

Abstract
The coronavirus spike (S) protein mediates infection of receptor-expressing host cells and is a critical target for antiviral neutralizing antibodies. Angiotensin-converting enzyme 2 (ACE2) is a functional receptor for the coronavirus (severe acute respiratory syndrome (SARS)-CoV) that causes SARS. Here we demonstrate that a 193-amino acid fragment of the S protein (residues 318–510) bound ACE2 more efficiently than did the full S1 domain (residues 12–672). Smaller S protein fragments, expressing residues 327–510 or 318–490, did not detectably bind ACE2. A point mutation at aspartic acid 454 abolished association of the full S1 domain and of the 193-residue fragment with ACE2. The 193-residue fragment blocked S protein-mediated infection with an IC50 of less than 10 nm, whereas the IC50 of the S1 domain was ∼50 nm. These data identify an independently folded receptor-binding domain of the SARS-CoV S protein.
A distinct coronavirus (SARS-CoV)1 has been identified as the etiological agent of SARS, an acute pulmonary syndrome characterized by an atypical pneumonia that results in progressive respiratory failure and death in close to 10% of infected individuals (1, 2, 3, 4). SARS-CoV does not belong to any of the three previously defined genetic and serological coronavirus groups; the SARS-CoV S protein, a surface glycoprotein that mediates coronavirus entry into receptor-bearing cells, is also distinct from those of other coronaviruses (5, 6). Reflecting this difference, SARS-CoV does not utilize any previously identified coronavirus receptors to infect cells. Rather, as we have recently demonstrated, angiotensin-converting enzyme 2 (ACE2) serves as a functional receptor for this coronavirus (7).
The S proteins of some coronaviruses, for example, that of mouse hepatitis virus (MHV), can be cleaved into two subunits (S1 and S2) (8, 9). The S proteins of other coronaviruses, such as those of human coronavirus 229E (HCoV-229E) and SARS-CoV, are not cleaved by the virus-producing cell (10). Nonetheless, S1 and S2 domains of these latter S proteins can be identified through their homology with the S1 and S2 subunits of cleaved coronavirus S proteins. The S1 domain of all characterized coronaviruses, including that of SARS-CoV, mediates an initial high affinity interaction with a cellular receptor (11, 12, 13).
Independently folded receptor-binding domains of two coronaviruses have been described. The first 330 amino acids of the 769-residue S1 subunit of the MHV S protein is sufficient to bind carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), the cellular receptor for MHV (13, 14, 15). A very different region of the S1 domain of HCoV-229E, between residues 407 and 547, is sufficient to associate with the cellular receptor for this coronavirus, aminopeptidase N (APN, CD13) (11, 12, 16). Here we show that a 193-amino acid fragment of the SARS-CoV S protein, residues 318–510, binds the SARS-CoV receptor ACE2 and blocks S protein-mediated infection more efficiently than does the full-length S1 domain. This region includes seven cysteines, five of which are essential for expression or ACE2 association. Point mutations within this domain, at glutamic acid 452 or aspartic acid 454, interfere with or abolish association with ACE2. These data identify a domain of the SARS-CoV S protein that may be a critical target for neutralizing antibodies against the virus.
EXPERIMENTAL PROCEDURES
Construction of S1-Ig, Truncation Variants, and Mutants—A plasmid encoding S1-Ig was generated by amplifying a region encoding residues 12–672 from an expression vector containing a codon-optimized form of the full-length S protein gene (7) and ligating this region into a previously described vector encoding the signal sequence of CD5 and the Fc domain of human IgG1 (17). Truncation variants were generated by inverse PCR amplification, using the S1-Ig plasmid as a template. Mutations within S1-Ig, or within a truncation mutant thereof expressing residues 318–510, were generated by site-directed mutagenesis using the QuikChange method (Stratagene). Two independent plasmids were generated for each variant, sequenced within their coding regions and assayed.
Purification of S1 Protein Variants—293T cells were transfected with plasmids encoding S1-Ig or S1-Ig variants. One day post-transfection, cells were washed in PBS and subsequently incubated in 293 SFM II medium (Invitrogen). Medium was harvested after 48 h and proteins precipitated with protein A-Sepharose beads at 4 °C for 16 h. Beads were washed in PBS, 0.5 M NaCl, eluted with 50 mm sodium citrate/50 mm glycine (pH 2), and neutralized with NaOH. Purified proteins were concentrated with Centricon filters (Amicon) and dialyzed in PBS.
