

Abstract
The severe acute respiratory syndrome (SARS) 3C-like protease consists of two distinct folds, namely the N-terminal chymotrypsin fold containing the domains I and II hosting the complete catalytic machinery and the C-terminal extra helical domain III unique for the coronavirus 3CL proteases. Previously the functional role of this extra domain has been completely unknown, and it was believed that the coronavirus 3CL proteases share the same enzymatic mechanism with picornavirus 3C proteases, which contain the chymotrypsin fold but have no extra domain. To understand the functional role of the extra domain and to characterize the enzyme-substrate interactions by use of the dynamic light scattering, circular dichroism, and NMR spectroscopy, we 1) dissected the full-length SARS 3CL protease into two distinct folds and subsequently investigated their structural and dimerization properties and 2) studied the structural and binding interactions of three substrate peptides with the entire enzyme and its two dissected folds. The results lead to several findings; 1) although two dissected parts folded into the native-like structures, the chymotrypsin fold only had weak activity as compared with the entire enzyme, and 2) although the chymotrypsin fold remained a monomer within a wide range of protein concentrations, the extra domain existed as a stable dimer even at a very low concentration. This observation strongly indicates that the extra domain contributes to the dimerization of the SARS 3CL protease, thus, switching the enzyme from the inactive form (monomer) to the active form (dimer). This discovery not only separates the coronavirus 3CL protease from the picornavirus 3C protease in terms of the enzymatic mechanism but also defines the dimerization interface on the extra helical domain as a new target for design of the specific protease inhibitors. Furthermore, the determination of the preferred solution conformation of the substrate peptide S1 together with the NMR differential line-broadening and transferred nuclear Overhauser enhancement study allows us to pinpoint the bound structure of the S1 peptide.
Introduction
A disease with overall fatality rates of 14-15%, characterized by high fever, malaise, rigor, headache, and nonproductive cough, suddenly appeared last year in southern China and then rapidly spread to other countries through Hong Kong (www.who.int/csr/sars/archive/2003_05_07a/en). The outbreak of this disease, now called severe acute respiratory syndrome (SARS),1 was not only a worldwide health hazard but also resulted in great damages to both the regional and global economies. To combat this unprecedented challenge, governmental agencies and scientists all over the world worked together to identify its causative agent and to develop effective strategies to halt SARS. Consequently, a novel coronavirus was identified as the pathogenic agent of SARS and was, thus, called SARS coronavirus (1, 2). On the other hand, neither an efficacious therapy nor a preventive treatment has been available to date despite tremendous efforts devoted to SARS-related research internationally. More seriously, the recent reports on several SARS-infected cases indicate that the new SARS outbreak in the future is not completely impossible. Therefore, there is an urgent demand to design potential therapeutic agents against SARS.
Coronaviruses are enveloped, positive-stranded RNA viruses with the largest single-stranded RNA genome (27-31 kilobases) among known RNA viruses (3). In general, the viral proteins required for genome replication and transcription are encoded by the large replicase gene. This gene encodes two very large replicative polyproteins, namely pp1a (∼450 kDa) and pp1b (∼750 kDa), that are subsequently processed by virus-encoded proteinases to release a group of functional subunits of the replication complex. The cleavage of the polyproteins is usually executed by two to three cysteine proteases, one with a chymotrypsin fold and the other two with a papain-like topology (3, 4). It is known that the central and C-proximal regions of pp1a and pp1b are cleaved by the 33-kDa viral protease with the chymotrypsin fold, which was called “main proteinase” or, alternatively, the “3C-like protease (3CLp)” to indicate the similarity with the picornavirus 3C protease in sharing the chymotrypsin fold and cleavage specificity. However, several features certainly separate the coronavirus 3CL proteases from the picornavirus 3C proteases, out of which the most distinguished one is that all coronavirus 3CL proteases gained an extra C-terminal α-helical domain during the evolution, although previously its function has remained totally unknown.
The genome sequence of the SARS coronavirus was cracked by several laboratories immediately after the outbreak (5, 6, 7). Sequence alignment with those of other coronaviruses indicated that the 3CL proteinase was conserved in the SARS coronavirus (8, 9). In this regard, the SARS 3CL protease might represent one of the most relevant targets validated so far for anti-SARS drug design (10). A homology model of the SARS 3CL protease was established (10), and recently the crystallographic structure of the SARS 3CL protease was published (11). The SARS 3CL protease, like other coronavirus 3CL proteases, comprises three domains with the first two β-barrel domains assembled into a chymotrypsin fold hosting the catalytic machinery and the third extra α-helical domain having unknown function (Fig. 1 ).
Fig. 1.
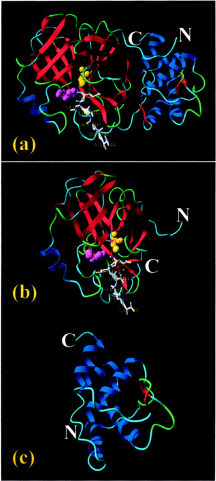
Ribbon diagram showing the domain organization of the coronavirus 3CL protease. The atomic coordinate used here is the crystallographic structure of transmissible gastroenteritis virus 3CL protease complexed with a peptide-like inhibitor (PDB code 1PAU). a, the full-length 3CL protease consisting of three domains. The N-terminal two β-barrel domains constitute the catalytic machinery that is a homologue of the 3C protease. The C-terminal α-helical domain is an extra domain unique in the coronavirus family without any known function. Two activesite residues, His-41 (purple) and Cys-144 (yellow), are highlighted in the ball-mode. The inhibitor bound to the active-site pocket is in stick mode. b, the first two β-barrel domains with the catalytic dyad His-41—Cys-144 embedded in the cleft correspond to the residues 1-196 of the SARS 3CL proteinase. c, the C-terminal extra α-helical domain corresponds to the residues 197-306 of the SARS 3CL protease.
