

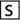
Abstract
Severe acute respiratory syndrome (SARS) coronavirus is a novel human coronavirus and is responsible for SARS infection. SARS coronavirus 3C-like proteinase (SARS 3CLpro) plays key roles in viral replication and transcription and is an attractive target for anti-SARS drug discovery. In this report, we quantitatively characterized the dimerization features of the full-length and N-terminal residues 1–7 deleted SARS 3CLpros by using glutaraldehyde cross-linking SDS-PAGE, size-exclusion chromatography, and isothermal titration calorimeter techniques. Glutaraldehyde cross-linking SDS-PAGE and size-exclusion chromatography results show that, similar to the full-length SARS 3CLpro, the N-terminal deleted SARS 3CLpro still remains a dimer/monomer mixture within a wide range of protein concentrations. Isothermal titration calorimeter determinations indicate that the equilibrium dissociation constant (Kd) of the N-terminal deleted proteinase dimer (262 μm) is very similar to that of the full-length proteinase dimer (227 μm). Enzymatic activity assay using the fluorescence resonance energy transfer method reveals that N-terminal deletion results in almost complete loss of enzymatic activity for SARS 3CLpro. Molecular dynamics and docking simulations demonstrate the N-terminal deleted proteinase dimer adopts a state different from that of the full-length proteinase dimer, which increases the angle between the two protomers and reduces the binding pocket that is not beneficial to the substrate binding. This conclusion is verified by the surface plasmon resonance biosensor determination, indicating that the model substrate cannot bind to the N-terminal deleted proteinase. These results suggest the N terminus is not indispensable for the proteinase dimerization but may fix the dimer at the active state and is therefore vital to enzymatic activity.
Between the end of year 2002 and June of year 2003, a highly contagious illness called severe acute respiratory syndrome (SARS)1 broke out in China and quickly spread to more than 30 other countries. A novel coronavirus, named SARS coronavirus (SARS-CoV), was soon identified as the etiological agent of the infection (1, 2, 3). It was revealed that the open reading frame of the replicase gene in SARS-CoV genome, similar to other coronaviruses (4, 5), features the sequence motifs of both papain-like proteinase and 3C-like proteinase (3CLpro). SARS 3CLpro is fully conserved among all released SARS coronavirus genome sequences and is highly homologous to other coronavirus 3CL proteinases (6, 7). Because of its functional indispensability in the coronavirus life cycle, SARS 3CLpro has become an attractive target in discovering new anti-SARS agents.
Recently, two crystal structures of 3CL proteinases from human coronavirus (strain 229E) and transmissible gastroenteritis virus (TGEV) have been determined and confirmed in a remarkable degree of conservation for the substrate-binding sites (8). The reported crystal structures of SARS 3CLpro and its complex with an inhibitor revealed substantially pH-dependent conformational changes and an unexpected model for inhibitor binding (9). All the reported crystal structures indicated that coronavirus 3CL proteinases are well conserved at the three-dimensional level. Structurally, SARS 3CLpro contains the following three domains: domains I and II (residues 1–184) have an antiparallel β-barrel structure forming a chymotrypsin-fold, the substrate-binding site is located in a cleft between these two domains; whereas residues 201–303 form a third compact α-helical domain (domain III) connecting to domain II by a long loop (residues 185–200) (8, 9, 10).
As the crystal structures of 3CL proteinases in different coronaviruses give similar dimeric structures (8, 9) and nearly all side chains of 3CLpro involved in the formation of the dimer are conserved, it has been proposed that the dimer might be the biological functional form of 3CLpro (8). From the dimeric structures of coronavirus 3CLpros, it has been also predicted that domain III might play a role in substrate recognition and be responsible for positioning the N terminus of one protomer (monomer) to interact with the active site of the other protomer (9, 10, 11). The critical role of domain III in dimerization and enzymatic activity of SARS 3CLpro has been presented by a recent study (12). In addition, Lai and co-workers (13) reported that SARS 3CLpro exists as a monomer/dimer mixture at a relatively high protein concentration (∼4 mg/ml) and is exclusively monomer at a lower protein concentration (∼0.2 mg/ml) in solution, and only the dimer was the active form of the proteinase.
Based on the crystal structures and biochemical studies of several coronavirus 3CLpros, one hypothesis was proposed that the N-terminal residues 1–7 might play an important role in both dimerization and enzymatic activity of SARS 3CLpro (9). However, our molecular dynamics simulations revealed that the N terminus was not necessary for the dimer formation of SARS 3CLpro, i.e. the N-terminal deleted SARS 3CLpro may possibly form a dimer with a new state different from the full-length proteinase. Therefore, further systematic analyses of the role of the N terminus in dimerization and enzymatic activity of SARS 3CLpro may contribute to understanding the biological function of 3CLpros.
In the following, we report our biochemical and biophysical studies of the dimerization and proteolytic activities of the full-length and N-terminal residues 1–7 deleted SARS 3CLpros using CD spectroscopy, fluorescence resonance energy transfer (FRET), glutaraldehyde cross-linking SDS-PAGE, size-exclusion chromatography (SEC), and isothermal titration calorimeter (ITC) techniques. The experimental results are consistent with the MD simulations on these two 3CLpros. It is demonstrated that the N terminus of SARS 3CLpro is necessary for its enzymatic activity but not for dimerization. The N-terminal deleted SARS 3CLpro can still form a dimer, and the binding affinity between the two protomers is almost equal to that of the full-length SARS 3CLpro. Compared with the full-length SARS 3CLpro, the N-terminal deleted SARS 3CLpro forms a dimer adopting a different conformational state, which reduces substrate-binding pocket, thereby abolishing the proteolytic activity of SARS 3CLpro. This conclusion is verified by molecular docking and surface plasmon resonance (SPR) biosensor determination, indicating that the N-terminal deleted SARS 3CLpro has almost completely lost the binding affinity to a model peptide substrate.
EXPERIMENTAL PROCEDURES
Materials—The restriction and modifying enzymes used in this work were purchased from Takara, and the vector pQE30 and the bacterial strain M15 were from Qiagen. TRIzol and Superscript II reverse transcriptase were bought from Invitrogen. DNase and RNase were from New England Biolabs. The chelating column and low molecular weight markers for SDS-PAGE were from Amersham Biosciences. All other chemicals were of reagent grade or ultra-pure quality and purchased from Sigma.
The Full-length and N-terminal Deleted SARS 3CLpros Preparations—The full-length SARS 3CLpro was obtained according to our published method (14). The proteinase was highly pure according to SDS-PAGE and dialyzed to 20 mm sodium phosphate, pH 7.5, containing 100 mm NaCl, 5 mm dithiothreitol (DTT), and 1 mm EDTA. The purified protein was further confirmed by N-terminal sequencing and mass spectrometry and concentrated by Centriprep (Millipore). The protein concentration used in all experiments was determined from the absorbance at 280 nm.
