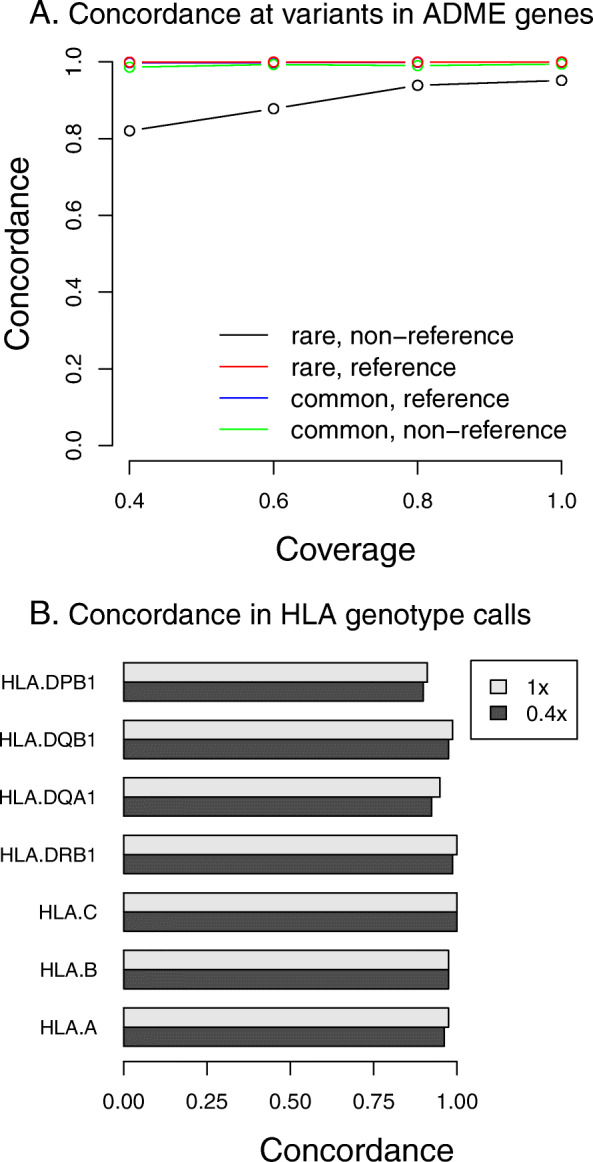
Genotype concordance across platforms at specific variants relevant to pharmacogenomics. a. Concordance at SNPs in ADME genes. Variants were classified as “rare” if the minor allele was present in five or fewer copies in the sample (corresponding to an allele frequency of about 3%. Concordance rates are split according to the genotype calls on the PRMA, which was considered “truth”—reference concordance is at variants where the PRMA is homozygous reference, and non-reference concordance is for all other sites. b. Concordance in HLA genotypes across platforms. Shown are the concordance rates between sequencing and genotyping array data in imputed HLA genotypes. Concordance is shown for 0.4x and 1x sequencing
Genotype concordance at HLA
Apart from ADME genes, another important locus in PGx is the MHC region. We imputed four digit HLA alleles from both the PMRA and sequencing data using HIBAG [18], and assessed the concordance across the two platforms at each of the seven HLA genes assessed by HIBAG. (Fig. 1b). There was little variation in imputed genotype concordance across levels of sequencing coverage, and with the exception of the gene DPB1, concordance was above 95%.
For samples where we saw consistent discordance for a given gene between the platforms, we then generated gold standard HLA genotype calls (Methods). A total of 15 HLA genotype calls in 12 samples were retested in this manner. The correct calls were obtained at 7/15 genotypes from the PMRA, and 6, 7, 7, and 8/16 genotypes after imputation from 0.4x, 0.6x, 0.8x, and 1x sequencing, respectively.
Imputation quality and comparison
Finally, an important metric of how well a technology assays known polymorphisms in the genome is the squared correlation between imputed genotype dosages and the true genotypes (known as “imputation r2”). Intuitively, if the researcher has a flat prior on where in the genome to look for an association between a genetic variant and a trait, the average squared correlation is a measure of the power of the study.
