

Abstract
Hidradenitis suppurativa is a chronic relapsing disease with multiple abscesses, nodules, and scars in the apocrine bearing areas. Dowling-Degos is a rare autosomal dominant genodermatosis characterized by multiple hyperpigmented macules or papules in reticulate pattern, affecting mainly the flexures. We report a case of coexisting hidradenitis suppurativa and Dowling-Degos disease in a 31-year-old male in whom PSENEN mutation analysis revealed a splice site mutation c.62-1G>T.
Keywords: Dowling-Dego, hidradenitis, PSENEN
Introduction
Hidradenitis suppurativa (HS) is a chronic relapsing disease with multiple abscesses, nodules, and scars in the apocrine bearing areas. The pathogenesis is attributed to follicular occlusion due to hyperkeratosis. It has been reported with genetic diseases like Dowling-Degos disease (DDD), pachyonychia congenita, and keratitis-ichthyosis-deafness syndrome.[1,2] Dowling-Degos disease is a rare autosomal dominant genodermatosis with multiple hyperpigmented macules or papules in a reticulate pattern, affecting the flexures, with prominent comedo-like lesions and pitted scars.[2] Genetic mutations in DDD have been identified in KRT5, POFUT1, and POGLUT1.[3,4,5] Mutations in the PSENEN gene have been reported in patients with the coexistence of HS and DDD and in familial HS.[6,7]
Case Report
A 31-year-old man presented with recurrent painful nodules and pus discharging sinuses on the axillae, back, and gluteal region for the past 15 years. He had asymptomatic hyperpigmented macules over the face, axillae, and trunk since childhood. There was family history of HS in his mother, maternal grandfather, and maternal uncle and the history of hyperpigmented lesions over the body in his maternal grandfather, mother, maternal aunts, and cousins.
His BMI was 25. There were pitted scars on the face [Figure 1a] and hyperpigmented macules over the axillae [Figure 1b]. There were multiple pus-discharging abscesses, nodules, and double comedones over the axillae and gluteal region [Figure 2a]. There were multiple comedones, scars, and pigmented macules over the back [Figure 2b]. His routine blood investigations were normal. His ESR was 41 mm/hr. Pus culture from a discharging abscess grew Citrobacter diversus. Histopathology from a nodule over the gluteal region showed lamellar hyperkeratosis with increased basal cell pigmentation [Figure 3] and a pan dermal moderate perivascular inflammatory infiltrate. Mycobacterial and fungal cultures from the tissue were negative.
Figure 1.
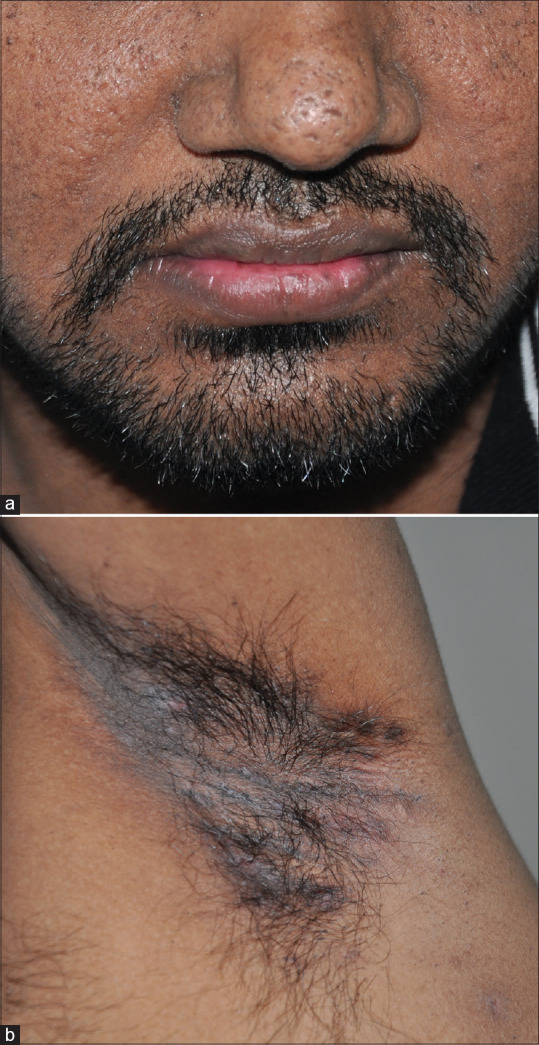
(a) Multiple pitted scars on the face. (b) Hyperpigmented macules over axilla and comedone-like lesions
Figure 2.