Design of protease inhibitors requires many parameters in addition to the three-dimensional structure of an enzyme. For example, it is very crucial to obtain the detailed knowledge of the domain interaction of the enzyme as well as enzyme-substrate interactions. In the present study we used circular dichroism (CD), dynamic light scattering, and NMR spectroscopy to accomplish two objectives; they are 1) to understand the functional role of the extra domain of the SARS 3CL protease by dissecting the enzyme into two parts, namely the N-terminal chymotrypsin fold and the C-terminal extra domain, and 2) to study the interactions of the substrate peptides with the full-length enzyme and its two dissected parts. Very strikingly, the results reveal that the extra domain contributes to the dimerization of the SARS 3CL protease, a mechanism to switch the 3CL protease from the inactive form (monomer) to the active form (dimer). Furthermore, determination of the preferred conformation of the substrate peptide S1 together with the NMR differential line-broadening and transferred NOE studies provide important insight into the interaction between the substrate and the enzyme. Therefore, the results reported here not only contribute to our fundamental understanding of the unique catalytic mechanism of the coronavirus 3CL proteases but also lead to defining the dimerization interface on the extra domain as a new target for design of the highly specific protease inhibitors.
MATERIALS AND METHODS
Dissection and Cloning of SARS 3CL Protease and Its Fragments—The full-length SARS 3CL protease designated as 3CLp was identified to consist of 306 residues corresponding to residues 3241-3546 of SARS coronavirus (strain TOR2) on the basis of the sequence comparison with existing 3CL proteases (10, 12). Further comparison with the crystallographic structure of the transmissible gastroenteritis virus revealed that SARS 3CL protease was also organized in a three-domain architecture (Fig. 1a), the first two domains forming a chymotrypsin fold (residues 1-196) designated as 3CLc (Fig. 1b) and the third domain (197-306) designated as 3CLh (Fig. 1c).
Molecular cloning was performed on the cDNA templates provided by Genome Institute of Singapore. Because the encoding region for the 3CL protease was found to be located over two Genome Institute of Singapore constructs (the first 5′-terminal 34 bp were on one construct, and the rest were on another), a two-step procedure was used to obtain the DNA fragment encoding 3CLp. First, a long forward primer (46 bp) with a sequence of 5′-AGT GGT TTT AGG AAA ATG GCA TTC CCG TCA GGC AAA GTT GAA GGG T-3′ and a reverse primer, 5′-CGC GCG CTC GAG CTA TTG GAA GGT AAC ACC-3′, were designed to replicate the DNA fragment encoding the entire 3CL proteinase. Second, a subsequent PCR reaction with two short primers, 5′-CGC GCG CGG ATC CAG TGG TTT TAG G-3′ (forward) and 5′-GCG CTC GAG CTA TTG GAA GGT AAC ACC-3′, was conducted on the above-obtained PCR product to introduce restriction enzyme sites. Similarly, the DNA fragment encoding 3CLc was replicated by using primers 5′-CGC GCG CGG ATC CAG TGG TTT TAG G-3′ (forward) and 5′-GGC GGC CTC GAG CTA TGT ACC TGC AG-3′ (reverse), and the DNA fragment encoding 3CLh was replicated with two primers, 5′-CGC GCG CGG ATC CGA CAC AAC CAT AAC-3′ (forward) and 5′-CGC GCG CTC GAG CTA TTG GAA GGT AAC ACC-3′ (reverse).
Construction of the GST Fusion Plasmids and Expression of Recombinant Proteins—The PCR products encoding the 3CLp, 3CLc, and 3CLh were cloned into the pGEX-4X-1 vector (Amersham Biosciences) using EcoR1/BamH1 restriction sites. DNA sequencing identified one nucleotide mutation on the constructs resulting in an amino acid change (Gly-278 to Asp-278), which was traced back to the original Genome Institute of Singapore vector by DNA sequencing. Because this amino acid was located on the loop region of the extra helical domain, it most likely did not affect the activity of the 3CL proteinase (13). No further effort was devoted to changing the mutation. The expression constructs were transformed into the Escherichia coli strain BL21 to overexpress the GST fusion proteins. Briefly, the cells were cultured at 37 °C until the absorbance at 600 nm reached 0.7. Then 0.5 mm isopropyl-1-thio-β-d-galactopyranoside was added into the cell culture medium to induce the foreign protein expression at 20 °C overnight. The cells were then centrifuged and sonicated in the cell lysis buffer to release GST proteins, which were subsequently purified using glutathione-Sepharose (Amersham Biosciences). The in-gel cleavage of the fusion proteins was performed at room temperature by incubating the fusion proteins attached to the Sepharose beads with bovine thrombin. The pure 3CLp, 3CLc, and 3CLh were obtained by reloading the supernatants to the new glutathione-Sepharose beads to remove minor GST and fusion proteins. The molecular weights of the recombinant proteins were measured using a matrix-assisted time-of-flight mass spectrometer (Voyager-DE™ STR Biospectrometry™ workstation). For heteronuclear NMR experiments, the proteins were prepared in 15N-labeled forms using a similar expression protocol except for growing E. coli cells in minimal M9 media instead of the 2YT media, with an addition of (15NH4)2SO4 for 15N labeling.
Substrate Design and Enzymatic Activity—A 14-mer peptide S1 with amino acid sequences of ITSAVLQSGFRKMA was designed to mimic N-terminal autocleavage sites of the SARS 3CL protease and, therefore, served as the substrate for SARS 3CL protease in all measurements reported in the present study. A 7-mer peptide, S2, with a sequence of ITSAVLQ and a 14-mer peptide, S3, with a sequence of SGFRKMAFPSGKVE were designed to mimic the N- and C-terminal products cleaved by the SARS 3CL protease. The peptides were synthesized using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry and subsequently purified by a reverse-phase HPLC C18 column. Their identities were verified by MALDI-TOF mass spectrometry and NMR resonance assignments. The enzymatic assay was performed in 50 mm phosphate buffer (pH 7.2) containing 5 mm dithiothreitol and 150 mm NaCl. Usually, the peptide concentrations ranged between 100 to 500 μm, and the protein concentrations ranged from 1 to 30 μm.
CD Spectroscopy—CD experiments were performed on a Jasco J-810 spectropolarimeter equipped with thermal controller. The samples were prepared in 20 mm phosphate buffer at pH 6.8, with protein concentrations ranging from 20 to 100 μm. Far-UV CD spectra from 190 to 260 nm were collected using 1-mm path length cuvettes with a 0.1-nm spectral resolution. Five independent scans were averaged for each sample. Thermal unfolding with a temperature range between 20 to 93 °C was monitored at 222 nm at several protein concentrations.
