

Abstract
One missing link in the coronavirus assembly is the physical interaction between two crucial structural proteins, the membrane (M) and envelope (E) proteins. In this study, we demonstrate that the coronavirus infectious bronchitis virus E can physically interact, via a putative peripheral domain, with M. Deletion of this domain resulted in a drastic reduction in the incorporation of M into virus-like particles. Immunofluorescent staining of cells coexpressing M and E supports that E interacts with M and relocates M to the same subcellular compartments that E resides in. E was retained in the pre-Golgi membranes, prior to being translocated to the Golgi apparatus and the secretory vesicles; M was observed to exhibit similar localization and translocation profiles as E when coexpressed with E. Deletion studies identified the C-terminal 6-residue RDKLYS as the endoplasmic reticulum retention signal of E, and site-directed mutagenesis of the −4 lysine residue to glutamine resulted in the accumulation of E in the Golgi apparatus. The third domain of E that plays a crucial role in virus budding is a putative transmembrane domain present at the N-terminal region, because deletion of the domain resulted in a free distribution of the mutant protein and in dysfunctional viral assembly.
- VLP
virus-like particle
- TGEV
transmissible gastroenteritis virus
- MHV
mouse hepatitis virus
- IBV
infectious bronchitis virus
- IC
intermediate compartment
- ER
endoplasmic reticulum
- PCR
polymerase chain reaction
- FITC
fluorescein isothiocyanate
Morphogenesis and assembly of mammalian enveloped RNA viruses are complex processes. During these processes, the viral core, consisting of viral RNA and core proteins, becomes wrapped in a membranous structure (viral envelope) derived from host cell membranes to form virion particles. The assembly process, which is referred to as budding, usually occurs at either the plasma or intracellular membranes. The formation of viral core, envelope, and the assembly of virus particles would involve interaction between viral proteins and host membrane components, among viral structural proteins and between viral proteins and viral RNA. Characterization of these events at the molecular level has been greatly facilitated by the understanding that viral structural proteins may contain all of the necessary information to dictate the assembly process. In fact, it was observed that coexpression of viral structural proteins would result in the formation of virus-like particles (VLPs)1 in many different viral systems (1, 2, 3, 4, 5, 6, 7, 8, 9). In this study, we exploit the coronavirus VLP system to study the interaction between two structural proteins during coronavirus assembly and the implication of this interaction in the assembly and release of coronavirus particles.
Coronavirus is the largest RNA virus known so far. It has a positive-sense, single-strand RNA genome of 27–30 kilobases in length. Despite of the huge genome size, coronavirus typically contains four structural proteins, i.e. a type I spike (S) glycoprotein required for infectivity, a phosphorylated nucleocapsid (N) protein that interacts with the viral genome to form a helical core, a major type III integral membrane (M) protein, and a minor type III envelope (E) protein (10, 11, 12, 13, 14, 15). A fifth protein, the hemagglutinin esterase glycoprotein (HE) is found in some but not all coronaviruses as short spikes (16). Extensive cellular studies on porcine coronavirus transmissible gastroenteritis virus (TGEV), murine coronavirus mouse hepatitis virus (MHV), and avian coronavirus infectious bronchitis virus (IBV) have demonstrated that coronaviruses assemble at the pre-Golgi membranes of the intermediate compartment (IC) early in infection and in the rough endoplasmic reticulum (ER) at late times of the infection (17, 18, 19, 20). Unlike most other enveloped RNA viruses, coronaviruses employ a nucleocapsid-independent strategy to drive assembly and budding (2). Coexpression of both M and E in intact cells was initially shown to be required for inducing the formation of VLPs, which are similar in size and appearance as the authentic MHV virions (2). More recently, it was demonstrated that expression of E alone resulted in the release of E-containing vesicles (15, 21). These particles were referred to as VLPs (15). The crucial role of E in viral assembly was also indicated by other studies on MHV and TGEV (23,24).
Previously, E was shown to be an integral membrane protein (22, 15). It expresses on the surface of the infected cell (25), in the ER and the IC compartment (22) and in the Golgi complex (15). In this study, the subcellular localization and the intracellular translocation of IBV E were studied in detail in intact cells by using specific organelle markers. Indirect immunofluorescence showed that the protein resided temporarily in the pre-Golgi compartments consisting of the ER and IC membranes for up to 7 h post-transfection, before it progressed down the secretory pathway. The signal that determines the temporal retention of E in the pre-Golgi compartments was mapped to the C-terminal extreme 6-residue RDKLYS, which may resemble the well characterized di-lysine ER targeting motif for membrane proteins. Interestingly, coexpression of E with IBV M, a typical Golgi-localizing protein, showed temporal retention of M in the pre-Golgi compartments. In fact, M was shown to be colocalized and cotranslocated to the same subcellular compartments when coexpressed with E in a time course experiment, suggesting a strong physical interaction between the two proteins. Coimmunoprecipitation and deletion analysis revealed that the two proteins could indeed form a heterogeneous complex and that a putative peripheral domain was required for the interaction with M. Deletion of this domain significantly reduced the assembly of M into VLPs. Furthermore, the membrane anchorage of E was demonstrated to be contributed by a stretch of hydrophobic residues at the N-terminal region of the protein and to be essential for the formation of VLP, because deletion of this putative transmembrane domain resulted in the relocation of the mutant protein to the cytosol and led to a drastic reduction in the release of VLPs. This study reveals that IBV E protein plays a fundamental role in the assembly of viral particles.
EXPERIMENTAL PROCEDURES
Viruses and Cells
The egg-adapted Beaudette strain of IBV (VR-22) was obtained from the American Type Culture Collection and was adapted to Vero cells as described previously (26). Vero cells and Cos-7 cells were grown at 37 °C in 5% CO2 and maintained in Glasgow's modified Eagle's medium supplemented with 10% fetal calf serum.
Transient Expression of IBV Sequence in Cos-7 Cells Using a Vaccinia Virus-T7 Expression System
IBV sequences were placed under the control of a T7 promoter and transiently expressed in mammalian cells using the system described by Fuerst et al.(27). Briefly, 60–80% confluent monolayers of Cos-7 cells grown on 35-mm dishes (Falcon) were infected with 10 plaque-forming units/cell of a recombinant vaccinia virus (vTF7–3) that expresses bacteriophage T7 RNA polymerase. The cells were then transfected with 5 μg of plasmid DNA (purified by Qiagen plasmid Midi kits) mixed with Lipofectin transfection reagent according to the instructions of the manufacturer (Life Technologies, Inc.). After incubation at 37 °C, 5% CO2 for 5 h, the cells were washed twice with methionine-free medium and labeled with 25 μCi/ml [35S]methionine. The radiolabeled cells and culture media were then harvested at 18 h post-transfection.
SDS-Polyacrylamide Gel Electrophoresis
Electrophoresis of viral polypeptides was performed on SDS-17.5% polyacrylamide gels (28). The 35S-labeled polypeptides were detected by autoradiography of the dried gels.