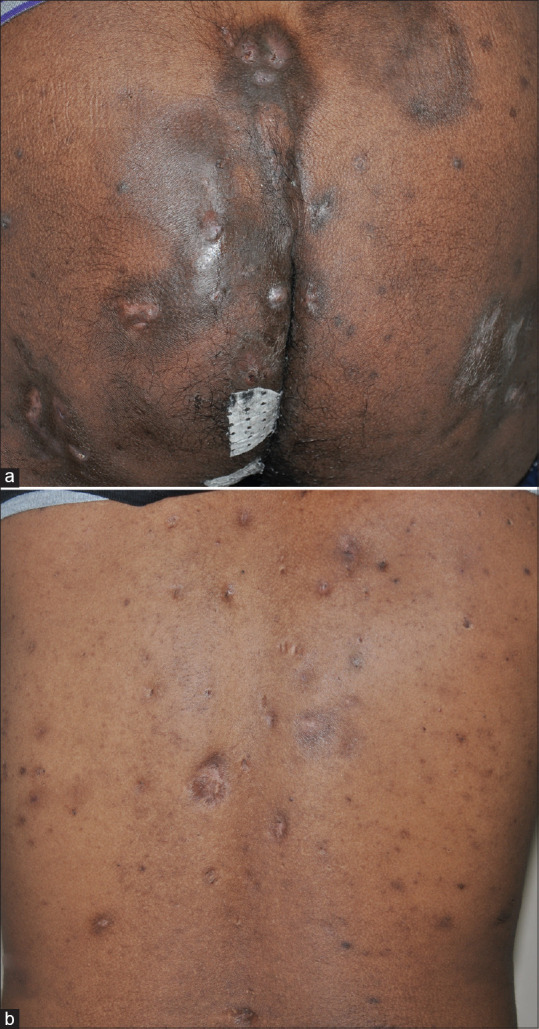
(a) Multiple pus discharging abscesses, scars, and comedones over gluteal region. (b) Multiple scars, comedones, and hyperpigmented macules over the back
Figure 3.
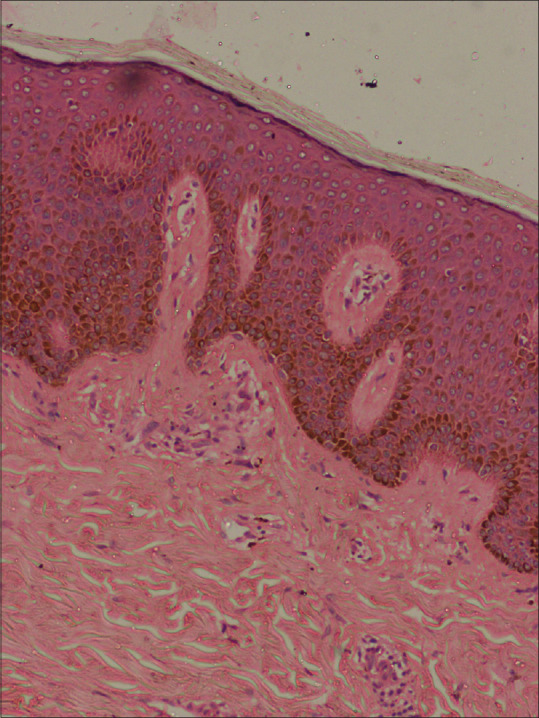
Increase in basal hyperpigmentation (H&E × 40)
He was diagnosed to have HS with coexisting DDD based on the history and clinical presentation. No pathogenic variants were identified in the KRT5 gene by DNA sequencing. However, PSENEN gene mutation analysis from genomic DNA by Sanger sequencing (at the University of Dundee) identified the splice site mutation c.62-1G>T.