We computed this metric for different levels of sequencing coverage by correlating the imputed allelic dosages with directly genotyped sites. We computed this same metric for the genotype data by using the leave-one-out r2 at genotyped sites computed by minimac2. At common variants (allele frequency > 5% in the cohort), the average r2 obtained from the genotyping array was 0.9 (Fig. 2), consistent with previous reports from a European population [13]. For the sequencing data, this metric varied from 0.91 (for 0.4x coverage) to 0.96 (for 1x coverage). When all variants across the frequency spectrum were considered, the difference in average r2 between assays was even more pronounced, with the average r2 for the genotyping array being 0.85 and the average r2 for the sequencing data ranging from 0.88 (for 0.4x coverage) to 0.93 (for 1x coverage).
Fig. 2.
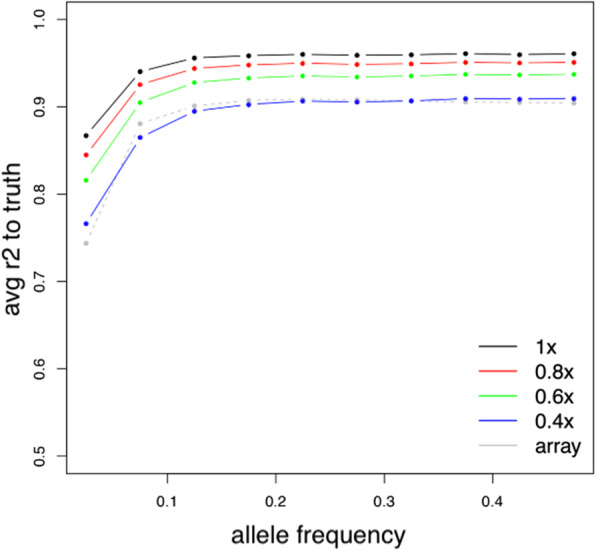
Comparison of imputation quality across platforms. Alleles were binned according to their minor allele frequency (as measured on the genotyping array) and imputation r2 averaged across all variants in the bin. For sequencing data, the array data was treated as ‘truth’ and imputation r2 computed by correlating imputed dosages to array genotypes. For the array data, imputation r2 for all genotyped variants was computed using a leave-one-out procedure implemented in minimac2
To investigate the effect of the choice of imputation reference panel on imputation performance on the sequencing data, we performed a head-to-head comparison between using the 1000 Genomes and a subset of the Haplotype Reference Consortium (HRC) haplotypes [12] as reference panels using the above methodology (i.e., by treating the array data as “truth” and comparing overlapping sites between the reference panel and the array sites). Using the HRC dataset as the imputation reference panel yielded marginal increases in average r2 values in all minor allele frequency (MAF) bins but the lowest, where it suffered a decrease of about 0.036 as compared to the 1000 Genomes imputed sites in the same bin (Supp. Tables S1, S2). The exact details and further discussion on this particular comparison can be found in the accompanying Supplementary Materials (Supp. Figs. S1, S2).
Discussion
In this paper, we performed a direct comparison between low-pass sequencing (combined with imputation) and a commonly-used genotyping array for the purposes of trait mapping in pharmacogenetics.
We observed that overall, genotype calls across the two platforms were highly concordant, with a positive percent agreement (PPA) of the imputed sequence data to the genotyping array calls ranging from 98.22% at 0.4x to 99.19% at 1x coverage.
At ADME genes, we observed qualitatively similar results, with a PPA ranging from 98.5 to 99.4% at sites of common variation (> 3% minor allele frequency in this cohort) and a PPA ranging from 82.1 to 95.2% at rarer variants (< 3%).
Four-digit HLA alleles in the MHC region imputed from sequencing data had high concordance with those imputed from the PMRA, with all concordances across the range of sequencing coverage observed to be above 95% with the exception of those in the DPB1 gene; further validation using a gold-standard assay of the HLA genotype calls resulted in similar concordance results between the imputed sequence or PMRA data and the resulting gold standards.
For the purposes of trait mapping, low-pass sequencing above a sequencing coverage of 0.4x had higher overall imputation accuracy as measured by imputation r2 than the genotyping array, indicating a corresponding increase in power.