The cloning of N-terminal deleted SARS 3CLpro (residues 8–303) was processed by a modified PCR method using pQE30-SARS 3CLpro as template. The nucleotide sequences of the primers used for N-terminal deletion mutagenesis are as follows: 5′-AATGGATCCACCATGTTCCCGTCAGGCAAAG-3′ (forward) and 5′-TATAAGCTTTTGGAAGGTAACACCAGAGCATTG-3′ (reverse). The pQE30-SARS 3CLpro plasmid encoding the N-terminal deleted form of SARS 3CLpro was verified by sequencing and then transformed into Escherichia coli M15 cells, and the recombinant protein was expressed and purified in a procedure similar to that of the full-length proteinase. The purity and structural integrity of the N-terminal deleted proteinase were analyzed by SDS-PAGE, N-terminal sequencing, and mass spectrometry. The concentrated proteinase was stored in 20 mm sodium phosphate, pH 7.5, 100 mm NaCl, 5 mm DTT, 1 mm EDTA, at -20 °C.
CD Spectroscopy—CD spectra were recorded on a Jasco-810 spectropolarimeter collected with thermal controller. The protein sample was prepared in 20 mm sodium phosphate, pH 7.5, 100 mm NaCl at 25 °C with concentration of 10 μm. Far-UV CD spectra from 195 to 250 nm were collected with 1 nm bandwidth using a 0.1-cm path length cuvette and normalized by subtracting the base line recorded for the buffer under identical conditions. Thermally induced unfolding with a temperature range from 30 to 75 °C was monitored at 222 nm by heating velocity of 2 °C/min. Each measurement was repeated three times, and the final result was the average of the three independent scans.
Enzymatic Activity Assay—The enzymatic activities of the full-length and N-terminal deleted SARS 3CLpros were measured by FRET-based assays using a 12-amino acid fluorogenic substrate, EDANS-VN-STLQSGLRK(Dabcyl)-M (supplemental Fig. S1), that was synthesized and characterized by using a modified procedure described by methods published previously (15, 16). Stock solution of the fluorogenic substrate was prepared in dimethyl sulfoxide (Me2SO) and stored at -20 °C. Subsequent dilutions were performed using the assay buffer (20 mm sodium phosphate, pH 7.5, 100 mm NaCl, 5 mm DTT, and 1 mm EDTA) with the final concentration of Me2SO less than 1% (v/v). Kinetic assays were performed continuously in a 1-ml quartz cuvette with a 1-cm path length at 25 °C. During the assay, the enzyme (final concentration 1 μm) was preincubated for 30 min at 25 °C in the cuvette containing the assay buffer, followed by the addition of the fluorogenic substrate stock solution to a final concentration of 10 μm. The increase in emission fluorescence intensity was recorded at a 10-min interval on a Hitachi F-2500 fluorescence spectrophotometer connected with a thermostat. During the fluorescent measurement, the instrument was first set to zero with the fluorogenic substrate itself in the assay buffer. Cleavage of the substrate as a function of time was followed by monitoring the emission fluorescence intensity at a 490 nm wavelength upon excitation at 340 nm (slit width 10 nm), and the initial reaction velocity (ν0) was determined from the linear portion of the progress curve. The final emission fluorescence intensity of the totally hydrolyzed substrate was determined by adding the full-length SARS 3CLpro until no emission fluorescence intensity change at 490 nm wavelength was recorded. Enzymatic activity was the average of three parallel assays, and the activity of the full-length SARS 3CLpro was taken as 100%.
Glutaraldehyde Cross-linking SDS-PAGE—For the full-length and N-terminal deleted SARS 3CLpros (final concentration from 1 to 7 mg/ml in 20 mm sodium phosphate, pH 7.5, 100 mm NaCl, 5 mm DTT, 1 mm EDTA), an aliquot of 25% (v/v) glutaraldehyde was added to make a final concentration of 0.01 or 0.1% glutaraldehyde. This sample was incubated at 25 °C for 30 min followed by quenching the cross-linking reaction by adding 1.25 μl of 1.4 m ethanolamine, pH 8.0. After centrifugation (12,000 rpm, 4 °C), the precipitate was redissolved in loading buffer and heated at 100 °C for 5 min. SDS-PAGE was run with 10% gels.
Size-exclusion Chromatography (SEC) Analysis—The dimer-monomer equilibria of the full-length and N-terminal deleted SARS 3CLpros were studied by size-exclusion chromatography on a HiLoad 16/60 Superdex 75 prep grade column through an AKTA FPLC system (Amersham Biosciences). Buffer used was 20 mm sodium phosphate, pH 7.5, 100 mm NaCl, 5 mm DTT, and 1 mm EDTA. The buffer was degassed, and the column was equilibrated with the buffer before injecting protein samples. Protein samples with a wide range of concentrations from 1 to 7 mg/ml were loaded on the column and then eluted with the buffer at a flow rate of 1 ml/min by detection of absorbance at 280 nm. The integrated area values of absorbance peaks were calibrated by AKTA FPLC evaluation software. The column was calibrated by using a low molecular weight gel filtration kit (Amersham Biosciences) with the following four marker proteins: ribonuclease A (15.6 kDa), chymotrypsinogen A (22.8 kDa), ovalbumin (48.9 kDa), and albumin (65.4 kDa). Standard calibration curves were obtained by plotting the ratio of peak elution volume (Ve) minus void volume (V 0) to total bed volume (Vt) minus void volume (Ve - V 0)/(Vt - V 0) against the logarithm of molecular mass.
ITC Determination—Dilution ITC analyses for monomer-dimer equilibrium were carried out at 25 °C using a VP-ITC titration calorimeter (Microcal, Northampton, MA). All solutions were thoroughly degassed before use. Before each experiment, the ITC sample cell was washed several times with doubly distilled water followed by loading with 1.43 ml of the assay buffer (20 mm sodium phosphate, pH 7.5, 100 mm NaCl, 1 mm EDTA), and the reference cell contained doubly distilled water. Titration was carried out using a 250-μl syringe filled with the concentrated full-length or N-terminal deleted SARS 3CLpro solution (20 mm sodium phosphate, pH 7.5, 100 mm NaCl, and 1 mm EDTA), with stirring at 300 rpm. The concentrations of protein were varied between 500 and 600 μm. A typical dilution ITC experiment involved 30 sequential injections of 10-μl volume of concentrated protein solution into the sample cell initially containing the assay buffer alone, with 20 s duration each and a 300-s interval between injections. This titration gave rise to a series of endothermic heat pulses which, after correction for appropriate buffer mixing control experiments under identical conditions, could be analyzed using Microcal ORIGIN software and purpose-made software in terms of a simple dimer-monomer dissociation model to give the dissociation constant of the dimer (Kd) and the enthalpy change of dimerization (
). Standard thermodynamic quantities were calculated using the general thermodynamic Equation 1.
