

Abstract
Long-term disability after stroke is common but the mechanisms of post-stroke recovery is unclear. Cerebral Ras-related C3 botulinum toxin substrate (Rac) 1 contributes to functional recovery after ischemic stroke in mice. As Rac1 plays divergent roles in individual cell types after central neural system injury, we herein examined the specific role of neuronal Rac1 in post-stroke recovery and axonal regeneration. Young male mice were subjected to 60-minutes middle cerebral artery occlusion (MCAO). Inducible deletion of neuronal Rac1 by daily intraperitoneal injection of tamoxifen (2 mg/40 g) into Thy1-creER/Rac1-floxed mice day 7-11 after MCAO worsened cognitive (assayed by novel object recognition test) and sensorimotor (assayed by adhesive removal and pellet reaching tests) recovery day 14-28 accompanied with the reduction of neurofilament-L (NFL) and myelin basic protein (MBP) and the elevation of glial fibrillary acidic protein (GFAP) in the peri-infarct zone assessed by immunostaining. Whereas the brain tissue loss was not altered assayed by cresyl violet staining. In another approach, delayed overexpression of neuronal Rac1 by injection of lentivirus encoding Rac1 with neuronal promotor into both the cortex and striatum (total 4 μl at 1×109 transducing units/mL) of stroke side in C57BL/6J mice day 7 promoted stroke outcome, NFL and MBP regrowth and alleviated GFAP invasion. Furthermore, neuronal Rac1 overexpression led to the activation of p21 activating kinases (PAK) 1, mitogen-activated protein kinase kinase (MEK) 1/2 and extracellular signal-regulated kinase (ERK) 1/2, and the elevation of brain-derived neurotrophic factor (BDNF) day 14 after stroke. Finally, we observed higher counts of neuronal Rac1 in the peri-infarct zone of subacute/old ischemic stroke subjects. This work identified a neuronal Rac1 signaling in improving functional recovery and axonal regeneration after stroke, suggesting a potential therapeutic target in the recovery stage of stroke.
Keywords: functional recovery, ischemic stroke, glia scar formation, neural plasticity, Rac1
Introduction
Stroke is one of the leading causes of death and disability worldwide, of which 87 percent is characterized as ischemic stroke (Liu et al. 2014a; Writing Group et al. 2016; Liu et al. 2018; Zhou et al. 2019). Although the mortality rate of stroke is decreasing, the incidence of stroke is rising, which leads to an increasing prevalence of patients living with persistent disability (Benowitz & Carmichael 2010), and no effective treatment for long-term recovery (Benowitz & Carmichael 2010). It has been well recognized enhancing axonal regeneration and brain remapping may improve recovery and reduce long-term disability after stroke (Lopez-Valdes et al. 2014; Uphaus et al. 2019).
Ras-related C3 botulinum toxin substrate (Rac) 1 is a Rho-related small GTPase. It is ubiquitously expressed in multiple cell types, including neurons, astrocytes, endothelial cells and oligodendrocytes (Etienne-Manneville & Hall 2002), throughout the brain (Stankiewicz & Linseman 2014) and plays a key role in cell differentiation and growth, for example axonal growth, during development (Lundquist 2003). In experimental stroke models, cerebral Rac1 was activated from hours to 2 weeks after ischemia—reperfusion injury (Choi et al. 2015). However, its role in brain remapping and recovery after stroke is far from clear. Our previous work suggested that Rac1 enhanced stroke recovery as pharmacological inhibition of Rac1 worsened behavior recovery in a mouse ischemic stroke model (Liu et al. 2018). It may be partially attributed to the beneficial effect of endothelial Rac1 to post-stroke recovery and angiogenesis (Bu et al. 2019). However, others suggested a detrimental function of Rac1 to the stroke recovery (Choi et al. 2015; Karabiyik et al. 2018; Ishii et al. 2017). For instance, pharmacological inhibition of cerebral Rac1 (Meng et al. 2015) or neuronal Rac1 ablation (Karabiyik et al. 2018) significantly ameliorated brain damage after stroke. The discrepancy seen in different studies may be attributed to the indiscriminate manipulation of Rac1 regardless its cell-specific roles, and the time of interventions considering early vs. late stage after stroke. This further argues for a specific role of neuronal Rac1 and the need to study its function in chronic survival after stroke. The role of neuronal Rac1 has never been reported in stroke model for stroke recovery, brain plasticity and regeneration.
Rac1 interacts with specific downstream substrates that coordinate activation of a multitude of signaling cascades and influence the potential pro-regenerative effects (Bustelo et al. 2007). Among the first described Rac1 effector protein was the family of p21 activating kinases (PAK) (Bustelo et al. 2007). Rac1 binds PAK1 in a GTP-dependent manner, resulting in the transportation of signals through several downstream kinases, including the mitogen-activated protein kinase kinase (MEK) 1/2 and the extracellular signal-regulated kinase (ERK) 1/2 (del Pozo et al. 2000; Frost et al. 1996; Fujita & Yamashita 2014). It is known that PAK1 expression increased in the peri-infarct area of human postmortem tissue and experimental stroke models (Mitsios et al. 2007). In addition, our previous study shows that delayed inhibition of Rac1 prevented MCAO-induced MEK1/2 and ERK1/2 activation in mice, which may be associated with the effect of Rac1 in axonal regrowth during stroke recovery (Liu et al. 2018). But whether the Rac1 signaling in mediating neurite regrowth after stroke is of neuronal original remains unknown.
In this study, we explored the function of neuronal Rac1 in post stroke recovery in mouse model from broad behavior outcomes to its role in axonal regeneration, and investigated its underlying mechanisms involving PAK1 signaling. To complement the data from the animal model, we studied the clinical profiles of Rac1 in postmortem brain tissue of older ischemic stroke subjects.
Method:
Animals and Study Design
All animal protocols were approved by the Center for Laboratory Animal Medicine and Care (CLAMC) at the Medical School of University of Texas Health Science Center in Houston (protocol AWC-18-0123) and were performed in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals. Mice were maintained in 12-h light/dark cycle and fed the food and water ad libitum. All animals were housed 3-5 per cage in specific pathogen-free conditions and were allowed to acclimate to the housing room for at least 1 week before use. All procedures for animals were carried out during daytime (light cycle). Mice used were young males at 7 – 8 week old and identified by ear-punch in the order of 1st as left ear up, 2nd as left ear down, 3rd as right ear up, 4th as right ear down and 5th nothing. For stroke surgery, it was decided that the 1st operated mouse would be assigned to the control group, the 2nd to the treatment group, 3rd to the control group and so on. For all other experiments, simple randomization were performed using Excel. Briefly, we input the animal code in column A, and the function “=RAND” in column B. Then we ranked the randomly generated number (column B) in column C. The number of animals/samples was picked up in the order from top downward from column C for each group respectively. Randomization and treatment were carried out by the same people, but blinded to the examiners and data analysts. Total of 71 mice were used in this study, including 48 C57BL/6J wild-type (WT) mice (Stock No: 000664, purchased in year 2018 and 2019, The Jackson Laboratories), 13 inducible Thy1-creER/Rac1-floxed (T-Rac1-floxed) mice and 10 Rac1-floxed mice as controls. T-Rac1-floxed (Cat# 012708, RRID: IMSR_JAX:012708, The Jackson Laboratories) and Rac1-floxed (Cat# 005550, RRID: IMSR_JAX:005550, The Jackson Laboratories) mice were fed and crossed in CLAMC. 23 mice were eliminated from the study due to death, no neurological deficits 24 hours after reperfusion, or failure to complete the pre-stroke behavior evaluation. A detailed description of the study design and animal numbers per experimental group are presented in Figure 1. The study was not pre-registered and exploratory.
Figure 1.
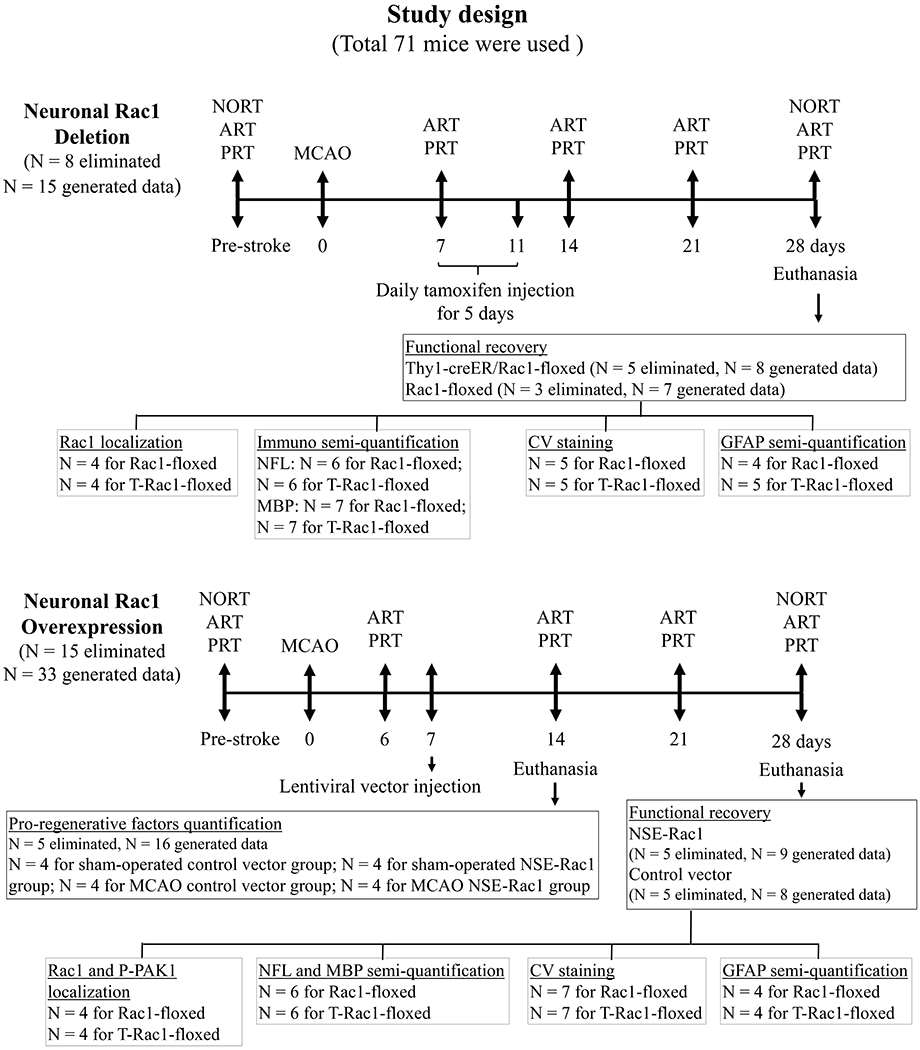
Schematic representation of the study design with number of animals used for each experimental set. NORT, novel object recognition test; ART, adhesive removal test; PRT, pellet reaching test. N = number of animals.