Binding and Flow Cytometry—293T cells were transfected with a previously described plasmid encoding ACE2 (7) or with vector (pcDNA3.1, Invitrogen) alone. Three days post-transfection, cells were detached in PBS, 5 mm EDTA and washed with PBS, 0.5% BSA. S1-Ig or variants thereof were added to 106 cells to a final concentration of 250 nm, and the mixture was incubated on ice for 1 h. Cells were washed three times with PBS, 0.5% BSA, then incubated for 30 min on ice with anti-human IgG FITC conjugate (Sigma; 1:50 dilution). Cells were again washed with PBS, 0.5% BSA. Binding of IgG-tagged viral proteins to 293T cells transfected with ACE2-expressing plasmid was detected by flow cytometry. The mean value of the binding of S1-Ig or variants with the ACE2-transfected cells was subtracted from that of the mock-transfected cells and normalized to that of S1-Ig.
Immunoprecipitation of Soluble ACE2—293T cells transfected with a previously described plasmid expressing soluble ACE2 (7) were metabolically labeled with [35S]cysteine and -methionine. Labeled medium was harvested 3 days post-transfection. 0.5 ml of soluble ACE2-containing medium was incubated for 15 min on ice with 25 pmol of purified S1-Ig or variants, to a final concentration of 50 nm. 20 μl of protein A-Sepharose was added to the mixture, which was then incubated for 1 h at room temperature. Protein A-Sepharose beads were washed three times with PBS, 0.1% Nonidet P-40 and once with PBS. Protein was analyzed by SDS-PAGE and quantified by phosphorimaging using ImageQuant software.
Infection Assay with S Protein-Pseudotyped Virus—–293T cells were transfected with a plasmid encoding SARS-CoV S protein or VSV-G, together with a previously described plasmid encoding the genome of simian immunodeficiency virus (SIV), modified by deletion of the env gene and by replacement of the nef gene with that for green fluorescent protein (GFP) (18). Supernatants of transfected cells were harvested, and viral reverse transcriptase activity was measured. Supernatants containing S protein- or VSV-G-pseudotyped SIV were added to ACE2- or mock-transfected 293T cells in the presence or absence of the indicated concentrations of S1-Ig or of the 12–327 or 318–510 variants thereof. Media were changed the following day and GFP expression in infected cells was measured 2 days later by flow cytometry.
RESULTS AND DISCUSSION
A protein in which the S1 domain of the SARS-CoV S protein was fused to the Fc region of human IgG1 has been shown to associate with ACE2-expressing cells and to precipitate ACE2 (7). To identify the receptor-binding domain of the S protein, this fusion protein, S1-Ig, was sequentially deleted at the N and C termini of the S1 domain to make a total of 12 additional variants. Each variant expressed efficiently and could be readily purified using protein A-Sepharose beads (Fig. 1A , top). S1-Ig and truncation variants thereof were used to precipitate a metabolically labeled and soluble form of ACE2. In contrast to an analogous MHV S protein truncation variant, which efficiently binds the MHV receptor CEACAM1 (13), the S1-Ig variant containing S1 domain residues 12–327 did not associate with ACE2. Neither did another expressing residues 12–481, whereas variants expressing residues 12–510 and 12–572 efficiently bound soluble ACE2 (Fig. 1A). These data indicate that residues 511–672 at the C terminus of the S1 domain do not contribute significantly to ACE2 association.
Fig. 1.