NMR Experiments and Structure Generation—The NMR samples of the 15N-labeled 3CLp, 3CLc, and 3CLh were prepared by exchanging the proteins into 20 mm sodium phosphate buffer (pH 6.8). Samples of the synthetic peptides S1, S2, and S3 were prepared by dissolving the lyophilized peptides in 400 μl of aqueous buffer containing 20 mm sodium phosphate (pH 6.8). The deuterium lock signal for the NMR spectrometers was provided by the addition of 40 μl of D2O.
For NMR line-broadening study, two identical samples with a concentration of 0.4 mm were prepared in the 20 mm phosphate buffer (pH 6.8) for each NMR experiments. For the titration with 3CLp, the concentrated protein was added to 1 S1 peptide sample to reach a molar ration of 1:40 (protein:peptide), and this sample was kept at room temperature overnight in order to have the S1 peptide completely cleaved by the 3CLp as monitored by HPLC. Titration was conducted later by adding more 3CLp into the samples. The contribution of the added 3CLp, 3CLc, and 3CLh to the NMR spectra was removed by subtracting the spectra of the proteins from the spectra of the peptides in the presence of the proteins. For the transferred NOE experiment, the S1 peptide (1.5 mm) was mixed with the 3CLp overnight at a molar ratio of 20:1 (peptide:protease).
All NMR experiments including two-dimensional NOESY (14), TOCSY (15), and 1H-15N HSQC (16) were collected on a Bruker Avance 500 MHz NMR spectrometer equipped with an actively shielded cryoprobe and pulse field gradient units at 25 °C. A mixing time of 250 ms was used for NOESY and 65 ms for TOCSY experiments. Spectral processing and analysis were carried out using the XwinNMR (Bruker) and NMRview (17) software. Sequence-specific assignments for the synthetic peptides were achieved through identification of spin systems in the TOCSY spectra combined with sequential NOE connectivities in the NOESY spectra (18, 19).
For structure modeling, NOE connectivities were collected from NOESY spectra of the S1 peptide and subsequently converted into a uniform upper-bound interproton distance of 5.0 Å. The sum of the van der Waals radii of 1.8 Å was set to be the lower distance bound. The solution structure of S1 peptide was calculated on a Linux-based PC station by using the simulated annealing protocol (20) in the CNS program (21). The structures were analyzed by using YASARA (22) and MolMol graphic softwares (23).
Dynamic Light Scattering and Size-exclusion FPLC Analysis—The dimerization of the 3CLp, 3CLc, and 3CLh proteins were studied by use of dynamic light scattering and size-exclusion FPLC on a KTA fast protein liquid chromatography (Amersham Biosciences). For dynamic light-scattering analysis, the measurements were performed at 25 °C on a DynaPro-MS/X instrument (Protein Solutions Inc.). The protein samples were dissolved in a phosphate buffer (pH 7.2) with the presence of 5 mm dithiothreitol to prevent the possible formation of the intermolecular disulfide bridge. Molecular mass values were calculated by the Protein Dynamics analysis software using the standard conditions molecular weight curve, which is the mean of five readings. For the size-exclusion FPLC analysis, protein samples with a wide spectrum of concentrations from 1 to 600 μm were loaded to a Hiload 16/60 Superdex 200 column (Amersham Biosciences) and then eluted with a phosphate buffer (pH 7.2) with a flow rate of 1 ml/min. The column calibration was conducted with a low molecular weight protein kit (Amersham Biosciences) with 4 proteins: ribonuclease A (15.6 kDa), chymotrypsinogen A (22.8 kDa), ovalbumin (48.9 kDa), and albumin (65.4 kDa).
RESULTS
Cloning and Expression of 3CLp, 3CLc, and 3CLh—We have cloned and expressed the entire SARS 3CL protease (3CLp) and its two dissected parts (3CLc and 3CLh) as GST fusion proteins in E. coli BL21 cells. The purified 3CLp, 3CLc, and 3CLh were successfully isolated from GST through in-gel thrombin cleavage (Fig. 2 ). The molecular weights of the purified proteins were characterized by MALDI-TOF MS. As seen in Table I , except for the entire enzyme, which could not be ionized under several MS conditions probably due to very high molecular weight and the dimerization, two dissected parts have molecular weights very close to predicted ones, indicating that no unspecific cleavage happened during the thrombin cleavage.
Fig. 2.
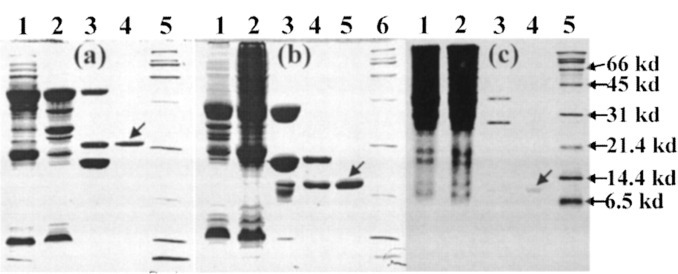
Expression and purification of the SARS 3CL protease (3CLp) and its two dissected fragments 3CLc and 3CLh.a, samples of the full-length 3CL protease on a 12% SDS-PAGE gel. Lane 1, total cell extract with a 0.5 mm isopropyl-1-thio-β-d-galactopyranoside induction at 20 °C overnight; lane 2, supernatant of the cell lysate by centrifugation; lane 3, mixture of the in-gel cleavage of the GST-3CL proteinase fusion protein by thrombin at room temperature for 4 h. Lane 4, purified 3CL proteinase; lane 5, protein molecular weight marker. b, samples of the dissected fragment 3CLc protein on a 12% SDS-PAGE gel. Lane 1, total cell extract with a 0.5 mm isopropyl-1-thio-β-d-galactopyranoside induction at 20 °C overnight; lane 2, supernatant of the cell lysate by centrifugation; lane 3, beads after the in-gel cleavage of the GST-3CLc by thrombin at room temperature for 4 h; lane 4, supernatant after the in-gel cleavage of the GST-3CLc by thrombin at room temperature for 4 h; lane 5, purified 3CLc protein; lane 6; protein molecular weight marker. c, samples of the dissected fragment 3CLh protein on a 15% SDS-PAGE gel. Lane 1, total cell extract with a 0.5 mm isopropyl-1-thio-β-d-galactopyranoside induction at 20 °C overnight; lane 2, supernatant of the cell lysate by centrifugation; lane 3, mixture after the in-gel cleavage of the GST-3CLc by thrombin at room temperature for 3 days; lane 4, purified 3CLc protein; lane 5, protein molecular weight marker.
