

Abstract
Of all known small molecules targeting human immunodeficiency virus (HIV) capsid protein (CA), PF74 represents by far the best characterized chemotype, due to its ability to confer antiviral phenotypes in both early and late phases of viral replication. However, the prohibitively low metabolic stability renders PF74 a poor antiviral lead. We report herein our medicinal chemistry efforts toward identifying novel and metabolically stable small molecules targeting the PF74 binding site. Specifically, we replaced the inter-domain-interacting, electron-rich indole ring of PF74 with less electron-rich isosteres, including imidazolidine-2,4-dione, pyrimidine-2,4-dione, and benzamide, and identified four potent antiviral compounds (10, 19, 20 and 26) with markedly improved metabolic stability. Compared to PF74, analog 20 exhibited similar submicromolar potency, and much longer (51-fold) half-life in human liver microsomes (HLMs). Molecular docking corroborated that 20 binds to the PF74 binding site, and revealed distinct binding interactions conferred by the benzamide moiety. Collectively, our data support compound 20 as a promising antiviral lead.
KEY WORDS: HIV-1, Capsid protein, PF74, Microsomal stability
Abbreviations: ART, antiretroviral therapy; CA, capsid protein; CACTD, CA C-terminal domain; CANTD, CA N-terminal domain; HBA, H-bond acceptor; HBD, H-bond donor; HIV, human immunodeficiency virus; HLM, human liver microsome; MLM, mouse liver microsome; PK, pharmacokinetic; SAR, structure‒activity relationship; TSA, thermal shift assay
Graphical abstract
HIV-1 capsid protein (CA)-targeting PF74 suffers from prohibitively low metabolic stability. Structural modifications have yet to produce analogs with balanced potency and metabolic stability. Replacing the inter-domain interacting indole ring with less electron-rich isosteres allowed the identification of compound 20 with drastically improved metabolic stability while largely maintaining antiviral potency.

1. Introduction
Human immunodeficiency virus type 1 (HIV-1) remains a global healthcare challenge despite the successful development of many antiviral drugs1. Until an HIV-1 cure2,3 is developed, current antiretroviral therapy (ART) is expected to be lifelong, and the virus will eventually select strains resistant to current drug classes. This necessitates the development of new drugs with novel molecular targets and distinct resistance profiles. Toward this end, HIV-1 drug discovery research targeting the multifunctional HIV-1 capsid protein (CA) has drawn increasing interest4, 5, 6. CA and its assembled product, the capsid core7, 8, 9, play critical roles in multiple steps within the HIV-1 replication cycle10, 11, 12 via CA‒CA interactions or CA‒host factor interactions. The capsid core provides a protected environment13 for viral reverse transcription and shields viral genome products from host nucleic acid sensing14,15. CA‒CA interactions drive capsid assembly7,9,16,17 and impact capsid disassembly and core stability18. Perturbation of core stability results in premature uncoating, impaired reverse transcription, and attenuated infection19,20. Importantly, HIV-1 core stability and uncoating are also tightly regulated via the interactions between CA and various cellular factors to enable most post entry steps such as cytoplasmic trafficking, nuclear entry, integration, and host restriction12,21. CA-interacting host factors include TRIM5α22,23, cleavage and polyadenylation specific factor 6 (CPSF6)24,25, nucleoporins 15326, 27, 28 and 35829,30 (NUP153 and NUP358), MxB31,32, and cyclophilin A33, 34, 35. Therefore, in addition to potentially inhibiting viral strains resistant to current drug classes, CA-targeting small molecules also have the advantage of conferring both early stage and late stage antiviral phenotypes by disrupting CA‒CA and CA‒host interactions.
HIV-1 CA consists of seven helices in the CANTD and four helices in the CACTD (Fig. 1A)36, 37, 38. A few small molecule-binding sites have been identified to accommodate distinct CA-targeting chemotypes4, of which the PF74 binding site36 represents a particularly attractive drug target. Located at the CA dimer interface, this binding site is formed by many residues (lemon surface) within H3 and H4 of the CANTD (green), and a few residues (gray surface) in the H8 and H9 of the adjacent CACTD (blue, Fig. 1A). The detailed CA-binding mode of PF74 is shown in Fig. 1B36. Key molecular interactions include: 1) three H-bonds by the phenylalanine core of PF74 (two with N57, and one with K70); 2) interactions of the aniline moiety with the N-methyl group N53, and the phenyl ring with A105, T107, and Y130; 3) hydrophobic interactions by the phenyl ring of the phenylalanine core with residues M66 and L69; and 4) inter-CA domain interactions by the indole moiety with the CANTD (via H-bond with Q63 and π−cation interaction with K70) and the adjacent CACTD (via Y169, R173, and K172)36. Significantly, the PF74 site also binds host co-factors, such as nucleoporin Nup15326, 27, 28 and CPSF624,25,39,40. The binding features and the competition against host factors likely account for the unique concentration-dependent dual antiviral mechanisms of PF74: competing against host factors for capsid binding at low concentrations, and inducing premature uncoating at high concentrations19.
Figure 1.

Design of novel analogs targeting the PF74 binding site of HIV-1 CA. (A) Structure of HIV-1 CA dimer. The PF74 binding pocket is lined by numerous residues (lemon surface) of CANTD, as well as a few residues (gray surface) of the adjacent CACTD. Dimer structure and PF74 binding were reproduced in PyMOL based on PDB ID: 4XFZ36. (B) Detailed binding interactions of PF74. The indole moiety of PF74 is involved in inter-CA domain interactions: with the CANTDvia H-bond to Q63 (dotted line) and π−cation interaction with K70 (arrow), and with the adjacent CACTD through interactions involving K182, Y169 and R173. (C) Novel analogs binding to the PF74 site were designed by replacing the indole ring with monocyclic rings with the aim to improve metabolic stability while retaining antiviral potency.
Despite all the favorable characteristics, including well-established binding mode, unique dual mechanism of action, good synthetic tractability and high amenability for optimization, PF74 as an antiviral lead is severely flawed due to its extremely poor metabolic stability41, 42, 43, 44. We have previously identified a compound showing much improved (44-fold) microsomal stability when compared to PF7443. However, the compound features an altered backbone which conferred moderately reduced antiviral potency. Recently reported efforts also involved replacing the indole moiety with a 1,2,3-triazole ring41,45 or benzenesulfonamide moiety46, though these studies resulted in compounds with only marginally improved (<3-fold) microsomal stability. Therefore, the need to identify novel small molecules targeting the PF74 binding site with potent antiviral activity and improved metabolic stability remains. Toward this end, our current work is centered around replacing the indole ring with a few distinct 5- and 6-membered scaffolds (Fig. 1C). The rationale is that the susceptibility to oxidative metabolism could be effectively mitigated by replacing the electron-rich indole ring with an electron-deficient isostere. Such a scaffold-hopping strategy has been widely used to address metabolic issues associated with electron-rich aromatic rings47. Our efforts led to the synthesis and testing of 26 analogs, of which 4 demonstrated potent anti-HIV-1 activity, significant effects on CA hexamer stability, and more importantly, substantially improved metabolic stability over PF74.
2. Results and discussion
2.1. Chemistry
Synthesis of analogs 1−15 and 17−26 is outlined in Scheme 1. Briefly, Boc-protected phenylalanine (28) was reacted with various amines under a well-established method using T3P to afford 29. After removal of Boc protecting group using TFA, intermediate 30 was obtained, which was further reacted with commercially available acid derivative 31 to produce 1−15 and 18−26. Meanwhile, analog 17 was produced from the reaction of 30a and acid 32. The synthetic procedure of 16 is similar to the synthesis of 1−15 and 17−26 except amino acid 33 was used in the first step (Scheme 2).
Scheme 1.

Synthesis of analogs 1−15 and 17−26. Reagents and conditions: (i) amine, T3P, DIPEA, DMF, rt, 12 h; (ii) TFA; DCM, rt, 4−6 h; (iii) HATU, DIPEA, DMF, rt, 12 h.
Scheme 2.

Synthesis of analog 16. Reagents and conditions: (i) N-methylaniline, T3P, DIPEA, DMF, rt, 12 h; (i) TFA; DCM, rt, 6 h; (iii) HATU, DIPEA, DMF, rt, 12 h.
The preparation of analog 27 is described in Scheme 3. Boc-protected phenylalanine 28 was treated with l-proline methyl ester to afford intermediate 37 which was converted to carboxylic acid intermediate 38 in the presence of lithium hydroxide. Intermediate 38 was reacted with aniline to afford 39 which was converted to 40 via a TFA-mediated hydrolysis. Finally, 40 was treated with acid intermediate 41 to produce analog 27.
Scheme 3.