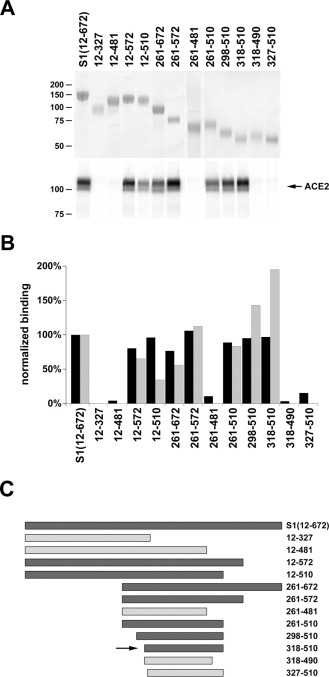
Residues 318–510 of the SARS-CoV S protein include the receptor-binding domain.A, S1-Ig, containing S protein residues 12–672 fused to the Fc region of human IgG1, or truncation variants of S1-Ig containing the indicated S protein residues, were purified from media of transfected 293T cells. S1-Ig and variants were normalized for expression, as shown by Coomassie staining (top panel), and used to precipitate soluble metabolically labeled ACE2 (bottom panel). Precipitates were analyzed by SDS-PAGE, and ACE2 was quantified by phosphorimaging. B, the indicated S1-Ig variants were incubated with ACE2-transfected 293T cells and analyzed by flow cytometry (black bars) or used, as in (A), to immunoprecipitate soluble ACE2 (gray bars). Bars indicate averages of two or more experiments normalized to results for S1-Ig. C, representation of truncation variants assayed in A and B. Dark gray indicates association with ACE2 greater than 25% of that observed for S1-Ig in both precipitation and flow-cytometry assays. Light gray indicates ACE2 association less than 10%, in both assays, of that for S1-Ig. The arrow indicates 318–510 variant, the smallest fragment observed to bind ACE2.
Removal of residues 12–260 from the S1-Ig N terminus had no effect on ACE2 association (Fig. 1A). Variants expressing residues 298–510 and 318–510 efficiently bound S protein. The 318–510 variant precipitated ACE2 more efficiently than did the full S1 domain. However, two variants expressing slightly smaller fragments of the S1 domain (residues 318–490 and 327–510) did not detectably precipitate ACE2. These data imply that some residues from 318 to 326 and from 491 to 509 contribute either directly to the association of the S1 domain with ACE2 or to the correct folding of the receptor-binding domain.
Fig. 1B compares the ability of each S1-Ig truncation variant to precipitate soluble ACE2 over several experiments (gray bars) with its ability to bind ACE2-expressing 293T cells, as measured by flow cytometry (black bars). A good correlation is observed between these two binding assays. We note that, under conditions used here, flow cytometry more sensitively detects low affinity associations with ACE2, whereas precipitation better reveals differences among efficiently binding variants. The truncation variants assayed in Fig. 1, A and B, are represented in Fig. 1C.
We further examined the ability of the S1-Ig variant containing residues 318–510 to bind ACE2 with higher affinity than does full-length S1-Ig. A 50 nm concentration of S1-Ig was compared with varying concentrations of the 318–510 variant. As shown in Fig. 2A , the same concentration (50 nm) of 318–510 precipitated more than twice as much ACE2 as did S1-Ig. A 25 nm concentration of 318–510 precipitated the same amount of soluble ACE2 as did 50 nm S1-Ig. The results of two such experiments are summarized in Fig. 2B. These data imply that the 318–510 variant binds ACE2 at least twice as efficiently as does S1-Ig.
Fig. 2.
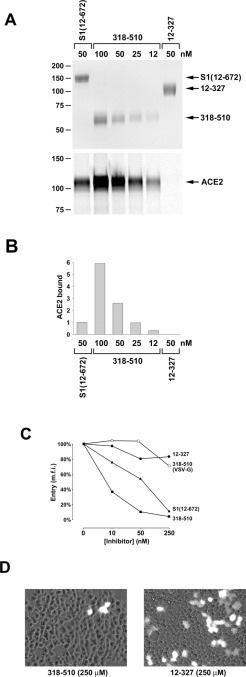
An S1-Ig variant containing residues 318–510 associates with ACE2 and blocks S protein-mediated entry better than does S1-Ig.A, S1-Ig, or variants containing residues 318–510 or 12–327, were purified from transfected 293T cells and quantified. An aliquot of each variant diluted to the indicated concentrations was visualized by SDS-PAGE and Coomassie staining (top panel) and used to precipitate soluble metabolically labeled ACE2. Precipitates were analyzed by SDS-PAGE, and ACE2 was quantified by phosphorimaging. B, ACE2 precipitated by the indicated concentrations of S1-Ig and the indicated variants. An average of two experiments is shown. C, the indicated concentrations of S1-Ig, or of the 318–510 or 12–327 variants, were incubated with 293T cells expressing ACE2, together with an SIV modified to express green fluorescent protein (SIV-GFP) and pseudotyped with S protein of SARS-CoV or with VSV-G. Infection with pseudotyped virus was quantified by measuring green fluorescence by flow cytometry and shown here as mean fluorescent intensity (m.f.i.). D, fluorescent microscopic fields of 293T cells transfected with ACE2 and incubated with SIV-GFP pseudotyped with S protein in the presence of a 250 nm concentration of the 12–327 (right) or the 318–510 (left) variants. Many microscopic fields of cells incubated with the 318–510 variant lacked observable fluorescing cells.