Middle Cerebral Artery Occlusion (MCAO)
Transient MCAO surgery was performed as described previously (Bu et al. 2019). Briefly, mice were anesthetized with isofiurane (5% induction, 1.5% during surgery), and a heating pad (TC-1000 System, CWE) was used during the surgery to monitor and maintain body temperature at 37±0.5°C. Isoflurane was chosen due to its short induction time and allows fast recovery in mice. To minimize animal suffering, 0.25% bupivacaine will be administered at the surgical site before an incision is made to the skin. The common carotid artery, external carotid artery and internal carotid artery were isolated. Then, an intraluminal filament (size 6-0, coating diameter 0.21±0.02 mm, coating length 5-6 mm, #602156PK5Re, Doccol Corporation) from the external carotid artery stump into the internal carotid artery. One hour after occlusion, mice were reperfused by suture withdrawal. The mouse was received softened chow and intraperitoneal injection of saline next 7 days.
Neurological deficit score
Neurological deficit score was performed at reperfusion using a previously described four-point scale (Liu et al. 2016; Liu et al. 2014a). The scores were assigned according to the following criteria: 0, no deficit; 1, forelimb weakness and torso turning to the ipsilateral side when animals were held by the tail; 2, circling to the affected side; 3, inability to bear weight on the affected side; and 4, no spontaneous locomotor activity or barrel rolling. Animals without neurological deficits after reperfusion were excluded.
Rac1 Intervention In Vivo
To minimize potential effects on acute injury size, we deliberately started Rac1 intervention at day 7 after the onset of stroke. This allowed us to focus on studying the role of Rac1 in brain plasticity because stroke infarction completes within a week in this model (Liu et al. 2009). For delayed deletion of neuronal Rac1, intraperitoneal injection of tamoxifen (2 mg/40 g/daily, T5648, Sigma, purchased in 2018) was performed on T-Rac1-floxed mice from day 7 to 11 after stroke as previous described (Nandan et al. 2014). Rac1-floxed mice received tamoxifen and served as control.
Lentiviral vectors carrying GFP-Rac1 with neuronal promotor NSE (Custom Mouse Rac1 Lentivirus (pLenti-GIII-Rat NSE Promoter-GFP-2A-Puro), #LV002-c(41218), Abm) were customized from Applied Biological Materals Inc (Purchased in 2018). The concentrations of the lentivirus were 1×109 transducing units/mL, which was titered prior to experiment. For overexpression of neuronal Rac1, vectors were stereotactically injected into both the cortex and striatum of WT mice at day 7 after MCAO surgery following established methods (Yuan et al. 2016). Briefly, a four-point injection was carried out at the following coordinates: 0.5 mm anterior to the bregma, 2.0 or 3.0 mm lateral to the sagittal suture at the ipsilateral side of MCAO (right), and 1.0 or 2.8 mm from the surface of the skull. A total of 1 μl of lentivirus was injected into each position at a rate of 0.5 μl/minutes with a 30-gauge needle on a 10-μl syringe (1701 RN, Hamilton). Vectors in the absence of Rac1 was used as control. The dose of lentiviral vector in our preparation has no toxic effect to tissue (Figure S1).
Behavior Measurements
Given the majority of clinical survivors suffered sensorimotor and cognitive disability after stroke (Patel et al. 2002; Smania et al. 2008), evaluation of functional outcome in animal models is a key component in improving the clinical relevance of experimental studies. In this study, the novel object recognition test (NORT) was used to evaluate recognition memory (Bevins & Besheer 2006), which was performed at day 7 and 28 after stroke. The adhesive removal (ART) (Bouet et al. 2009) and pellet reaching (PRT) tests (Liu et al. 2014b) were used to evaluate sensorimotor disability. They were performed at day 7, 14, 21 and 28 on inducible gene deletion mice. The same time window was carried on WT mice except day 6 but not day 7 for behavior.
Novel object recognition test
Experiment was carried out as previously described with minor modification (Bevins & Besheer 2006; Antunes & Biala 2012). Briefly, animals were allowed to explore the arena with 2 identical objects for 5 minutes (trial 1). After five-minutes interval in feeding cage, mice were placed into the same arena where one of the objects was replaced with a novel one for another 5-minutes exploration (trial 2). The time spending in seconds on every object was recorded and reflected by a discrimination index (DI) calculating using the equation: DI = (TNtrial2/TFtrial2)/(TFtrial1/TFtrial1). TN = time spending on the novel object. TF = time spending on the familiar objects. Index/ratio was used to minimize the influence of potential changes in locomotion. Pre-tests were performed to exclude animals if they showed lack of exploration activity.
Adhesive removal test
As previously described (Bouet et al. 2009; Shen et al. 2007), one small piece of adhesive-backed paper dots (25 mm2) was used as unilateral tactile stimuli occupying the distal–radial region on the wrist of contralateral forelimb of stroke. The mice were then returned to its home-cage. The time to remove each stimulus was recorded during three trials per day. The animals were pre-trained for 3 days. Pre-tests were performed to exclude animals if they were unable to remove the dots within 10 seconds.
Pellet reaching test
The pellet reaching test measures the ability of skilled forepaw use. As previously described (Liu et al. 2014b; Shen et al. 2007), the mice were placed in a Plexiglas box with a vertical slot on the front wall. Animals were trained for 3 days before the first test to use their left forepaw to extract 14 mg food pellets (#F05684, BioServ) through the slot. If the animal was able to extract the pellet and bring it to the mouth, we scored it as success. If the animal dropped the pellet inside the box before eating or knocked the pellet off the shelf, fail was given. Performance of individuals was defined after 10 attempts by mice. The success rate was calculated according to the following equation: number of successful reaches/total number of reaches × 100. Pre-tests were performed to exclude animals if they showed lack of interest in grasping the pellet food.
Immunohistochemistry
Mice were deeply anesthetized with 5% isoflurane and transcardially perfused by 4% paraformaldehyde 28 days after stroke. 30 μm thick coronal sections were cut and incubated in 0.1% Trition×100 for 15-minutes permeabilization followed by 1-hour blocking in 1% BSA at room temperature (RT). The following primary antibodies were then used: rabbit anti-NeuN (1:500, #12943, CST, RRID: AB_2630395), mouse anti-NeuN (1:250, #MAB377, Millipore, RRID: AB_2298772), mouse anti-Rac1 (1:500, #610650, BD Biosciences, RRID: AB_397977), mouse anti-neurofilament-L (NFL, 1:500, #MA1-2010, Thermo Fisher, RRID: AB_347003), rabbit anti-myelin basic protein (MBP, 1:100, #78896S, CST, RRID: AB_2799920), rabbit anti-phospho T212 PAK1 (1:200, #PA5-104983, Thermo Fisher, RRID: AB_2816456), rabbit anti-glial fibrillary acidic protein (GFAP, 1:500, #12389, CST, RRID: AB_2631098), goat anti-Olig2 (1:200, #AF2418, R&D, RRID: AB_2157554) and rat anti-CD31 (1:25, #550274, B&D, RRID: AB_393571) overnight at 4℃, followed by incubating with the fluorescently-labeled secondary antibodies: anti-rabbit 594 (1:500, #A11072, Thermo Fisher, RRID: AB_2534116), anti-mouse 647 (1:500, #A31571, Life Tech, RRID: AB_162542), anti-goat 594 (1:1000, #150136, Abcam, RRID: AB_2782994) and anti-rat 594 (1:1000, #A21209, Thermo Fisher, RRID: AB_2535795) for 1 hour at RT. Nuclear was stained using DAPI (H-1500, Vector Lab, RRID: AB_2336788).
The mice tissue imaging was recorded using a Zeiss Axiovert 200M microscope (Carl Zeiss, Germany) with an X-Cite 120Q fluorescence illumination system (Lumen Dynamics Group Inc., Canada). Identical digital imaging acquisition parameters were acquired from 3 random fields in the penumbra of the ischemic hemisphere at 40× objective magnification and analyzed using ImageJ (V1.52o). NFL, MBP, GFAP and CD31 were semi-quantified as the integrated optical density (IOD), in which results were normalized by dividing each value by the mean IOD of the control groups. NeuN, Rac1, PAK1 and Olig2 positive cells were quantified as the cell number per mm2.