Synthesis of analog 27. Reagents and conditions: (i) l-proline methyl ester hydrochloride, T3P, DIPEA, DMF, rt, 12 h; (ii) LiOH, MeOH/H2O, rt, overnight; (iii) aniline, HATU, DIPEA, DMF, rt, 12 h; (iv) TFA; DCM, rt, 6 h; (v) HATU, DIPEA, DMF, rt, 12 h.
2.2. Biological assays and SAR
All final compounds were first tested in a thermal shift assay which measures the change of protein melting point (ΔTm) upon compound binding. A positive ΔTm value indicates a stabilizing effect and a negative value indicates a destabilizing effect on the protein. All analogs were also subjected to a cell-based antiviral screening at 20 μmol/L against HIV-1. Active compounds were further tested at 2 μmol/L, and those with significant inhibition were selecetd for dose response antiviral testing. In parallel, cytotoxicity was also measured for all final compounds. PF74 was used as a control in these assays and the values were consistent with previous reports43,44.
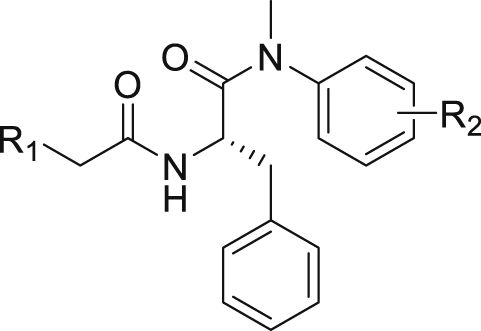
The 26 analogs synthesized in this work belong to three distinct subtypes, each featuring a monocyclic ring (R1) to engage in the inter-CA domain interactions in lieu of the indole ring. Within each subtype, we explored the effect of the aniline substituent (R2), typically including 4-Me, 4-Cl, 3-Cl, 4-F, 3-F, 3-Br, and 3-CF3 (Table 1) The first subtype contains the 5-imidazolidine-2,4-dione which has multiple H-bond donors (HBDs) and H-bond acceptors (HBAs). Compounds of this subtype (2−7) did not exhibit significant antiviral activity against HIV-1, with the exception of 4, which inhibited HIV-1 weakly at 2 μmol/L and almost completely at 20 μmol/L. The next series bears a 1-pyrimidine-2,4-dione ring as the indole replacement (8−17). Of these analogues, the ones closely mimicking PF74 (8−15) all strongly inhibited HIV-1 at 20 μmol/L (85%–100%). A few (8, 10, 11, 14 and 15) also exhibited inhibition at 2 μmol/L. The most potent analog of this series, compound 10, inhibited HIV-1 with an EC50 of 1.7 μmol/L. When the benzyl of the phenylalanine core was changed to a phenyl ring with reversed stereochemistry, the resulting compound 16 did not inhibit HIV-1. The lack of antiviral activity was observed also with compound 17, which features the same pyrimidine-2,4-dione, albeit with a different linker and a different site of connection (C5 vs. N1). In the thermal shift assay, analogs of the pyrimidine-2,4-dione subtype did not significantly impact the stability of CA hexamer (ΔTm = 0−1.7 °C).
Table 1.
Anti-HIV-1 activity, cytotoxicity, and CA hexamer stability profiles of all analogs.
| Compd. | R1 | R2 | EC50a (μmol/L) | Inhibition at 2 μmol/L:20 μmol/L (%) | CC50b (μmol/L) | TSA ΔTmc (°C) |
|---|---|---|---|---|---|---|
| PF74d (1) | 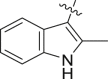 |
H | 0.61±0.2 | – | 76±9 | 7.4 |
| 2 | 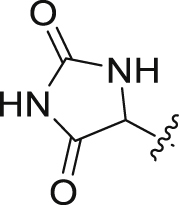 |
H | >20 | – | >50 | 0 |
| 3 | |
4-Me | – | 2/62 | >50 | 1.0 |
| 4 | |
4-Cl | – | 17/96 | >50 | ‒1.2 |
| 5 | |
3-Cl | >20 | – | >50 | 0 |
| 6 | |
4-F | >20 | – | >50 | 0 |
| 7 | |
3-F | >20 | – | >50 | 0 |
| 8 | 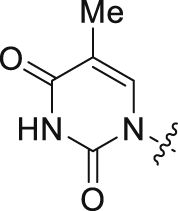 |
H | – | 35/95 | >50 | 0.5 |
| 9 | |
4-Me | – | 7/85 | >50 | 0.6 |
| 10 | |
4-Cl | 1.7±0.2 | – | >50 | 1.7 |
| 11 | |
3-Cl | – | 37/95 | >50 | 0.6 |
| 12 | |
4-F | – | 6/94 | >50 | 0 |
| 13 | |
3-F | – | 0/74 | >50 | 0.9 |
| 14 | |
3-Br | – | 22/100 | >50 | 0.5 |
| 15 | |
3-CF3 | – | 28/95 | >50 | 0 |
| 16 | 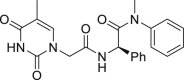 |
>20 | – | >50 | 1.3 | |
| 17 | 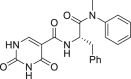 |
>20 | – | >50 | 0 | |
| 18 | 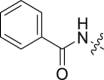 |
H | – | 15/96 | >50 | 1.3 |
| 19 | |
4-Me | 1.6±0.02 | – | >50 | 5.0 |
| 20 | |
4-Cl | 0.88±0.02 | – | >50 | 4.1 |
| 21 | |
3-Cl | – | 40/95 | >50 | 2.0 |
| 22 | |
4-F | – | 12/97 | >50 | 2.2 |
| 23 | |
3-F | – | 3/94 | >50 | 0 |
| 24 | |
3-Br | – | 38/97 | >50 | 2.2 |
| 25 | |
3-CF3 | – | 19/89 | >50 | 1.2 |
| 26 | 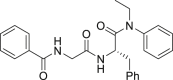 |
2.5±0.03 | – | >50 | −2.5 | |
| 27 | 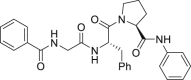 |
>20 | – | >50 | 1.1 | |
Half maximal effective concentration: mean ± standard deviation (SD) from at least two independent experiments.
50% cytotoxic concentration: mean ± SD from at least two independent experiments.
ΔTm: melting point change of CA hexamer in presence of a compound compared to DMSO control.
Values for PF74 also reported in Refs. 43 and 44. −Not applicable.
By far the most interesting series is the benzamide subtype (18−27). With the exception of 23 and 27, all analogs of this subtype demonstrated discernible HIV-1 inhibition at 2 μmol/L and complete inhibition at 20 μmol/L. Three analogs potently inhibited HIV-1 in dose response fashion (19: EC50 = 1.6 μmol/L; 20: EC50 = 0.88 μmol/L; 26: EC50 = 2.5 μmol/L) and moderately impacted the stability of CA hexamer (19: ΔTm = 5.0; 20: ΔTm = 4.1; 26: ΔTm = −2.5). It is not clear why 26 destabilized while 19 and 20 stabilized CA hexamer. Notably compound 27 has a proline insert between the phenylalanine core and the aniline terminus, rendering a substantially different backbone, which evidently does not favor binding to HIV-1 CA. Overall, scaffold-hopping on the indole ring allowed us to identify four analogs (10, 19, 20 and 26) of two different subtypes with low to sub micromolar antiviral activities (EC50 = 0.88−2.5 μmol/L). Particularly important is the benzamide subtype, which generally conferred significant antiviral activity, with compound 20 (EC50 = 0.88 μmol/L) exhibiting submicromolar potency comparable to PF74 (EC50 = 0.61 μmol/L).
2.3. Metabolic stability in liver microsomes
A serious drawback of PF74 is the lack of metabolic stability41,42. Cytochrome P450 (CYPs)-mediated oxidative metabolism constitutes a major pharmacokinetic barrier for oral drugs48. Since the vast majority of metabolizing enzymes are found in the liver, measuring in vitro metabolic stability in liver microsomes can reliably predict drug oral bioavailability49. Low stability in liver microsomes, as signified by short half-life (t1/2), predicts excessive first pass liver metabolism and poor oral bioavailability. To assess the metabolic stability of our analogs, we tested the four most potent antiviral compounds (10, 19, 20 and 26), along with PF74 and two other analogs (18 and 23), in both human liver microsomes (HLMs) and mouse liver microsomes (MLMs). In addition, we also measured the microsomal stability of the four best compounds in the presence of cobicistat (Cobi)50, a CYP3A inhibitor. CYP3A is a liver metabolizing enzyme responsible for the metabolism of more than half of all drugs51. Peptidomimetics, such as PF74, are well-known substrates for CYP3A52 and are highly susceptible to oxidative metabolism. By inhibiting CYP3A, Cobi acts as a pharmacokinetic (PK) enhancer53 to boost the plasma concentration of peptidomimetic drugs, such as HIV-1 protease inhibitors53. The metabolic stability results are summarized in Table 2. In our assays, the half-life of PF74 was less than 1 min in both HLMs and MLMs, consistent with previous reports41,42. Remarkably, all six of our tested analogs demonstrated hugely improved metabolic stability in both HLMs (t1/2 = 12−36 min, 17−50-fold increase over PF74) and MLMs (t1/2 = 2.4−13 min, 4−22-fold increase over PF74). These results indicate that scaffold-hopping of the indole ring with more electron-deficient moieties could lead to resistance to phase I metabolism. When measured in the presence of Cobi, the half-lives were drastically increased, except for compound 19 in HLMs. Based on our metabolic stability studies, compound 20, with half-lives of 36 min (51-fold over PF74) in HLMs and 13 min (22-fold over PF74) in MLMs, is predicted to have favorable oral bioavailability.
