

Abstract
Cardiac fibrosis is mediated by the activation of resident cardiac fibroblasts, which differentiate into myofibroblasts in response to injury or stress. While myofibroblast formation is a physiological response to acute injury, such as myocardial infarction, myofibroblast persistence, as occurs in heart failure, contributes to maladaptive remodeling and progressive functional decline. While traditional pathways of activation, such as transforming growth factor beta (TGFβ) and angiotensin II (AngII), have been well characterized, less understood are the alterations in mitochondrial function and cellular metabolism that are necessary to initiate and sustain myofibroblast formation and function. In this review, we highlight recent reports detailing the mitochondrial and metabolic mechanisms that contribute to myofibroblast differentiation, persistence and function with the hope of identifying novel therapeutic targets to treat, and potentially reverse, tissue organ fibrosis.
Keywords: Fibrosis, Myofibroblast, Metabolism, Mitochondria, Epigenetics, Myocardial
Introduction
Cardiac fibrosis occurs in ischemic and non-ischemic heart failure, genetic cardiomyopathies, diabetes, and aging1, 2. A hallmark of fibrosis is the excessive remodeling and accumulation of extracellular matrix (ECM), a three-dimensional network of macromolecules including collagens and glycoproteins that provide structural and biochemical support to neighboring cells3–5. Based on correlative expression of ECM proteins, the primary cell-type implicated in the myocardial fibrotic response is the cardiac fibroblast. Cardiac fibroblasts are abundant in the myocardium, comprising ~10–20% of the cell population in the heart6. In response to injury, cardiac fibroblasts differentiate into highly specialized synthetic and contractile myofibroblasts, increasing fibrillar collagen, matricellular protein production/secretion, as well as the expression of smooth muscle ⍺-actin (⍺SMA, ACTA2 gene)7. Myofibroblasts are essential for maintaining the structural integrity of the myocardium by contracting surrounding tissue and remodeling the ECM scaffold8–12. As proof, experimental elimination of activated myofibroblasts following myocardial infarction (MI) results in substantial mortality due to inefficient scar formation and ventricular rupture13, highlighting their importance in cardiac injury repair. However, excessive fibrosis driven by sustained and unremitting myofibroblast activation results in a progressive decline in tissue compliance, reduced nutrient and oxygen delivery, and increased cardiomyocyte atrophy and cell death, resulting in progressive left ventricular dilation and dysfunction14–16. This is important as nearly 6M Americans suffer from heart failure, an epidemic projected to encumber the nation with a cost of care of ~$70 billion by 203017. On the bright side, a recent report demonstrated that ablation of myofibroblasts under chronic stress was cardioprotective and that removal of this stress stimuli following myofibroblast activation permitted these cells to revert back to a quiescent state13. This suggests it’s possible to target myofibroblasts to reduce fibrosis and lessen disease severity. Therefore, it is imperative we identify the molecular mechanisms initiating and sustaining myofibroblast activation to generate novel therapeutics for heart failure and other fibrotic diseases.
Numerous mitochondrial and metabolic mechanisms have recently been reported to be necessary for myofibroblast formation. For example, changes in mitochondrial ROS (mtROS) generation and oxidative phosphorylation appear essential for myofibroblast formation18, 19. Further, our lab recently identified that myofibroblast differentiation was dependent on a reduction in mitochondrial calcium (Ca2+) uptake to initiate specific and highly coordinated metabolic changes to activate the differentiation gene program20. In this article, we review what defines a cardiac fibroblast and the evidence implicating mitochondrial signaling and cellular metabolism in myofibroblast differentiation, which may represent novel therapeutic targets for treating fibrosis.
The Cardiac Myofibroblast
Defining the myofibroblast
Fibroblasts mediate tissue repair of nearly every organ. In the heart, fibroblasts have been identified and characterized in the uninjured, infarcted, and pressure overloaded myocardium animal models and human patients21–24. During embryonic development, fibroblast progenitors infiltrate the myocardium at embryonic day (E) 13.5, developing into a molecularly distinct fibroblast population by E17.5–18.521, 25–28. While the expression of numerous epicardial genes, such as transcription factor 21 (TCF21)21, Wilms tumor protein (WT1), and T-box transcription factor 18 (TBX18) define this developmental fibroblast population, only TCF21 reliably labels most all adult cardiac fibroblasts13, 21, 27, 29, denoting its utility as a lineage tracing marker (see review by Tallquist and Molkentin28). In the quiescent state, fibroblasts are small, spindle-shaped cells with a reduced amount of rough endoplasmic reticulum (ER). Cardiac fibroblasts contribute to myocardial homeostasis by synthesizing and maintaining the ECM network critical for structural and functional integrity (Fig. 1)30. Fibroblasts also provide insulation of the electrical conduction system, potentiate blood vessel formation, and secrete paracrine signals that control muscle growth31, 32. During activation, fibroblasts express cytoplasmic actin and adhesion complexes, permitting migration to sites of tissue injury33. Additionally, activated fibroblasts undergo rapid proliferation dependent on the activation of p38-MAPK34 and ERK1/235 signaling pathways and the upregulation of cell cycle proteins13. Following stress or injury, fibroblasts reach a maximum proliferation rate in 2 to 4 days36. Once localized to sites of injury fibroblasts secrete various ECM proteins, such as collagens and fibronectin (FN), to remodel the ECM and initiate wound healing37. In addition, the de novo expression of periostin (POSTN), a matricellular protein important in development and wound healing, is a robust and early marker of fibroblast activation (Fig. 1)13, 38, 39.