| (Eq. 1) |
Molecular Dynamics Simulation—The molecular dynamics (MD) simulations were performed using the GROMACS package version 3.1.4 (17) with the GROMOS87 force field (18). The models for the MD simulations were built up based on the x-ray crystal structure of SARS 3CLpro at a 1.9-Å resolution (Protein Data Bank entry code 1UJ1) (9). To construct the three-dimensional model of the full-length SARS 3CLpro dimer (model 1) for the MD simulations, the coordinates of the missing residues, Ser-A1 and Gly-A2 in protomer A, were repaired using the molecular modeling software Sybyl 6.8 (molecular modeling package, version 6.8. St. Louis, MO, Tripos Associates). The initial three-dimensional model (model 2) of the N-terminal deleted proteinase dimer was constructed by deleting residues 1–7 from model 1. These two models were simulated in a water environment. Water was described by the simple point charge model (19). All bond lengths including hydrogen atoms were constrained by the LINCS algorithm (20). The simulation cell was a rectangular periodic box with the minimum distance between the protein and the box walls set to 0.75 nm, so that the protein did not directly interact with its own periodic image given the cut off. Numerical integration of the equations of motion used a time step of 2 fs, with atomic coordinates saved every 10 ps for analysis. To neutralize the modeled system and simulate the physiological conditions, 210 water molecules were replaced by 102 Cl- and 108 Na+ ions in model 1, and 222 water molecules were replaced by 106 Cl- and 116 Na+ ions in model 2; these ions were located at positions of the chosen water oxygen atoms. At the end, model 1 contained 5988 protein atoms and 38,091 solvent molecules in a 9.1 × 8.7 × 15.8-nm3 box, and model 2 contained 5804 solute atoms and 40,289 solvent molecules in a 9.3 × 9.7 × 14.7-nm3 box. All water molecules and ions with the whole fixed protein were energy-minimized with steepest descent until reaching the convergence value of 100 kJ/mol·nm. Afterward, minimizations were performed continuously on the protein by fixing the main chain and C-α atoms, respectively. After heating the systems (solute and solvent) to 300 K, two 5-ns MD simulations were performed on models 1 and 2, respectively.
Surface Plasmon Resonance (SPR) Biosensor Analysis—A 6-amino acid peptide (TSAVLQ) representing the N-terminal P6–P1 sequence of a reported best peptide substrate (TSAVLQSGFRK) for SARS 3CLpro (13) was synthesized as a model substrate. The binding affinities of the full-length and N-terminal deleted SARS 3CLpros to the substrate peptide were determined using the SPR biosensor technology (21). The measurements were performed using the dual flow cell Biacore 3000 instrument (Biacore AB, Uppsala, Sweden). The full-length or N-terminal deleted proteinase was immobilized to one of the four serial measurement cells of the sensor chip surface by standard Biacore procedure using HBS-EP buffer (10 mm HEPES, 150 mm NaCl, 3.4 mm EDTA, and 0.005% (v/v) surfactant P20, pH 7.4) and a flow rate of 5 μl/min. The protein was coupled covalently to the carboxymethylated dextran of a CM5 sensor chip by amino coupling. Carboxyl groups in the immobilization matrix were first activated by treatment with a fresh mixture of 0.2 m 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride and 50 mm N-hydroxysuccinimide for 7 min. To ensure an identical amount of the full-length and N-terminal deleted proteinases immobilized to the sensor chip, the protein at the same concentration (10 mg/ml in 10 mm sodium acetate, pH 5.2) was then injected over the surface until a desired immobilization level (4000 RU) was reached. Finally, unreacted N-hydroxysuccinimide esters were blocked by 1 m ethanolamine, pH 8.5, for 7 min.
Binding affinity measurements were performed in a continuous flow of 20 μl/min HBS-EP as the running buffer. The 6-amino acid peptide substrate was diluted in the running buffer and automatically injected in a series of increasing concentrations (20–500 μm). The binding responses were recorded continuously in resonance units (RU) at a frequency of 1 Hz as sensorgrams and presented as a function of time. Sensorgrams were processed by using automatic correction for nonspecific bulk refractive index effects. The equilibrium constants (KD) were estimated by the steady-state binding affinity fitting model encoded in the Biacore analysis software.
RESULTS
Folding Behavior and Thermal Stability of the N-terminal Deleted Proteinase—With the similar preparation strategy for the full-length SARS 3CLpro (14), the N-terminal deleted SARS 3CLpro was also successfully cloned and expressed in E. coli M15 cells. The majority of the proteins could be obtained in the soluble fraction of the cell lysate after introduction with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 25 °C for 5 h. The molecular weights of the purified proteins were characterized by LCQ-DECA MS (Thermofinnigan, San Jose, CA) (data not shown). The measured masses of the full-length and N-terminal deleted proteinases are 35,831 and 35,055 daltons, respectively, which are well in agreement with the values calculated from the protein sequences.
Fig. 1 shows the CD spectra of the full-length and N-terminal deleted proteinases. Far-UV spectra of the two proteins give a positive peak at 196 nm characteristic of a β-sheet structure, and dual negative peaks at 209 and 222 nm, respectively, typical of an α-helical structure (Fig. 1a), indicating that the secondary structure of the N-terminal deleted SARS 3CLpro is similar to that of the full-length proteinase, and also similar to those of TGEV and HCoV 229E 3CLpros (8). This excludes the possibility of structural misfolding caused by N-terminal deletion (residues 1–7). Thermal induced unfolding of the two proteins was performed by monitoring the continuous changes of CD signals at 222 nm within temperatures ranging from 30 to 75 °C, the results are shown in Fig. 1b. As Fig. 1b indicates, the N-terminal deletion does not dramatically affect the denaturation behavior of the proteinase, and the N-terminal deleted proteinase adopts a thermal induced unfolding profile similar to that of the full-length proteinase. The transition temperature (Tm) of the N-terminal deleted proteinase was estimated as 60.5 °C, which is very close to that of the full-length proteinase, 61.4 °C. All these results imply that N-terminal deletion has not changed the folding manner and thermal stability of the proteinase.
Fig. 1.
CD spectra of the full-length and N-terminal deleted SARS 3CLpros.a, far-UV CD spectra of the full-length (▪) and N-terminal deleted (○) SARS 3CLpros at 25 °C. b, thermal induced unfolding monitored at 222 nm with a temperature range from 30 to 75 °C for the full-length (▪) and N-terminal deleted (○) SARS 3CLpros. Protein concentrations used in CD experiments were 10 μm, and all protein samples were prepared in 20 mm sodium phosphate, pH 7.5, 100 mm NaCl.
Enzymatic Activity—Anand et al. (10) found that the N-terminal residues 1–5 deleted mutant of TGEV 3CLpro is almost completely inactive. Based on this and the x-ray crystal structure, Yang et al. (9) predicted that the N terminus is also indispensable for enzymatic activity of SARS 3CLpro. To verify this prediction, we determined the enzymatic activities of both the full-length and N-terminal residues 1–7 deleted SARS 3CLpros using the FRET method. Fluorescence-based assay is a convenient and sensitive method for quantitative proteinase activity analysis. In this respect, the FRET method has been successfully used to develop spectrophotometric assays for proteinases (22, 23, 24). Cleavage of the fluorogenic substrate produces an increase in fluorescence as the donor and quencher separate, which can thus be used to follow the kinetics of the enzymatic reaction. Recently a fluorogenic 14-amino acid peptide has been reported for measurement of the enzymatic activity of SARS 3CLpro (25). In this study, we used a 12-amino acid fluorogenic substrate with an EDANS/Dabcyl fluorescence/quenching pair to detect the enzymatic activities of the full-length and N-terminal deleted SARS 3CLpros (16).
Hydrolysis of the fluorogenic substrate by the full-length SARS 3CLpro results in an appreciable increase in emission fluorescence intensity at the wavelength of 490 nm. The fluorescence profile following hydrolysis of the substrate is depicted in Fig. 2 . As a control, the incubation of the substrate in the assay buffer in the absence of 3CLpro shows no fluorescence intensity change over time (data not shown). The result implicates that the fluorogenic substrate could be efficiently hydrolyzed by the full-length SARS 3CLpro. The initial reaction velocity (ν0) was determined as 0.14 μm/min from the linear portion of the progress curve. In addition, we also measured the effect of pH on the enzymatic activity for the full-length proteinase, and we found that SARS 3CLpro exhibits a relatively stable proteolytic activity at pH 7.0–9.0 (see supplemental Fig. S2). This is in agreement with the published data (13, 26), further indicating that our fluorogenic method for determining SARS 3CLpro enzymatic activity is reliable.