Beyond simply comparing concordance between assays, it is important to consider other, orthogonal considerations when deciding between low-pass sequencing and genotyping arrays for PGx purposes. For instance, due to the sheer number of measurements (reads) made during a sequencing run (even at very low coverages), sequence data allows far more sensitive detection of copy number and structural variation [5, 20]. As intuition, consider that sequencing a human genome at a depth of 0.1x using 150 bp reads yields 2.2 million reads corresponding to measurements at 330Mbp, compared to a typical genotyping array which generates point measurements at only a few hundreds of thousands to a couple million sites.
Similarly, sequencing affords the capability to perform metagenomic profiling and analysis of the microbiome via analysis of non-human sequencing reads deriving from a DNA sample, as well as analysis of mitochondrial count [2, 17].
Logistical and budgetary considerations are also essential in real-world project planning. In a number of scenarios with different outcomes and study designs, low-pass sequencing has been shown to increase performance, for example by increasing the effective sample size of a genome-wide association study [14] or increasing the accuracy of polygenic risk scoring [10].
One of the drawbacks of low-pass sequencing compared to genotyping arrays is that while the average performance over the entire genome is consistently higher with sequencing, there may occasionally be a subset of specific SNPs or genes that, for a given study design, must be assayed with a level of accuracy and precision which low-coverage sequencing is simply unable to provide. In order to address this, it may be useful to develop an assay which combines low-pass whole genome sequencing with higher-depth coverage of pre-selected target regions on the genome, such that a single sequencing run simultaneously yields low-coverage sequence data across the genome at the same time as assaying specific SNPs, variants, or genes of interest with clinical grade accuracy.
Conclusion
As research into the effects of genetics on drug response continues to accelerate, it will become increasingly important for assays used in pharmacogenetics to provide reliable measurements both across the entire genome and at specific PGx focused variants. Our results demonstrate that low-pass sequencing and imputation provide a competitive alternative to genotyping arrays in both of these applications.
It is worth noting that the cost of sequencing is declining rapidly; if sequencing a human genome to 30x coverage costs $1000, then the cost of sequencing a human sample to 0.4x coverage is around $13. The key components of cost in a low-pass sequencing assay then become sequencing library preparation and analysis. As the costs of sequencing continue to drop, the importance of these latter costs will continue to grow.
Methods
Genotyping
Samples were from study TMT109167: A study to collect blood samples from members of the Clinical Unit Cambridge, UK (CUC) volunteer panel for DNA extraction and storage, for investigation of prospective genotype-phenotype relationships and stratification of subjects for recruitment into future clinical trials.
DNA samples were genotyped by BioStorage Technologies/Bioprocessing Solutions Alliance, Brooks Life Sciences (Piscataway, NJ, USA) using the Affymetrix Axiom PMRA.
Prior to genotype imputation, variants in each GWAS dataset were excluded using standard Affymetrix QC thresholds for the PMRA, if there were deviations from Hardy-Weinberg proportions within subgroups of any given ancestry or showed gross and irreconcilable differences in alleles or allele frequency with reference panel genotypes from the HapMap or 1000 Genome projects. Standard Affymetrix array QC sample level thresholds were also applied prior to imputation.
(http://www.affymetrix.com/support/downloads/manuals/axiom_genotyping_solution_analysis_guide.pdf)
Imputation of PMRA data
Genotype imputation for genetic variants that were not directly genotyped (“untyped variants”) was performed using a cosmopolitan haplotype reference panel from the 1000 Genomes Project [The 1000 Genomes Project Consortium, 2015], and using Hidden Markov Model methods as implemented in MaCH and minimac [8, 9].
HLA genotyping
High resolution HLA genotyping was performed at BioStorage Technologies/Bioprocessing Solutions Alliance, Brooks Life Sciences (Piscataway, NJ, USA) using the Thermo Fisher AllSet+ Gold SSP High-Resolution HLA kit for HLA-A, HLA-B, HLA-DRB1, HLA-DQB1 and HLA-DPB1 following the manufacturer’s instructions.