Polymerase Chain Reaction
Complementary DNA templates for PCR were prepared from purified IBV virion RNA by using a first strand cDNA synthesis kit (Roche Molecular Biochemicals). Amplification of the respective template DNAs with appropriate primers was performed with Pfu DNA polymerase (Stratagene) under the standard buffer conditions with 2 mm MgCl2. The reaction conditions used were 30 cycles of 95 °C for 45 s, X °C for 45 s, and 72 °C for X min. The annealing temperature (55 °C) and the extension time (4 min) were subjected to adjustments according to the melting temperature of the primers employed and the length of PCR fragments synthesized.
Radioimmunoprecipitation
Media of transfected Cos-7 cells were collected and mixed with 5× RIPA buffer (50 mmTris-HCl, pH 7.5, 150 mm NaCl, 0.5% sodium deoxycholate, 0.5% Nonidet P-40, 0.05% SDS) and precleared by centrifugation at 4,000 × g for 30 min at 4 °C in a microcentrifuge. Cells were lysed with 1× RIPA buffer and precleared by centrifugation at 12,000 rpm. Immunoprecipitation with anti-E and anti-M rabbit polyclonal antisera (14, 29) and anti-T7 monoclonal antiserum (Novagen) was carried out as previously described (30).
Immunofluorescence and Confocal Microscopy
IBV sequences were transiently expressed in Cos-7 cells grown on 4-well chamber slides (IWAKI). At 5 h post-transfection (or otherwise stated), cells were rinsed with phosphate-buffered saline and subjected to fixation using 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100. Fluorescence staining was performed by incubating cells with either an antibody or a mixture of both primary antibodies (rabbit anti-M (1:30) or mouse anti-T7 (1:200)) for 1 h at room temperature, followed by FITC- or tetramethyl rhodamine isocyanate-conjugated secondary antibodies for 1 h at 4 °C. Goat anti-rabbit antibody was used at 1:400 (Sigma) and goat anti-mouse IgG at 1:20 (DAKO). Images were viewed and collected with a Zeiss confocal microscope connected to a Bio-Rad MRC1024 laser scanner.
Construction of Plasmids
Plasmid pIBVM-1, which covers the IBV sequence between nucleotides 24498 and 25159, was constructed by cloning a PvuII/SacI-digested PCR fragment intoPvuII/SacI digested pKTO vector (31). The PCR fragment was generated using primers LDX59 (5′-CAGCAACAGCTGAAGATGCCCAACG-3′) and LDX60 (5′-CTACACACGAGCTCTTATGTGTAAAGA-3′).
Plasmid pIBVE was constructed as follows. A 735-base pair fragment, obtained by PCR using LDX55 (5′-GATTGTTCAGGCCATGGTGAATTTATTGAA-3′) and XIANG8 (5′-GCACCATTGGCACACTC-3′), was digested with NcoI andBamHI and ligated intoNcoI/BamHI-digested pKTO, resulting in a plasmid containing the IBV sequence between nucleotides 24205 and 24795. Plasmid pT7E was constructed by fusing the 11-amino acid T7 tag (MASMTGGQQMG) to the N terminus of E.
RESULTS
Subcellular Localization of IBV E in Cells Overexpressing the Protein
In a previous study using immunoaffinity-purified antibodies specific for IBV E, the protein was demonstrated to be localized to intracellular membrane structures as well as on cell surface in IBV-infected cells (25). Although some evidence of polarization of the fluorescence into structures resembling the Golgi apparatus was observed, the reticular staining patterns suggest that the majority of the protein may be localized to pre-Golgi compartments at the time of observation (25). More recently, Raamsman et al. (22) reported that E colocalizes with Rab-1, a marker for the IC and the ER, supporting that the protein may be localized to the pre-Golgi compartments. However, Corse and Machamer (15) have recently presented data showing that IBV E may be colocalized with M to the Golgi apparatus. To address this issue further, IBV E was fused to an 11-amino acid T7 Tag (Novagen), and a highly specific monoclonal antibody against the T7 tag was used to study the subcellular distribution of E in intact cells. This strategy would also facilitate dual labeling of cells expressing both M and E in subsequent studies. Subcellular localization of E in Cos-7 cells was examined by indirect immunofluorescence microscopy. A reticular staining pattern (Fig.1 A) colocalizes with the staining profile of R6 (rhodamine B hexyl-ester chloride, Molecular Probes) (Fig. 1 B), a short chain carbocyanine dye known to stain specifically the ER of mammalian cells, as indicated by the merged image (Fig. 1 C). In contrast, IBV M, which was previously shown to be localized to the Golgi apparatus (32), does not colocalize with the R6 marker (Fig. 1, D–F); instead, it coaligns with the staining pattern of the fluorescence vital dye BODIPY TR-ceramide (Molecular Probes) (Fig. 1, G–I). This dye was shown to stain the Golgi apparatus specifically (Molecular Probes).
Figure 1.

Subcellular localization of IBV E and M proteins in transfected Cos-7 cells. The T7-tagged E and M expressed in Cos-7 cells were detected using monoclonal anti-T7 (A) and polyclonal anti-M antibodies (D andG), respectively. The proteins were then labeled with the FITC-conjugated secondary antibodies. Immunofluorescent staining of E (A) and M (D and G) gives a reticular staining pattern and a perinuclear, Golgi-like staining pattern, respectively. B and E refer to cells stained with R6, a dye for the ER, and H refers to a cell stained with BODIPY TR-ceramide (TR-C), a vital dye for the Golgi apparatus. The green images represent the FITC-derived green fluorescence, and red images represent the rhodamine and Texas Red-derived red fluorescence. Colocalization of viral proteins with the organelle markers is represented by theyellow region within each cell in the merged images (C, F, and I). The fluorescence was viewed using a confocal scanning Zeiss microscope.
To further clarify the subcellular distribution pattern of E, transfected cells were incubated in the presence of 100 μg/ml of cycloheximide for 30 min at 3.5 h post-transfection to stop new protein synthesis. The expressed viral proteins were then chased and fixed at 4, 7, 10, and 16 h post-transfection, respectively, and the subcellular localization patterns were viewed by indirect immunofluorescent staining. E was observed to display the reticular staining profiles overlapping with the R6 staining patterns up to 7 h post-transfection (Fig. 2 ,A–F). A more perinuclear staining pattern was observed at 10 h post-transfection (Fig. 2 G). It overlaps with the staining pattern of the fluorescence vital dye BODIPY TR-ceramide (Fig.2, G–I). Granular fluorescent aggregates scattering in the cytoplasm of the transfected cells were observed at 16 h post-transfection (Fig. 2 J). This fluorescence pattern overlaps with the staining pattern of LysoTrackerTM Red DND-99 (Molecular Probes), a biotinylated acidotropic probe that stains acidic compartments including lysosomes, trans-Golgi vesicles, and secretory vesicles.