Table 2.
Half-life (t1/2, min) of selected compounds in liver microsomes.
2.4. Molecular modeling
To study the potential binding mode of our analogs and understand the SAR, we conducted molecular docking for a few representative compounds, including potent compounds (10 and 20), and less potent (4) or inactive (2 and 17) analogs. The modeling used the co-crystal structure of PF74-bound HIV-1 CA (PDB code: 4XFZ36). Compound 2 (yellow, Fig. 2A) which bears an imidazolidine-2,4-dione in place of indole ring, was completely inactive, probably because of the potential loss of key H-bonds with N57 and Q63 and the cation‒π interaction with K70, resulting a very poor docking score (Glide score: −2.7 for 2 vs. −5.8 for PF74). Compound 4 (pink, Fig. 2A), the aniline 4-Cl congener of 2, adopted a conformation and binding mode similar to 2, except that the 4-Cl formed a halogen-bond with N74, which likely contributed to the activity observed with 4. Significantly, a 6-membered pyrimidine-2,4-dione in compound 10 is predicted to restore key interactions with N57 and Q63 (Fig. 2B). These interactions also allowed compound 10 to fit in the PF74 pocket and interact with all other key residues, which could explain the observed improved potency (17% inhibition at 2 μmol/L for 4 vs. EC50: 1.7 μmol/L for 10). However, when the same pyrimidine-2,4-dione scaffold is connected to the phenylalanine core via the C5 site (vs. the N-1 site in compound 10) and the methylene linker deleted, the resulting compound 17 (Fig. 2C) appeared to be forced out of the PF74 pocket and bound away from the preferred site (circled). This may explain why compound 17 was completely inactive. Finally, the most interesting modeling results were observed with compound 20 which features a benzamide moiety as the indole replacement. Important docking observations for 20 (Fig. 2D) included 1) the aniline moiety and the phenylalanine core appear bound in a very similar way as in PF74; 2) the 4-Cl of compound 20 formed a halogen bond54 with N74; 3) the benzamide moiety adopted an orientation similar to the indole ring of PF74 and formed a similar H-bond with Q63 (via the amide NH); 4) the interaction with K70 was via an H-bond by the amide carbonyl in 10 as opposed to the π−cation interaction in PF74. This potential binding mode for compound 20 is likely to contribute to the observed potent antiviral activity and increased CA hexamer stabilization (Table 1).
Figure 2.

Docking of selected compounds into PF74-bound HIV-1 CA (PDB ID: 4XFZ36). Predicted binding modes of: (A) compounds 2 (yellow sticks) and 4 (pink sticks). Glide scores (kcal/mol): −2.7 for 2 and −3.0 for 4; (B) compound 10. Glide score: −6.4 kcal/mol; (C) compound 17, away from the binding site (circled). Glide score: −4.7 kcal/mol; (D) compound 20. Glide score: −6.7 kcal/mol. Hydrogen-bonding, halogen-bonding, and cation‒π interactions are depicted as black dotted lines, pink dashed lines, and double-headed arrows, respectively. CA is shown in cyan cartoon with key residues around binding site shown as sticks, and ligands 10, 17, and 20 are shown as pink sticks. The nitrogen, oxygen, and chlorine atoms are colored blue, red, and green, respectively.
3. Conclusions
Although PF74 is a potent and well-characterized CA-targeting antiviral small molecule, it lacks metabolic stability to be an antiviral lead. The indole ring of PF74 is a particularly interesting handle for exploring both the potency and metabolic stability for two reasons: 1) it interacts with both the CANTD and the adjacent CACTD; 2) it is highly electron-rich, and thus could be susceptible to oxidative metabolism. In this work, we used a scaffold-hopping approach to replace the inter-CA domain-interacting and electron-rich indole ring with electron-deficient moieties, including imidazolidine-2,4-dione, pyrimidine-2,4-dione, and benzamide, and identified four potent antiviral compounds (10, 19, 20 and 26) with dramatically improved metabolic stability. Our best compound, analog 20, exhibited submicromolar potency comparable to PF74 and much longer half-lives than PF74 in HLMs (51-fold) and MLMs (22-fold). Considerably longer half-life was also observed when metabolic stability was measured in the presence of PK enhancer, Cobi. Molecular modeling corroborated the binding of 20 to the PF74 pocket and revealed interesting potential binding interactions conferred by the benzamide moiety. Altogether, these data suggest that analog 20 can be a potentially viable antiviral lead.
4. Experimental
4.1. Chemistry
All commercial chemicals were used as supplied unless indicated otherwise. Compounds were purified via flash chromatography using a Combiflash RF-200 (Teledyne ISCO, Lincoln, NE, USA) with RediSep columns (silica) and indicated mobile phase. 1H and 13C NMR spectra were recorded on a Varian 600 MHz (Agilent Technologies, Santa Clara, CA, USA) or Bruker 400 spectrometer (Bruker, Billerica, MA, USA). Diastereomeric ratio was determined by 1H NMR analysis. Mass data were acquired using an Agilent 6230 TOF LC/MS spectrometer (Agilent Technologies). PF74 was synthesized according to reported procedures43,44.
4.1.1. General procedure for synthesis of 2−27
HATU (2 equiv.) and DIPEA (3 equiv.) were added to a solution of an acid derivative (1 equiv.) in DMF. After the mixture was stirred for 20 min at room temperature, amine (1.2 equiv.) was added, and the resulting mixture was further stirred overnight at room temperature. To the reaction mixture was added H2O and the mixture was extracted with ethyl acetate three times. The organic layers were combined, washed with brine, dried over anhydrous MgSO4, and concentrated in vacuum. The crude mixture was purified using Combi-flash on silica gel (EtOAc in hexanes as eluent).
4.1.1.1. (2S)-2-(2-(2,5-Dioxoimidazolidin-4-yl)acetamido)-N-methyl-N,3-diphenylpropanamide (2)
Yield 70%, dr 1.1:1. 1H NMR (600 MHz, CD3OD) δ 8.18−8.15 (m, 1H), 7.28−7.24 (m, 3H), 7.11−7.09 (m, 3H), 6.93−6.76 (m, 4H), 4.58−4.52 (m, 1H), 4.23−4.18 (m, 1H), 3.10−3.07 (m, 3H), 2.87−2.83 (m, 1H), 2.70−2.58 (m, 2H), 2.47−2.41 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 177.4, 177.2, 173.5, 173.2, 171.0, 170.5, 159.9, 159.7, 144.0, 143.9, 138.1, 130.8, 130.3, 130.2, 129.5, 129.3, 128.7, 128.6, 127.9, 56.9, 56.6, 53.8, 53.4, 39.4, 39.1, 38.3, 38.2, 38.1, 38.0; HRMS-ESI (−) m/z Calcd. for C21H21N4O4 [M−H]− 393.1568, Found 393.1573.
4.1.1.2. (2S)-2-(2-(2,5-Dioxoimidazolidin-4-yl)acetamido)-N-methyl-3-phenyl-N-(p-tolyl)propanamide (3)
Yield 65%, dr 1.3:1. 1H NMR (600 MHz, CDCl3) δ 9.52−9.36 (m, 1H), 7.52−7.36 (m, 1H), 7.22−7.14 (m, 4H), 6.91−6.78 (m, 4H), 4.85−4.77 (m, 1H), 4.35−4.32 (m, 1H), 2.92−2.81 (m, 2H), 2.74−2.70 (m, 1H), 2.51−2.45 (m, 1H), 2.37−2.36 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 174.7, 174.4, 172.1, 171.7, 169.1, 168.5, 157.3, 157.2, 139.6, 139.5, 138.4, 138.3, 136.2, 130.4, 129.4, 129.2, 128.5, 128.4, 126.9, 126.9, 55.8, 55.5, 51.9, 51.3, 39.1, 38.5, 38.0, 37.9, 37.7, 37.4, 21.1; HRMS-ESI (−) m/z Calcd. for C22H23N4O4 [M−H]− 407.1725, Found 407.1730.