Table I.
Molecular weights (Mr) of the entire SARS 3CL protease (3CLp) and two dissected parts, 3CLc and 3CLh, as measured by MALDI-TOF MS and dynamic light scattering (DLS)
| Predicted Mr | Measured by MS | Measured by DLS | |
|---|---|---|---|
| kDa | |||
| 3CLp | 34047.88 | ND | 63.0 ± 1.5 |
| 3CLc | 21732.84 | 21755.81 | 22.0 ± 1.0 |
| 3CLh | 12745.53 | 12769.08 | 21.0 ± 1.2 |
The enzymatic activities of 3CLp, 3CLc, and 3CLh were measured by using a 14-mer S1 peptide as a substrate. With a protein concentration of 20 μm, the full-length 3CL protease was able to cleave the peptide S1 (0.3 mm) very rapidly. As shown in Fig. 3a , only 10 min after initiating the reaction by mixing 3CLp with the peptide S1, about 15% of the peptides were cut into two short peptides. After 1 h, the S1 peptide was completely cleaved. On the other hand, as shown in Fig. 3b, although the dissected part 3CLc contained the complete catalytic machinery homologous to the picornavirus 3C protease, it showed a very weak catalytic activity on the S1 peptide. After 20 h of the initiation, only ∼20% peptide was cleaved, and ∼35% peptide was cleaved after 80 h. The cleavage experiment failed to detect any catalytic activity of 3CLh on the peptide S1 (data not shown).
Fig. 3.
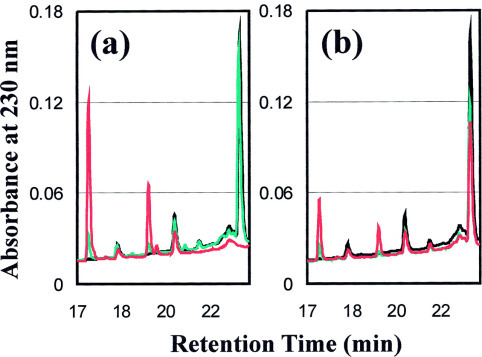
Catalytic activities of the SARS 3CL protease on the substrate peptide S1 as monitored by HPLC chromatography on an RP-18 column (Vydac). Cleavage reactions were carried out at room temperature in a 50 mm phosphate buffer (pH 7.2) with 5 mm dithiothreitol and 150 mm NaCl containing 0.3 mm S1 peptide and 20 μm recombinant proteins. At different time points a volume of 80 μl of reaction mixture was removed and mixed with 40 μl of 10% trifluoroacetic acid to stop the reaction. Subsequently the acidified mixture was analyzed by HPLC. a, HPLC profile of the cleavage reaction by 3CLp. Black, 0 min; green, 10 min; red, 1 h. b, HPLC profile of the cleavage reaction by 3CLc. Black, 0 min; green, 20 h; red, 80 h.
Structural Characterization by CD and NMR Spectroscopy—Far-UV CD spectroscopy was used to measure the secondary structures of the 3CLp, 3CLc, and 3CLh at a 30 μm protein concentration. As shown in Fig. 4a , all three proteins had well formed secondary structures. The CD spectrum of the full-length 3CL protease indicated that it assumed an α/β structure as detailed previously (24). On the other hand, the 3CLc had a spectrum typical of a β-sheet structure with a negative peak at ∼221 nm and a positive peak at 199 nm, whereas 3CLh had a spectrum characteristic of an α-helical structure with dual negative peaks at ∼221 and 209 nm. The thermal stability of all three proteins was assessed by monitoring the continuous changes in the ellipticity at 222 nm during the temperature range from 20 to 93 °C. The results shown in Fig. 4b indicated that both 3CLp and 3CLc started to precipitate at 48 and 56 °C, respectively. For the extra domain 3CLh, the conformation underwent an unfolding, but no precipitation was observed. Interestingly, even at 93 °C the structure of 3CLh was not completely denatured, and its CD spectrum appeared to resemble that of a β-sheet protein (data not shown).
Fig. 4.
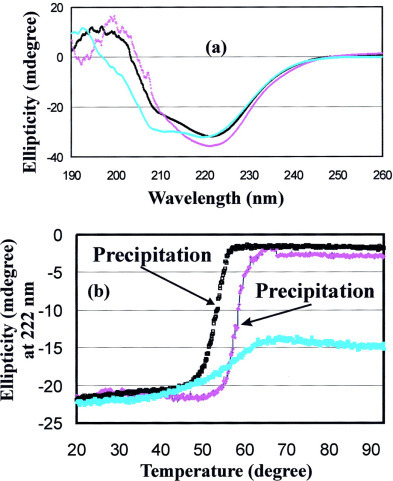
CD characterization of the structure and stability of 3CLp, 3CLc, and 3CLh.a, far-UV CD spectra of three recombinant proteins at a 50 μm concentration in 20 mm phosphate buffer (pH 6.8) at 20 °C. Black, full-length SARS 3CL protease 3CLp; purple, 3CLc and cyan, 3CLh. b, thermal unfolding followed at 222 nm with a temperature range between 20 to 93 °C. Black, 3CLp; purple, 3CLc; cyan, 3CLh.