Discussion
Hidradenitis suppurativa is a chronic relapsing inflammatory disease characterized by painful nodules, abscesses, and sinus tract formation in the apocrine areas. Dowling-Degos disease is an autosomal-dominant disorder of skin pigmentation. The exact pathogenesis of HS is not known and is considered to be multifactorial. Follicular occlusion due to hyperkeratosis is the primary event. Genetic and hormonal factors have been described as the etiology of the disease. The main risk factors for HS are obesity, smoking, and positive family history. There are case reports of genetic disorders including DDD, pachyonychia congenita and keratosis-ichthyosis-deafness syndrome being associated with HS. Like HS, they also have disruptive follicular keratinization.[1]
Dowling-Degos disease is associated with mutations in keratin 5 (KRT5), protein O-fucosyltransferase 1 (POFUT1), or protein O-glucosyltransferase 1(POGLUT1).[3,4,5] POGLUT1 adds O-linked glucose to serine residues in epidermal growth factor-like repeats of Notch receptors, and is an essential regulator of Notch signalling. The loss of Notch1 and Notch2 in mice results in abnormal pigmentation; these and other studies suggest that mutations in POGLUT1 resulting in aberrations in Notch signalling would lead to abnormal pigmentation and keratinocyte morphology.[8,9] The Notch pathway in the skin is thought to mediate the interactions between melanocytes and keratinocytes, thereby regulating the delicate balance between the proliferation and differentiation of these cells.[8] Disturbance in Notch pathway can lead to pigmentary disorders and cause epidermal and follicular hyperkeratosis and epidermal cyst formation. PSENEN encodes presenilin enhancer protein 2 (PEN-2), a member of the γ-secretase enzyme complex. The other members of this complex include presenilin, nicastrin, and anterior pharynx defective. The γ-secretase enzyme complex is responsible for the intracellular cleavage of Notch receptors that are necessary for the activation of Notch signaling.[10]
Globally, there are a few reports of mutations in the γ-secretase subunit–encoding PSENEN gene in patients with the coexistence of HS and DDD and in familial HS. Ralser et al. hypothesized that PSENEN mutation carriers primarily present with DDD; however, nicotine abuse and obesity may predispose them to develop HS.[6] The coexistence of hidradenitis suppurativa and Dowling-Degos disease points towards a common defect of follicular occlusion. Although the coexistence of HS and DDD with PSENEN gene mutation in Indian patients has been reported before, this report is of a different splice site mutation in the same position in PSENEN, c.62-1G>C.[6] A report of a different mutation in the same position of the PSESEN gene could indicate that this position is a mutation hotspot for Indian patients but studies in larger number of patients are needed. It also points to the genetic heterogeneity of DDD as KRT5 mutations were not found in this case. Genetic mutation studies will help in genetic counselling of families and also help us implement preventive measures to avoid obesity and nicotine use in those with the mutation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Fimmel S, Zouboulis CC. Comorbidities of hidradenitis suppurativa (acne inversa) Dermatoendocrinol. 2010;2:9–16. doi: 10.4161/derm.2.1.12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choudhary SV, Jain D, Agrawal P, Singh A. Dowling-Degos disease and hidradenitis suppurativa: Co occurrence or association? Indian Dermatol Online J. 2013;4:191–4. doi: 10.4103/2229-5178.115514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betz RC, Planko L, Eigelshoven S, Hannekan S, Pastermack SM, Bussow H, et al. Loss-of-function mutationsin the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510–9. doi: 10.1086/500850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li M, Cheng R, Liang J, Yan H, Zhang H, Yang L, et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am J Hum Genet. 2013;92:895–903. doi: 10.1016/j.ajhg.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135–43. doi: 10.1016/j.ajhg.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ralser DJ, Basmanav FBÜ, Tafazzoli A, Wititsuwannakul J, Delker S, Danda S, et al. Mutations in γ-secretase subunit–encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485–90. doi: 10.1172/JCI90667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pink AE, Simpson MA, Brice GW, Smith CH, Desai N, Mortimer PS, et al. PSENEN and NCSTN mutations in familial hidradenitis suppurativa (Acne Inversa) J Invest Dermatol. 2011;131:1568–70. doi: 10.1038/jid.2011.42. [DOI] [PubMed] [Google Scholar]
- 8.Aubin-Houzelstein G, Djian-Zaouche J, Bernex F, Gadin S, Delmas V, Larue L, et al. Melanoblasts’ proper location and timed differentiation depend on Notch/RBP-J signaling in postnatal hair follicles. J Invest Dermatol. 2008;128:2686–95. doi: 10.1038/jid.2008.120. [DOI] [PubMed] [Google Scholar]
- 9.Kumano K, Masuda S, Sata M, Saito T, Lee SY, Sakata Yanagimoto M, et al. Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res. 2008;21:70–8. doi: 10.1111/j.1755-148X.2007.00423.x. [DOI] [PubMed] [Google Scholar]
- 10.Bergmans BA, De Strooper B. γ-secretases: From cell biology to therapeutic strategies. Lancet Neurol. 2010;9:215–26. doi: 10.1016/S1474-4422(09)70332-1. [DOI] [PubMed] [Google Scholar]