4.1.1.3. (2S)-N-(4-Chlorophenyl)-2-(2-(2,5-dioxoimidazolidin-4-yl)acetamido)-N-methyl-3-phenylpropanamide (4)
Yield 82%, dr 2.3:1. 1H NMR (600 MHz, CD3OD) δ 7.34−7.24 (m, 6H), 6.98−6.86 (m, 3H), 4.61−4.55 (m, 1H), 4.33−4.30 (m, 1H), 3.14−3.12 (m, 3H), 2.98−2.92 (m, 1H), 2.79−2.72 (m, 2H), 2.58−2.54 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 177.5, 173.4, 173.1, 170.9, 170.4, 160.0, 142.6, 142.5, 138.0, 137.9, 134.9, 130.7, 130.4, 130.3, 129.6, 128.1, 56.8, 56.7, 53.6, 53.2, 39.7, 39.4, 38.2, 38.1, 37.9; HRMS-ESI (−) m/z Calcd. for C21H20ClN4O4 [M−H]− 427.1179, Found 427.1183.
4.1.1.4. (2S)-N-(3-Chlorophenyl)-2-(2-(2,5-dioxoimidazolidin-4-yl)acetamido)-N-methyl-3-phenylpropanamide (5)
Yield 83%, dr 1.2:1. 1H NMR (600 MHz, CD3OD) δ 7.34−7.25 (m, 6H), 6.97−6.65 (m, 3H), 4.61−4.54 (m, 1H), 4.35−4.32 (m, 1H), 3.14−3.12 (m, 3H), 2.97−2.92 (m, 1H), 2.80−2.71 (m, 2H), 2.58−2.53 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 177.4, 177.3, 173.3, 173.1, 171.0, 170.5, 159.9, 159.8, 145.1, 137.9, 137.8, 135.9, 135.8, 131.8, 130.3, 129.7, 129.4, 128.7, 128.2, 127.3, 56.8, 56.7, 53.8, 53.4, 39.7, 39.4, 38.2, 38.1, 37.9; HRMS-ESI (−) m/z Calcd. for C21H20ClN4O4 [M−H]− 427.1179, Found 427.1184.
4.1.1.5. (2S)-2-(2-(2,5-Dioxoimidazolidin-4-yl)acetamido)-N-(4-fluorophenyl)-N-methyl-3-phenylpropanamide (6)
Yield 66%, dr 1.6:1. 1H NMR (600 MHz, CD3OD) δ 7.26−7.23 (m, 3H), 7.06−6.93 (m, 6H), 4.61−4.54 (m, 1H), 4.33−4.29 (m, 1H), 3.15−3.12 (m, 3H), 2.98−2.93 (m, 1H), 2.79−2.69 (m, 2H), 2.58−2.53 (m, 1H); HRMS-ESI (−) m/z Calcd. for C21H20FN4O4 [M−H]− 411.1474, Found 411.1475.
4.1.1.6. (2S)-2-(2-(2,5-Dioxoimidazolidin-4-yl)acetamido)-N-(3-fluorophenyl)-N-methyl-3-phenylpropanamide (7)
Yield 75%, dr 1.7:1. 1H NMR (600 MHz, CD3OD) δ 7.36−7.24 (m, 5H), 7.10−7.06 (m, 1H), 6.96−6.81 (m, 3H), 4.64−4.58 (m, 1H), 4.35−4.31 (m, 1H), 3.16−3.14 (m, 3H), 2.98−2.93 (m, 1H), 2.80−2.70 (m, 2H), 2.58−2.52 (m, 1H); HRMS-ESI (−) m/z Calcd. for C21H20FN4O4 [M−H]− 411.1474, Found 411.1479.
4.1.1.7. (S)-N-Methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-N,3-diphenylpropanamide (8)
Yield 75%. 1H NMR (600 MHz, CDCl3) δ 9.35 (s, 1H), 7.50 (s, 1H), 7.38−7.35 (m, 3H), 7.19−7.17 (m, 3H), 6.94−6.86 (m, 4H), 4.84 (q, J = 7.8 Hz, 1H), 4.43 (d, J = 15.8 Hz, 1H), 4.21 (d, J = 15.8 Hz, 1H), 3.26 (s, 3H), 2.92 (dd, J = 13.4, 6.8 Hz, 1H), 2.74 (dd, J = 13.4, 7.6 Hz, 1H), 1.89 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 171.2, 166.0, 164.1, 151.1, 142.2, 140.4, 136.0, 129.9, 129.3, 128.4, 128.4, 127.2, 126.9, 110.9, 51.5, 49.6, 38.9, 37.8, 12.4; HRMS-ESI (−) m/z Calcd. for C23H23N4O4 [M−H]− 419.1725, Found 419.1730.
4.1.1.8. (S)-N-Methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-3-phenyl-N-(p-tolyl)propanamide (9)
Yield 75%. 1H NMR (600 MHz, CDCl3) δ 9.47 (s, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.19−7.14 (m, 5H), 6.95−6.89 (m, 3H), 6.80 (brs, 1H), 4.87–4.83 (m, 1H), 4.45 (d, J = 15.8 Hz, 1H), 4.20 (d, J = 15.8 Hz, 1H), 3.23 (s, 3H), 2.92 (dd, J = 13.4, 6.9 Hz, 1H), 2.74 (dd, J = 13.4, 7.5 Hz, 1H), 2.36 (s, 3H), 1.89 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 171.4, 166.0, 164.2, 151.1, 140.5, 139.6, 138.3, 136.1, 130.4, 129.4, 128.4, 126.9, 126.8, 110.9, 51.4, 49.5, 39.0, 37.9, 21.1, 12.4; HRMS-ESI (−) m/z Calcd. for C24H25N4O4 [M−H]− 433.1881, Found 433.1890.
4.1.1.9. (S)-N-(4-Chlorophenyl)-N-methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-3-phenylpropanamide (10)
Yield 79%. 1H NMR (600 MHz, CDCl3) δ 9.48 (s, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.28 (d, J = 8.6 Hz, 2H), 7.23–7.22 (m, 3H), 6.98–6.97 (m, 1H), 6.94–6.92 (m, 2H), 6.73 (brs, 1H), 4.78 (dd, J = 15.3, 8.0 Hz, 1H), 4.43 (d, J = 15.8 Hz, 1H), 4.24 (d, J = 15.8 Hz, 1H), 3.20 (s, 3H), 2.93 (dd, J = 13.2, 8.0 Hz, 1H), 2.78 (dd, J = 13.2, 6.9 Hz, 1H), 1.91 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 171.3, 166.1, 164.2, 151.1, 140.6, 140.4, 135.8, 134.1, 129.9, 129.4, 128.6, 128.5, 127.0, 111.0, 51.5, 49.6, 39.2, 37.8, 12.4; HRMS-ESI (−) m/z Calcd. for C23H22ClN4O4 [M−H]− 453.1335, Found 453.1338.
4.1.1.10. (S)-N-(3-Chlorophenyl)-N-methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-3-phenylpropanamide (11)
Yield 70%. 1H NMR (600 MHz, CD3OD) δ 7.33–7.26 (m, 7H), 6.96 (m, 3H), 4.59 (t, J = 7.6 Hz, 1H), 4.40 (q, J = 16.4 Hz, 2H), 3.13 (s, 3H), 2.98 (dd, J = 13.0, 8.7 Hz, 1H), 2.79 (dd, J = 13.1, 6.6 Hz, 1H), 1.86 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 172.9, 168.9, 167.0, 153.0, 145.0, 143.6, 137.7, 135.9, 131.9, 130.3, 129.7, 129.4, 128.7, 128.2, 127.3, 111.0, 53.5, 50.5, 39.6, 38.0, 12.2; HRMS-ESI (−) m/z Calcd. for C23H22ClN4O4 [M−H]− 453.1335, Found 453.1340.
4.1.1.11. (S)-N-(4-Fluorophenyl)-N-methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-3-phenylpropanamide (12)
Yield 90%. 1H NMR (600 MHz, CD3OD) δ 7.25–7.23 (m, 4H), 7.03–6.95 (m, 6H), 4.58 (t, J = 7.5 Hz, 1H), 4.41–4.35 (m, 2H), 3.14 (s, 3H), 2.98 (dd, J = 13.2, 8.1 Hz, 1H), 2.77 (dd, J = 13.2, 7.0 Hz, 1H), 1.86 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 173.1, 168.9, 167.0, 163.4 (d, JCF = 247.1 Hz), 153.0, 143.6, 139.9 (d, JCF = 3.0 Hz), 137.9, 130.7, 130.4, 129.6, 128.1, 117.4 (d, JCF = 23.0 Hz), 111.0, 53.4, 50.5, 39.4, 38.1, 12.2; HRMS-ESI (−) m/z Calcd. for C23H22FN4O4 [M−H]− 437.1631, Found 437.1635.