The structural properties of the three proteins were further investigated by use of NMR 1H-15N HSQC spectroscopy, which is very sensitive to both the secondary structure and tertiary packing. For the full-length 3CL proteinase, the attempt to collect a HSQC spectrum failed probably due to the relaxation properties or/and aggregation commonly observed for large proteins (25). However, its one-dimensional proton NMR spectrum presented in Fig. 5a indicated that 3CLp would be structured with tightly packed tertiary structure, as evidenced from the several very up-field NMR resonance peaks at -0.25 and -0.76 ppm, although these NMR lines were very broad, again mostly due to a very large molecular mass (68 kDa after dimerization) or/and further aggregation (25). However, it is very hard to exclude the possibility that the entire 3CL protease (3CLp) might have some properties of the molten globule in solution (26), although it was shown that the SARS 3CLp adopted a well packed structure in the crystal (11). Strikingly, well dispersed HSQC spectra were obtained for 3CLc and 3CLh (Fig. 5, b and c), unquestionably indicating that two isolated fragments had not only well formed secondary structures shown by CD spectra but also well packed tertiary structures. As shown in Fig. 5b, many downfield resonance peaks (>8.5 ppm) were observed, indicating the 3CLc adopted a β-sheet structure, consistent with the CD observation. On the other hand, as seen in Fig. 5c, only a small portion of downfield resonance peaks (>8.5 ppm) were found, clearly suggesting that 3CLh assumed an α-helical structure.
Fig. 5.
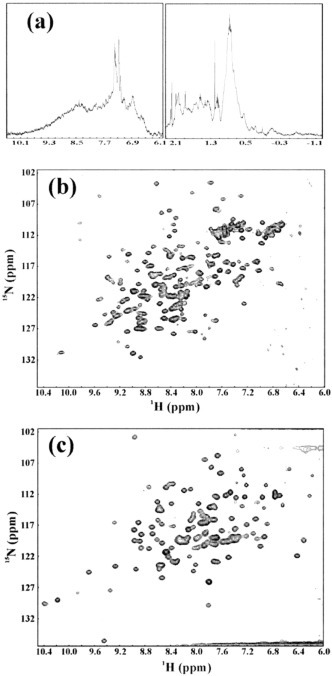
NMR characterization of the 15N isotope-labeled 3CLp, 3CLc, and 3CLh.a, a one-dimensional NMR spectrum of the full-length SARS 3CL proteinase at 25 °C in 20 mm phosphate buffer (pH 6.8). b, a two-dimensional 1H-15N HSQC spectrum of the 3CLc protein at 25 °C in 20 mm phosphate buffer (pH 6.8). c, a two-dimensional 1H-15N HSQC spectrum of the 3CLh protein at 25 °C in 20 mm phosphate buffer (pH 6.8).
Dimerization of the Extra Helical Domain 3CLh—As seen in Table I, the molecular weights of the 3CLc and 3CLh proteins measured by MALDI-TOF MS were very close to those predicted from their amino acid sequences, indicating that both dissected domains had no unwanted truncation. Interestingly, when measured by the dynamic light scattering, the molecular mass of the entire 3CLp was estimated to be around 63.0 kDa, indicating the dimerization of the entire enzyme occurred to some extent. On the other hand, the molecular masses of the dissected 3CLc and 3CLh were estimated to be around 22.0 and 21.0 kDa, respectively. The results clearly indicated that the dissected catalytic domain existed as a monomer, whereas the dissected helical domain existed as a dimer. Because the dynamic light-scattering measurement requires a narrow range of the protein concentration to achieve good results, we therefore further followed the concentration dependence of the dimerization by use of FPLC chromatography. As showed in Fig. 6 , only the full-length 3CLp showed a concentration-dependent dimerization, and it existed mostly as a monomer at a protein concentration less than 0.2 mg/ml, consistent with the recent report (27). It is also worthwhile to note that the catalytic domain remained as a monomer even at a very high protein concentration (14.8 mg/ml). Very interestingly, the isolated extra domain 3CLh existed as a dimer even at a very low protein concentration (0.12 mg/ml) and showed no concentration dependence. The observation that the chymotrypsin fold 3CLc existed as a monomer at a very high concentration whereas the extra helical domain 3CLh existed as a dimer even at a very low concentration strongly implies that the extra domain contributes significantly to the dimerization of the SARS 3CL protease.
Fig. 6.
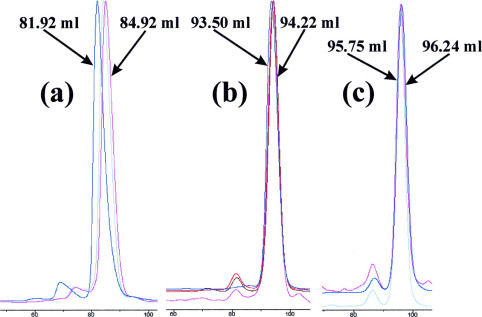
Concentration-dependent dimerization of 3CLp, 3CLc, and 3CLh as monitored by FPLC chromatography.a, FPLC profiles for 3CLp. Blue, 3CLp with a protein concentration of 9.2 mg/ml and elution volume of 81.92 ml; gray, 3CLp with a protein concentration of 0.98 mg/ml and elution volume of 84.12 ml; pink, 3CLp with a protein concentration of 0.8 mg/ml and elution volume of 84.92 ml. b, FPLC profiles for 3CLc. Blue, 3CLc with a protein concentration of 14.88 mg/ml and elution volume of 93.5 ml; red, 3CLc with a protein concentration of 1.28 mg/ml and elution volume of 94.07 ml; brown, 3CLc with a protein concentration of 0.48 mg/ml and elution volume of 93.97 ml; pink, 3CLc with a protein concentration of 0.1 mg/ml and elution volume of 94.22 ml. c, FPLC profiles for 3CLh. Pink, 3CLh with a protein concentration of 2.63 mg/ml and elution volume of 96.07 ml; gray, 3CLh with a protein concentration of 0.56 mg/ml and elution volume of 95.73 ml; blue, 3CLh with a protein concentration of 0.12 mg/ml and elution volume of 96.24 ml.