Figure 2.
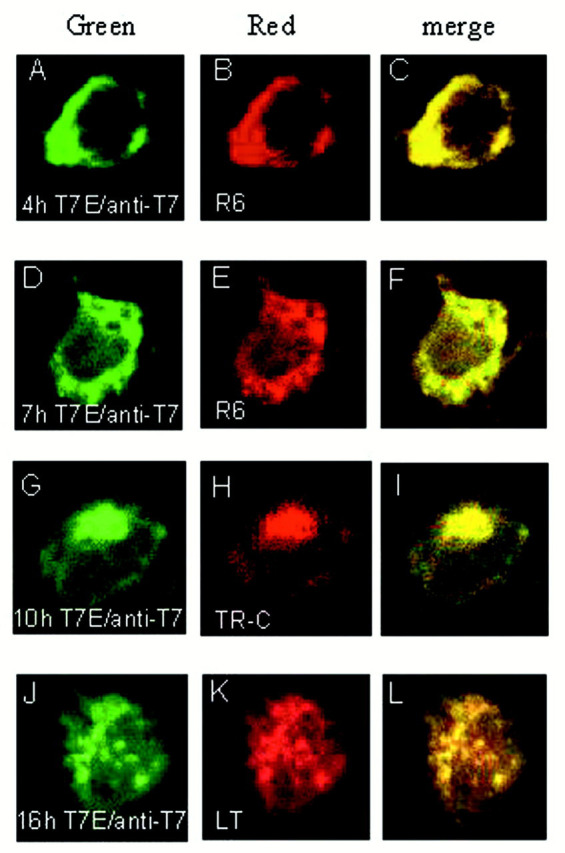
Subcellular localization and translocation of E protein. Cos-7 cells expressing E were incubated in the presence of 100 μg/ml cycloheximide for 30 min at 3.5 h post-transfection. The cells were fixed at 4, 7, 10, and 16 h and subjected to indirect immunofluorescent staining with anti-T7 monoclonal antibody followed by incubating with FITC-conjugated anti-mouse antiserum. Immunostaining of E is presented in A,D, G, and J. B andE show the ER staining with R6; H shows the BODIPY TR-ceramide (TR-C) staining pattern; and Kshows the LysoTrackerTM Red DND-99 (LT) staining pattern. The green images represent the FITC-derived green fluorescence, and red images represent the Rhodamine and Texas Red-derived red fluorescence. Colocalization of viral proteins with the organelle markers is represented by theyellow region within each cell in the merged images (C, F, I, and L).
The Effects of Deletion on the Subcellular Distribution of IBV E and Delineation of Its ER Retention Signal
The amino acid sequence of E was then subjected to computer analysis using the PSORT program for prediction of subcellular localization signals (33) and the TMPRED proteomics tools for prediction of transmembrane regions and membrane topology of proteins (34). As shown in Fig.3 a, a putative transmembrane domain is predicted for residues between 17 and 33, a putative peripheral domain is located between amino acids 37 to 53, and a potential ER retention signal is predicted to be present at the C-terminal extreme end of the protein.
Figure 3.

a, diagram indicating the presence of four putative domains in IBV E. E adopts a type III topology. Theboxed regions refer to the putative domains. Irefers to a potential N-linked glycosylation site which is not utilized (15), II refers to a putative transmembrane domain between residues 17 and 33, III refers to a putative peripheral domain located between amino acids 37 and 53, and IV refers to a potential ER retention signal. b, summary of the effects of deletion and mutation on the subcellular localization of IBV E, the interaction with M, VLP release, and the incorporation of M into VLPs. The 109 amino acids of E are represented by white boxes, and the putative transmembrane (TM) and peripheral domains are indicated in gray boxes. The deleted regions are represented in black boxes, and a single mutation is represented by a stippled box. RDKLYSstands for the cytoplasmic tail sequence of E containing the potential ER retention signal. Also included is a summary of the position of deletion, subcellular localization of each mutant, coimmunoprecipitation with M, VLP release, and the incorporation of M into the wild type and mutant E-induced VLPs.
To determine the effects of each putative domain on the subcellular distribution of E, several deletion constructs were constructed. EΔ1 deletes the N-terminal sequence from amino acids 1 to 14, EΔ2 deletes the putative transmembrane domain (residues 18–33), EΔ3 contains a deletion of residues 33–51 in the putative peripheral membrane domain, EΔ4 deletes amino acids 50–64, EΔ5 deletes amino acids 67–83, and EΔ6 deletes 26 residues from the C-terminal end (thus removing the potential ER retention signal) (Fig. 3 b). The effects of each deletion on the subcellular distribution of E are summarized in Fig. 3 b. As shown in Fig. 4 , the two deletions that resulted in an altered subcellular distribution profile of E are EΔ2 and EΔ6. Deletion of the putative transmembrane domain (EΔ2) resulted in a diffuse staining pattern (Fig. 4 G), which does not overlap with the staining pattern of R6 (Fig. 4, H and I). Deletion of the C-terminal 26 residues led to the detection of the mutant protein with a staining profile similar to that of M (Fig. 4, S–U). This result indicates that the C-terminal region of E may contain sufficient information for its ER retention. Other deletion constructs including deletion of ∼95% of the putative peripheral domain of E rendered no obvious effects on the subcellular localization of E (Fig. 4).
Figure 4.

The effects of deletions on the subcellular localization of E protein. Cos-7 cells expressing the wild type and mutant E were fixed at 7 h post-transfection. The expressed proteins were detected with anti-T7 monoclonal antibody against the T7 tag fused to the N terminus of the wild type E (A), EΔ1 (D), EΔ2 (G), EΔ3 (J), EΔ4 (M), EΔ5 (P), and EΔ6 (S).B, E, H, K, N,Q, and T refer to cells stained with R6. Thegreen images represent the FITC-derived green fluorescence, and red images represent the Rhodamine and Texas Red-derived red fluorescence. The merged images (C, F, I, L, O, R, andU; yellow) represent colocalization of proteins with the ER marker.
Further deletion of the C-terminal most six amino acids was carried out, giving rise to EΔ8. Similar to EΔ6, expression of EΔ8 showed that the protein exhibits a Golgi-like staining profile (Fig. 5 A), which does not overlap with the R6 staining pattern (Fig. 5, B andC). Instead, it overlaps with the staining pattern of dye BODIPY TR-ceramide (Fig. 5, D–F), indicating that the mutant protein may be localized to the Golgi apparatus. Furthermore, when the −4 lysine residue was mutated to a nonconservative glutamine, the mutant protein accumulates at the Golgi apparatus, as it colocalizes with the staining pattern of dye BODIPY TR-ceramide (Fig.5, G and I).
Figure 5.