Tissue loss were measured by as previously described (Liu et al. 2018). Generally, total 8 coronal 30 μm-thick brain sections was picked throughout the cerebrum for CV staining. After staining, SigmaScan Pro 5 (Version 5.0.0) was applied to measure the tissue loss determined following this calculation: % brain tissue lost = 100% × [(contralateral hemisphere area − contralateral ventricular area) − (ipsilateral hemisphere area − ipsilateral ventricular area)] / (contralateral hemisphere area − contralateral ventricular area). Freehand drawing was performed along the edge of brain tissue of slices. The actual density of CV staining was not needed in this case. At the 28 day time point, there was no more measurable infarcted tissue in the brain in this model. The appropriate assessment of brain injury is tissue loss.
All human brain samples were obtained from the University of Pittsburgh neurodegenerative brain bank with appropriate ethics committee approval. All slides were analyzed by an investigator blinded to the database search case and demographics. 20 male cases from postmortem brain were used including 10 older subjects with subacute to old ischemic stroke, and 10 age-matched subjects, who died because of no stroke pathology as “controls”. Samples were fixed in formalin and embedded in paraffin. The staining process was performed as previously described (Ritzel et al. 2018). Briefly, after antigen retrieval, the sections were treated with 3% H2O2 for 10 minutes at RT to quench endogenous peroxidase, and incubated with 3% BSA for 30 minutes at RT. The sections were then exposed to primary antibodies mice anti-Rac1 (1:500, #610650, BD Biosciences, RRID: AB_397977) and rabbit anti-NeuN (1:50, #12943) overnight at 4°C, followed by secondary antibodies anti-mouse labeled with horseradish peroxidase and anti-rabbit labeled with alkaline phosphatase according to the instructions (ADI-950-100-0001, Enzo). All slides were counterstained with hematoxylin. We considered the tissue area within 1.5 mm around the infarct core as the peri-infarct area in mice (Agulla et al. 2013) as well as in human tissue (below). The regions in contralateral hemispheres were anatomically selected as control.
The human tissue images were captured using a Leica DM2000 LED microscope with a Leica DFC310 FX digital camera system (Leica, Germany). Infarcted area was delimitated by an experienced neuropathologist and the contiguous tissue around infarct core was considered as peri-infarct area. The number of NeuN and Rac1 positive cells per mm2 was averaged from three random fields in peri-infarct area at 20× objective magnification.
Western Blot Analysis
All animals in this study were decapitated under 5% isoflurane anesthesia. Brain tissues from the ischemic hemisphere were used for Western blot analysis. After measuring protein concentration, proteins (30 μg) were loaded onto a 10% resolving gel for electrophoresis. Proteins were then transblotted to polyvinylidene fluoride membrane and blocked with 5% skim milk for 60 minutes at RT. Next, the membranes were exposed to primary antibodies including anti-phospho T212 PAK1 (1:1000, #PA5-104983), anti-phospho S217/221 MEK1/2 (1:1000, #9154, CST, RRID: AB_2138017), anti-phospho T202/T204 ERK1/2 (1:1000, #4370, CST, RRID: AB_2315112) or anti-brain-derived neurotrophic factor (BDNF, 1:1000, #ab108319, Abcam, RRID: AB_10862052) overnight at 4°C. Once excess primary antibody was washed off by TBST, membranes were incubated with horseradish peroxidase-linked secondary antibody (1:1000, #7074, CST, RRID:AB_2099233) for 1 hour at RT. β-actin was employed as a loading control (1:1000, #4970, CST, RRID:AB_2223172). Each data was divided by β-actin for comparison.
Statistical Analyses
The results are presented as mean ± SEM. Normal distribution (tested by Kolmogorov-Smirnov Test) and statistical analysis were done using GraphPad Prism 7.0 software. No sample size calculations were performed in this study. Instead, the sample size was based on our previous similar experiments using power analysis (Liu et al. 2018). A Mann-Whitney test was used for comparisons between two individual groups in animal experiments. Unpaired t-test comparisons was used for comparison between two individual groups in human sample assay. Two-way ANOVA with subsequent Bonferonni test was applied for comparisons across groups. Linear correlation analyses were performed using Pearson correlation analyses. Values graphed as box and whiskers, in which the box extended from the 25th to 75th percentiles, whiskers ranged from minimal to maximal and the line in the box was plotted at the median. Significant differences were set when p < 0.05.
Result
Delayed deletion of neuronal Rac1 led to poorer functional recovery
Tamoxifen-induced neuronal Rac1 deletion was validated by immunohistochemistry 21 days after induction, the number of Rac1 in neurons (NeuN) but not in endothelial cells (CD31) (Figure S2. A) was significantly decreased in T-Rac1-floxed mice, compared with control (Rac1-floxed) group (132.5 ± 9.06/mm2 vs 78.56 ± 8.85/mm2, *p < 0.05, Figure 2A). Similarly with previous study (Blasi et al. 2014), stroke led to cognitive impairment assessed by NORT in the control group (2.08 ± 0.2 pre-stroke vs 1.16 ± 0.11 day 7 after stroke , ***p < 0.001, Figure 2B). Delayed deletion of Rac1 in T-Rac1-floxed mice worsened behavioral deficits 28 days after stroke compared with the control group (1.51 ± 0.09 vs 1.08 ± 0.05, *p < 0.05, Figure 2B). Neuronal Rac1 elimination also prevented sensorimotor recovery after stroke evidenced by longer time spent in ART (48.33 ± 4.74 seconds vs 80.17 ± 8.69 seconds starting at day 14, *p < 0.05, Figure 2C) and lower success rate in PRT (47.14 ± 2.86% vs 30 ± 2.67% starting at day 21, *p < 0.05, Figure 2D).
Figure 2.
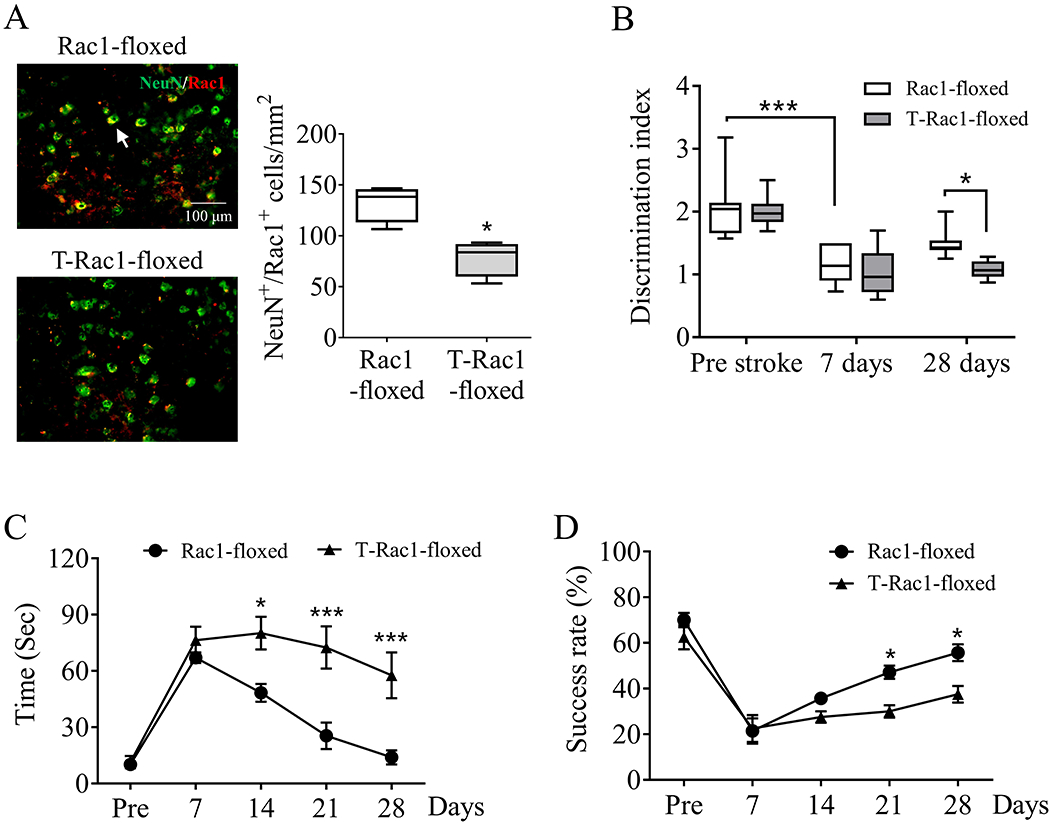
Delayed deletion of neuronal Rac1 led to poorer functional recovery after ischemic stroke in mice. Tamoxifen (2 mg/40 g/daily) was intraperitoneally injected to Thy1-creER/Rac1-floxed (T-Rac1-floxed) mice from day 7 to day 11 after stroke. Same process was performed on Rac1-floxed mice as control. (A) Immunohistochemistry assessment of cerebral Rac1 expression (red, Alexa Fluor 647) on neurons (NeuN, green, Alexa Fluor 594) 28 days after stroke (N = 4/each group). Arrows indicated the Rac1 positive neurons (yellow). Functional recovery assessed by (B) novel object recognition test, (C) adhesive removal test and (D) pellet reaching test before (Pre) up to 28 days after stroke (N = 7 in the Rac1-floxed group, N = 8 in the T-Rac1-floxed group). N = number of animals. *p < 0.05, ***p < 0.001. Mann-Whitney test was used for immune-staining. Two-way ANOVA with subsequent Bonferonni test was applied for behavioral tests.