Binding Interactions of 3CLp, 3CLc, and 3CLh with the Substrate Peptides—The NMR differential line-broadening and transferred NOE experiments were powerful probes for characterizing protein-peptide interactions in the fast exchange regime on the NMR time scale (28, 29, 30, 31). Fig. 7 presents the aliphatic and aromatic regions of the one-dimensional NMR spectra of the 14-mer S1 peptide in the absence and presence of 3CLp, 3CLc, and 3CLh. Fig. 7a showed the spectral region with NMR resonance peaks from the side chains of the residues Ile-1, Val-5, and Leu-6 in the absence and presence of the full-length 3CL protease at different ratios. It can be seen that with the addition of the full-length 3CL protease at a ratio of 1:40 (protein:S1 peptide), most resonance peaks started to become broadened. This broadening process was demonstrated by the peaks resulting from the methyl group of the residue Leu-5, which was well separated from the other peaks (Fig. 7a). The line-broadening became more severe when more 3CL protease was added to reach a ratio of 20:1 (Fig. 7a). This observation indicated the binding interactions occurred between the 3CL protease and the side chains of Val-4 and Leu-5 in the fast-exchange regime. Interestingly, the similar line-broadening of the aromatic resonance peaks from the residue Phe-10 were also observed (Fig. 7b), indicating that both the N-terminal half represented by residues Val-5 and Leu-6 and C-terminal half carrying residue Phe-9 interacted with the full-length 3CL protease even after the cleavage of the S1 peptide at Gln-6—Ser-7 bond was completed.
Fig. 7.
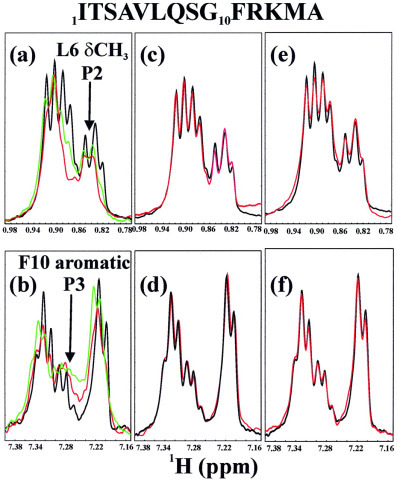
Binding interactions of the substrate peptide S1 with 3CLp, 3CLc, and 3CLh as probed by NMR differential line-broadening. All spectra were collected at 25 °C in a 20 mm phosphate buffer (pH 6.8) in the absence or presence of recombinant proteins at different molar ratios. Two NMR spectral regions of the S1 peptide were presented; the 0.77-0.99 ppm region, resulting from resonance peaks of the aliphatic side chains of the residues Ile-1, Val-5, and Leu-6, and 7.15-7.40 ppm region, resulting from the aromatic protons of the residue Phe-10. a and b, two spectral regions in the presence of 3CLp at different ratios. Black, in the absence of 3CLp; green, in the presence of 3CLp at a molar ratio of 1:40 (protein/peptide); red, in the presence of 3CLp protein at a molar ratio of 1:20 (protein/peptide). c and d, two spectra regions in the presence of the 3CLc. Black, in the absence of 3CLc; red, in the presence of 3CLc at a molar ratio of 1:20 (protein/peptide). e and f, two spectral regions in the presence of the 3CLh. Black, in the absence of 3CLc; red, in the presence of 3CLh at a molar ratio of 1:20 (protein/peptide).
Similar experiments were carried out with 3CLc and 3CLh, and the results were shown in Fig. 7, c-f. Surprisingly, additions of 3CLc and 3CLh resulted in no significant line-broadening of the NMR resonances, indicating that S1 peptide had no detectable binding interaction with both dissected fragments. This conclusion was further confirmed by HSQC titrations of the 15N-labeled 3CLc and 3CLh with S1 peptide. No significant HSQC peak shift was observed for both 3CLc and 3CLh even in the presence of S1 peptide at up to a ×20 excess (data not shown).
To confirm the interaction between the 3CL protease and the two cleaved fragments of the S1 peptide, the NMR differential line-broadening experiments were further conducted on a 7-mer S2 and a 14-mer S3 peptides. As seen in Fig. 8a , line-broadening and peak shifts were observed when the 3CL protease was added to the 7-mer N-terminal half of the cleaved fragments. As far as the 14-mer C-terminal half was concerned, very unbelievably, the addition of 3CLp even induced the line-broadening and peak shifts for the residue Val-13 of the S3 peptide. These results suggested that the amino acids C-terminal to the cleavage site also have extensive interactions with the 3CL protease.
Fig. 8.

Binding interactions of the S2 and S3 peptides with 3CLp as followed by differential NMR line-broadening. All spectra were collected at 25 °C in a 20 mm phosphate buffer (pH 6.8) in the absence or presence of the 3CLp at different molar ratios. a, NMR spectrum of the S2 peptide over 0.76-0.99 pm. The well separated NMR peaks from the methyl groups of the residue Leu-6 were labeled. NMR spectra of S3 peptide over two spectral regions were presented; they are, the 0.87-0.97 ppm region, resulting from resonance peaks from the methyl groups of the residues Val-13 (b), and the 7.20-7.42 ppm region, resulting from the overlapped aromatic protons of Phe-3 and Phe-10 (c). Spectra in black, in the absence of 3CLp protein; green, in the presence of 3CLp at a molar ratio of 1:40 (protein/peptide); red, in the presence of 3CLp at a molar ratio of 1:20 (protein/peptide).
The Preferred Conformations of the S1 Peptide—The structural properties of the S1 peptide in the absence and presence of the full-length 3CL protease were further addressed by two-dimensional NMR experiments. As seen in Fig. 9a , in the free state, the aromatic ring of the residue Phe-10 had extensive long-range contacts with the side chains of the residues Val-5 and Leu-6. These NOE connectivities summarized in Fig. 9b indicated the formation of a β-turn conformation over the residues Val-5 to Phe-10. Interestingly, even after the S1 peptide was cleaved into two fragments by the 3CL proteinase at Gln-6—Ser-7 bond, similar transferred NOE patterns could still be observed, although most NOE resonances were too broadened to be discriminated from the noise. For example, there were still NOEs between the HG protons of the residue Gln-7 and the HD protons of the residue Phe-10 and between HD/HE/HZ protons of the residue Phe-10 and HG/HD protons of the residue Leu-6. This observation strongly suggests that although S1 peptide was cleaved into two fragments at the Gln-6-Ser-7 bond, the two parts were still bound to the 3CL protease and the spatial relationship between two fragments might still remain similar to that adopted by the uncleaved S1 peptide in the free state.