Figure 1. Progression and characterization of cardiac fibroblast to myofibroblast conversion.

Resident fibroblasts contribute to cardiac homeostasis under normal physiological conditions. Upon increased mechanical tension and pro-fibrotic mediators (e.g. TGFβ, AngII), these resident cardiac fibroblasts become activated, at which time they infiltrate and expand at the site of injury as well as begin to remodel the ECM. Upon sustained and unremitting activation signaling, these fibroblasts differentiate into myofibroblasts characterized by the de novo expression of αSMA and the excessive production of ECM proteins (FN, collagens). Myofibroblasts are also resistant to cell death, resulting in their persistence in the injured heart, eventually leading to maladaptive tissue remodeling. Recent evidence suggest that myofibroblasts are capable of de-differentiating upon removal of stress stimuli, however the mechanisms by which they do so, and the potential for targeted mechanistic interventions, require further investigation. ECM – extracellular matrix; TGFβ – transforming growth factor beta; AngII – angiotensin II; Mechano – mechanical stress; POSTN – periostin; αSMA – α-smooth muscle actin; FN – fibronectin.
Upon differentiation fibroblasts become a phenotypically distinct cell referred to as the myofibroblast. Transmission electron microscopy of myofibroblasts in the pressure-overloaded heart identified several morphological features including: multiple dendritic processes, elongated and serrated nuclei, extensive rough ER, and the formation of an extensive stress-fiber network40. Other defining features are copious production and secretion of ECM proteins such as POSTN, collagens (e.g. COL1 and COL3), and a specialized isoform (splice-variant) of fibronectin (FN-EDA)41. The most defining characteristic is the de novo expression of the globular microfilament protein, alpha-smooth muscle actin (⍺SMA, ACTA2 gene)8 (see Fig. 1). In addition, myofibroblasts increase the expression of cadherin-2 and −11 for the formation of cell-to-cell adhesions critical for force generation and transduction42, 43. Experimentally, the ability to contract a collagen gel-matrix is commonly used to demonstrate myofibroblast formation44. Upon the formation of a fully matured scar 7–10 days after MI, myofibroblasts were recently reported to morph into a new differentiated state recently referred to as a matrifibrocyte36.
In addition to these classical myofibroblast characteristics, emerging evidence point to mitochondrial alterations and metabolic remodeling as initiators of differentiation. For instance, alterations in mitochondrial morphology45 and mtROS production46 are reported to occur early in myofibroblast formation. Remodeling of cellular metabolism is universally observed and is necessary to support the biomass required for cellular growth and differentiation, as well as the energetic demand of the newly acquired contractile phenotype. Compared to the quiescent state, myofibroblasts increase both aerobic glycolysis and oxidative metabolism20. Interestingly, recent work from our lab identified changes in mitochondrial Ca2+ uptake as a key mediator linking metabolism to the epigenetic modifications required for the differentiation program20. While the phenotypic characteristics of the myofibroblast are well defined, our understanding of the signaling and molecular events necessary to initiate the differentiation program is evolving.
Activation signaling pathways
Myofibroblast differentiation is initiated by various cytokines, hormones and ligands (e.g. TGFβ, AngII, ET-1)47, 48, ions (e.g. Ca2+)49, and environmental mechanical forces8, 12, 49–51. In addition, recent studies have brought to light the importance of mitochondrial signaling and function, as well as alterations in cellular metabolism, as key regulators of myofibroblast formation and persistence. Here we will briefly highlight well-established signaling pathways initiating myofibroblast differentiation, reviewed in more detail elsewhere47, 48, with a special emphasis on the mitochondrial and metabolic underpinnings that instigate and promote myofibroblast differentiation.