Delayed overexpression of neuronal Rac1 promoted functional recovery
To evaluate whether overexpression of neuronal Rac1 could promote post-stroke recovery, lentiviral vectors encoding Rac1 with neuronal promotor NSE was injected into mice brain 7 days after MCAO. The approach was confirmed by that the number of Rac1 in neurons (NeuN), but not in endothelial cells (CD31) (Figure S2. B) was significantly increased 21 days after injection (116.6 ± 18.35/mm2 vs 213.3 ± 17.21/mm2, *p < 0.05, Figure 3A) compared with the control vector group. Stroke produced a cognitive impairment in the control group (2.1 ± 0.19 pre-stroke vs 0.95 ± 0.19 day 6 after stroke, ***p < 0.001, Figure 3B), which is consistent with our previous study (Figure 2B). As expected, delayed overexpression of neuronal Rac1 promoted the cognitive recovery (1.33 ± 0.26 vs 2.09 ± 0.26 at day 28, **p < 0.01, Figure 3B). In addition, NSE-Rac1 also produced an improved performance in ART (57.27 ± 13.18 seconds vs 25.22 ± 6.37 seconds starting at day 14, **p < 0.01, Figure 3C) and PRT (45 ± 4.23% vs 61.11 ± 3.09% starting at day 21, *p < 0.05, Figure 3D) after stroke compared with the control groups.
Figure 3.
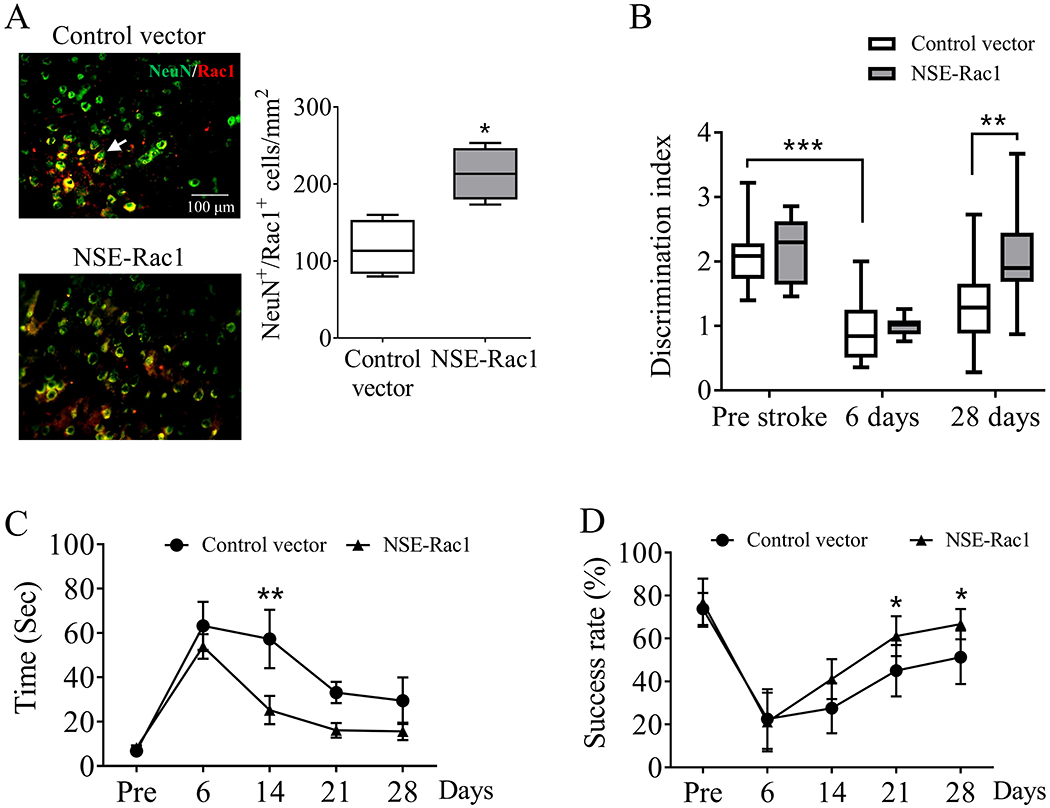
Delayed overexpression of neuronal Rac1 promoted functional recovery after ischemic stroke in mice. Highly concentrated lentivirus encoding Rac1 with neuronal promotor NSE was injected into both the cortex and striatum of WT mice at day 7 after stroke. Same process was performed using lentivirus without Rac1 sequence as controls. (A) Immunohistochemistry assessment of cerebral Rac1 expression (red, Alexa Fluor 647) on neurons (NeuN, green, Alexa Fluor 594) 28 days after stroke (N = 4/each group). Arrows indicated the Rac1 positive neurons (yellow). Functional recovery assessed by (B) novel object recognition test, (C) adhesive removal test and (D) pellet reaching test before (Pre) up to 28 days after stroke (N = 8 in the control vector group, N = 9 in the NSE-Rac1 group). N = number of animals. *p < 0.05, **p < 0.01, ***p < 0.001. Mann-Whitney test was used for immune-staining. Two-way ANOVA with subsequent Bonferonni test was applied for behavioral tests.
Delayed deletion of neuronal Rac1 reduced axonal regeneration and myelination without altering tissue loss
NFL levels have been considered as a predictive marker for neurite density and long-term outcome after ischemic stroke both in animal models and patients (Uphaus et al. 2019). Consistently with previous study (Mages et al. 2018), ischemic stroke led to a reduction in NFL staining in the peri-infarct zone 28 days after stroke compared with the contralateral hemisphere (159 ± 7.02% vs 100 ± 5.01%, ***p < 0.001, Figure 4A) in the control group (Rac1-floxed). Delayed deletion of neuronal Rac1 (T-Rac1-floxed) further reduced NFL density compared with control (100 ± 5.01% vs 44.16 ± 5.68%, ***p < 0.001, Figure 4A). There was no significant change in NFL density in the contralateral hemisphere between groups.
Figure 4.
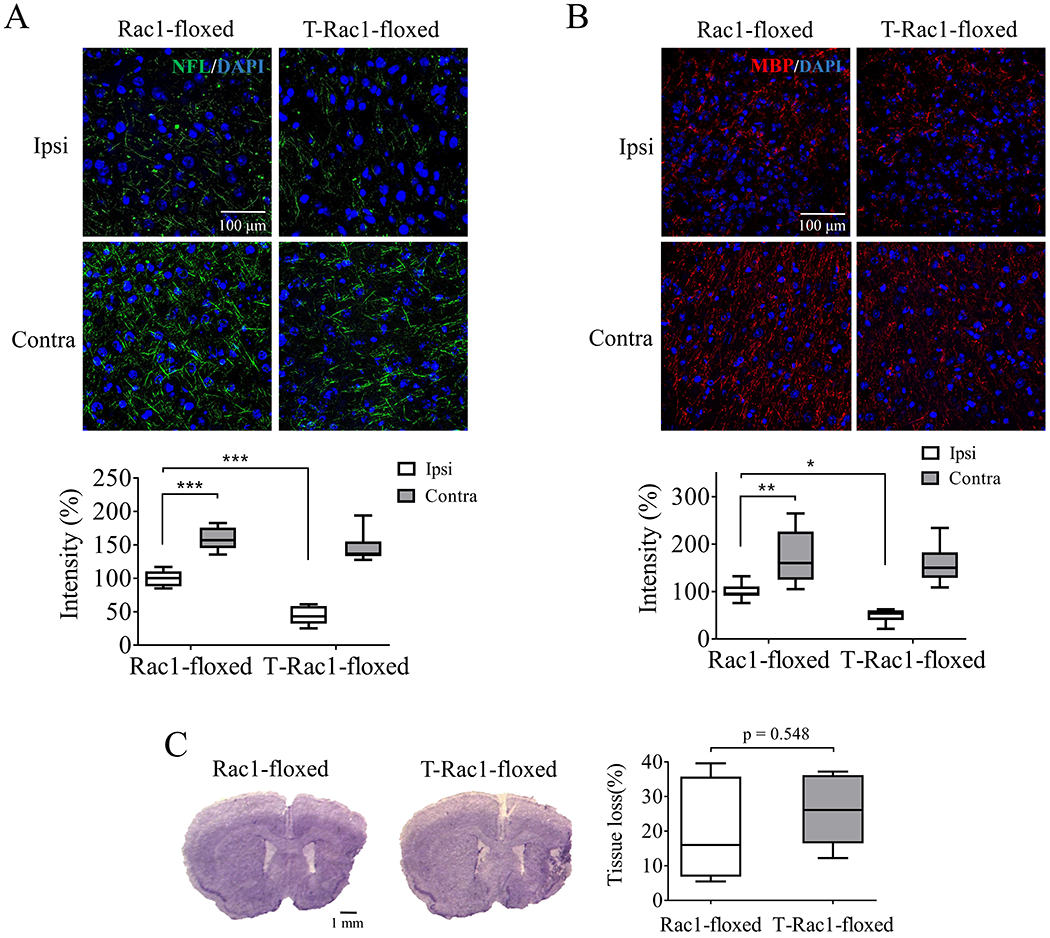
Tamoxifen-induced deletion of neuronal Rac1 reduced axonal regeneration with no effect on tissue loss after ischemic stroke in mice. Density assessment of (A) neurofilament-L (NFL, green, Alexa Fluor 647, N = 6/each group) and (B) myelin basic protein (MBP, red, Alexa Fluor 594, N = 7/each group) in the ipsilateral (ipsi) and contralateral (contra) hemisphere 28 days after unilateral stroke. Blue indicated cell nucleus staining using DAPI. (C) Quantification of brain tissue loss 28 days after stroke was assayed by CV staining (N = 5/each group). N = number of animals. *p < 0.05, **p < 0.01, ***p < 0.001. Two-way ANOVA with subsequent Bonferonni test was applied for immune-staining. Mann-Whitney test was used for CV staining.