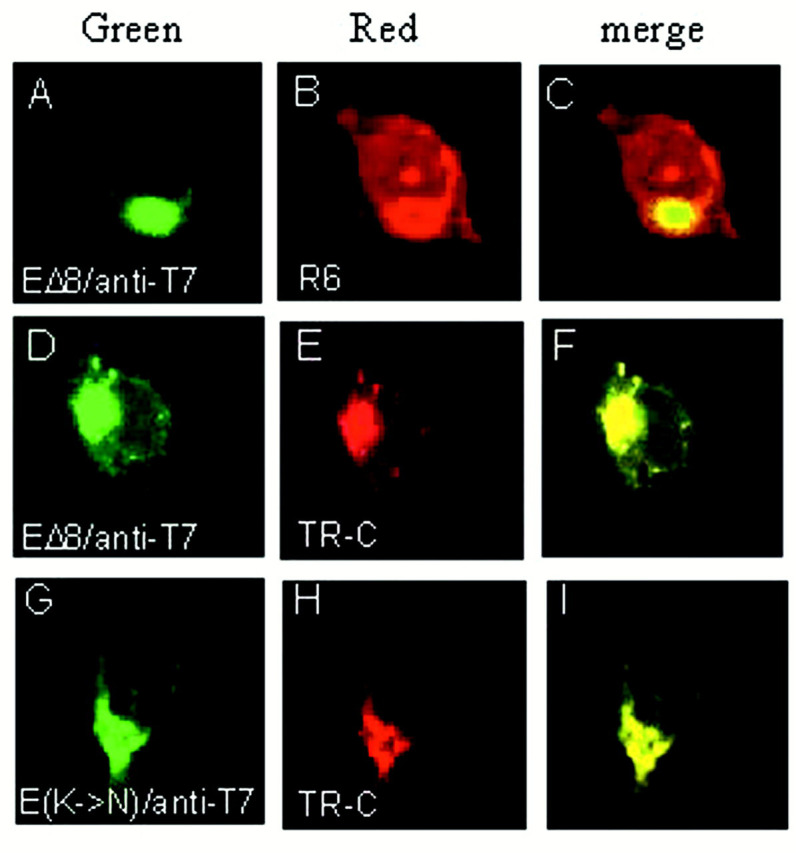
Subcellular localization of E proteins lacking the putative ER retention signal. Cos-7 cells transiently expressing EΔ8 (A and D) and E(K→N) (G) were fixed at 7 h post-transfection. Proteins are detected with anti-T7 monoclonal antibody. B refers to cells stained with R6, and E and H show cells stained with BODIPY TR-ceramide (TR-C). The green images represent the FITC-derived green fluorescence, and redimages represent the Rhodamine and Texas Red-derived red fluorescence. The merged images (yellow) represent colocalization of proteins with the organelle markers (C, F, andI).
Physical Interaction of IBV E with M and Temporary Retention of M in the ER via the Interaction
Because E has been shown to be the sole component for VLP production (15), it was speculated that incorporation of other viral structural proteins into VLPs would involve the interaction between E and those proteins. In fact, Maedaet al. (21) demonstrated that anti-E antibody is able to coimmunoprecipitate structural proteins E, M, and N in MHV-infected cells, indicating the interactions of E with other structural proteins. Because M was also shown to be an essential component for VLP formation, we tested the interaction between E and M by coexpressing them in Cos-7 cells. Fig. 6 refers to coimmunoprecipitation of cells expressing both proteins. Fig.6 a shows that M alone cannot be precipitated by anti-T7 antiserum (lane 6). In the presence of E (lane 4), a protein species corresponding to the pre-Golgi form of M was precipitated (lane 5). This form of M is likely to be a mixture of unglycosylated and pre-Golgi modified M and was referred to as pre-Golgi form of M in this report. Similarly, E cannot be immunoprecipitated by anti-M when expressed alone (lane 1). However, when M was coexpressed, E could be precipitated by anti-M (lane 2). Similar forms of the physical interaction between E and M were observed in IBV-infected Vero cells. As shown in Fig.6 b, E and both pre- and post-Golgi forms of M were coimmunoprecipitated by anti-M (lane 1) and anti-E (lane 5) antisera, respectively, in cell lysates prepared from IBV-infected Vero cells harvested at 18 h post-infection. Coimmunoprecipitation of both proteins were also observed from the viral particles released into the culture medium (lanes 2and 6).
Figure 6.
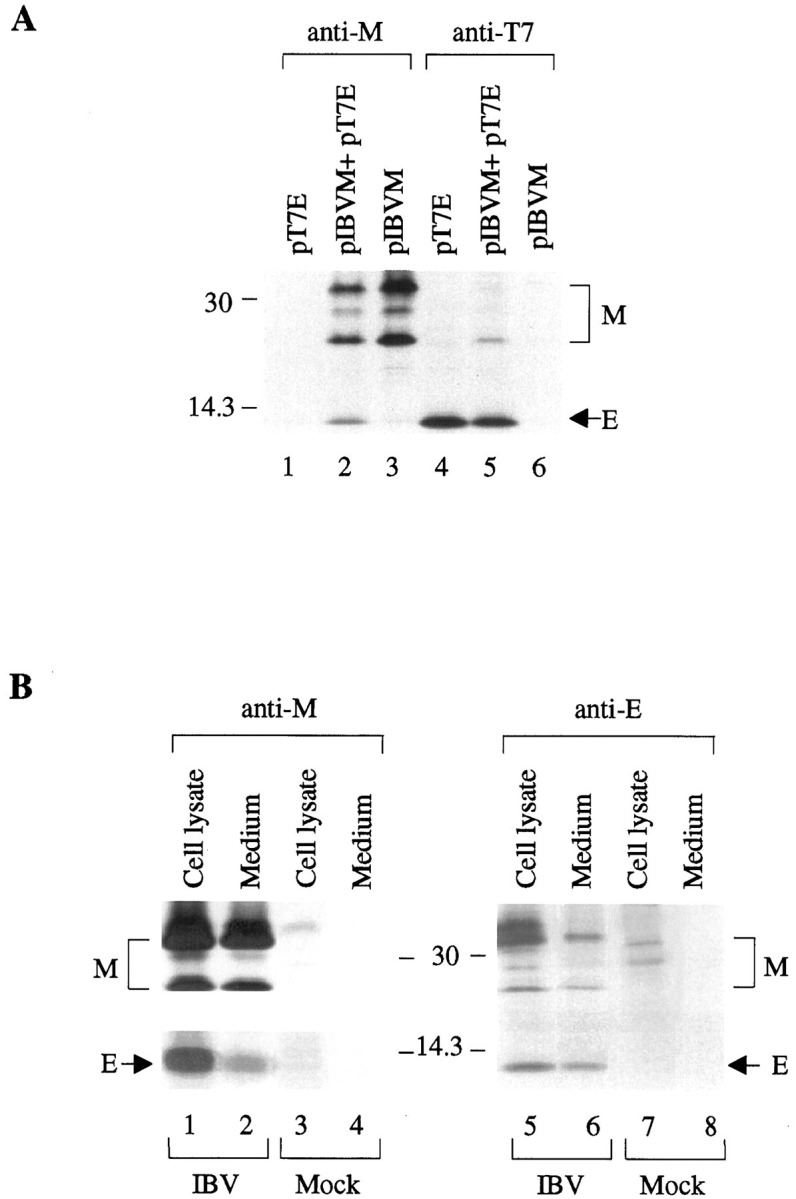
a, coimmunoprecipitation of M and E proteins in Cos-7 cells expressing the two proteins. Cos-7 cells expressing E (lanes 1 and 4), M (lanes 3 and 6), or coexpressing both proteins (lanes 2 and 5) were lysed and subjected to immunoprecipitation before analysis on SDS-17.5% polyacrylamide gel. The expressed proteins were immunoprecipitated with anti-M (lanes 1–3) and anti-T7 (lanes 4–6) antisera, respectively.Numbers on the left indicate molecular masses in kilodaltons. b, coimmunoprecipitation of M and E proteins in IBV-infected Vero cells. Cell lysates were prepared from Vero cells harvested at 18 h post-infection, and the collected culture media were precleared by centrifugation. The viral proteins in the cell lysates (lanes 1, 3, 5, and7) and in the culture medium (lanes 2,4, 6, and 8) were immunoprecipitated with anti-M (lanes 1–4) and anti-E (lanes 5–8), before analysis on SDS-17.5% polyacrylamide gel. The upper part of the left panel was prepared from a gel exposed for 1 day, and the lower part was prepared from the same gel exposed for 3 days. Numbers on the left indicate molecular masses in kilodaltons.