We also investigated the ability of S1-Ig and the 318–510 variant to block S protein-mediated infection. To do so, we utilized a system we recently developed in which a lentivirus expressing green fluorescent protein and pseudotyped with the SARS-CoV S protein infects 293T cells stably expressing ACE2.2 Incubation of 293T cells with the 12–327 variant had no effect on infection, consistent with the inability of this variant to bind ACE2 (Fig. 2C). In this assay, S1-Ig inhibited infection by S protein-pseudotyped lentivirus with an IC50 of ∼50 nm, whereas the 318–510 variant blocked infection by the same virus with an IC50 of less than 10 nm (Fig. 2C). The 318–510 variant did not substantially interfere with infection of lentivirus pseudotyped with the VSV-G protein, which mediates entry independently of ACE2 (Fig. 2C). Fig. 2D displays fluorescent microscopic fields of view in the presence of 250 nm of the 12–327 or 318–510 variants. Many fields lacked observable green cells in the presence of the 318–510 variant.
We asked whether the difference in the abilities of the 318–510 and 327–510 variants to bind ACE2 was a consequence of the loss of cysteine 323 in the latter variant. Fig. 3A demonstrates that this is not the case. A series of point mutations was made in which each of the seven cysteines within 318–510 was altered to alanine. The variant in which cysteine 323 was altered bound ACE2 as efficiently as 318–510 itself. Alteration of cysteine 378 also had little effect on binding; however, a combination of mutations at residues 323 and 378 resulted in a construct with decreased ability to bind ACE2 (Fig. 3A, right panel). Alteration of cysteine 366 or 419 substantially impaired expression of the 318–510 variant. Similar alterations of cysteines 348, 467, and 474 prevented efficient precipitation of ACE2 without a major effect on expression. These data indicate that determinants between 318 and 326 other than cysteine 323 contribute directly or indirectly to ACE2 association.
Fig. 3.
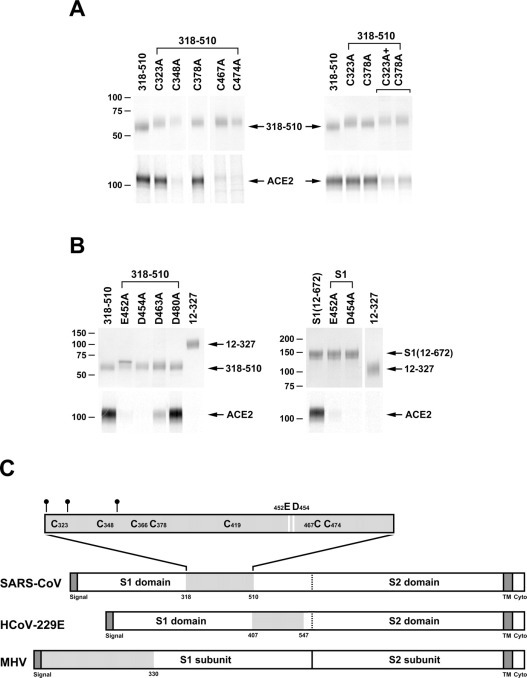
Analysis of point mutations of S1-Ig and the 318–510 variant.A, the 318–510 S1-Ig truncation variant, or variants thereof in which each of seven cysteines was altered individually to alanine, were analyzed as described in the legend to Fig. 1. Variants in which cysteine 366 or 419 was altered to alanine did not express and were not further analyzed. A variant containing alterations of both cysteines 323 and 378 was also analyzed (right panel). B, 318–510 variants (left panel) or S1-Ig variants (right panel) in which glutamic acid 452 or aspartic acids 454, 463, or 480 were altered individually to alanine were analyzed as in Fig. 1. C, representation of the S proteins of SARS-CoV, HCoV-229E, and MHV, aligned by their S2 domains. Dark gray indicates leader and transmembrane sequences. Light gray indicates receptor-binding domain. The receptor-binding domain of SARS-CoV is shown with N-glycosylation sites (small circles) and cysteines indicated. Residues that make a substantial contribution to ACE2 association (glutamic acid 452 and aspartic acid 454) are shown as white bars.