4.1.1.12. (S)-N-(3-Fluorophenyl)-N-methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-3-phenylpropanamide (13)
Yield 82%. 1H NMR (600 MHz, CDCl3) δ 9.77 (s, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.32−7.29 (m, 1H), 7.25−7.21 (m, 3H), 7.04−7.00 (m, 2H), 6.93−6.92 (m, 2H), 6.77 (s, 1H), 6.37 (s, 1H), 4.84−4.80 (m, 1H), 4.46−4.28 (m, 2H), 3.21 (s, 3H), 2.94 (dd, J = 13.2, 8.1 Hz, 1H), 2.80 (dd, J = 13.2, 6.9 Hz, 1H), 1.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.4, 166.2, 164.3, 162.8 (d, JCF = 249.4 Hz), 151.3, 143.5 (d, JCF = 9.4 Hz), 140.6, 135.9, 130.9 (d, JCF = 9.1 Hz), 129.3, 128.5, 127.1, 123.2, 115.5 (d, JCF = 20.9 Hz), 114.7 (d, JCF = 22.3 Hz), 111.0, 51.7, 49.6, 39.2, 37.7, 12.4; HRMS-ESI (−) m/z Calcd. for C23H22FN4O4 [M−H]− 437.1631, Found 437.1632.
4.1.1.13. (S)-N-(3-Bromophenyl)-N-methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-3-phenylpropanamide (14)
Yield 70%. 1H NMR (600 MHz, CD3OD) δ 7.47 (d, J = 7.9 Hz, 1H), 7.28−7.22 (m, 6H), 7.01−6.95 (m, 3H), 4.60–4.57 (m, 1H), 4.40 (q, J = 16.5 Hz, 2H), 3.13 (s, 3H), 2.97 (dd, J = 13.1, 8.7 Hz, 1H), 2.79 (dd, J = 13.1, 6.6 Hz, 1H), 1.86 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 172.8, 168.9, 167.0, 153.0, 145.1, 143.6, 137.6, 132.4, 132.1, 131.5, 130.3, 129.7, 128.2, 127.7, 123.6, 111.0, 53.5, 50.5, 39.6, 38.0, 12.2; HRMS-ESI (−) m/z Calcd. for C23H22BrN4O4 [M−H]− 497.0830, Found 497.0835.
4.1.1.14. (S)-N-Methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-3-phenyl-N-(3-(trifluoromethyl)phenyl)propanamide (15)
Yield 70%. 1H NMR (600 MHz, CD3OD) δ 7.61 (d, J = 7.9 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.34–7.22 (m, 6H), 6.95–6.93 (m, 2H), 4.55 (t, J = 8.1 Hz, 1H), 4.40 (q, J = 16.5 Hz, 2H), 3.16 (s, 3H), 2.98 (dd, J = 13.0, 8.7 Hz, 1H), 2.79 (dd, J = 13.1, 6.5 Hz, 1H), 1.87 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 172.9, 168.9, 167.0, 153.0, 144.5, 143.6, 137.6, 132.9 (q, JCF = 33 Hz), 132.8, 131.7, 130.2, 129.7, 128.2, 127.4 (q, JCF = 3.5 Hz), 125.4, 124.9 (q, JCF = 271.7 Hz), 111.0, 53.5, 50.5, 39.6, 38.0, 12.2; HRMS-ESI (−) m/z Calcd. for C24H22F3N4O4 [M−H]− 487.1599, Found 487.1604.
4.1.1.15. (R)-N-Methyl-2-(2-(5-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)acetamido)-N,2-diphenylacetamide (16)
Yield 69%. 1H NMR (600 MHz, CDCl3) δ 8.73 (s, 1H), 7.63 (t, J = 7.1 Hz, 1H), 7.33−7.28 (m, 3H), 7.23−7.15 (m, 3H), 6.99 (s, 1H), 6.92−6.88 (m, 3H), 5.51 (d, J = 7.1 Hz, 1H), 4.43−4.25 (m, 2H), 3.27 (s, 3H), 1.88 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 169.5, 165.5, 163.9, 150.9, 141.7, 140.7, 136.6, 129.7, 128.6, 128.5, 128.2, 127.9, 127.8, 110.7, 55.0, 49.6, 38.1, 12.3; HRMS-ESI (−) m/z Calcd. for C22H21N4O4 [M−H]− 405.1569, Found 405.1568.
4.1.1.16. (S)-N-(1-(Methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (17)
Yield 59%. 1H NMR (600 MHz, DMSO-d6) δ 11.64 (s, 1H), 11.62 (s, 1H), 9.05 (d, J = 7.6 Hz, 1H), 8.00 (s, 1H), 7.42–7.38 (m, 3H), 7.17–7.14 (m, 5H), 6.81 (d, J = 6.6 Hz, 2H), 4.65–4.62 (m, 1H), 3.11 (s, 3H), 2.88 (dd, J = 13.6, 5.6 Hz, 1H), 2.63 (dd, J = 13.6, 8.5 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 170.8, 164.5, 161.7, 150.8, 148.3, 143.2, 137.3, 130.1, 129.3, 128.8, 128.4, 127.9, 127.1, 104.2, 51.5, 38.7, 37.6; HRMS-ESI (−) m/z Calcd. for C21H19N4O4 [M−H]− 391.1413, Found 391.1415.
4.1.1.17. (S)-N-(2-((1-(Methyl(phenyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (18)
Yield 79%. 1H NMR (600 MHz, CDCl3) δ 7.80 (d, J = 7.4 Hz, 2H), 7.50 (t, J = 7.3 Hz, 1H), 7.42 (t, J = 7.7 Hz, 1H), 7.37–7.35 (m, 3H), 7.20–7.18 (m, 3H), 6.91–6.90 (m, 3H), 6.83 (brs, 1H), 4.85 (q, J = 7.4 Hz, 1H), 4.15–4.07 (m, 2H), 3.24 (s, 3H), 2.94 (dd, J = 13.3, 7.2 Hz, 1H), 2.76 (dd, J = 13.3, 7.1 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 171.1, 168.0, 167.4, 142.3, 136.0, 133.7, 131.7, 129.8, 129.3, 128.5, 128.5, 128.3, 127.3, 127.1, 127.0, 51.4, 43.2, 39.2, 37.7; HRMS-ESI (−) m/z Calcd. for C25H24N3O3 [M−H]− 414.1824, Found 414.1823.
4.1.1.18. (S)-N-(2-((1-(Methyl(p-tolyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (19)
Yield 82%. 1H NMR (600 MHz, CDCl3) δ 7.80 (d, J = 7.4 Hz, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.43−7.41 (m, 2H), 7.21−7.20 (m, 3H), 7.14 (d, J = 8.0 Hz, 2H), 6.94−6.90 (m, 4H), 6.76 (brs, 1H), 4.86 (q, J = 7.3 Hz, 1H), 4.14−4.08 (m, 2H), 3.22 (s, 3H), 2.95 (dd, J = 13.3, 7.2 Hz, 1H), 2.77 (dd, J = 13.3, 7.0 Hz, 1H), 2.37 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 171.2, 167.9, 167.3, 139.7, 138.2, 136.1, 133.8, 131.7, 130.4, 129.3, 128.5, 128.4, 127.1, 127.0, 126.9, 51.3, 43.1, 39.2, 37.8, 21.1; HRMS-ESI (−) m/z Calcd. for C26H26N3O3 [M−H]− 428.1988, Found 428.1989.
4.1.1.19. (S)-N-(2-((1-((4-Chlorophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (20)
Yield 90%. 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 7.2 Hz, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.7 Hz, 2H), 7.28−7.22 (m, 5H), 7.03–6.96 (m, 4H), 6.69 (s, 1H), 4.79 (dd, J = 14.7, 8.2 Hz, 1H), 4.14 (d, J = 7.4 Hz, 2H), 3.18 (s, 3H), 2.96 (dd, J = 13.1, 8.4 Hz, 1H), 2.81 (dd, J = 13.1, 6.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 171.2, 168.1, 167.4, 140.8, 135.9, 134.0, 133.7, 131.8, 129.8, 129.4, 128.7, 128.6, 128.6, 127.1, 127.1, 51.4, 43.2, 39.5, 37.7; HRMS-ESI (−) m/z Calcd. for C25H23ClN3O3 [M−H]− 448.1433, Found 448.1440.