Sequencing
Sequencing libraries were prepared from DNA using the KAPA Library Preparation Kit by Roche and sequenced on an lllumina HiSeq 4000 instrument. Sequencing reads for each sample were aligned to the genome using bwa mem [9], and sequencing reads were randomly sampled to obtain different levels of sequencing coverage. Imputation of genotypes from sequencing data was done using loimpute v. 0.18 by Gencove, Inc. (New York, NY) to a reference panel comprising a subset of the 1000 Genomes Phase 3 (described in more detail in the Supplementary Materials).
Imputation of sequencing data
Imputation was performed using an implementation of the Li and Stephens model [11], described in more detail in the Supplementary Note.
Supplementary Information
Additional file 1: Supplementary note. Details of the model underlying loimpute, the software used to impute the low-pass sequencing data analysed in this study.
Additional file 2: Supplementary materials. Description of imputation performance using the HRC as a haplotype reference panel rather than the 1000 Genomes Phase 3 release. Figure S1. Comparison of imputation r2 across allele frequency bins for the 1000 Genomes panel. Figure S2. Comparison of imputation r2 across allele frequency bins for the HRC panel.
Acknowledgements
The abstract from an early version of this study was presented at the 51st European Society of Human Genetics Conference (see https://www.nature.com/articles/s41431-019-0407-4#Sec270).
Abbreviations
- PGx
Pharmacogenetics
- PMRA
Precision Medicine Research Array
- HLA
Human leukocyte antigen
- ADME
Absorption, distribution, metabolism, and excretion
- SNP
Single nucleotide polymorphism
- MHC
Major histocompatibility complex
- PPA
Positive percent agreement
- NPA
Negative percent agreement
- MAF
Minor allele frequency
- HRC
Haplotype Reference Consortium
Authors’ contributions
KW, TB, JKP, and CC designed the study. KW, DJF, and KK performed the experiments. TB, JKP, JHL, DJF, KK, and CC analyzed the data. All authors reviewed the manuscript. The authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
The datasets generated and/or analysed during the current study are proprietary and are therefore not publicly available. The data were not consented for use outside the scope of the present study, and as such, dataset access cannot be applied for.
Details on the 1000 Genomes Phase 3 release dataset and instructions on how to download the data can be found at the following URL: https://www.internationalgenome.org/category/phase-3/
Details on the Haplotype Reference Consortium datasets and instructions on how to apply for and access the data can be found at the following URL: http://www.haplotype-reference-consortium.org/data-access
Declarations
Ethics approval and consent to participate
Ethical approval (LREC ref.: 08/H0302/100, UK) and written informed consent were obtained prior to conducting this study. Namely, ethics approval was granted by National Research Ethic Service, Cambridgeshire 2 Research Ethics Committee, Victoria House, Capital Park, Fulbourn, Cambridge. CB21 5XB on 2nd Dec 2008. They are part of The Research Ethics Service (RES) affiliated with the Health Research Authority (HRA), based in the UK.
Consent for publication
Not applicable.
Competing interests
K. W., T.B., J.K.P., and J.H.L were employees of Gencove, Inc. at the time of writing.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai N, Li Y, Chang S, Liang J, Lin C, Zhang X, Liang L, Hu J, Chan W, Kendler KS, Malinauskas T, Huang GJ, Li Q, Mott R, Flint J, et al. Genetic control over mtDNA and its relationship to major depressive disorder. Curr Biol. 2015;25(24):3170–3177. doi: 10.1016/j.cub.2015.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, Gardina P, Hubbard J, Turpaz Y, Langaee TY, Eby C, King CR, Brower A, Schmelzer JR, Glurich I, Vidaillet HJ, Yale SH, Qi Zhang K, Berg RL, Burmester JK, et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111(8):4106–4112. doi: 10.1182/blood-2007-11-122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CONVERGE consortium. Cai N, Bigdeli TB, Kretzschmar W, Li Y, Liang J, Song L, Hu J, Li Q, Jin W, et al. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–591. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong Z, Zhang J, Hu P, Chen H, Xu J, Tian Q, Meng L, Ye Y, Wang J, Zhang M, Li Y, Wang H, Yu S, Chen F, Xie J, Jiang H, Wang W, Choy KW, Xu Z. Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med. 2016;18(9):940–948. doi: 10.1038/gim.2015.199. [DOI] [PubMed] [Google Scholar]