Fig. 2.

Representative fluorescence profiles of hydrolysis of the fluorogenic substrate by SARS 3CLpros. The fluorogenic substrate at a concentration of 10 μm was incubated with 1 μm full-length (▪) or N-terminal deleted (○) SARS 3CLpro in 20 mm sodium phosphate, pH 7.5, 100 mm NaCl, 5 mm DTT, 1 mm EDTA, at 25 °C. Increase of emission fluorescence intensity at 490 nm wavelength was recorded at 10-min intervals, λEX = 340 nm. The emission spectrum was recorded for more than 4 h. The initial reaction velocity (ν0) was determined from the linear portion of the progress curve, which corresponded to between 2 and 10% hydrolysis of the substrate. The activity of the full-length SARS 3CLpro was taken as 100%.
As expected, the fluorescence increase following hydrolysis of the substrate by the N-terminal deleted SARS 3CLpro is below the detection limit of the activity assay (Fig. 2), suggesting that the N-terminal deleted 3CLpro is almost completely inactive. This result strongly demonstrates the indispensability of the N terminus for enzymatic activity of SARS 3CLpro.
Dimerization of the Full-length and N-terminal Deleted Proteinases—The recent research has revealed that SARS 3CLpro, similar to 3CL proteinases of HCoV 229E and TGEV (8), forms a dimer in the crystal structure and exists as a mixture of monomer and dimer in solution (9, 12, 13). Because the crystal structures of 3CLpros in different coronaviruses exhibit the same dimeric structures and nearly all the side chains of 3CLpro involved in the formation of the dimer are well conserved, it has been proposed that the dimeric form of 3CLpro might be responsible for its proteolytic activity (8, 9). On the other hand, the crystal structure of SARS 3CLpro suggests that the N-terminal residues 1–7 are squeezed into between domains II and III of the parent protomer and domain II of the other protomer to make a number of very specific interactions with the other protomer in the dimer. Based on this fact, one hypothesis was proposed that the N-terminal residues 1–7 might play an important role in both proteinase dimerization and enzymatic activity (9).
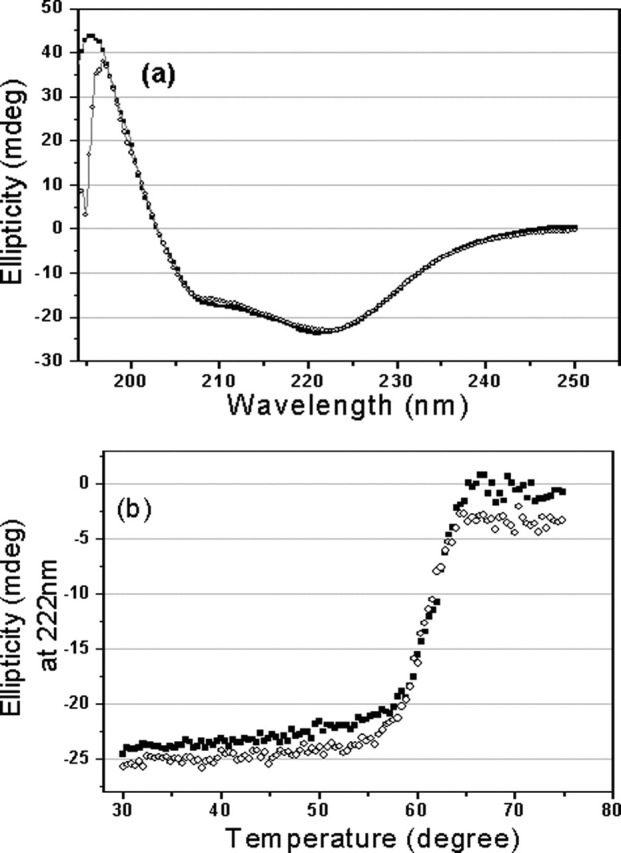
To better characterize dimerization of SARS 3CLpro and the role of the N terminus in dimer formation, we have performed SDS-PAGE analyses using a glutaraldehyde cross-linking method (27). The results are shown in Fig. 3 . When incubated with 0.01% glutaraldehyde, the full-length proteinase at a concentration of 1 mg/ml displayed a form of monomer with one other species around 66.0 kDa (Fig. 3a, lane 4a), corresponding to the dimer of the proteinase. At the higher concentration of glutaraldehyde (0.1%), the full-length SARS 3CLpro (1 mg/ml) showed a similar cross-linking pattern (Fig. 3a, lane 4b), indicating that the amount of glutaraldehyde was sufficient throughout and no artificial cross-linking occurred. With protein concentration increasing, the dimeric form increased and was almost equivalent to the amount of the monomeric form at higher concentration (7 mg/ml) (Fig. 3a, lanes 2 and 3). These results suggest that dimerization of the full-length SARS 3CLpro is concentration-dependent, consistent with the previous results (12, 13).
Fig. 3.
SDS-PAGE (10% acrylamide) profiles of glutaraldehyde cross-linked SARS 3CLpros.a, cross-linking analyses of the full-length SARS 3CLpro. Lane 1, untreated 3CLpro (7 mg/ml); lane 2a, 3CLpro (2 mg/ml) cross-linked by 0.01% glutaraldehyde; lane 2b, 3CLpro (2 mg/ml) cross-linked by 0.1% glutaraldehyde; lanes 3a and 3b, 3CLpro (7 mg/ml) cross-linked by 0.01 and 0.1% glutaraldehyde, respectively; lanes 4a and 4b, 3CLpro (1 mg/ml), 0.01 and 0.1% glutaraldehyde; lane 5, molecular weight SDS calibration kit protein standards are as follows: phosphorylase b (97.0 kDa), albumin (66.0 kDa), ovalbumin (45.0 kDa), carbonic anhydrase (30.0 kDa), trypsin inhibitor (20.1 kDa), α-lactalbumin (14.4 kDa). b, cross-linking analyses of the N-terminal deleted SARS 3CLpro. Lane 1, protein standards; lane 2 untreated 3CLpro (7 mg/ml); lanes 3a and 3b, 3CLpro (1 mg/ml), 0.01 and 0.1% glutaraldehyde, respectively; lanes 4a and 4b, 3CLpro (2 mg/ml), 0.01 and 0.1% glutaraldehyde; lanes 5a and 5b, 3CLpro (7 mg/ml), 0.01 and 0.1% glutaraldehyde.
Most strikingly, similar results were obtained with the N-terminal deleted proteinase. The dimeric form of the N-terminal deleted SARS 3CLpro also existed within a wide range of protein concentrations. When incubated with 0.01 or 0.1% glutaraldehyde, the N-terminal deleted 3CLpro at a concentration of 1 mg/ml also displayed forms that migrated as monomer and dimer (Fig. 3b, lanes 3a and 3b), whereas the monomer was the predominant species at a relatively low protein concentration. When the protein concentration increased to 2 mg/ml, the amount of the dimeric species was fortified (Fig. 3b, lanes 4a and 4b). At higher concentration (7 mg/ml), the band corresponding to the monomer of the N-terminal deleted 3CLpro was only slightly thicker than that of the dimer (Fig. 3b, lanes 5a and 5b). These results indicate that the N-terminal deletion seems to have no impact on the concentration-dependent dimerization of 3CLpro. Obviously, this finding is not fully consistent with the conclusion reported previously (9), which supposed that the N terminus plays an important role in both dimerization and formation of the active site of SARS 3CLpro.