Finally, we explored the ability of some acidic residues between 318 and 510 to contribute to ACE2 association, focusing on a region highly divergent among coronavirus S proteins. Glutamic acid 452 and aspartic acids 454, 463, and 480 were individually altered to alanine in the 318–510 variant (E452A, D454A, D463A, and D480A, respectively). These 318–510 variants were assayed for their ability to bind ACE2 (Fig. 3B, left panel). No effect was observed with the D480A alteration. The D454A and E452A 318–510 variants precipitated ∼1 and 10%, respectively, of the ACE2 precipitated by the wild-type 318–510 variant. The full S1 domain, when mutated at E452, precipitated ACE2 with an efficiency similar to that of the 318–510 variant bearing the same mutation (Fig. 3B, right panel). The D454A alteration completely abolished ACE2 association both in the 318–510 variant (Fig. 3B, left panel) and in the full-length S1 domain (Fig. 3B, right panel), without affecting expression of either protein. These data suggest that ACE2 interacts with the SARS-CoV S domain in the vicinity of aspartic acid 454.
Fig. 3C represents the 318–510 region within the SARS-CoV S protein, aligned with the S proteins of HCoV-229E and MHV. As is apparent from this figure, each of the receptor-binding domains of these S proteins is found in a different region of the S1 domain, consistent with the fact that each of these coronaviruses belongs to a distinct serological and genetic group.
The studies described here localize the SARS CoV S protein receptor-binding domain. A series of truncation variants of the S1 domain, fused to the Fc region of human IgG1, were assayed for their ability to associate with ACE2 on the surface of transfected cells and to immunoprecipitate soluble ACE2. The smallest fragment that retained ACE2 association was composed of residues 318–510 and bound ACE2 more efficiently than did the full-length S1 domain, whereas slightly smaller fragments did not. The higher affinity of the 193-residue fragment raises the possibility that other regions of the S protein partially mask this receptor-binding domain. Alternatively, the receptor-binding domain described here may simply be more soluble or better folded than the S1 protein, which includes regions that may contact the S2 domain or other S proteins in the trimeric complex. The 193-amino acid receptor-binding region also more efficiently blocked S protein-mediated infection of ACE2-expressing cells than did the full S1 domain, presumably due to its greater affinity for ACE2. Further study of this fragment may therefore provide insight into development of therapeutics that block SARS-CoV infection.
We also investigated the role of cysteines and some acidic residues within the 193-residue fragment. We found that most of the seven cysteines contributed to expression or to ACE2 association and were unable to immediately identify non-essential or unpaired cysteines within this variant. Work in this direction continues. We did, however, identify two acidic residues, glutamic acid 452 and aspartic acid 454, that appear to make an important contribution to S1 protein interaction with ACE2. Although conformational changes due to alteration of these residues cannot be excluded, the observations that variants containing these mutations expressed as efficiently as those bearing wild-type sequences, and that these mutations had nearly identical effects on the 318–510 variant and the full-length S1 domain, suggest that one or both of these residues contribute directly to ACE2 association.
At this time, public health measures have successfully controlled transmission of SARS-CoV, but it remains unclear whether SARS will reemerge as a threat to human health. Fortunately, several observations suggest that the development of a vaccine against this virus will be less challenging than, for example, the development of an anti-HIV-1 vaccine. SARS-CoV is transmitted more rapidly than an anti-viral antibody response can develop; this suggests that, in contrast to the HIV-1 envelope glycoprotein, the S protein may do little to cloak its receptor-binding domain. Consistent with this relative exposure of the ACE2-binding region, SARS-CoV-neutralizing antibodies that compete for S protein association with ACE2 have already been identified.3 Also, again in contrast to HIV-1, and due either to the fidelity of the RNA polymerase or to the rate of transmission, surprisingly little variation has been observed in S protein genes obtained from separate patients. Together, these observations suggest that a subunit vaccine that includes the S protein receptor-binding domain described here may be effective in the control of virus transmission.