4.1.1.20. (S)-N-(2-((1-((3-Chlorophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (21)
Yield 79%. 1H NMR (600 MHz, CD3OD) δ 7.87 (d, J = 7.4 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.46 (t, J = 7.7 Hz, 2H), 7.33–7.22 (m, 6H), 6.96–6.94 (m, 3H), 4.62 (t, J = 7.8 Hz, 1H), 4.04 (s, 2H), 3.13 (s, 3H), 2.96 (dd, J = 13.1, 8.5 Hz, 1H), 2.79 (dd, J = 13.1, 6.6 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 173.0, 171.0, 170.4, 145.1, 137.7, 135.9, 135.0, 132.9, 131.8, 130.3, 129.7, 129.6, 129.4, 128.7, 128.5, 128.1, 127.3, 53.4, 43.7, 39.6, 38.0; HRMS-ESI (−) m/z Calcd. for C25H23ClN3O3 [M−H]− 448.1433, Found 448.1439.
4.1.1.21. (S)-N-(2-((1-((4-Fluorophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (22)
Yield 79%. 1H NMR (600 MHz, CD3OD) δ 7.86 (d, J = 7.2 Hz, 2H), 7.55 (t, J = 8.4 Hz, 1H), 7.48–7.45 (m, 3H), 7.22–7.21 (m, 3H), 7.06–7.03 (m, 3H), 6.95–6.94 (m, 2H), 4.62 (t, J = 7.5 Hz, 1H), 4.02 (s, 2H), 3.14 (s, 3H), 2.97 (dd, J = 13.2, 7.9 Hz, 1H), 2.77 (dd, J = 13.2, 7.0 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 173.2, 171.0, 170.4, 163.4 (d, JCF = 246.9 Hz), 140.0, 137.9, 135.0, 132.9, 130.7 (d, JCF = 7.5 Hz), 130.3, 129.6, 129.5, 128.5, 128.0, 117.3 (d, JCF = 23.1 Hz), 53.2, 43.7, 39.4, 38.1; HRMS-ESI (−) m/z Calcd. for C25H23FN3O3 [M−H]− 432.1729, Found 432.1734.
4.1.1.22. (S)-N-(2-((1-((3-Fluorophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (23)
Yield 80%. 1H NMR (600 MHz, CDCl3) δ 7.82 (d, J = 7.6 Hz, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.44−7.42 (m, 2H), 7.32−7.28 (m, 1H), 7.25−7.21 (m, 3H), 7.04−6.95 (m, 5H), 6.73 (s, 1H), 6.33 (s, 1H), 4.85−4.81 (m, 1H), 4.17−4.11 (m, 2H), 3.20 (s, 3H), 2.96 (dd, J = 13.1, 8.4 Hz, 1H), 2.82 (dd, J = 13.1, 6.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 171.2, 168.2, 167.5, 162.8 (d, JCF = 249.3 Hz), 143.7 (d, JCF = 9.5 Hz), 135.9, 133.7, 131.8, 130.8 (d, JCF = 9.1 Hz), 129.3, 128.6, 128.6, 127.2, 123.2 (d, JCF = 2.9 Hz), 115.4 (d, JCF = 20.9 Hz), 114.7 (d, JCF = 22.2 Hz), 51.5, 43.2, 39.5, 37.6; HRMS-ESI (−) m/z Calcd. for C25H23FN3O3 [M−H]− 432.1729, Found 432.1732.
4.1.1.23. (S)-N-(2-((1-((3-Bromophenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (24)
Yield 80%. 1H NMR (600 MHz, CD3OD) δ 7.87 (d, J = 7.3 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.48–7.45 (m, 3H), 7.25–7.22 (m, 5H), 6.98–6.95 (m, 3H), 4.62 (t, J = 7.8 Hz, 1H), 4.04 (s, 2H), 3.13 (s, 3H), 2.96 (dd, J = 13.1, 8.6 Hz, 1H), 2.79 (dd, J = 13.1, 6.6 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 173.0, 171.0, 170.4, 145.2, 137.7, 135.0, 132.9, 132.4, 132.1, 131.5, 130.3, 129.7, 129.6, 128.5, 128.2, 127.7, 123.6, 53.4, 43.7, 39.6, 38.0; HRMS-ESI (−) m/z Calcd. for C25H23BrN3O3 [M−H]− 492.0928, Found 492.0933.
4.1.1.24. (S)-N-(2-((1-(Methyl(3-(trifluoromethyl)phenyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (25)
Yield 89%. 1H NMR (600 MHz, CD3OD) δ 7.87 (d, J = 7.5 Hz, 2H), 7.63 (d, J = 7.7 Hz, 1H), 7.56–7.46 (m, 5H), 7.24–7.19 (m, 4H), 6.94 (d, J = 6.8 Hz, 2H), 4.60–4.57 (m, 1H), 4.04 (s, 2H), 3.17 (s, 3H), 2.97 (dd, J = 13.0, 8.6 Hz, 1H), 2.80 (dd, J = 13.1, 6.6 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 173.1, 171.1, 170.4, 144.6, 137.7, 135.1, 132.9, 132.8, 131.7, 130.2, 129.7, 129.6, 128.5, 128.2, 126.0, 125.4, 124.9 (q, JCF = 272.6 Hz), 124.0, 53.4, 43.7, 39.6, 38.0; HRMS-ESI (−) m/z Calcd. for C26H23F3N3O3 [M−H]− 482.1697, Found 482.1700.
4.1.1.25. (S)-N-(2-((1-(Ethyl(phenyl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoethyl)benzamide (26)
Yield 70%. 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 7.6 Hz, 2H), 7.49−7.45 (m, 2H), 7.40−7.34 (m, 5H), 7.26−7.25 (m, 1H), 7.19−7.16 (m, 3H), 6.93−6.92 (m, 2H), 4.73 (q, J = 7.4 Hz, 1H), 4.13−4.08 (m, 2H), 3.85−3.79 (m, 1H), 3.66−3.60 (m, 1H), 2.97 (dd, J = 13.3, 7.1 Hz, 1H), 2.78 (dd, J = 13.3, 7.3 Hz, 1H), 1.09 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.8, 168.3, 167.4, 140.7, 136.3, 133.8, 131.6, 129.6, 129.4, 128.5, 128.4, 128.4, 127.2, 126.9, 51.8, 44.8, 43.1, 39.1, 12.8; HRMS-ESI (−) m/z Calcd. for C26H26N3O3 [M−H]− 428.1988, Found 428.1990.
4.1.1.26. (S)-1-(Benzoylglycyl-L-phenylalanyl)-N-phenylpyrrolidine-2-carboxamide (27)
Yield 68%. 1H NMR (600 MHz, CDCl3) δ 9.73 (s, 1H), 8.31 (d, J = 9.1 Hz, 1H), 7.82 (d, J = 7.3 Hz, 2H), 7.59 (d, J = 7.7 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.47−7.44 (m, 2H), 7.29−7.27 (m, 2H), 7.12−7.07 (m, 3H), 7.04−7.00 (m, 3H), 5.13−5.10 (m, 1H), 4.85−4.83 (m, 1H), 4.49−4.45 (m, 1H), 4.14–4.11 (m, 1H), 3.85–3.81 (m, 1H), 3.59–3.56 (m, 1H), 3.12 (dd, J = 13.7, 6.0 Hz, 1H), 2.85 (dd, J = 13.7, 7.8 Hz, 1H), 2.27–2.17 (m, 3H), 2.02–1.98 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 170.7, 169.6, 168.1, 167.9, 138.7, 135.8, 133.5, 132.0, 129.6, 129.0, 128.7, 128.4, 127.1, 126.9, 123.9, 119.6, 61.0, 51.9, 47.9, 43.4, 39.0, 29.2, 24.9; HRMS-ESI (−) m/z Calcd. for C29H29N4O4 [M−H]− 497.2194, Found 497.2192.