In order to gain more insight into the dimer-monomer equilibrium of SARS 3CLpro in solution and to exactly evaluate the role of the N-terminal deletion in 3CLpro dimerization, SEC analyses on a HiLoad 16/60 Superdex 75 prep grade column were carried out by using the method published previously (28). The result is represented in Fig. 4 . The column was first calibrated with four marker proteins for evaluation of the elution profiles of the protein samples. In the gel filtration experiments under conditions similar to those for chemical cross-linking SDS-PAGE data collection, the physical states corresponding to native dimeric and monomeric 3CLpros were observed within a wide range of protein concentration. For the full-length 3CLpro (1 mg/ml) at neutral pH (7.5), two peaks centered at 44.57 and 62.14 ml were observed (Fig. 4a). The fact that the peak at 62.14 ml is between the retention volumes of 57.22 and 67.89 ml for the two molecular mass marker proteins of 48.9 and 22.8 kDa under similar conditions suggests that this peak corresponds to the monomer state of SARS 3CLpro (35.8 kDa). The other peak centered at 44.57 ml is lower than the retention volume of 51.72 ml for the molecular mass marker protein of 65.4 kDa, which is well attributed to the dimeric species of 3CLpro. This observation thus indicates that the full-length 3CLpros under the conditions studied have both forms of monomer and dimer. The amount of the dimer and monomer forms could be quantified by the integrated area value of two corresponding peaks. According to the integrated areas of the peaks, the dimer/monomer ratios for the concentrations of 1, 2, and 7 mg/ml are estimated as 0.68, 0.71, and 0.84, respectively, indicating again that 3CLpro dimerization is concentration-dependent. This is in agreement with the results obtained from glutaraldehyde cross-linking SDS-PAGE determinations and literature reports (12, 13).
Fig. 4.

Analyses of monomer-dimer equilibria of SARS 3CLpros by SEC.a, elution profiles of the full-length SARS 3CLpro at neutral pH (7.5) and concentrations of 1 (○), 2 mg/ml (▵), and 7 mg/ml (□). Elution profiles of four marker proteins, ribonuclease A (15.6 kDa), chymotrypsinogen A (22.8 kDa), ovalbumin (48.9 kDa), and albumin (65.4 kDa), are also shown as dashed lines. The dimer/monomer ratios for the concentrations of 1, 2, and 7 mg/ml are 0.68, 0.71, and 0.84, respectively. b, elution profiles of the N-terminal deleted SARS 3CLpro at neutral pH (7.5) and concentrations of 1 (○), 2 (▵), and 7 mg/ml (□). Elution profiles of marker proteins are shown as dashed lines. The dimer/monomer ratios for the concentrations of 1, 2, and 7 mg/ml are 0.65, 0.66, and 0.80, respectively. In the SEC-FPLC experiment, each protein sample was loaded to a HiLoad 16/60 Superdex 75 prep grade column pre-equilibrated with the buffer and then eluted at a flow rate of 1 ml/min with a detection of absorbance at 280 nm. The buffer used was 20 mm sodium phosphate, pH 7.5, 100 mm NaCl, 5 mm DTT, 1 mm EDTA. Standard calibration curves were used to determine the molecular masses of monomeric and dimeric form of the full-length (c) and N-terminal deleted (d) SARS 3CLpros. Elution volumes of the full-length and N-terminal deleted proteinases are indicated by open circles (○) and for marker proteins by filled circles (•). Ve and V0 are peak elution volume and column void volume, respectively, and Vt is the total bed volume.
For the N-terminal deleted SARS 3CLpro under the identical conditions, two peaks centered at 44.91 and 62.67 ml were observed and were slightly higher than the corresponding elution volumes of the full-length proteinase (Fig. 4b), indicative of a slightly less molecular mass perhaps attributing to the N-terminal deletion. By comparing with the elution volumes of the marker proteins, we found that the N-terminal deleted proteinase also existed as a mixture of dimers and monomers, and the dimer/monomer ratios for the concentrations of 1, 2, and 7 mg/ml are estimated as 0.65, 0.66, and 0.80, respectively. Similar to the full-length 3CLpro, dimerization of the N-terminal deleted 3CLpro is also concentration-dependent. These observations convincingly support the results obtained from glutaraldehyde cross-linking SDS-PAGE measurement that the N-terminal residues 1–7 contribute little to dimerization of SARS 3CLpro. In addition, molecular mass analyses were carried out from standard calibration curves to exactly evaluate the elution profiles of the full-length and N-terminal deleted 3CLpros, as shown in Fig. 4, c and d, respectively. The experimentally measured elution volumes of monomeric and dimeric 3CLpro agree well with the theoretic values.
Thermodynamics of Dimerization—Both chemical cross-linking SDS-PAGE and SEC analyses have revealed that both the full-length and N-terminal deleted SARS 3CLpro might form dimers at varied concentrations in solution. To evaluate quantitatively the influence of N-terminal deletion on dimerization of 3CLpro, the thermodynamic parameters (Kd and
) for the monomer-dimer equilibria of the full-length and N-terminal deleted proteinases were determined using the dilution ITC method (29, 30).
In the present study, dilution of a concentrated solution of the full-length or N-terminal deleted SARS 3CLpro was performed by injecting small aliquots into the calorimeter sample cell containing a larger volume of buffer. The typical calorimetric dilution profile of the full-length 3CLpro is shown in Fig. 5a, which indicates that the dilution process is endothermic. This is in agreement with the phenomenon derived from the chemical cross-linking SDS-PAGE and SEC analyses that 3CLpro dimerization is concentration-dependent (Figs. 3 and 4). The sequence of dilution injections produces a series of endothermic heat pulses which, after integration and correction for buffer mixing control experiments, gives the absolute heat uptake per injection. In such a dilution series, successive injections become progressively less endothermic as the 3CLpro concentration increases in the sample cell. The resulting thermal dilution profile is consistent with a simple dimer-monomer dissociation model (31). In a similar way, the calorimetric dilution profile of the N-terminal deleted 3CLpro was obtained, which is shown in Fig. 5, c and d, indicating that dimerization of the N-terminal deleted proteinase is also endothermic and concentration-dependent.
Fig. 5.

The dimer dissociation constants (Kd values) and enthalpy changes (
values) for dimerization of the two proteinases can be determined from their calorimetric dilution profiles, and thus the free energies (
values) and entropy changes (
values) for dimerization can be derived by using Equation 1. All these thermodynamic parameters are listed in Table I . Most interestingly, N-terminal deletion did not essentially affect the binding affinity between two protomers of the proteinase (the Kd values are 227 ± 34 and 262 ± 15 μm, respectively), and the binding free energies for dimerization of the full-length and N-terminal deleted proteinases are -4.968 ± 0.083 and -4.879 ± 0.033 kcal/mol, respectively (Table I). These results demonstrate thermodynamically that the N terminus is not substantially necessary for the proteinase dimerization. In addition, the enthalpy changes (
values) and entropy changes (
values) for dimerization shown in Table I indicate that, for dimerization of SARS 3CLpro, the N-terminal deletion decreases the enthalpy and entropy changes by ∼1.4 kcal/mol and ∼5.3 cal/mol·K, respectively.