To view this form of interaction in intact cells, M and E proteins were coexpressed in Cos-7 cells. At 3.5 h post-transfection, new protein synthesis was stopped by incubation of the cells in the presence of 100 μg/ml of cycloheximide for 30 min. The expressed viral proteins were then chased and fixed at 4, 7, 10, and 16 h post-transfection, respectively, and were viewed by immunofluorescent staining. Representative cells are shown in Fig.7 . E was observed to show a reticular staining pattern similar to that in Figs. 1 A and2 D up to 7 h post-transfection (Fig. 7, Aand D). At 10 and 16 h post-transfection, the protein accumulates in the Golgi apparatus and the secretory vesicles, respectively (Fig. 7, G–J), as shown in Fig. 2(G and J). Interestingly, dual labeling of M in cells coexpressing both E and M showed that M colocalizes with E to the ER up to 7 h post-transfection (Fig. 7, A–F). M was then translocated with E to the Golgi apparatus and the secretory vesicles at 10 and 16 h post-transfection, respectively (Fig. 7,G–L). These results suggest that interaction of E with M may occur in intact cells, because E can retain and translocate M to the compartments it resides in. The detection of both E and M in the ER, the Golgi and the secretory vesicles may represent the route that M-E protein complexes take to exit the host cells.
Figure 7.

The effects of coexpression of E with M on the subcellular localization of M. M was coexpressed with E in Cos-7 cells, incubated in the presence of 100 μg/ml of cycloheximide for 30 min, and subjected to indirect immunofluorescent staining. The cells were fixed at 4 (A–C), 7 (D–F), 10 (G–I), and 16 (J–L) h, respectively, and the subcellular localization of proteins were examined by dual labeling with a mixture of anti-T7 (mouse) and anti-M (rabbit) antisera, followed by incubating with a mixture of FITC-conjugated anti-mouse and tetramethyl rhodamine isocyanate-conjugated anti-rabbit antisera. The staining patterns of E are indicated by the green images (A, D, G, and J), and the staining patterns of M are indicated by the red images (B, E, H, K, N, and Q). M and P refer to cells expressing EΔ2 and EΔ7, respectively. Merged images (yellow) represent colocalization of the two proteins (C, F, I, L, O, and R).
To further confirm this observation, M was coexpressed with EΔ2 and EΔ7, respectively, and the cells were stained at 7 h post-transfection. EΔ7 contains a deletion of residues 37–57, the putative peripheral domain. Once again, EΔ2 exhibits a diffuse staining pattern (Fig. 7 M). Instead of the typical Golgi localization profile (Fig. 1 D), coexpression of EΔ2 with M forces M to adapt the same diffuse staining pattern of EΔ2 (Fig. 7,M–O). Coexpression of EΔ7 and M showed a reticular staining pattern of EΔ7 (Fig. 7 P), similar to the staining pattern of EΔ3, which contains a deletion considerably overlapping with that in EΔ7 (Fig. 4 J). However, the staining pattern of M did not overlap with that of EΔ7 (Fig. 7, P–R). As can be seen, the majority of M was accumulated in the Golgi region, with a faint reticular staining (Fig. 7 Q).
Subcellular Localization of M and E in IBV-infected Vero Cells
The subcellular distribution patterns of E and M in IBV-infected cells were analyzed by indirect immunofluorescence with polyclonal antisera against E and M, respectively. At 4.5 h post-infection, Vero cells were incubated in the presence of 100 μg/ml of cycloheximide for 30 min to stop new protein synthesis and were chased and fixed at 5, 7, 9, and 12 h post-infection, respectively. Both E and M were observed to display the reticular staining profiles overlapping with the R6 staining patterns up to 7 h post-infection (Fig. 8 ,A–F and M–R). E and M proteins were then observed to accumulate at the perinuclear region at 10 h post-infection (Fig. 8, G and S), overlapping with the staining pattern of the fluorescence vital dye BODIPY TR-ceramide (Fig. 8, G–I and S–U). Granular fluorescent aggregates of both E and M were detected in the cytoplasm at 12 h post-infection (Fig. 8, J and V), overlapping with the staining pattern of LysoTrackerTM Red DND-99 (Molecular Probes) (Fig. 8, J–L andV–X). These distribution patterns resemble the staining patterns observed in cells coexpressing the T7-tagged E and M, suggesting that the T7 tag does not obviously affect the subcellular distribution patterns of E. However, we were unable to dual label the same cells for detailed analysis of the distribution profiles of the two proteins, because both anti-E and anti-M were raised in rabbits.
Figure 8.

Subcellular localization of IBV E and M proteins in IBV infected cells. Vero cells infected with IBV were incubated in the presence of 100 μg/ml cycloheximide for 30 min at 4.5 h post-infection and subjected to indirect immunofluorescent staining. The cells were fixed at 5 (A–C andM–O), 7 (D–F and P–R), 9 (G–I and S–U), and 12 (J–L andV–X) h post-infection, respectively. Viral proteins were detected with anti-E (A, D, G, andJ) and anti-M (M, P, S, andV) antisera. B, E, N, andQ refer to ER staining with R6; H andT show cells stained with BODIPY TR-ceramide (TR-C); and K and W show cells stained with LysoTrackerTM Red DND-99 (LT). Thegreen images represent the FITC-derived green fluorescence, and red images represent the rhodamine and Texas Red-derived red fluorescence. The merged images (yellow) represent colocalization of the proteins with the organelle markers (C, F, I, L, O,R, U, and X).