Fig. 9.
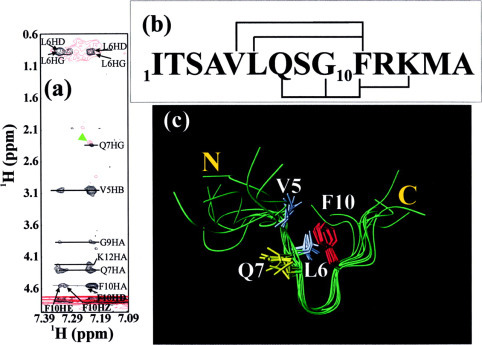
Solution conformation of the S1 substrate peptide.a, the aromatic-aliphatic regions of NOESY spectra of the S1 peptide in 20 mm phosphate buffer (pH 6.8) in the absence of the 3CLp (black) and in the presence of the 1/20 3CLp (red). The key NOE connectivities are labeled to indicate the presence of a β-turn conformation. b, the amino acid sequence of the S1 peptide with key NOE connectivities defining the β-turn over the residues Val-5 to Phe-10. c, solution conformation of the S1 peptide calculated from experimental NOEs.
The NOE connectivities were used to model the preferred conformation of the S1 peptide. Fig. 9c presents the 8 lowest-energy structures of the S1 peptide superimposed over residues Val-5 to Phe-10. It is interesting to note that the β-turn region is well defined, whereas the rest of the molecule such as the N- and C-terminal tails is flexible. It appears that this β-turn conformation is stabilized by the interactions between the aromatic ring of the residue Phe-10 and the hydrophobic side chains of the residues Val-5 and Leu-6, as previously observed for small peptide fragments and partially folded intermediates (32, 33). The relatively flexible β-turn conformation observed in the free state should allow the local conformational rearrangements to take place when the S1 peptide was bound to the enzyme.
DISCUSSION
The central role of the SARS 3CL protease in the replication cycle of the virions ranks this enzyme as a top target for design of anti-SARS drugs. If compared with picornavirus 3C proteases, SARS 3CL protease, like other coronavirus 3CL proteases, gained an extra C-terminal helical domain during the evolution. Previously no clue has been available regarding the functional role of this extra domain, and it was extensively believed that the coronavirus 3CL proteases and picornavirus 3C proteases shared a similar enzymatic mechanism and substrate specificity. On the other hand, several previous attempts have utilized the fragment deletion approach (12, 34, 35) to assess the role of the extra domain and demonstrated that the extra domain was indispensable for the activity of the coronavirus 3CL protease. However, because no structural property of truncated fragments was acquired, the possibility still existed that the loss of the activity might be simply the consequence of the misfolding of truncated forms. In the present study, we first demonstrated that the significant activity loss of the catalytic fold 3CLc, a structural homologue of the picornavirus 3C protease, was not due to the misfolding. Most importantly, we have discovered that the extra helical domain contributed to the dimerization of the enzyme. Considering the observations that only the dimer form is the active form of the 3CL proteases (11, 12, 27, 36), we therefore propose that one key role of the extra domain unique for 3CL proteases is to regulate the activity and specificity of the 3CL proteases by controlling the association-dissociation equilibrium of the enzyme. Our results also strongly indicate that the catalytic mechanism of the coronavirus 3CL proteases is totally different from that of the picornavirus 3C proteases, which only require the presence of the chymotrypsin fold. The observation that the isolated helical domain was fully folded implies that this extra domain may further interact with other proteins/biomacromolecules to link the activity and specificity of the 3CL proteases to other signaling networks, as previously proposed for other viral proteases (37). Consequently, our current study defines the dimerization interface on the extra domain as a new target for design of specific inhibitors for the SARS 3CL protease.
In the present study other features unique for the SARS 3CL protease were also illustrated. Our results clearly indicate that only the entire enzyme is capable of interacting with the substrate peptide S1. Furthermore, to our surprise, unlike other proteases such as hepatitis C virus N3 protease, which only binds to the N-terminal part after the substrate cleavage (38), the SARS 3CL protease showed binding interactions with both the N- and C-terminal parts of the cleaved substrate peptide S1. These observations indicate that the interaction interface between the SARS 3CL protease and its in vivo substrates might be very large, and the enzymatic activity and specificity are under regulation by many factors.
It is also worthwhile to note that the substrate peptide S1 adopts a preferred β-turn conformation over residue Val-5 to Phe-10 that might remain similar after bound to the enzyme, as implied by the transferred NOE study. Indeed, the conformational similarity between the free state and the bound state was observed for a thrombin inhibitor previously (39). These results are particularly enlightening in light of the recent CD study on the conformations of the 11 substrate peptides for the SARS 3CL protease which implied that substrates with more turn-like conformation tended to react with the enzyme faster (27). These results together offer a novel possibility for design of active-site inhibitors differing from the current design by which the inhibitors were only derived from the N-terminal half of the cleaved substrate. Certainly it is worth to give an attempt to design active site inhibitors based on the β-turn conformation obtained in our study (Fig. 9c).
Our results only not bear fundamental implications, but they offer valuable clues for the design of inhibitors for the SARS 3CL protease. Considering the pivotal role of the helical domain in controlling dimerization of the enzyme, it is logically anticipated that the disruption of the dimerization interface could be a promising strategy to develop novel inhibitors targeting at 3CL proteases. For example, phage-display or natural product library could be used to screen ligands binding to the dimerization interface on the extra domain. Consequently, a promising strategy might be established in which two separate inhibitors, one binding to the active site and another disrupting the dimerization interface on the extra domain, are linked together to create a bifunctional inhibitor with significantly enhanced binding affinity and specificity as previously demonstrated on another proteinase (40, 41, 42).
Acknowledgments
We thank Prof. C. L. Hew for providing initial funding for the current study under the Structural Biology Program and for continuous encouragement, Prof. B. Halliwell for continuous encouragement and for providing funding under the Office of Life Sciences Neurobiology program, Dr. H. W. Song for accessing the dynamic light-scattering instrument, and M. S. Wu for assistance of measurements. We also thank the Genome Institute of Singapore for the generous gift of the two SARS cDNA constructs we used for cloning the 3CL protease.