MBP is a major component of myelin sheath that wraps nerve fibers and is associated with the functional sufficiency of signal transduction (Bunge 1968). We proposed that the reduction of neurite after neuronal Rac1 deletion is accompanied with the disassembly of MBP. Indeed, we found that the intensity of MBP in the peri-infarct zone decreased compared with the contralateral hemisphere (171.8 ± 21.73% vs 100 ± 6.72%, **p < 0.01, Figure 4B) in the control group. Delayed deletion of neuronal Rac1 further decreased MBP staining compared with control (100 ± 6.72% vs 48.95 ± 5.39%, *p < 0.05, Figure 4B), whereas it did not alter MBP in the contralateral hemisphere. In addition, we examined oligodendrocytes population, as Rac1 plays a key role in the differentiation of oligodendrocytes (Liang et al. 2004). We found that Olig2, an oligodendrocyte marker initiating cell differentiation, was not altered after delayed deletion of neuronal Rac1 (Figure S3. A), suggesting that the changes of MBP was due to neuronal manipulations rather than changes of in oligodendrocytes.
We further performed CV staining to evaluate tissue loss. We found that the brain tissue loss between groups were similar (20.25 ± 6.67% vs 26.3 ± 4.62%, p = 0.548, Figure 4C), suggesting that the decrease of neurite regeneration after neuronal Rac1 deletion was not due to tissue loss.
Delayed overexpression of neuronal Rac1 promoted axonal regeneration and myelination without altering tissue loss
The contribution of neuronal Rac1 to the axonal regeneration was confirmed using lentiviral vector encoding Rac1 with NSE promotor. Similar as previous study (Figure 4A), MCAO led to a reduction of NFL (198.3 ± 16.8% vs 100 ± 8.36%, ***p < 0.001, Figure 5A) and MBP (244.6 ± 27.5% vs 100 ± 12.21%, ***p < 0.001, Figure 5B) staining compared with their contralateral hemisphere in the control group. Lentiviral NSE-Rac1 improved NFL (100 ± 8.36% vs 195.1 ± 20.15%, **p < 0.01, Figure 5A) and MBP (100 ± 12.21% vs 184 ± 14.36%, *p < 0.05, Figure 5B) staining in compared with the control groups. Whereas the intensity of contralateral NFL or MBP was not altered between groups after treatment. Olig2 was not altered after over-expression of neuronal Rac1 (Figure S3. B). No differences in cavity sizes were seen after treatment assayed by tissue loss (26.55 ± 4.24% vs 30.81 ± 2.67%, p = 0.383, Figure 5C).
Figure 5.
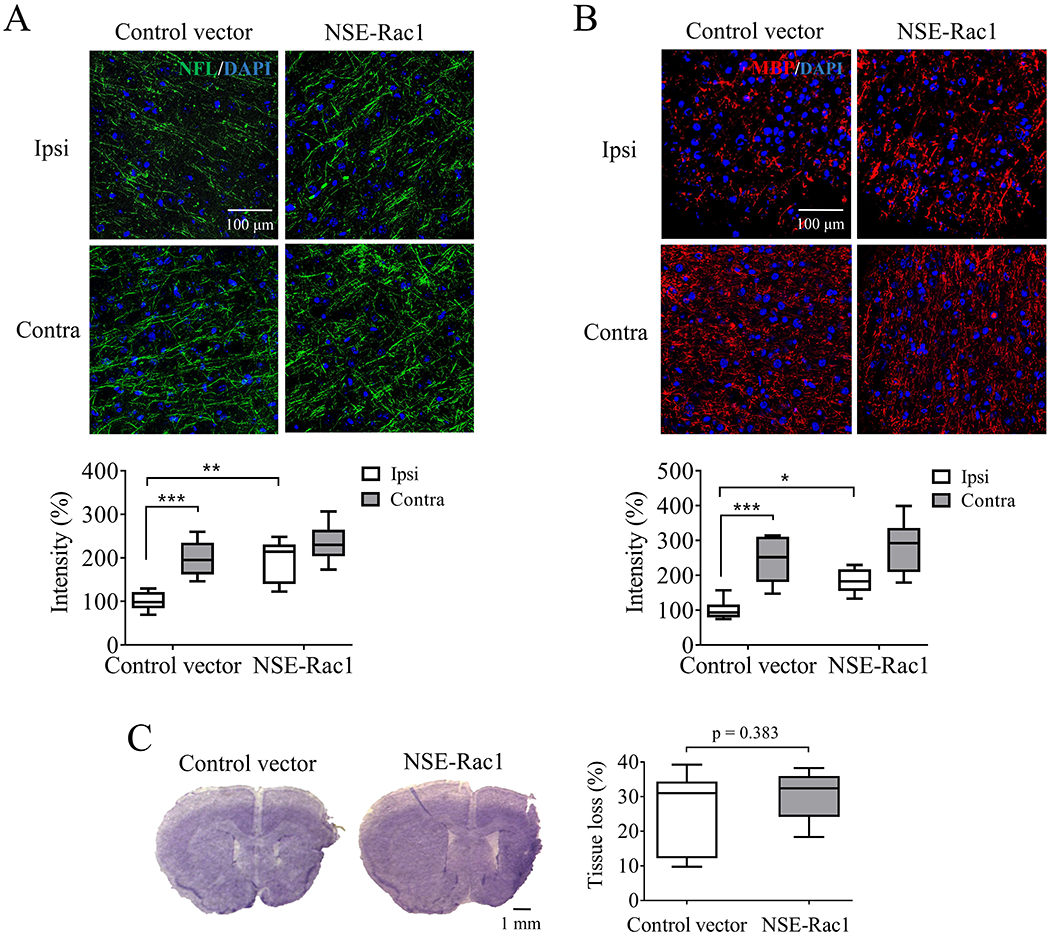
Delayed overexpression of neuronal Rac1 increased axonal density with no effect on tissue loss 28 days after ischemic stroke in mice. Density assessment of (A) neurofilament-L (NFL, green, Alexa Fluor 647, N = 6/each group) and (B) myelin basic protein (MBP, red, Alexa Fluor 594, N = 6/each group) in the ipsilateral (ipsi) and contralateral (contra) hemisphere 28 days after unilateral stroke. Blue indicated cell nucleus staining using DAPI. (C) Quantification of brain tissue loss 28 days after stroke was assayed by CV staining (N = 7/each group). N = number of animals. *p < 0.05, **p < 0.01, ***p < 0.001. Two-way ANOVA with subsequent Bonferonni test was applied for immune-staining. Mann-Whitney test was used for CV staining.
Neuronal Rac1 activated pro-regenerative pathways
We then moved on to study the mechanisms of neuronal Rac1 in stimulating axonal regeneration after stroke. PAK1 has been considered as one of the major downstream regulators of Rac1 (Kumar et al. 2006; Shutes et al. 2007), which is known to further activate MEK1/2 and ERK1/2 signaling, promote BDNF production enhancing the cell proliferation (Liu et al. 2006; Kichina et al. 2010; Bu et al. 2019). We first examined the activation of PAK1 on neurons after delayed overexpression of neuronal Rac1. We found that the phospo-PAK1 (T212) cell population on NeuN positive cells significantly increased 21 days after NSE-Rac1 vector injection compared with the control vector group (129.7 ± 14.75% vs 232.8 ± 16.74%, *p < 0.05, Figure 6A). We then assessed these pro-regenerative signaling as early as day 14 after stroke when delayed overexpression of neuronal Rac1 took effect to the functional recovery. We found that MCAO stroke significantly increased the phosphorylation of PAK1 (T212) (**p < 0.01), MEK1/2 (S217/221) (*p < 0.05), ERK1/2 (T202/T204) (***p < 0.001) and the total level of BDNF (*p < 0.05) (sham-operated with control vector group vs MCAO with control vector group, Figure 6B). Importantly, neuronal Rac1 expression enhanced the phosphorylation of PAK1 (1.52 ± 0.14 vs 1.99 ± 0.17, *p < 0.05), p-MEK1/2 (0.85 ± 0.05 vs 1.1 ± 0.04, **p < 0.01), p-ERK1/2 (1.11 ± 0.06 vs 1.43 ± 0.06, **p < 0.01) and the total level of BDNF (1.48 ± 0.07 vs 1.77 ± 0.06, *p < 0.05, Figure 6B) compared with vector control. However, the total level of PAK1, MEK1/2 and ERK1/2 were not altered after treatment.
Figure 6.