Determination of the IBV E Sequences Responsible for the Physical Interaction with M
To determine the domain(s) required for the interaction with M, immunoprecipitation of lysates prepared from cells coexpressing M and an E deletion mutant was performed. Most mutants can be coimmunoprecipitated with M, except for EΔ3 and EΔ7 (Fig.9 ). As shown in Fig. 9, only trace amounts of EΔ3 and EΔ7 were coimmunoprecipitated by anti-M (lanes 4 and 15), and no M was coimmunoprecipitated by anti-T7 (lanes 11 and16). Taken together with the colocalization data present in Fig. 7P-R, these results suggest that the predicted peripheral domain (amino acids 37–57) may be responsible for the interaction of E with M.
Figure 9.

Coimmunoprecipitation of M protein and E deletion mutants from transfected Cos-7 cells. Cos-7 cells were transfected with plasmids expressing M and the wild type and mutant E, as indicated above each lane. The cells were lysed and subjected to immunoprecipitation before analysis with SDS-17.5% polyacrylamide gel.Lanes 1–7 and 15 refer to products precipitated with anti-M, and lanes 8–14 and 16 refer to products precipitated with anti-T7. Numbers on theleft indicate molecular masses in kilodaltons.
The Effects of Deletion of IBV E on the Release of VLPs and on the Assembly of M into VLPs
The effects of deletion of E on the release of VLPs were tested by expression of E and the deletion constructs in Cos-7 cells and by detection of the expressed proteins in the culture medium. As shown in Fig.10 a, the wild type and deletion constructs EΔ2 and EΔ7 were efficiently detected in lysates prepared from cells transfected with the corresponding constructs (lanes 6–8). The wild type and EΔ7 were also efficiently detected from the culture media by immunoprecipitation with anti-T7 antibody (Fig. 10 a, lanes 14 and16), indicating that VLPs were efficiently released from cells expressing the two constructs. Because EΔ7 contains a deletion of the putative peripheral domain of E, this result implies that this putative domain may be not essential for the release of VLPs. Interestingly, no release of VLPs was observed from cells transfected with EΔ2 (Fig. 10 a, lane 15), suggesting that the transmembrane domain may be essential for the release of VLPs. Because deletion of the putative transmembrane domain resulted in the detection of E in the cytoplasm (Fig. 4 G), these results demonstrate that insertion of E into the intracellular membrane structures is essential for the release of VLPs. As a control experiment, M was detected in transfected cells by immunoprecipitation with anti-M (Fig. 10 a, lane 1); however, it was undetectable in the culture media (Fig. 10 a, lane 2). The effects of other deletions on the release of VLPs are summarized in Fig. 3 b.
Figure 10.

a, the effects of deletion of the putative transmembrane and the peripheral membrane domains of E on the release of VLPs. Cos-7 cells were transfected with plasmids as indicated above each lane. Cell lysates were prepared and subjected to immunoprecipitation with anti-M (lanes 1–4) and anti-T7 (lanes 5–8), and protein expression efficiency was analyzed on SDS-17.5% polyacrylamide gel. The release of VLP was analyzed by immunoprecipitation of the culture medium from each transfection with anti-M (lanes 9–12) and anti-T7 (lanes 13–16) after the medium was centrifuged at low speed (4,000 ×g) to preclear the cell debris. The numbersbetween the two panels indicate molecular masses in kilodaltons.b, the effects of deletion of the putative transmembrane and the peripheral membrane domains of E on the incorporation of M into VLPs. Cos-7 cells were transfected with plasmids as indicated above each lane. Cell lysates were prepared and subjected to immunoprecipitation with anti-M (lanes 1–3) and anti-T7 (lanes 4–6), and protein expression efficiency was analyzed on SDS-17.5% polyacrylamide gel. The release of VLP was analyzed by immunoprecipitation of the culture medium from each transfection with anti-M (lanes 7–9) and anti-T7 (lanes 10–12) after the medium was centrifuged at low speed (4,000 ×g) to preclear the cell debris. The numbersbetween the two panels indicate molecular masses in kilodaltons.
We next tested the effects of the deletions of the putative transmembrane domain and the peripheral domain on the release of VLPs and on the assembly of M into VLPs by coexpression of E and the deletion mutants with M in Cos-7 cells. Efficient expression of M and E constructs were observed in cells transfected with the corresponding constructs (Fig. 10 b, lanes 1–6). In the culture medium, wild type E was readily immunoprecipitated by either anti-M or anti-T7 when coexpressed with M (Fig. 10 b, lanes 7 and 10). Anti-M was also able to immunoprecipitate at least two different types of M from cells coexpressing M and the wild type E (Fig. 10 b, lane 7), whereas anti-T7 could predominantly immunoprecipitate the pre-Golgi form of M only (Fig.10 b, lane 10). Once again, neither EΔ2 nor M was detectable by either antiserum in the culture medium when two proteins were coexpressed (Fig. 10 b, lanes 8 and11). Coexpression of EΔ7 and M resulted in the detection of only EΔ7 from the culture medium by anti-T7 (Fig. 10 b,lane 12), whereas anti-M could weakly coimmunoprecipitate both types of M and trace amounts of EΔ7 (Fig. 10 b,lane 9). These results suggest that a strong physical interaction between M and E via the putative peripheral domain of E protein may facilitate the incorporation of the pre-Golgi form of M into VLPs. The detection of both types of M in EΔ7-induced VLPs suggested that although the ability of EΔ7 to interact with M was severely diminished, EΔ7-induced VLPs could passively incorporate M when they form at the pre-Golgi membranes and progress through the Golgi complex for maturation.
DISCUSSION
Increasing evidence has shown that coronavirus E protein may play a pivotal role in viral morphogenesis and virion assembly. Direct evidence of the involvement of E in coronavirus morphogenesis is derived from a recent study using clustered charged-to-alanine mutagenesis and targeted RNA recombination to mutate the hydrophilic tail of E (23). One of the two mutant viruses generated exhibits aberrant morphology, with many virions showing pinched and elongated shapes (23). The essential role of E in virion assembly was revealed by studies showing the formation and release of VLPs from cells expressing E alone (21, 15). It suggests that E may be the driving force in determining the budding process, including selection of the budding site, the formation of viral envelope, and the assembly of virion particles. In this study, we provide evidence that IBV E resides in the pre-Golgi compartments consisting of the ER and IC membranes for up to 7 h post-transfection. The signal determined that this localization pattern was mapped to a di-lysine-like ER targeting signal located in the C-terminal extreme 6-residue RDKLYS. The protein was able to retain M in the same compartments by forming a heterogeneous complex with M, thereby facilitating the assembly of M into VLPs.