Table I.
Thermodynamic parameters for SARS 3CLprodimerization at 25 °C and pH 7.5
| SARS 3CLpro | Kda |
|
|
|
|||
|---|---|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | cal/mol·K | ||||
| Full-length | 227 ± 34 | -8.283 ± 0.103 | -4.968 ± 0.083 | -11.124 ± 0.163 | |||
| N-terminal deleted | 262 ± 15 | -6.893 ± 0.250 | -4.879 ± 0.033 | -6.758 ± 0.729 | |||
From nonlinear regression of calorimetric dilution data in Fig. 5, fitted by a simple dimer-monomer dissociation model
Dynamic Behavior of the Dimers—To gain insights into the detailed influence of the N-terminal deletion on dimerization of SARS 3CLpro and to interpret why the N terminus is indispensable for proteolytic activity but not for enzyme dimerization at the molecular and atomic levels, 5-ns MD simulations were performed on both the full-length (model 1) and N-terminal deleted (model 2) SARS 3CLpros.
The energetic properties of the systems with respect to time give an indication of the overall stability of the MD trajectory. The profiles of interaction energies between the two protomers (monomers) of models 1 and 2 and their electrostatic and van der Waals contributions versus simulation time are shown in Fig. 6a and supplemental Fig. S3. For the full-length proteinase dimer (model 1), the interaction energy between the two protomers is relatively stable; after an ∼3-ns simulation, the total interaction energy fluctuates slightly around -250 kcal/mol. For the N-terminal deleted proteinase (model 2), because of N-terminal deletion, the interaction energy and its electrostatic and van der Waals contributions between the two protomers decrease dramatically at the beginning of the simulation. However, along with the MD simulation, the two protomers of model 2 may adjust their positions, and the dimer tends to be more and more stable; after ∼3 ns, both the electrostatic and van der Waals interactions have been stable, and the total interaction energy fluctuates slightly around -240 kcal/mol. The MD simulation results indicate that the N-terminal deletion only reduces slightly the interaction energy, which is in agreement with the thermodynamic parameters for dimerization (Table I).
Fig. 6.

Interaction energy and dimerization state changes from the full-length SARS 3CLpro to the N-terminal deleted SARS 3CLpro.a, total interaction energies for the full-length (thick line) and N-terminal deleted (thin line) SARS 3CLpros versus MD simulation time (shown as 10 ps average). b, the angles between the two protomers of the full-length (thick line) and N-terminal deleted (thin line) SARS 3CLpros versus MD simulation time (shown as 10 ps average). The angle is defined by two vectors, and each vector points from the center of mass of domain III to the center of mass of domains I and II in one protomer.
Moreover, we monitored the hydrogen bond (H-bond) number between the two protomers of the two models versus simulation time. The result is shown in supplemental Fig. S4 a. The H-bond number between the two protomers for the N-terminal deleted proteinase decreases dramatically at the beginning of the MD simulation but fluctuates around 12 after ∼3 ns; the H-bond number between the two protomers for the full-length proteinase fluctuates around 16. The important residues involved in the hydrogen bonding at the interfaces for both full-length and N-terminal deleted proteinases are listed in supplemental Table S1, which also indicates that the important H-bond number does not change a lot because of the N-terminal deletion, since some new H-bonds are formed during the breaking of some old H-bonds. Accordingly, the N-terminal deletion does not seriously weaken the hydrogen bonding between the two protomers.
To address the dynamics behaviors of the two SARS 3CLpros, we monitored the angles between two protomers for the two models. The angle between two protomers was defined as the cross-angles of two vectors, each vector in one protomer points from the center of mass of domain III to the center of mass of domains I and II. The result is shown in Fig. 6b. For the full-length proteinase, the angle between two protomers fluctuates around 80° during the 5-ns MD simulation; for the N-terminal deleted proteinase, this angle increases from 80 to 95°, indicating that the two protomers of the N-terminal deleted proteinase form a dimer adopting a state different from that of the full-length proteinase. The superposition of one equilibrium structure of the N-terminal deleted proteinase dimer with that of the full-length proteinase dimer is shown in supplemental Fig. S5, which demonstrates the dimerization state changes from the full-length proteinase dimer to the N-terminal deleted proteinase dimer: domains I and II of protomer A rotate upwards and domain III rotates downwards, as indicated by the arc arrows.
The interface area of dimer is an important criterion for the stability of the protein-protein interaction. The profiles of the interface areas between the two protomers of models 1 and 2 versus MD simulation time were monitored, which are shown in supplemental Fig. S4 c. The interface area for the full-length SARS 3CLpro remains relatively stable, fluctuating around ∼1450 Å2 during the simulation time. Because of the N-terminal deletion, the interface area decreases to ∼500 Å2 at the beginning of the MD simulation. Because of the relative movement between the two protomers, the interface area finally increases to ∼1400 Å2. As the x-ray structure indicated that the interface of the 3CLpro is hydrophobic, the dimer of the N-terminal deleted proteinase adopts a different state from the full-length proteinase, producing an interface area almost equal to that of the full-length proteinase. Complementary to the interface area measurement, the hydrophobic interaction pairs (HIPs) between the two protomers for both the full-length and N-terminal deleted proteinases were also monitored versus the MD simulation time, and the result is shown in supplemental Fig. S4 b. Although the N-terminal deletion dramatically decreases the HIPs at the beginning of MD simulation, however, the HIPs for the N-terminal deleted proteinase increase along the MD simulation and reach the HIPs for the full-length proteinase. Therefore, the N-terminal deletion does not dramatically decrease the hydrophobic interaction between the two protomers.
To investigate further the effect of the N-terminal deletion on the proteolytic activity, we monitored the profile of the volumes of the substrate-binding pockets of models 1 and 2 versus the MD simulation time, and the results are shown in supplemental Fig. S6 (a and b). The substrate-binding pockets of both the full-length and N-terminal deleted proteinases fluctuate dramatically during the MD simulation. The average binding pocket volume of the full-length proteinase is ∼220 Å3, whereas that of the N-terminal deleted proteinase is ∼200 Å3, a decrease of ∼10%.
To demonstrate the significance of the ∼10% decrease of the binding pocket volume for the N-terminal deleted SARS 3CLpro to the substrate binding, we performed an additional calculation. A model substrate (TSAVLQ) representing the N-terminal P6–P1 sequence of a reported best peptide substrate (TSAVLQSGFRK) for SARS 3CLpro (13) was devised as a model substrate to dock into the binding pockets of a series of conformations isolated from the MD trajectories of models 1 and 2, respectively, using the Autodock3.03 program (32), and the binding free energy of the substrate to each conformation was calculated using the Autodock3.03 scoring function. The docking procedure is described in the Supplemental Data. The binding free energy profiles of the substrate to models 1 and 2 versus the MD simulation time are shown in supplemental Fig. S6c. The average binding free energies of the substrate to the two proteinases are -5.33 and -2.50 kcal/mol, respectively, indicating that the binding affinity of the N-terminal deleted SARS 3CLpro to the substrate is much lower than that of the full-length SARS 3CLpro to the substrate. This may be one of the reasons why the N-terminal deleted proteinase is inactive in proteolytic catalytic reaction, because the peptide substrate cannot bind well into the binding pocket. This calculation is in good agreement with the SPR determination, as shown below.