- 6.Gilly, A., Kuchenbaecker, K., Southam, L., Suveges, D., Moore, R., Melloni, G., Hatzikotoulas, K., Farmaki, A.-E., Ritchie, G., Schwartzentruber, J., et al. (2017). Very low depth whole genome sequencing in complex trait association studies. [DOI] [PMC free article] [PubMed]
- 7.Hovelson DH, Xue Z, Zawistowski M, Ehm MG, Harris EC, Stocker SL, Gross AS, Jang I-J, Ieiri I, Lee J-E, et al. Characterization of ADME gene variation in 21 populations by exome sequencing: Pharmacogenet. Genomics. 2017;27:89–100. doi: 10.1097/FPC.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li JH, Mazur CA, Berisa T, Pickrell JK. Low-pass sequencing increases the power of GWAS and decreases measurement error of polygenic risk scores compared to genotyping arrays. Genome Res. 2021. gr-266486. [DOI] [PMC free article] [PubMed]
- 11.Li N, Stephens M. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics. 2003;165(4):2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Min Kang H, Fuchsberger C, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson, S.C., Romm, J.M., Doheny, K.F., Pugh, E.W., and Laurie, C.C. (2017). Imputation-based genomic coverage assessments of current genotyping arrays: Illumina HumanCore, OmniExpress, multi-ethnic global array and sub-arrays, global screening array, Omni2.5M, Omni5M, and Affymetrix UK biobank.
- 14.Pasaniuc B, Rohland N, McLaren PJ, Garimella K, Zaitlen N, Li H, Gupta N, Neale BM, Daly MJ, Sklar P, Sullivan PF, Bergen S, Moran JL, Hultman CM, Lichtenstein P, Magnusson P, Purcell SM, Haas DW, Liang L, Sunyaev S, Patterson N, de Bakker PIW, Reich D, Price AL, et al. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat Genet. 2012;44(6):631–635. doi: 10.1038/ng.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.SEARCH Collaborative Group, Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med 2008;359(8):789–799. doi: 10.1056/NEJMoa0801936. Epub 2008 Jul 23. PMID: 18650507. [DOI] [PubMed]
- 16.Shuldiner AR, O’Connell JR, Bliden KP, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of Clopidogrel therapy. JAMA. 2009;302(8):849–857. doi: 10.1001/jama.2009.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood DE, Lu J, Langmead B. Improved metagenomic analysis with kraken 2. Genome Biol. 2019;20(1):257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR, Weir BS. HIBAG—HLA genotype imputation with attribute bagging. Pharma J. 2014;14(2):192–200. doi: 10.1038/tpj.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.U.S. Food & Drug Administration. (2018) Considerations for Design, Development, and Analytical Validation of Next Generation Sequencing (NGS) – Based In Vitro Diagnostics (IVDs) Intended to Aid in the Diagnosis of Suspected Germline Diseases. Retrieved from https://www.fda.gov/media/99208/download
- 20.Zhou B, Ho SS, Zhang X, Pattni R, Haraksingh RR, Urban AE. Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J Med Genet 2018;55(11):735–743. doi: 10.1136/jmedgenet-2018-105272. Epub 2018 Jul 30. PMID: 30061371. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplementary note. Details of the model underlying loimpute, the software used to impute the low-pass sequencing data analysed in this study.
Additional file 2: Supplementary materials. Description of imputation performance using the HRC as a haplotype reference panel rather than the 1000 Genomes Phase 3 release. Figure S1. Comparison of imputation r2 across allele frequency bins for the 1000 Genomes panel. Figure S2. Comparison of imputation r2 across allele frequency bins for the HRC panel.
Data Availability Statement
The datasets generated and/or analysed during the current study are proprietary and are therefore not publicly available. The data were not consented for use outside the scope of the present study, and as such, dataset access cannot be applied for.
Details on the 1000 Genomes Phase 3 release dataset and instructions on how to download the data can be found at the following URL: https://www.internationalgenome.org/category/phase-3/
Details on the Haplotype Reference Consortium datasets and instructions on how to apply for and access the data can be found at the following URL: http://www.haplotype-reference-consortium.org/data-access