Coronavirus M protein, being the most abundant membrane component of the coronavirus virion, is able to laterally interact with itself to form homogeneous complexes (35). It could also form heterogeneous complexes with S, HE, and N proteins (36, 37, 38). The interaction of these structural proteins with M may facilitate their assembly into the virion particles (2, 36, 39). However, M must also interact with E to be assembled into the virion, as expression of M alone could not induce the formation of VLP. The demonstration of strong physical interaction between IBV E and M via the putative peripheral domain of E in this report connects the missing link between M and the E-induced envelope components. By this interaction, E provides a temporary anchor to relocate M in the pre-Golgi compartments, where it “prepares” the membranes for budding (22). Meanwhile, a certain proportion of M may be passively incorporated into virions during envelopment, because considerable amounts of M were detected in VLPs induced by coexpression of EΔ7 and M. Upon accumulation of sufficient essential structural proteins, VLPs assemble and bud at the budding site, transport through a functional Golgi stack, and are released out of the host cells by the exocytic pathway. These VLP assembly and release processes, as revealed by the immunofluorescence microscopy and time course experiments, resemble the assembly and maturation processes of the coronavirus virion as demonstrated by extensive cellular studies on coronavirus-infected cells (17, 18, 19, 20, 22, 40, 41).
IBV E was previously shown to be an integral protein with an Nexo-Cendo orientation (15). The putative peripheral domain is therefore exposed on the cytoplasmic face of the intracellular membranes and is required for interaction with M. In a recent study, the C-terminal cytoplasmic tail of MHV M was shown to have more detrimental effects on VLP assembly and release than domains within its N-terminal region (42). One possible explanation is that this region may be required for the interaction with E, because it is exposed on the same cytoplasmic face as the putative peripheral domain of E.
In a study on the intracellular assembly of TGEV (17), virions formed in the perinuclear region were observed to be large and have a clear center (immature virion), dissimilar to the smaller extracellular particles (mature virion) containing compact internal cores with polygonal contours. The mature virions were observed only at or after the trans-Golgi network, indicating that maturation starts in the late Golgi compartments. The molecular mechanisms underlining this maturation process are unknown, but one reason may be the differential glycosylation of M, as suggested by Risco et al. (43). The coimmunoprecipitation of E with the pre-Golgi form but not the post-Golgi form of M supports that E interacts with M in the pre-Golgi compartments. Because this interaction facilitates the assembly of M into the virion, it is likely that the immature virions may contain mostly the pre-Golgi form of M. How and where do the immature virions acquire the post-Golgi form of M? One possibility is that the pre-Golgi form of M that has already assembled into the virion undergoes modification when the particle goes through the Golgi apparatus. The occurrence and significance of this modification in the maturation of the immature virion are yet to be understood. Alternatively, incorporation of M carrying complex sugar chains into the immature virion may occur when the particle goes through the Golgi complex, contributing to the maturation of the virus. In this case, the incorporation of M must be independent of E, because the post-Golgi form of M does not interact with E. The observation that certain amounts of M was still incorporated into the EΔ7-induced VLPs supports that M could be assembled into the virion by mechanisms independent of E.
The deletion analysis mapped the ER retention signal of E to the C-terminal extreme 6-residue RDKLYS, a di-lysine-like ER targeting motif. Many membrane proteins carry a di-lysine signal in their cytosolic domain that confers localization of a membrane protein to the ER. For example, a type I ER membrane protein encoded by theE19 gene of adenovirus 3 contains a 6-residue di-lysine motif DEKKMP located at the extreme C terminus that was necessary and sufficient for retention of the protein to the ER of mammalian cells (44). Mutagenesis study showed that the lysine residue at the −4 position of E is likely to be crucial, because a single mutation resulted in the accumulation of the protein in the Golgi complex. To support this observation, sequence analysis of IBV isolates showed that this residue is conserved in six out of seven strains, despite considerable variation in the total number of amino acid residues and the primary sequences of E among different isolates (14, 45, 46). Likewise, the leucine residue at the −3 position was also conserved in these six isolates (14, 45, 46). However, the di-lysine ER retention signal for all ER resident membrane proteins known so far contains a lysine residue at the −3 position (47). Mutation of this residue destroyed the targeting motif. We do not know whether the change to a leucine residue at this crucial position for IBV E might suggest a different targeting mechanism. Interestingly, neither the −3 leucine nor the −4 lysine residues were found to be conserved in the counterpart E protein sequences of other coronaviruses, including MHV, human coronavirus, and TGEV (23, 48, 49). Because MHV E was also localized to the ER and IC, a different retention signal may exist in this protein.
Further assessment of the di-lysine-like RDKLYS motif on the retention of E is complicated by the fact that E is retained only temporarily in the pre-Golgi membranes of the ER and IC. Immunofluorescence microscopy and time course experiments present in this report demonstrated that E was retained in the pre-Golgi membranes for up to 7 h before it was translocated to the Golgi apparatus and down the secretory pathway. Is this temporary retention of E due to the weakness of the di-lysine-like RDKLYS signal or due to the formation of VLPs that triggers the translocation of the protein? It is very likely that the latter is the case. After assembling into VLPs, the protein was translocated to the Golgi apparatus together with VLPs for the maturation and finally release of VLPs out of the cells. Systematic mutation of E to create a mutant E protein that maintains the same subcellular distribution pattern as the wild type protein but loses the ability to induce the formation of VLPs would be of help to study this retention signal further.
Acknowledgments
We thank Lisa F. P. Ng and H. Y. Xu for excellent technical assistance.