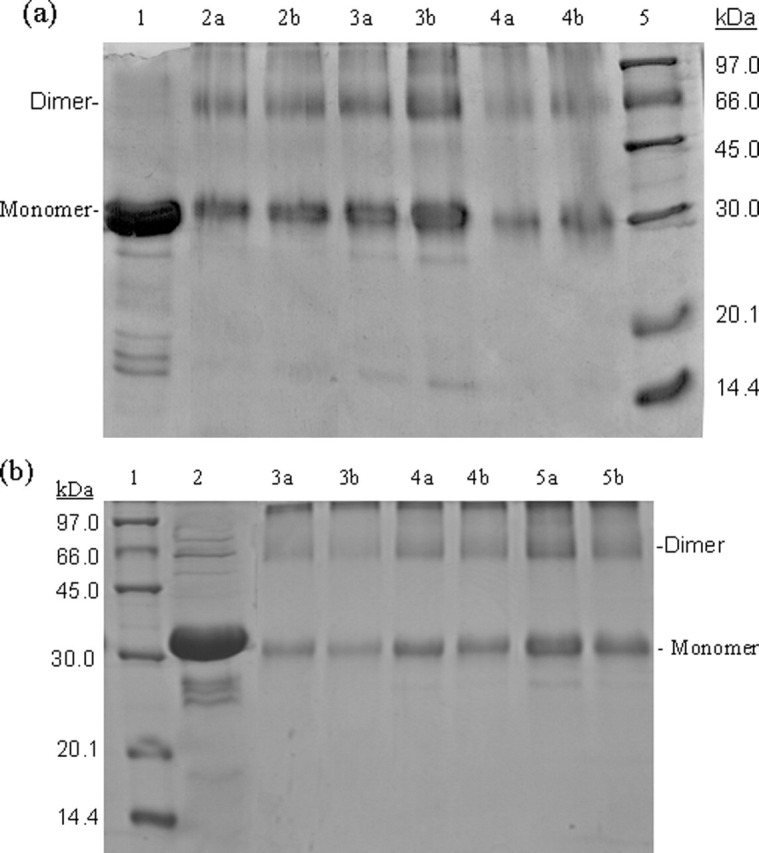
Binding of the Model Peptide Substrate to the Full-length and N-terminal Deleted Proteinases—To verify the conclusions reached from MD simulations that the N-terminal deletion reduces the volume of the binding pocket of SARS 3CLpro and makes the peptide substrate unable to bind into the small binding pocket, the model peptide substrate (TSAVLO) was synthesized to determine the proteinase-substrate binding affinity. The binding affinity of the peptide to the full-length and N-terminal deleted SARS 3CLpros was determined by using SPR biosensor technology. For kinetic analysis on the Biacore 3000 instrument, various concentrations of the peptide were injected for 60s at a flow rate of 20 μl/min to allow for binding with the proteinase immobilized on the chip surface. The interactions of the full-length and N-terminal deleted proteinases with the peptide are shown in Fig. 7 . For the full-length proteinase (Fig. 7a), the peptide resulted in a significant and dose-dependent increase in SPR response units and presented characteristic fast binding and slow dissociation curves, indicating a rapidly formed and relatively stable proteinase-peptide complex. The peptide concentration series were fitted to a steady-state binding model encoded in the Biacore 3000 evaluation software for binding affinity determination. The dissociation constant (KD value) between the substrate peptide and the full-length proteinase was determined as ∼152 μm. For the N-terminal deleted proteinase, a negligible and concentration-independent RU increase was obtained (Fig. 7b), indicating that almost no binding exists between the substrate peptide and the N-terminal deleted proteinase. This result is consistent with the results of the above MD simulation and docking calculation (supplemental Fig. S6).
Fig. 7.
Binding affinity of a 6-amino acid peptide (TSAVLQ) to the full-length and N-terminal deleted SARS 3CLpros determined by SPR. Real time binding affinity measurements of the peptide to the full-length SARS 3CLpro (a) and the N-terminal deleted proteinase (b) using Biacore 3000. Representative sensorgrams were obtained from injections of the peptide at concentrations of 500, 250, 100, 50, and 20 μm (curves from top to bottom). The peptides were injected for 60s, and dissociation was monitored for more than 120s.
DISCUSSION
In this work, the thermal stabilities, proteolytic activities, and dimerization of both the full-length and N-terminal deleted SARS 3CLpros were fully studied by using CD spectroscopy, FRET, glutaraldehyde cross-linking SDS-PAGE, SEC, ITC, SPR techniques, and complementary MD simulations.
Recently, 3CLpros of several coronaviruses have been appreciated because of their important roles in virus replication (8, 9, 33). Although SARS-CoV is a novel coronavirus discovered last year during the outbreak of SARS, sequence alignment indicated that SARS 3CLpro shares high homology with the 3CLpros of other coronaviruses such as TGEV and human 229E (8, 34). The recently determined x-ray crystal structures of the 3CLpros of TGEV, human 229E, and SARS revealed that the three-dimensional structures of 3CL proteinases are more conserved than their sequences (8, 9). Therefore, it can be deduced that the biochemical and biophysical properties and biological functions of coronavirus 3CLpros should be similar.
Based on the biochemical data and crystal structures of three coronavirus 3CLpros (TGEV, human 299E, and SARS), a functional role has been proposed for the N terminus of the coronavirus 3CLpros in both the enzymatic activity and dimerization, namely the N terminus is indispensable for both the proteolytic activity and enzyme dimerization (8, 9). In our present study, CD spectra indicated that the N-terminal residues 1–7 deleted SARS 3CLpro adopted a thermal induced unfolding profile that is similar to the full-length proteinase (Fig. 1b). This implies that the N-terminal deletion does not affect the folding manner and structural stability of the proteinase. Like the 3CLpro of TGEV, the N-terminal deletion almost completely abolished the enzymatic activity of SARS 3CLpro (Fig. 2). This verified the prediction proposed by Yang et al. (9).
However, our glutaraldehyde cross-linking SDS-PAGE result questions the hypothesis that N-terminal residues 1–7 play an important role in dimerization of SARS 3CLpro and 3CLpros of other coronaviruses (9, 10), because SDS-PAGE analyses clearly indicated that the N-terminal deleted SARS 3CLpro can also form dimer and the concentration dependence of dimerization is not essentially affected by N-terminal deletion (Fig. 3). Furthermore, SEC analyses revealed that, like the full-length proteinase, dimer-monomer equilibrium also exists in the solution of the N-terminal deleted SARS 3CLpro (Fig. 4). Both the chemical cross-linking SDS-PAGE and SEC analyses indicated that the N terminus contributes little to formation of SARS 3CLpro dimer.
The dilution ITC analyses provided quantitative evidence that N-terminal residues 1–7 deletion does not affect dimerization of SARS 3CLpro (Fig. 5). Dissociation constant determinations for the proteinase dimer surprisingly revealed that the binding affinity between the two protomers of the N-terminal deleted proteinase (Kd = 261 ± 15 μm) is almost equal to that between the two protomers of the full-length proteinase (Kd = 227 ± 34 μm) (Table I). MD simulations indicate that the N-terminal deletion slightly decreases the total interaction energy between the two protomers (Fig. 6a), which was reflected by the decrease of the enthalpy change for dimerization of the N-terminal deleted proteinase (
= -6.893 ± 0.250 kcal/mol) in comparison with that for dimerization of the full-length proteinase (
= -8.283 ± 0.103 kcal/mol). On the other hand, the N-terminal deletion decreased the flexibility of the proteinase and thereby decreased the entropy loss during the time the two protomers were aggregated together, which compensated for the enthalpy drop for dimerization of the N-terminal deleted proteinase. This may be the major reason why the N-terminal deletion almost did not reduce the binding affinity of the two protomers.