Footnotes
- SARS
- severe acute respiratory syndrome
- CoV
- coronavirus
- S
- spike
- ACE2
- angiotensin-converting enzyme 2
- CEACAM
- carcinoembryonic antigen-related cell adhesion molecule
- MHV
- mouse hepatitis virus
- HCoV
- human coronavirus
- VSV
- vesicular stomatitis virus
- SIV
- simian immunodeficiency virus
- GFP
- green fluorescent protein
- HIV-1
- human immunodeficiency virus, type 1
- PBS
- phosphate-buffered saline
- BSA
- bovine serum albumin.
M. J. Moore, T. Dorfman, W. Li, S. K. Wong, T. C. Greenough, M. Farzan, and H. Choe, manuscript in preparation.
J. Sui, personal communication.
References
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Kuiken T., Fouchier R.A., Schutten M., Rimmelzwaan G.F., van Amerongen G., van Riel D., Laman J.D., de Jong T., van Doornum G., Lim W., Ling A.E., Chan P.K., Tam J.S., Zambon M.C., Gopal R., Drosten C., van der Werf S., Escriou N., Manuguerra J.C., Stohr K., Peiris J.S., Osterhaus A.D. Lancet. 2003;362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fouchier R.A., Kuiken T., Schutten M., van Amerongen G., van Doornum G.J., van den Hoogen B.G., Peiris M., Lim W., Stohr K., Osterhaus A.D. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 6.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 7.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzeriaga C., Greenough T.C., Choe H., Farzan M. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sturman L.S., Holmes K.V. Adv. Exp. Med. Biol. 1984;173:25–35. doi: 10.1007/978-1-4615-9373-7_3. [DOI] [PubMed] [Google Scholar]
- 9.Jackwood M.W., Hilt D.A., Callison S.A., Lee C.W., Plaza H., Wade E. Avian Dis. 2001;45:366–372. [PubMed] [Google Scholar]
- 10.Gallagher T.M., Buchmeier M.J. Virology. 2001;279:371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonavia A., Zelus B.D., Wentworth D.E., Talbot P.J., Holmes K.V. J. Virol. 2003;77:2530–2538. doi: 10.1128/JVI.77.4.2530-2538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breslin J.J., Mork I., Smith M.K., Vogel L.K., Hemmila E.M., Bonavia A., Talbot P.J., Sjostrom H., Noren O., Holmes K.V. J. Virol. 2003;77:4435–4438. doi: 10.1128/JVI.77.7.4435-4438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubo H., Yamada Y.K., Taguchi F. J. Virol. 1994;68:5403–5410. doi: 10.1128/jvi.68.9.5403-5410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dveksler G.S., Pensiero M.N., Cardellichio C.B., Williams R.K., Jiang G.S., Holmes K.V., Dieffenbach C.W. J. Virol. 1991;65:6881–6891. doi: 10.1128/jvi.65.12.6881-6891.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dveksler G.S., Dieffenbach C.W., Cardellichio C.B., McCuaig K., Pensiero M.N., Jiang G.S., Beauchemin N., Holmes K.V. J. Virol. 1993;67:1–8. doi: 10.1128/jvi.67.1.1-8.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeager C.L., Ashmun R.A., Williams R.K., Cardellichio C.B., Shapiro L.H., Look A.T., Holmes K.V. Nature. 1992;357:420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farzan M., Mirzabekov T., Kolchinsky P., Wyatt R., Cayabyab M., Gerard N.P., Gerard C., Sodroski J., Choe H. Cell. 1999;96:667–676. doi: 10.1016/s0092-8674(00)80577-2. [DOI] [PubMed] [Google Scholar]
- 18.Bannert N., Schenten D., Craig S., Sodroski J. J. Virol. 2000;74:10984–10993. doi: 10.1128/jvi.74.23.10984-10993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]