Footnotes
Published, JBC Papers in Press, February 8, 2001, DOI 10.1074/jbc.M009731200
REFERENCES
- 1.Brautigam S., Snezhkov E., Bishop D.H. Virology. 1993;192:512–524. doi: 10.1006/viro.1993.1067. [DOI] [PubMed] [Google Scholar]
- 2.Vennema H., Godeke G.-J., Rossen J.W.A., Voorhout W.F., Horzinek M.C., Opstelten D.-J., E., Rottier P.J.M. EMBO J. 1996;15:2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li T.C., Yamakawa Y., Suzuki K., Tatsumi M., Razak M.A., Uchida T., Takeda N., Miyamura T. J. Virol. 1997;71:7207–7213. doi: 10.1128/jvi.71.10.7207-7213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White L.J., Hardy M.E., Estes M.K. J. Virol. 1997;71:8066–8072. doi: 10.1128/jvi.71.10.8066-8072.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumert T.F., Ito S., Wong D.T., Liang T.J. J. Virol. 1998;72:3827–3836. doi: 10.1128/jvi.72.5.3827-3836.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang B., Barniak V., Smith R.P., Sharma R., Corsaro B., Hu B., Madore H.P. Biotechnol. Bioeng. 1998;60:369–374. doi: 10.1002/(sici)1097-0290(19981105)60:3<369::aid-bit14>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 7.Garbutt M., Chan H., Hobman T.C. Virology. 1999;261:340–346. doi: 10.1006/viro.1999.9857. [DOI] [PubMed] [Google Scholar]
- 8.Neumann G., Watanabe T., Kawaoka Y. J. Virol. 2000;74:547–551. doi: 10.1128/jvi.74.1.547-551.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haglund K., Forman J., Krausslich H., G., Rose J.K. Virology. 2000;268:112–121. doi: 10.1006/viro.1999.0120. [DOI] [PubMed] [Google Scholar]
- 10.Holmes K.V., Doller E.W., Sturman L.S. Virology. 1981;115:334–344. doi: 10.1016/0042-6822(81)90115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogue B.G., Brian D.A. Virus Res. 1986;5:131–144. doi: 10.1016/0168-1702(86)90013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmas B., Laude H. J. Virol. 1990;64:5367–5375. doi: 10.1128/jvi.64.11.5367-5375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Locker J.K., Rose J.K., Horzinek M.C., Rottier P.J.M. J. Biol. Chem. 1992;267:21911–21918. doi: 10.1016/S0021-9258(19)36699-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D.X., Inglis S.C. Virology. 1991;185:911–917. doi: 10.1016/0042-6822(91)90572-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corse E., Machamer C.E. J. Virol. 2000;74:4319–4326. doi: 10.1128/jvi.74.9.4319-4326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dea S., Tijssen P. Arch. Virol. 1988;99:173–186. doi: 10.1007/BF01311068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Risco C., Muntion M., Enjuanes L., Carrascosa J.L. J. Virol. 1998;72:4022–4031. doi: 10.1128/jvi.72.5.4022-4031.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salanueva I.J., Carrascosa J.L., Risco C. J. Virol. 1999;73:7952–7964. doi: 10.1128/jvi.73.10.7952-7964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tooze J., Tooze S.A., Warren G. Eur. J. Cell Biol. 1984;33:281–293. [PubMed] [Google Scholar]
- 20.Chen B.Y., Itakura C. Avian Pathol. 1996;25:675–690. doi: 10.1080/03079459608419174. [DOI] [PubMed] [Google Scholar]
- 21.Maeda J., Maeda A., Makino S. Virology. 1999;263:265–272. doi: 10.1006/viro.1999.9955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raamsman M.J.B., Locker J.K., de Hooge A., de Vries A.A.F., Griffiths G., Vennema H., Rottier P.J.M. J. Virol. 2000;74:2333–2342. doi: 10.1128/jvi.74.5.2333-2342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer F., Stegen C.F., Masters P.S., Samsonoff W.A. J. Virol. 1998;72:7885–7894. doi: 10.1128/jvi.72.10.7885-7894.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baudoux P., Carrat C., Besnardeau L., Charley B., Laude H. J. Virol. 1998;72:8636–8643. doi: 10.1128/jvi.72.11.8636-8643.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith A.R., Boursnell M.E.G., Binns M.M., Brown T.D.K., Inglis S.C. J. Gen. Virol. 1990;71:3–11. doi: 10.1099/0022-1317-71-1-3. [DOI] [PubMed] [Google Scholar]
- 26.Liu D.X., Shen S., Xu H.Y., Wang S.F. Virology. 1998;246:288–297. doi: 10.1006/viro.1998.9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuerst T.R., Niles E.G., Studier F.W., Moss B. Proc. Natl. Acad. Sci. U. S. A. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laemmli U.K. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 29.Ng L.F.P., Liu D.X. Virology. 2000;272:27–39. doi: 10.1006/viro.2000.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu D.X., Cavanagh D., Green P., Inglis S.C. Virology. 1991;184:531–544. doi: 10.1016/0042-6822(91)90423-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu D.X., Brierley I., Tibbles K.W., Brown T.D.K. J. Virol. 1994;68:5772–5780. doi: 10.1128/jvi.68.9.5772-5780.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Machamer C.E., Mentone S.A., Rose J.K., Farquhar M.G. Proc. Natl. Acad. Sci. U. S. A. 1990;87:6944–6948. doi: 10.1073/pnas.87.18.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakai K., Kanehisa M. Genomics. 1992;14:897–911. doi: 10.1016/S0888-7543(05)80111-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hofmann K., Stoffel W. Biol. Chem. Hoppe-Seyler. 1993;347:166. doi: 10.1515/bchm3.1992.373.1.187. [DOI] [PubMed] [Google Scholar]
- 35.de Haan C.A.M., Vennema H., Rottier P.J.M. J. Virol. 2000;74:4967–4978. doi: 10.1128/jvi.74.11.4967-4978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Haan C.A.M., Smeets M., Vernooij F., Vennema H., Rottier P.J.M. J. Virol. 1999;73:7441–7452. doi: 10.1128/jvi.73.9.7441-7452.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen V-P., Hogue B., G. J. Virol. 1997;71:9278–9284. doi: 10.1128/jvi.71.12.9278-9284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narayanan K., Maeda A., Maeda J., Makino S. J. Virol. 2000;74:8127–8134. doi: 10.1128/jvi.74.17.8127-8134.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Budzilowicz C.J., Weiss S.R. Virology. 1987;157:509–515. doi: 10.1016/0042-6822(87)90293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alonso-Caplen F.V., Matsuoka Y., Wilcox G.E., Compans R.W. Virus Res. 1984;2:153–167. doi: 10.1016/0168-1702(84)90070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klumperman J., Locker J.K., Meijer A., Horzinek M., C., Geuze H.J., Rottier P.J.M. J. Virol. 1994;68:6523–6534. doi: 10.1128/jvi.68.10.6523-6534.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Haan C.A., Kuo L., Masters P., Vennema H., Rottier P.J.M. J. Virol. 1998;72:6838–6850. doi: 10.1128/jvi.72.8.6838-6850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Risco C., Anton I.M., Enjuanes L., Carrascosa J.L. J. Virol. 1996;70:4773–4777. doi: 10.1128/jvi.70.7.4773-4777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nilsson T., Jackson M., Peterson P.A. Cell. 1989;58:707–718. doi: 10.1016/0092-8674(89)90105-0. [DOI] [PubMed] [Google Scholar]
- 45.Lee C.-W., Jackwood M.W. Avian Dis. 2000;44:650–654. [PubMed] [Google Scholar]
- 46.Sutou S., Sato S., Okabe T., Nakai M., Sasaki N. Virology. 1988;165:589–595. doi: 10.1016/0042-6822(88)90603-4. [DOI] [PubMed] [Google Scholar]
- 47.Teasdale R.D., Jackson M.R. Annu. Rev. Cell Dev. Biol. 1996;12:27–54. doi: 10.1146/annurev.cellbio.12.1.27. [DOI] [PubMed] [Google Scholar]
- 48.Mounir S., Talbot P.J. Virology. 1993;192:355–360. doi: 10.1006/viro.1993.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Almanzan F., Gonzalez J.M., Penzes Z., Izeta A., Calvo E., Plana-Duran J., Enjuanes L. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5516–5521. doi: 10.1073/pnas.97.10.5516. [DOI] [PMC free article] [PubMed] [Google Scholar]