5-ns MD simulations indicated that the two protomers of the N-terminal deleted proteinase might reach a new dimerization state. Indirect evidence for the N-terminal deleted proteinase adopting a different dimerization state can be seen from the cross-angle change between the two protomers, which increased from ∼80° for the full-length proteinase dimer to ∼95° for the N-terminal deleted proteinase dimer (Fig. 6b). The new dimerization state of the N-terminal deleted proteinase maximized the interface area and thereby enhanced the interaction between the two protomers. Another phenomenon recognized by MD simulations is that the binding pocket of the N-terminal deleted proteinase tends to become smaller, which is not beneficial to the substrate binding (supplemental Fig. S6). This may be one of the reasons why the N-terminal deleted proteinase loses its proteolytic activity. This prediction has been verified by indirect proteinase substrate docking simulation (supplemental Fig. S6 c) and by direct proteinase substrate binding assay (Fig. 7).
The x-ray crystal structure of SARS 3CLpro indicated that the N terminus of one protomer is squeezed between domains II and III of another protomer (9). We hypothesize that the interactions between the N terminus of one protomer and domains II and III of another protomer appear to be the consequence rather than the cause of dimerization, so the N terminus contributes little to the formation of the dimer, but it can stabilize the dimeric structure to an active state. Nevertheless, this hypothesis should be further verified by the x-ray crystallographic determination, which will be a future study.
Footnotes
The abbreviations used are: SARS, severe acute respiratory syndrome; SARS-CoV, severe acute respiratory syndrome coronavirus; 3CLpro, 3C-like proteinase; SEC, size-exclusion chromatography; ITC, isothermal titration calorimeter; FRET, fluorescence resonance energy transfer; MD, molecular dynamics; SPR, surface plasmon resonance; TGEV, transmissible gastroenteritis virus; DTT, dithiothreitol; FPLC, fast protein liquid chromatography; RU, resonance units; HIPs, hydrophobic interaction pairs; EDANS, 5-[(2′-aminoethyl)-amino]naphthelenesulfonic acid; Dabcyl, 4-[[4-(dimethylamino)phenyl]azo]benzoic acid.
Supplementary Material
References
- 1.Kathryn V.H. J. Clin. Investig. 2002;111:1605–1609. [Google Scholar]
- 2.Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fouchier R.A., Kuiken T., Schutten M., Amerongen VanG., Van Doornum G.J., Van Den Hoogen B.G., Peiris M., Lim W., Stohr K., Osterhaus A.D. Nature. 2003;423:240. doi: 10.1038/423240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herold J., Raabe T., Schelle-Prinz B., Siddell S.G. Virology. 1993;195:680–691. doi: 10.1006/viro.1993.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee H.J., Shieh C.K., Gorbalenya A.E., Koonin E.V., La Monica N., Tuler J., Bagdzhadzhyan A., Lai M.M. Virology. 1991;180:567–582. doi: 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 7.Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L., Guan Y., Rozanov M., Spaan W.J., Gorbalenya A.E. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anand K., Plam G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Higenfeld R. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziebuhr J., Snijder E.J., Gorbalenya A.E. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 12.Shi J., Wei Z., Song J. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun H., Luo H., Yu C., Sun T., Chen J., Peng S., Qin J., Shen J., Yang Y., Xie Y., Chen K., Wang Y., Shen X., Jiang H. Protein Expression Purif. 2003;32:302–308. doi: 10.1016/j.pep.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Echeverria C., Rich D.H. FEBS Lett. 1992;297:100–102. doi: 10.1016/0014-5793(92)80336-f. [DOI] [PubMed] [Google Scholar]
- 16.Chen S., Chen L.L., Luo H.B., Sun T., Chen J., Ye F., Cai J.H., Shen J.K., Shen X., Jiang H.L. Acta Pharmacol Sin. 2005;26:99–107. doi: 10.1111/j.1745-7254.2005.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Berendsen H.J.C., Spoel D., van Drunen A.R. Comp. Phys. Comm. 1995;91:43–56. [Google Scholar]
- 18.Van Gunsteren W.F., Berendsen H.J.C. Gromos-87 Manual. Biomos BV; Nijenborgh 4 9747 AG Groningen, The Netherlands: 1987. [Google Scholar]
- 19.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J. In: Intermolecular Forces. Pullman B., editor. D. Reidel Publishing Company; Dordrecht, The Netherlands: 1981. pp. 331–342. [Google Scholar]
- 20.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 21.Spiga O., Bernini A., Scarselli M., Ciutti A., Bracci L., Lozzi L., Lelli B., Maro DiD., Calamandrei D., Niccolai N. FEBS Lett. 2002;511:33–35. doi: 10.1016/s0014-5793(01)03274-4. [DOI] [PubMed] [Google Scholar]
- 22.Kinght C.G., Willenbrock F., Murphy G. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
- 23.Angliker H., Neumann U., Molloy S.S., Thomas G. Anal. Biochem. 1995;224:409–412. doi: 10.1006/abio.1995.1058. [DOI] [PubMed] [Google Scholar]
- 24.Mittoo S., Sundstrom L.E., Bradley M. Anal. Biochem. 2003;319:234–238. doi: 10.1016/s0003-2697(03)00324-5. [DOI] [PubMed] [Google Scholar]
- 25.Kuo C.J., Chi Y.H., Hsu J.T., Liang P.H. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang C., Wei P., Fan K., Liu Y., Lai L. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- 27.Prakash K., Prajapati S., Ahmad A., Jain S.K., Bhakuni V. Protein Sci. 2002;11:46–57. doi: 10.1110/ps.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akhtar M.S., Ahmad A., Bhakuni V. Biochemistry. 2002;41:3819–3827. doi: 10.1021/bi0116700. [DOI] [PubMed] [Google Scholar]
- 29.Lovatt M., Cooper A., Camilleri P. Eur. Biophys. J. 1996;24:354–357. doi: 10.1007/BF00180377. [DOI] [PubMed] [Google Scholar]
- 30.Burrows S.D., Doyle M.L., Murphy K.P., Franklin S.G., White J.R., Brooks I., McNulty D.E., Scott M.O., Knutson J.R., Porter D., Young P.R., Hensley P. Biochemistry. 1994;33:12741–12745. doi: 10.1021/bi00209a002. [DOI] [PubMed] [Google Scholar]
- 31.McPhail D., Cooper A. J. Chem. Soc. Faraday Trans. 1997;93:2283–2289. [Google Scholar]
- 32.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 33.Ziebuhr J., Heusipp G., Siddell S.G. J. Virol. 1997;71:3992–3997. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong B., Gui C.S., Xu X.Y., Luo C., Chen J., Luo H.B., Chen L.L., Li G.W., Sun T., Yu C.Y., Yue L.D., Duan W.H., Shen J.K., Qin L., Shi T.L., Li Y.X., Chen K.X., Luo X.M., Shen X., Shen J.H., Jiang H.L. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.