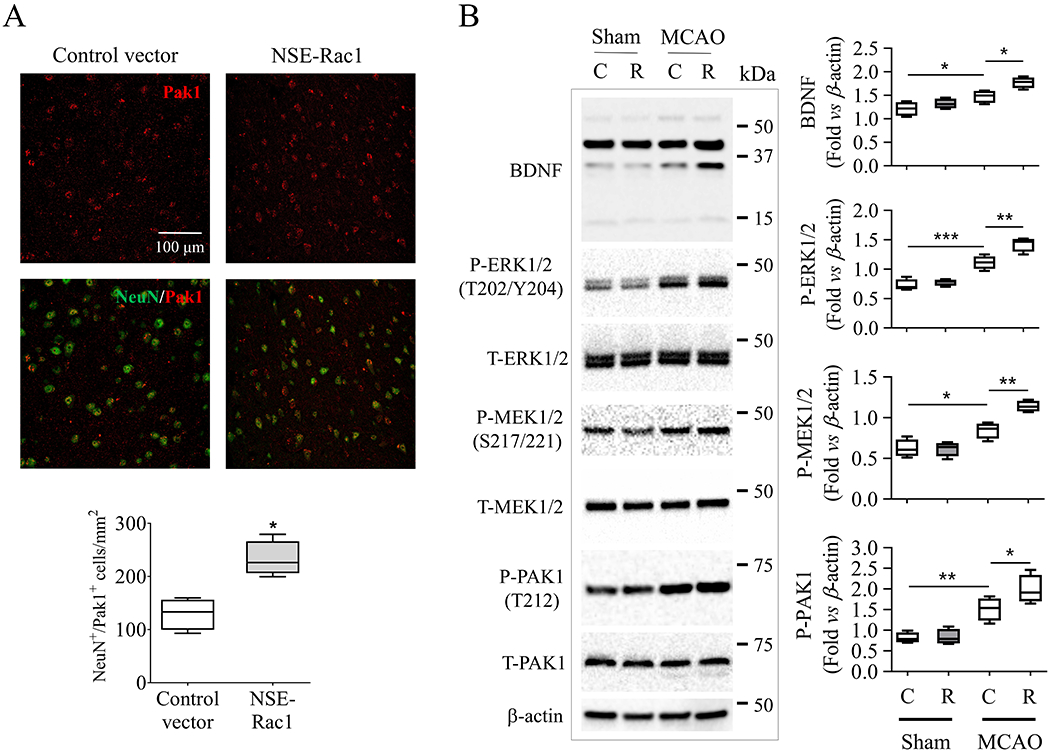
Modulation of pro-regenerative pathways by neuronal Rac1. (A) Delayed overexpression of neuronal Rac1 promoted the expression of phosphorylated (T212) PAK1 (red, Alexa Fluor 594) on neurons (NeuN, Alexa Fluor 647) at peri-infarct area 28 days after MCAO. (B) Delayed overexpression of neuronal Rac1 by cerebral injection of NSE-Rac1 vector (R) promoted phosphorylation of PAK1 (T212), MEK1/2 (S217/221), ERK1/2 (T202/Y204) and promoted BDNF production in the ipsilateral hemisphere 14 days after MCAO compared with control vector (C) group assayed by western blotting. Quantitative analysis of relative intensity of interest proteins by normalizing individuals to β-actin. N (number of animals) = 4/each group. The intensity of all bands was calculated for ERK1/2 and BDNF in individual sample. *p < 0.05, **p < 0.01, ***p < 0.001. Mann-Whitney test was used for immune-staining. Two-way ANOVA with subsequent Bonferonni test was applied for western blotting.
Delayed intervention of neuronal Rac1 affects the expression of GFAP after stroke
Our previous study showed that systematic inhibition of Rac1 counteracted the glial scar formation, one of the leading reason for growth cone collapse after stroke (Yiu & He 2006; Liu et al. 2018). In this study, we further evaluate the cell-specific role of neuronal Rac1 in gliosis. As expected, we found that GFAP, a marker of glial scar formation and a signal of axonal growth inhibition, had elevated expression in the peri-infarct zone after delayed deletion of neuronal Rac1 compared with control (100 ± 7.09% vs 130.7 ± 5.99%, *p < 0.05, Figure 7A). In turn, delayed overexpression of neuronal Rac1 by NSE-Rac1 vector significantly counteracted the GFAP density assessed 28 days after ischemia (100 ± 1.99% vs 48.46 ± 5.23%, *p < 0.05; Figure 7B). The GFAP intensity was negatively correlated with the NFL intensity in both neuronal Rac1 deletion (R = 0.642, p = 0.005, Figure 7A) and overexpression (R = 0.697, p = 0.005, Figure 7B) cohorts.
Figure 7.
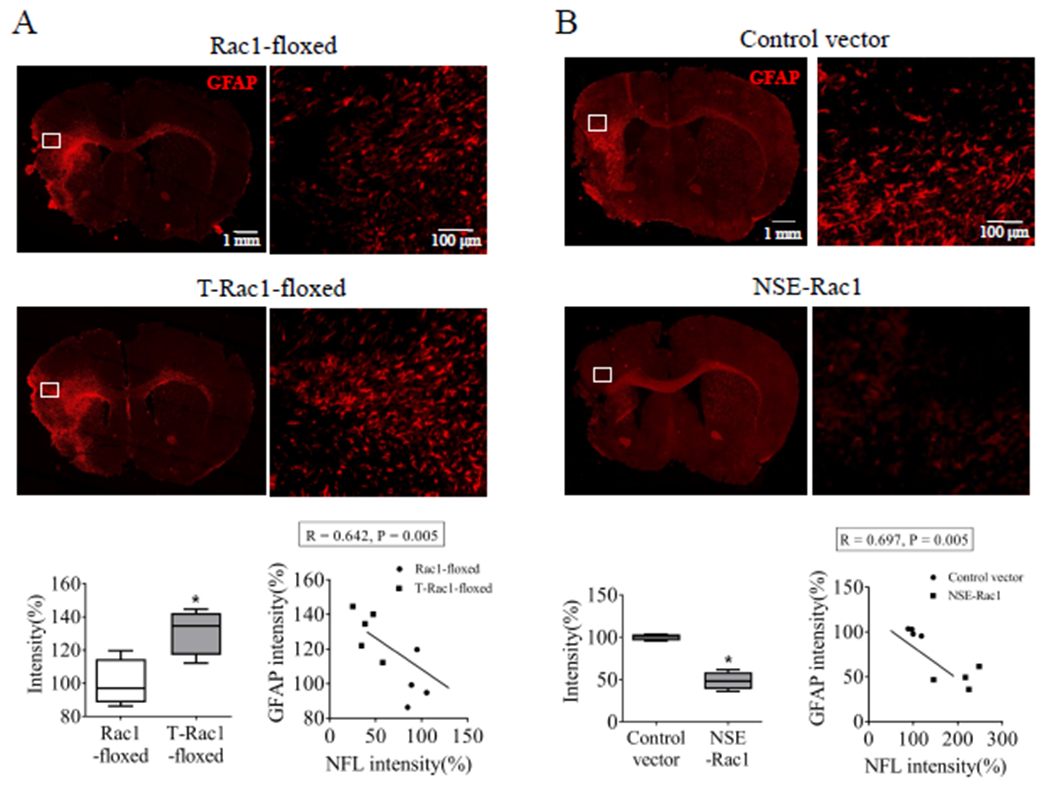
Delayed deletion of neuronal Rac1 enhanced the glial fibrillary acidic protein (GFAP) intensity, whereas delayed overexpression of neuronal Rac1 reduced its intensity 28 days after stroke in mice. Assessment of GFAP density (red, Alexa Fluor 594) in the peri-infarct zone (A) after neuronal Rac1 gene deletion (control mice: Rac1-floxed, N = 4; Rac1 deletion mice: T-Rac1-floxed, N = 5) and (B) after neuronal Rac1 overexpression (control vector: N = 4; Rac1 overexpression: NSE-Rac1, N = 4). N = number of animals. *p < 0.05. Mann-Whitney test was used for immune-staining. Pearson correlation was used for linear correlation analyses.
Rac1 increased in the postmortem brain tissue of older ischemic stroke subjects
Immunohistochemistry was performed to assess the expression of Rac1 in human samples. Postmortem brain tissue was acquired from older male, age-matched controls and subacute/old ischemic stroke individuals. There was no significant difference in the mean age between control (75.8 ± 3.34 years) and stroke (81.8 ± 2.08 years) groups (Table S1). We found stroke induced elevation of Rac1 levels in the peri-infarct zone of stroke group compared to the control group (92.88 ± 12.82/mm2 vs 249.4 ± 41.91/mm2, **p < 0.01, Figure 8A). In addition, the protein level of Rac1 in neurons (NeuN+) was significantly higher than that in the control group (61.42 ± 4.61/mm2 vs 138.7 ± 11.4/mm2, ***p < 0.001, Figure 8B). This finding supports our mice experiments, suggesting a critical role of neuronal Rac1 in post-stroke recovery.
Figure 8.