4.2. Thermal shift assays (TSAs)
TSAs used purified covalently-crosslinked hexameric CAA14C/E45C/W184A/M185A (CA121). CA121 cloned in a pET11a expression plasmid was kindly provided by Dr. Owen Pornillos (University of Virginia, Charlottesville, VA, USA). CA121 was expressed in Escherichia coli BL21(DE3)RIL and purified according to reported protocols37. The TSAs were conducted as previously described55, 56, 57, with each reaction containing 7.5 μmol/L CA121 in 50 mmol/L sodium phosphate buffer (pH 8.0), 1× Sypro Orange Protein Gel Stain (Life Technologies, Carlsbad, CA, USA), and either 1% DMSO (control) or 20 μmol/L compound (1% DMSO final). The plate was heated from 25 to 95 °C with a heating rate of 0.2 °C every 10 s in the PikoReal real-time PCR system (Thermo Fisher Scientific, Waltham, MA, USA) or the QuantStudio 3 real-time PCR system (Thermo Fisher Scientific). The fluorescence intensity was measured with an Ex range of 475–500 nm and Em range of 520–590 nm. The difference in the melting temperature (ΔTm) of CA121 in DMSO (T0) versus in the presence of compound (Tm) were calculated using the following Eq. (1):
| ΔTm (°C)=Tm−T0. | (1) |
4.3. Virus production
The wild-type laboratory HIV-1 strain, HIV-1NL4-358, was produced using a pNL4-3 vector (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, Bethesda, MD, USA). HIV-1NL4-3 was generated by transfecting HEK 293FT cells with 10 μg of pNL4-3 vector and FuGENE®HD Transfection Reagent (Promega, Madison, WI, USA) in a T75 flask. Supernatant was harvested 48–72 h post-transfection and transferred to MT2 cells for viral propagation. Virus was harvested upon observation of syncytia formation, typically after 3–5 days. The viral supernatant was then concentrated using 8% w/v PEG 8000 overnight at 4 °C, followed by centrifugation for 40 min at 3500 rpm (Sigma 4-16S, Sigma Laborzentrifugen, Osterode am Harz, Germany). The resulting viral-containing pellet was concentrated 10-fold by resuspension in DMEM without FBS and stored at −80 °C.
4.4. Anti-HIV-1 and cytotoxicity assays
Anti-HIV-1 activity of PF74 and related analogs was examined in TZM-GFP cells. The potency of HIV-1 inhibition was determined based on the inhibition of viral LTR-activated GFP expression in the presence of compounds compared to DMSO controls. Briefly, TZM-GFP cells were plated at density of 1 × 104 cells per well in a 96-well plate. After 24 h, media was replaced with increasing concentrations of compound. Cells were exposed to HIV-1NL4-3 (MOI = 1) 24 h post treatment. After 48 h incubation, anti-HIV-1 activity was determined by counting the amount of GFP positive cells on a Cytation™ 5 Imaging Reader (BioTek, Winooski, VT, USA) and 50% effective concentration (EC50) values were determined.
The cytotoxicity of each compound was also determined in TZM-GFP cells. Cells plated at a density of 1 × 104 cells per well in a 96-well plate were continuously exposed to increasing concentrations compounds over a period of 72 h. The number of viable cells in each well was determined using an XTT Cell Proliferation Kit (R&D Systems, Inc., Minneapolis, MN, USA), and 50% cytotoxicity concentration (CC50) values were determined. All cell-based assays were conducted in duplicate and in at least two independent experiments.
To obtain EC50 and CC50 dose response curves, values were plotted in GraphPad Prism 5 and analyzed with the loginhibitor vs. normalized response–variable slope equation as Eq. (2):
| Y = 100/(1+10(((LogIC50)‒X)×HillSlope)) | (2) |
where Y is the percent response, X is the logarithm of the concentration (μmol/L) of a compound, IC50 is the concentration (μmol/L) of the compound that gives a response half-way between the top and bottom of the curve, and HillSlope describes the steepness of the curve. Final values were calculated for each independent assay and average values for all assays were calculated. Statistical analysis (calculation of standard deviation) was performed using Microsoft Excel.
4.5. Microsomal stability assay
The in vitro microsomal stability assay was performed using commercially available human and mouse liver microsomes (Sekisui XenoTech, Kansas City, KS, USA). Briefly, the test compound (1 μmol/L final concentration) was pre-incubated with or without 0.5 μmol/L cobicistat (medchemexpress.com, Monmouth Junction, NJ, USA) in a 0.1 mol/L potassium phosphate buffer solution (pH 7.4, 37 °C) containing liver microsomal protein (0.5 mg/mL final concentration) and MgCl2 (1 mmol/L final concentration). After a 15 min pre-incubation, the enzyme cofactor nicotinamide adenine dinucleotide phosphate (NADPH, 1 mmol/L final concentration) was added to initiate the reaction. All reactions were carried out in duplicate and a negative control without NADPH was performed in parallel to assess any chemical instability or non-NADPH dependent enzymatic degradation. A 50 μL of reaction mixture was taken at different time points (0, 5, 15, 30, and 60 min), and quenched with 150 μL of acetonitrile containing an appropriate internal standard and 0.1% formic acid. The samples were then vortexed and centrifuged at 15,000 rpm (Eppendorf centrifuge 5424R, Enfield, CT, USA) for 5 min at 4 °C. The supernatants were collected and analyzed by a LC−MS/MS system consisting of an Agilent 1260 Infinity HPLC (Agilent Technologie) and an AB Sciex QTrap 5500 mass spectrometer (AB Sciex LLC., Toronto, Canada) to determine the in vitro metabolic half-life (t1/2).
4.6. Molecular modeling
PF74-bound full length native HIV-1 CA (PDB ID: 4XFZ)36 was utilized to perform molecular modeling using the Schrödinger small molecule drug discovery suite 2019-159 (Schrödinger Inc., New York, NY, USA). Maestro60 (Schrödinger Inc.) was employed to analyze the above crystal structure. A standard docking protocol entailed preparation of the protein of interest, grid generation, ligand preparation, and docking as key steps. Preparation of the protein involved refinement of the crystal structure using the protein preparation wizard61 (Schrödinger Inc.), in which the missing hydrogen atoms, side chains, and loops were added using prime, followed by minimization using the OPLS 3e force field62 to optimize the hydrogen bonding network and converge the heavy atoms to an rmsd of 0.3 Å. The binding site around the native ligand PF74, covering all the key residues within 12 Å, was defined using the receptor grid generation tool in Maestro (Schrödinger Inc.). After drawing compounds in Maestro, different conformers of those compounds were generated using LigPrep63 at pH of 7±2 to serve as input for docking process. Finally, Glide XP64(Glide, version 8.2, New York, NY, USA) was used to perform the docking with the van der Waals radii of nonpolar atoms for each of the ligands scaled by a factor of 0.8. Protein flexibility was accounted by post docking refinement and minimization under implicit solvent. Numbering of residues of HIV-1 CA, used in the discussion and the figures, was based on the full length native HIV-1 CA.
Acknowledgments
This research was supported by the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, USA, grant number R01AI120860 (to Stefan G. Sarafianos and Zhengqiang Wang). We thank the Minnesota Supercomputing Institute (Minneapolis, MN, USA) for molecular modeling resources.