Footnotes
The abbreviations used are: SARS, severe acute respiratory syndrome; GST, glutathione S-transferase; HPLC, high performance liquid chromatography; FPLC, fast performance liquid chromatography; CD, circular dichroism; MALDI-TOF MS, matrix-assisted time-of-flight mass spectrometer; NOE, nuclear Overhauser enhancement; NOESY, NOE spectroscopy; TOCSY, total correlation spectroscopy; HSQC, heteronuclear single-quantum coherence spectroscopy.
REFERENCES
- 1.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J, Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Lai M.M.C., Cavanagh D. Adv. Virus Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziebuhr J., Snijder E.J., Gorbalenya A.E. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 5.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 6.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 7.Ruan Y.J., Wei C.L., Ee A.L., Vega V.B., Thoreau H., Su S.T., Chia J.M., Ng P., Chiu K.P., Lim L., Zhang T., Peng C.K., Lin E.O., Lee N.M., Yee S.L., Ng L.F., Chee R.E., Stanton L.W., Long P.M., Liu E.T. Lancet. 2003;361:1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 9.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 11.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anand K., Palm G.J., Mesters J.R, Siddell S.G., Ziebuhr J., Hilgenfeld R. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hegyi A., Friebe A., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2002;83:581–593. doi: 10.1099/0022-1317-83-3-581. [DOI] [PubMed] [Google Scholar]
- 14.Jeener J., Meier B.H., Bachmann P., Ernst R.R. J. Chem. Phys. 1979;71:4546–4553. [Google Scholar]
- 15.Bax A., Davis D.G. J. Magn. Reson. 1985;65:355–360. [Google Scholar]
- 16.Sattler M., Schleucher J., Griesinger C. Prog. NMR Spectrosc. 1999;34:93–201. [Google Scholar]
- 17.Johnson B.A., Blevins R.A. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 18.Wagner G., Wuthrich K. J. Mol. Biol. 1982;155:347–366. doi: 10.1016/0022-2836(82)90009-2. [DOI] [PubMed] [Google Scholar]
- 19.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons, Inc.; New York: 1986. [Google Scholar]
- 20.Song J., Gilquin B., Jamin N., Drakopoulou E., Guenneugues M., Dauplais M., Vita C., Menez A. Biochemistry. 1997;36:3760–3766. doi: 10.1021/bi962720h. [DOI] [PubMed] [Google Scholar]
- 21.Brunger A.T., Adams P.D., Clore G.M., Delano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. Acta Crystallogr. Sect. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Krieger E., Vriend G. Bioinformatics. 2002;18:315–318. doi: 10.1093/bioinformatics/18.2.315. [DOI] [PubMed] [Google Scholar]
- 23.Koradi R., Billeter M., Wüthrich K. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 24.Venyaminov S.Y., Yang J.T. In: Circular Dichroism and the Conformational Analysis of Biomolecules. Fasman G.D., editor. Plenum Press; New York: 1996. pp. 69–107. [Google Scholar]
- 25.Wagner G. J. Biomol. NMR. 1993;3:375–385. doi: 10.1007/BF00176005. [DOI] [PubMed] [Google Scholar]
- 26.Song J., Bai P., Luo L., Peng Z.Y. J. Mol. Biol. 1998;280:167–174. doi: 10.1006/jmbi.1998.1826. [DOI] [PubMed] [Google Scholar]
- 27.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2003;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birdsall B., Bevan A.W., Pascual C., Roberts G.C., Feeney J., Gronenborn A., Clore G.M. Biochemistry. 1984;23:4733–4742. doi: 10.1021/bi00315a032. [DOI] [PubMed] [Google Scholar]
- 29.Sykes B.D. Curr. Opin. Biotechnol. 1993;4:392–396. doi: 10.1016/0958-1669(93)90003-f. [DOI] [PubMed] [Google Scholar]
- 30.Ni F. Prog. NMR Spectrosc. 1994;26:517–606. [Google Scholar]
- 31.Zuiderweg E.R. Biochemistry. 2002;41:1–7. doi: 10.1021/bi011870b. [DOI] [PubMed] [Google Scholar]
- 32.Dyson H.J., Rance M., Houghten R.A., Lerner R.A., Wright P.E. J. Mol. Biol. 1988;201:161–200. doi: 10.1016/0022-2836(88)90446-9. [DOI] [PubMed] [Google Scholar]
- 33.Song J., Jamin N., Gilquin B., Vita C., Menez A. Nat. Struct. Biol. 1999;6:129–134. doi: 10.1038/5815. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y., Denison M.R. Virology. 1997;230:335–342. doi: 10.1006/viro.1997.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziebuhr J., Heusipp G., Siddell S.G. J. Virol. 1997;71:3992–3997. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng L.F., Liu D.X. Virology. 2000;272:27–39. doi: 10.1006/viro.2000.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrette-Ng I.H., Ng K.K., Mark B.L., Van Aken D., Cherney M.M., Garen C., Kolodenko Y., Gorbalenya A.E., Snijder E.J., James M.N. J. Biol. Chem. 2002;277:39960–39966. doi: 10.1074/jbc.M206978200. [DOI] [PubMed] [Google Scholar]
- 38.LaPlante S.R., Cameron D.R., Aubry N., Lefebvre S., Kukolj G., Maurice R., Thibeault D., Lamarre D., Llinas-Brunet M. J. Biol. Chem. 1999;274:18618–18624. doi: 10.1074/jbc.274.26.18618. [DOI] [PubMed] [Google Scholar]
- 39.Song J., Xu P., Koutychenko A., Ni F. Biopolymers. 2002;65:373–386. doi: 10.1002/bip.10253. [DOI] [PubMed] [Google Scholar]
- 40.DiMaio J., Gibbs B., Munn D., Lefebvre J., Ni F., Konishi Y. J. Biol. Chem. 1990;265:21698–21703. [PubMed] [Google Scholar]
- 41.Song J., Ni F. Biochem. Cell Biol. 1998;76:177–188. doi: 10.1139/bcb-76-2-3-177. [DOI] [PubMed] [Google Scholar]
- 42.Shuker S.B., Hajduk P.J., Meadows R.P., Fesik S.W. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]