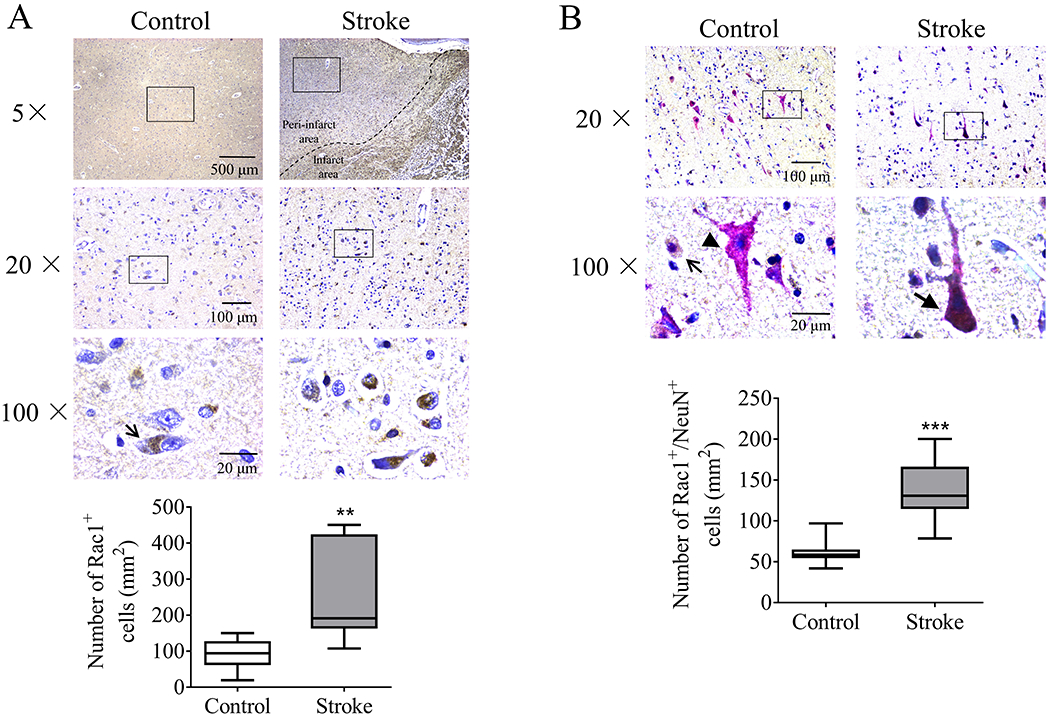
Protein level of Rac1 increased in the peri-infarct area of postmortem tissue of older ischemic stroke subjects. (Upper graphs in figure A and B) Representative immunohistochemical microphotographs showing Rac1 (brown) and NeuN positive cells (magenta) from age-matched control and subacute/old ischemic stroke subjects. Open arrows indicated representative Rac1+ cells. Arrow without bar indicated representative NeuN+ cells. Arrow with bar indicated representative Rac1+/NeuN+ cells. Hematoxylin (blue) was used for nucleus staining. (Lower charts in figure A and B) Quantification of Rac1+ and Rac1+/NeuN+ cells per mm2. N (number of humans) = 10/each group. **p < 0.01, ***p < 0.001. Unpaired t-test comparisons was used for comparison between two individual groups.
Discussion
In this study, we used multiple approaches to demonstrate the critical role of neuronal Rac1 in stroke recovery in animal model and human samples. The key findings are (i) neuronal Rac1 contributes to axonal regeneration and functional recovery after stroke (ii) overexpression of neuronal Rac1 activated pro-regenerative molecules such as p-PAK1, p-MEK1/2, p-ERK1/2, increased the levels of BDNF and counteracted the growth inhibitory signaling from glial scar and (iii) increased expression of neuronal Rac1 was seen in the peri-infarct area of postmortem tissue of ischemic stroke subjects.
Our work demonstrates that neuronal Rac1 promotes axonal outgrowth after stroke, which is supported by the findings that delayed deletion of neuronal Rac1 prevents axonal regeneration, whereas delayed overexpression of neuronal Rac1 promotes axonal regeneration. Further mechanism study shows that overexpression of neuronal Rac1 enhances the colocalization of neuron and phosphorylated PAK1, one of the critical downstream targets of Rac1 (Bustelo et al. 2007), which is known to be responsible for the axonal regeneration through phosphorylating MEK and ERK in the injured central nervous system (Koth et al. 2014). Indeed, our data further showed increased pMEK 1/2 and pERK 1/2 levels in the ischemic brain after Rac1 overexpression.
In addition to the weak intrinsic growth capacity in neurons, the inhibitory factors from extrinsic glial environments is another major contributing factor to regenerative failure in the CNS after injury (Di Giovanni 2009; Fujita & Yamashita 2014; Liu et al. 2018), in which reactive astrocytes are the primary contributors to the formation of glial scar (Sofroniew 2009). In this study, either neuronal specific overexpression of Rac1 by lentivirus or inducible deletion in T-Rac1-floxed mice were performed to explore its effect to astrocytic activity. We found the GFAP intensity, which indicates the activity of astrocytes, is negatively correlated with the levels of neuronal Rac1. The potential mechanisms could be through modulating BDNF levels. It is known that Rac1 can enhance release of BDNF (Kato et al. 2018). Exogenous delivery of BDNF into the stroke cavity shows a small gain of function from after stroke (Clarkson et al. 2015), and reduce GFAP positive reactive astrocytes in the late phase after spinal cord injury (Jain et al. 2006). Indeed, our study shows that overexpression of neuronal Rac1 elevates the protein level of BDNF in the ipsilateral hemisphere (Figure 6D), which raises a possibility that neuronal Rac1 negatively regulates astrocytic activity via releasing BDNF during stroke recovery. Interestingly, when we activated Rac1 selectively in astrocytes using lentivirus, no improvement in functional outcome was seen (Figure S4).This suggested the Rac1-PAK1-BDNF pathway is cell-specific during recovery in ischemic brain, highlighting the importance of targeting Rac1 in cell specific manner to enhance brain plasticity.
It is worth noting that we show increased expression of neuronal Rac1 in stroke brains in elderly, the subpopulation at greatest risk of stroke (Hollander et al. 2003). It is likely that neuronal Rac1 signaling is responding to the call of regeneration after stroke in aged. This is especially important as reduced expression of regeneration associated molecules and increased astrogliosis occur in aged, both of which are associated with worse recovery in animal models (Manwani et al. 2011). Enhancing Rac1 signaling in aged subjects may promote regeneration and recovery post-stroke as Rac1 targets these two major components of regeneration. Nevertheless, we are well aware of that the increase of neuronal Rac1 in human sample was not linked to the post-stroke outcomes due to the lack of human patient outcome data and it was therefore merely observational. Investigating the role of Rac1 in post-stroke regeneration in aged animal models is further required and in fact is an on-going project in our lab as the elderly population is the greatest risk of stroke. Another limitation of this study is that we only used males in our experiments, however, stroke is a sexually dimorphic disease. Future studies will also need to investigate the role of this molecule in females for recovery after stroke.
It is known that vascular smooth muscle specific deletion of Rac1 causes hypertension. Endothelial Rac1 also lowers blood pressure by enhancing the producing eNOS (Andre et al. 2014). Inhibition of Rac1 at brainstem NTS (nucleus tractus solitarii) decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats (Nozoe et al. 2007). In the present study, our over-expression approach is neuronal specific and focused on cerebrum Rac1 by local injection of vector into cortex and striatum. However, system treatment of tamoxifen in Rac1-floxed mice, reducing Rac1, may indeed affect NTS Rac1 in the deletion model and might have an effect at reducing blood pressure via changes in ROS (reactive oxygen species) production (Nozoe et al. 2007). Additionally, the potential effect of blood pressure change on axonal regeneration remains unclear. Our deletion model, however, produced consistent results in stroke outcome and axonal density with the over-expression experiment, which supposedly had no effect on central or peripheral regulation of blood pressure. Any potential impact of the deletion treatment on blood pressure may be investigated in future experiment.
In summary, this study utilized multiple approaches to improve our understanding of cell-specific Rac1 in mediating functional recovery after stroke. Our experiments in mice shows that neuronal Rac1 affords robust improved outcome post stroke through several signaling pathways including enhancing axonal plasticity and reducing the astrocytic barrier. These findings are also partially mirrored by our human data. Targeting neuronal Rac1 may offer a potential therapeutic target for promoting brain remapping and functional recovery after stroke.
Supplementary Material
Acknowledgements
F B performed the majority of the experiments, analyzed data and drafted manuscript; Y M and J W F performed experiments; J W M, L Q, A P and Z R S provided technical support; J K K provided human brain samples; A U and L D M edited manuscript and helped with data analysis; J L conceived the research project, helped design the experiments and finalized the paper.
Sources of Funding:
This work was supported by National Institutes of Health grant R01 NS099628 (J.Li) and by American Heart Association 20POST35180172 (F. Bu). NIH funding RF1 AG069466-01A1 (J.Li & L.D. McCullough).