Author contributions
Stefan G. Sarafianos and Zhengqiang Wang conceptualized the research. Lei Wang and Sanjeev Kumar V. Vernekar designed, synthesized and characterized all compounds. Rajkumar Lalji Sahani conducted molecular docking. Jiashu Xie performed the metabolic stability assays. Mary C. Casey, Karen A. Kirby, Haijuan Du, Huanchun Zhang, and Philip R. Tedbury performed TSA, antiviral and cytotoxicity assays. Lei Wang and Zhengqiang Wang wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2020.07.016.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.AIDSinfo . March 2020. FDA-approved HIV medicines.https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets/21/58/fda-approved-hiv-medicines Available from: [Google Scholar]
- 2.Ndung'u T., McCune J.M., Deeks S.G. Why and where an HIV cure is needed and how it might be achieved. Nature. 2019;576:397–405. doi: 10.1038/s41586-019-1841-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davenport M.P., Khoury D.S., Cromer D., Lewin S.R., Kelleher A.D., Kent S.J. Functional cure of HIV: the scale of the challenge. Nat Rev Immunol. 2019;19:45–54. doi: 10.1038/s41577-018-0085-4. [DOI] [PubMed] [Google Scholar]
- 4.Carnes S.K., Sheehan J.H., Aiken C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr Opin HIV AIDS. 2018;13:359–365. doi: 10.1097/COH.0000000000000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thenin-Houssier S., Valente S.T. HIV-1 capsid inhibitors as antiretroviral agents. Curr HIV Res. 2016;14:270–282. doi: 10.2174/1570162x14999160224103555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J.Y., Liu X.Y., de Clercq E. Capsid (CA) protein as a novel drug target: recent progress in the research of HIV-1 CA inhibitors. Mini Rev Med Chem. 2009;9:510–518. doi: 10.2174/138955709787847958. [DOI] [PubMed] [Google Scholar]
- 7.Ganser B.K., Li S., Klishko V.Y., Finch J.T., Sundquist W.I. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 8.Li S., Hill C.P., Sundquist W.I., Finch J.T. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature. 2000;407:409–413. doi: 10.1038/35030177. [DOI] [PubMed] [Google Scholar]
- 9.Freed E.O. HIV-1 assembly, release and maturation. Nat Rev Microbiol. 2015;13:484–496. doi: 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fassati A. Multiple roles of the capsid protein in the early steps of HIV-1 infection. Virus Res. 2012;170:15–24. doi: 10.1016/j.virusres.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 11.Le Sage V., Mouland A.J., Valiente-Echeverria F. Roles of HIV-1 capsid in viral replication and immune evasion. Virus Res. 2014;193:116–129. doi: 10.1016/j.virusres.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 12.Novikova M., Zhang Y., Freed E.O., Peng K. Multiple roles of HIV-1 capsid during the virus replication cycle. Virol Sin. 2019;34:119–134. doi: 10.1007/s12250-019-00095-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell E.M., Hope T.J. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol. 2015;13:471–483. doi: 10.1038/nrmicro3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao D., Wu J., Wu Y.T., Du F., Aroh C., Yan N. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lahaye X., Satoh T., Gentili M., Cerboni S., Conrad C., Hurbain I. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Ganser-Pornillos B.K., Yeager M., Sundquist W.I. The structural biology of HIV assembly. Curr Opin Struct Biol. 2008;18:203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sundquist W.I., Krausslich H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2012;2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byeon I.J., Meng X., Jung J., Zhao G., Yang R., Ahn J. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell. 2009;139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rankovic S., Ramalho R., Aiken C., Rousso I. PF74 reinforces the HIV-1 capsid to impair reverse transcription-induced uncoating. J Virol. 2018;92 doi: 10.1128/JVI.00845-18. e00845-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forshey B.M., von Schwedler U., Sundquist W.I., Aiken C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol. 2002;76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita M., Engelman A.N. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 2017;25:741–755. doi: 10.1016/j.tim.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sayah D.M., Sokolskaja E., Berthoux L., Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 23.Stremlau M., Owens C.M., Perron M.J., Kiessling M., Autissier P., Sodroski J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 24.Achuthan V., Perreira J.M., Sowd G.A., Puray-Chavez M., McDougall W.M., Paulucci-Holthauzen A. Capsid-CPSF6 interaction licenses nuclear HIV-1 trafficking to sites of viral DNA integration. Cell Host Microbe. 2018;24:392–404 e8. doi: 10.1016/j.chom.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bejarano D.A., Peng K., Laketa V., Borner K., Jost K.L., Lucic B. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. Elife. 2019;8 doi: 10.7554/eLife.41800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodward C.L., Prakobwanakit S., Mosessian S., Chow S.A. Integrase interacts with nucleoporin NUP153 to mediate the nuclear import of human immunodeficiency virus type 1. J Virol. 2009;83:6522–6533. doi: 10.1128/JVI.02061-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matreyek K.A., Yucel S.S., Li X., Engelman A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buffone C., Martinez-Lopez A., Fricke T., Opp S., Severgnini M., Cifola I. Nup153 unlocks the nuclear pore complex for HIV-1 nuclear translocation in nondividing cells. J Virol. 2018;92 doi: 10.1128/JVI.00648-18. e00648-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dharan A., Talley S., Tripathi A., Mamede J.I., Majetschak M., Hope T.J. KIF5B and Nup358 cooperatively mediate the nuclear import of HIV-1 during infection. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meehan A.M., Saenz D.T., Guevera R., Morrison J.H., Peretz M., Fadel H.J. A cyclophilin homology domain-independent role for Nup358 in HIV-1 infection. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1003969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fricke T., White T.E., Schulte B., de Souza Aranha Vieira D.A., Dharan A., Campbell E.M. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology. 2014;11:68. doi: 10.1186/s12977-014-0068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu B., Pan Q., Liang C. Role of MxB in alpha interferon-mediated inhibition of HIV-1 infection. J Virol. 2018;92 doi: 10.1128/JVI.00422-18. e00422-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franke E.K., Yuan H.E., Luban J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature. 1994;372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 34.Thali M., Bukovsky A., Kondo E., Rosenwirth B., Walsh C.T., Sodroski J. Functional association of cyclophilin A with HIV-1 virions. Nature. 1994;372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- 35.Kim K., Dauphin A., Komurlu S., McCauley S.M., Yurkovetskiy L., Carbone C. Cyclophilin A protects HIV-1 from restriction by human TRIM5α. Nat Microbiol. 2019;4:2044–2051. doi: 10.1038/s41564-019-0592-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gres A.T., Kirby K.A., KewalRamani V.N., Tanner J.J., Pornillos O., Sarafianos S.G. STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science. 2015;349:99–103. doi: 10.1126/science.aaa5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pornillos O., Ganser-Pornillos B.K., Kelly B.N., Hua Y., Whitby F.G., Stout C.D. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao G., Perilla J.R., Yufenyuy E.L., Meng X., Chen B., Ning J. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013;497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharya A., Alam S.L., Fricke T., Zadrozny K., Sedzicki J., Taylor A.B. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A. 2014;111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ning J., Zhong Z., Fischer D.K., Harris G., Watkins S.C., Ambrose Z. Truncated CPSF6 forms higher-order complexes that bind and disrupt HIV-1 capsid. J Virol. 2018;92 doi: 10.1128/JVI.00368-18. e00368-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu G., Zalloum W.A., Meuser M.E., Jing L., Kang D., Chen C.H. Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur J Med Chem. 2018;158:478–492. doi: 10.1016/j.ejmech.2018.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu J.P., Francis A.C., Meuser M.E., Mankowski M., Ptak R.G., Rashad A.A. Exploring modifications of an HIV-1 capsid inhibitor: design, synthesis, and mechanism of action. J Drug Des Res. 2018;5:1070. [PMC free article] [PubMed] [Google Scholar]
- 43.Vernekar S.K.V., Sahani R.L., Casey M.C., Kankanala J., Wang L., Kirby K.A. Toward structurally novel and metabolically stable HIV-1 capsid-targeting small molecules. Viruses. 2020;12:452. doi: 10.3390/v12040452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L., Casey M.C., Vernekar S.K.V., Do H.T., Sahani R.L., Kirby K.A. Chemical profiling of HIV-1 capsid-targeting antiviral PF74. Eur J Med Chem. 2020;200:112427. doi: 10.1016/j.ejmech.2020.112427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun L., Huang T., Dick A., Meuser M.E., Zalloum W.A., Chen C.H. Design, synthesis and structure−activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur J Med Chem. 2020;190:112085. doi: 10.1016/j.ejmech.2020.112085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun L., Dick A., Meuser M.E., Huang T., Zalloum W.A., Chen C.H. Design, synthesis, and mechanism study of benzenesulfonamide-containing phenylalanine derivatives as novel HIV-1 capsid inhibitors with improved antiviral activities. J Med Chem. 2020;63:4790–4810. doi: 10.1021/acs.jmedchem.0c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazzara P.R., Moore T.W. Scaffold-hopping as a strategy to address metabolic liabilities of aromatic compounds. RSC Med Chem. 2020;11:18–29. doi: 10.1039/c9md00396g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guengerich F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 49.Kirchmair J., Goller A.H., Lang D., Kunze J., Testa B., Wilson I.D. Predicting drug metabolism: experiment and/or computation?. Nat Rev Drug Discov. 2015;14:387–404. doi: 10.1038/nrd4581. [DOI] [PubMed] [Google Scholar]
- 50.Xu L.H., Liu H.T., Murray B.P., Callebaut C., Lee M.S., Hong A. Cobicistat (GS-9350): a potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer. ACS Med Chem Lett. 2010;1:209–213. doi: 10.1021/ml1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zanger U.M., Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 52.Wacher V.J., Silverman J.A., Zhang Y., Benet L.Z. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 1998;87:1322–1330. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- 53.Xu L., Desai M.C. Pharmacokinetic enhancers for HIV drugs. Curr Opin Invest Drugs. 2009;10:775–786. [PubMed] [Google Scholar]
- 54.Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G. The halogen bond. Chem Rev. 2016;116:2478–2601. doi: 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lo M.C., Aulabaugh A., Jin G., Cowling R., Bard J., Malamas M. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 56.Miyazaki Y., Doi N., Koma T., Adachi A., Nomaguchi M. Novel in vitro screening system based on differential scanning fluorimetry to search for small molecules against the disassembly or assembly of HIV-1 capsid protein. Front Microbiol. 2017;8:1413. doi: 10.3389/fmicb.2017.01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pantoliano M.W., Petrella E.C., Kwasnoski J.D., Lobanov V.S., Myslik J., Graf E. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 58.Adachi A., Gendelman H.E., Koenig S., Folks T., Willey R., Rabson A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schrödinger . Schrödinger LLC.; New York, NY, USA: 2019. Schrödinger small-molecule drug discovery suite 2019-1. [Google Scholar]
- 60.Schrödinger . Maestro, Schrödinger LLC.; New York, NY, USA: 2019. Schrödinger release 2019-1. [Google Scholar]
- 61.Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 62.Jorgensen W.L., Maxwell D.S., TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 63.Schrödinger . LigPrep, Schrödinger LLC.; New York, NY, USA: 2019. Schrödinger release 2019-1. [Google Scholar]
- 64.Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.