Abbreviations used:
- ART
adhesive removal test
- CLAMC
center for laboratory animal medicine and care
- CV
cresyl violet
- ERK
extracellular signal-regulated kinase
- GFAP
glial fibrillary acidic protein
- MBP
myelin basic protein
- MCAO
middle cerebral artery occlusion
- MEK
mitogen-activated protein kinase kinase
- NFL
neurofilament-L
- NORT
novel object recognition test
- PAK
p21 activating kinases
- PRT
pellet reaching test
- Rac
Ras-related C3 botulinum toxin substrate
- RRID
research resource identifier
- RT
room temperature
- WT
wild-type
Footnotes
Disclosures
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- Agulla J, Brea D, Campos F, Sobrino T, Argibay B, Al-Soufi W, Blanco M, Castillo J and Ramos-Cabrer P (2013) In vivo theranostics at the peri-infarct region in cerebral ischemia. Theranostics 4, 90–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre G, Sandoval JE, Retailleau K, Loufrani L, Toumaniantz G, Offermanns S, Rolli-Derkinderen M, Loirand G and Sauzeau V (2014) Smooth muscle specific Rac1 deficiency induces hypertension by preventing p116RIP3-dependent RhoA inhibition. J Am Heart Assoc 3, e000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes M and Biala G (2012) The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process 13, 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI and Carmichael ST (2010) Promoting axonal rewiring to improve outcome after stroke. Neurobiol Dis 37, 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins RA and Besheer J (2006) Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat Protoc 1, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Blasi F, Wei Y, Balkaya M, Tikka S, Mandeville JB, Waeber C, Ayata C and Moskowitz MA (2014) Recognition memory impairments after subcortical white matter stroke in mice. Stroke 45, 1468–1473. [DOI] [PubMed] [Google Scholar]
- Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P and Freret T (2009) The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc 4, 1560–1564. [DOI] [PubMed] [Google Scholar]
- Bu F, Min JW, Munshi Y, Lai YJ, Qi L, Urayama A, McCullough LD and Li J (2019) Activation of endothelial ras-related C3 botulinum toxin substrate 1 (Rac1) improves post-stroke recovery and angiogenesis via activating Pak1 in mice. Exp Neurol 322, 113059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunge RP (1968) Glial cells and the central myelin sheath. Physiol Rev 48, 197–251. [DOI] [PubMed] [Google Scholar]
- Bustelo XR, Sauzeau V and Berenjeno IM (2007) GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29, 356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DH, Kim JH, Lee KH, Kim HY, Kim YS, Choi WS and Lee J (2015) Role of neuronal NADPH oxidase 1 in the peri-infarct regions after stroke. PLoS One 10, e0116814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson AN, Parker K, Nilsson M, Walker FR and Gowing EK (2015) Combined ampakine and BDNF treatments enhance poststroke functional recovery in aged mice via AKT-CREB signaling. J Cereb Blood Flow Metab 35, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pozo MA, Price LS, Alderson NB, Ren XD and Schwartz MA (2000) Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J 19, 2008–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S (2009) Molecular targets for axon regeneration: focus on the intrinsic pathways. Expert Opin Ther Targets 13, 1387–1398. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S and Hall A (2002) Rho GTPases in cell biology. Nature 420, 629–635. [DOI] [PubMed] [Google Scholar]
- Frost JA, Xu S, Hutchison MR, Marcus S and Cobb MH (1996) Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol Cell Biol 16, 3707–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y and Yamashita T (2014) Axon growth inhibition by RhoA/ROCK in the central nervous system. Front Neurosci 8, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander M, Koudstaal PJ, Bots ML, Grobbee DE, Hofman A and Breteler MM (2003) Incidence, risk, and case fatality of first ever stroke in the elderly population. The Rotterdam Study. J Neurol Neurosurg Psychiatry 74, 317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Ueyama T, Shigyo M et al. (2017) A Novel Rac1-GSPT1 Signaling Pathway Controls Astrogliosis Following Central Nervous System Injury. J Biol Chem 292, 1240–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Kim YT, McKeon RJ and Bellamkonda RV (2006) In situ gelling hydrogels for conformal repair of spinal cord defects, and local delivery of BDNF after spinal cord injury. Biomaterials 27, 497–504. [DOI] [PubMed] [Google Scholar]
- Karabiyik C, Fernandes R, Figueiredo FR, Socodato R, Brakebusch C, Lambertsen KL, Relvas JB and Santos SD (2018) Neuronal Rho GTPase Rac1 elimination confers neuroprotection in a mouse model of permanent ischemic stroke. Brain Pathol 28, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Fogaca MV, Deyama S, Li XY, Fukumoto K and Duman RS (2018) BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol Psychiatry 23, 2007–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichina JV, Goc A, Al-Husein B, Somanath PR and Kandel ES (2010) PAK1 as a therapeutic target. Expert Opin Ther Targets 14, 703–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koth AP, Oliveira BR, Parfitt GM, Buonocore Jde Q and Barros DM (2014) Participation of group I p21-activated kinases in neuroplasticity. J Physiol Paris 108, 270–277. [DOI] [PubMed] [Google Scholar]
- Kumar R, Gururaj AE and Barnes CJ (2006) p21-activated kinases in cancer. Nat Rev Cancer 6, 459–471. [DOI] [PubMed] [Google Scholar]
- Liang X, Draghi NA and Resh MD (2004) Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J Neurosci 24, 7140–7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Schafer DP and McCullough LD (2009) TTC, fluoro-Jade B and NeuN staining confirm evolving phases of infarction induced by middle cerebral artery occlusion. J Neurosci Methods 179, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HQ, Li WB, Li QJ et al. (2006) Nitric oxide participates in the induction of brain ischemic tolerance via activating ERK1/2 signaling pathways. Neurochem Res 31, 967–974. [DOI] [PubMed] [Google Scholar]
- Liu L, McCullough L and Li J (2014a) Genetic deletion of calcium/calmodulin-dependent protein kinase kinase beta (CaMKK beta) or CaMK IV exacerbates stroke outcomes in ovariectomized (OVXed) female mice. BMC Neurosci 15, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yuan H, Denton K, Li XJ, McCullough L and Li J (2016) Calcium/calmodulin-dependent protein kinase kinase beta is neuroprotective in stroke in aged mice. Eur J Neurosci 44, 2139–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yuan H, Yi Y et al. (2018) Ras-Related C3 Botulinum Toxin Substrate 1 Promotes Axonal Regeneration after Stroke in Mice. Transl Stroke Res 9, 506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li Y, Cui Y, Roberts C, Lu M, Wilhelmsson U, Pekny M and Chopp M (2014b) Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia 62, 2022–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Valdes HE, Clarkson AN, Ao Y, Charles AC, Carmichael ST, Sofroniew MV and Brennan KC (2014) Memantine enhances recovery from stroke. Stroke 45, 2093–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist EA (2003) Rac proteins and the control of axon development. Curr Opin Neurobiol 13, 384–390. [DOI] [PubMed] [Google Scholar]
- Mages B, Aleithe S, Altmann S et al. (2018) Impaired Neurofilament Integrity and Neuronal Morphology in Different Models of Focal Cerebral Ischemia and Human Stroke Tissue. Front Cell Neurosci 12, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwani B, Liu F, Xu Y, Persky R, Li J and McCullough LD (2011) Functional recovery in aging mice after experimental stroke. Brain Behav Immun 25, 1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng S, Su Z, Liu Z, Wang N and Wang Z (2015) Rac1 contributes to cerebral ischemia reperfusion-induced injury in mice by regulation of Notch2. Neuroscience 306, 100–114. [DOI] [PubMed] [Google Scholar]
- Mitsios N, Saka M, Krupinski J et al. (2007) A microarray study of gene and protein regulation in human and rat brain following middle cerebral artery occlusion. BMC Neurosci 8, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandan MO, Ghaleb AM, Liu Y, Bialkowska AB, McConnell BB, Shroyer KR, Robine S and Yang VW (2014) Inducible intestine-specific deletion of Kruppel-like factor 5 is characterized by a regenerative response in adult mouse colon. Dev Biol 387, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozoe M, Hirooka Y, Koga Y, Sagara Y, Kishi T, Engelhardt JF and Sunagawa K (2007) Inhibition of Rac1-derived reactive oxygen species in nucleus tractus solitarius decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats. Hypertension 50, 62–68. [DOI] [PubMed] [Google Scholar]
- Patel MD, Coshall C, Rudd AG and Wolfe CD (2002) Cognitive impairment after stroke: clinical determinants and its associations with long-term stroke outcomes. J Am Geriatr Soc 50, 700–706. [DOI] [PubMed] [Google Scholar]
- Ritzel RM, Lai YJ, Crapser JD et al. (2018) Aging alters the immunological response to ischemic stroke. Acta Neuropathol 136, 89–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen LH, Li Y, Chen J et al. (2007) Therapeutic benefit of bone marrow stromal cells administered 1 month after stroke. J Cereb Blood Flow Metab 27, 6–13. [DOI] [PubMed] [Google Scholar]
- Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F and Der CJ (2007) Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem 282, 35666–35678. [DOI] [PubMed] [Google Scholar]
- Smania N, Picelli A, Gandolfi M, Fiaschi A and Tinazzi M (2008) Rehabilitation of sensorimotor integration deficits in balance impairment of patients with stroke hemiparesis: a before/after pilot study. Neurol Sci 29, 313–319. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32, 638–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz TR and Linseman DA (2014) Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci 8, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uphaus T, Bittner S, Groschel S et al. (2019) NfL (Neurofilament Light Chain) Levels as a Predictive Marker for Long-Term Outcome After Ischemic Stroke. Stroke 50, 3077–3084. [DOI] [PubMed] [Google Scholar]
- Writing Group M, Mozaffarian D, Benjamin EJ et al. (2016) Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 133, e38–360. [DOI] [PubMed] [Google Scholar]
- Yiu G and He Z (2006) Glial inhibition of CNS axon regeneration. Nat Rev Neurosci 7, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Denton K, Liu L, Li XJ, Benashski S, McCullough L and Li J (2016) Nuclear translocation of histone deacetylase 4 induces neuronal death in stroke. Neurobiol Dis 91, 182–193. [DOI] [PubMed] [Google Scholar]
- Zhou Q, An Y and Tang Y (2019) Long noncoding RNA-regulator of reprogramming alleviates hypoxia-induced cerebral injury in mice model and human via modulating apoptosis complexes. J Integr Neurosci 18, 431–437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.