TGFβ-mediated canonical and non-canonical signaling
The prototypical signaling effector of myofibroblast differentiation is the cytokine TGFβ. Secreted by macrophages, infiltrating immune cells, and fibroblasts themselves, TGFβ acts in both an autocrine and paracrine fashion. TGFβ binds to a heterodimeric transmembrane receptor composed of type I (TGFBR1) and type II (TGFBR2) proteins, for both canonical and non-canonical signaling52. The canonical TGFβ pathway involves TGFBR1-mediated phosphorylation of SMAD2/3, permitting its association with SMAD4 and subsequent nuclear translocation for transcriptional activation of the fibrotic gene program. Experimental evidence confirms the importance of canonical signaling in myofibroblast differentiation, as deletion of Tgfβr1 prevented TGFβ-mediated differentiation53, 54 and a constitutively-active TGFΒR1 mutant promoted myofibroblast formation53. Furthermore, Verrecchia et al.55 demonstrated Col1a2, Col3a1, Col6a1, Col6a3, and Timp1 as TGFβ/SMAD3 gene targets, reporting that expression of a dominant negative SMAD3 construct, or overexpression of inhibitory SMAD7, prevented TGFβ-mediated increases in promoter activity. Similar results were obtained in Smad3-null cardiac fibroblasts, which were resistant to TGFβ-mediated induction of collagens and FN in vitro, and demonstrated reduced collagen deposition in response to MI 56. SMAD3 influences ⍺SMA expression56, and not surprisingly, there are several SMAD transcription factor binding motifs within the ⍺SMA (ACTA2) promoter(add Lombardi Ref)57. In contrast to SMAD3, recent genetic studies suggest SMAD2 is likely not a mediator of this process58, 59.
Non-canonical signaling by TGFΒR2 (SMAD-independent) also appears to be required for myofibroblast differentiation. Deletion of fibroblast Tgfbr2 in mice resulted in reduced fibrosis in a bleomycin-induced lung injury60 model and in dermal wound healing61. The most well-characterized non-canonical (SMAD-independent) pathway is the MAPK/JNK/p38 pathway. Genetic deletion of mitogen-activated protein kinase 2 (Mapk2) prevented TGFβ-induced expression of ⍺SMA62. p38 has been implicated as a key effector of MAPK, as pharmacologic inhibition of p38 prevented myofibroblast formation, whereas gain-of-function experiments resulted in upregulation of phenotypic markers of myofibroblast activation in cardiac fibroblasts54. Furthermore, conditional deletion of p38 in vivo blocked myofibroblast formation and fibrosis following ischemic injury or chronic neurohormonal stimulation63. Although canonical and non-canonical pathways act through independent signaling cascades, they are also likely interconnected since p38 can also phosphorylate SMAD364. Further, TGFβ specifically binds to TGFΒR2 in the heterodimeric complex, which in turn catalyzes the phosphorylation and activation of TGFΒR165, providing a mechanism to coordinate canonical and non-canonical signaling at the receptor level. Other non-canonical pathways, such as Rho-GTPase-Actin, PI3K/Akt, NF-kB, and TNF receptor-associated factors, are discussed elsewhere47, 48, 66.
G protein-coupled receptor (GPCR) signaling
Cardiac fibroblasts transcriptionally express ~190 GPCRs67, including putative receptors for angiotensin II (AngII) and endothelin 1 (ET-1). AngII stimulation is sufficient for fibroblast proliferation, collagen synthesis, and inhibition of collagen degradation68, 69. AngII induces fibroblast expression of TGFβ70 and its receptors71, suggesting AngII is a pioneering signal to initiate differentiation through a positive-feedback mechanism50, 69. To this point, cardiac fibroblasts treated with anti-TGFβ1 antibody were resistant to AngII-mediated increases in collagen expression72. However, other studies have shown proximal activation of canonical and non-canonical TGFβ pathways, suggesting AngII signals independently of TGFβ. For example, AngII increases the phosphorylation of SMAD2/3 and the expression of SMAD4 72, independent of TGFβR2 activation 73. Furthermore, knockdown of Tgfbr1 in cardiac fibroblasts did not prevent the upregulation of ⍺SMA and gel contraction in response to AngII stimulation54, a response associated with p38 activation and binding of serum response factor (SRF) transcription factor54 to the ⍺SMA (Acta2) promoter74. Collectively, these studies suggest an important role for AngII in both canonical and non-canonical signaling in myofibroblast activation.
Endothelin-1 (ET-1) is an endothelium-derived vasoactive peptide secreted in response to mechanical and hemodynamic stress75–79 and is associated with unfavorable outcomes post-MI80. In vitro, ET-1 increases fibroblast proliferation81, collagen synthesis82, and the ability to contract a collagen gel matrix83. Myofibroblast formation in response to ET-1 appears, at least in part, to be mediated through the activation of nuclear factor of activated T-cells (NFAT)84. In addition to endothelial-derived ET-1, AngII has been shown to induce the expression of ET-1 in cardiac fibroblasts85, further intertwining these signaling effectors and pathways. To date, few studies have rigorously investigated the effects of ET-1 in vivo, likely because ET-1 is not exclusively produced by coronary endothelial cells, making genetic targeting of this pathway in cardiovascular disease difficult.
Mechanical and Ca2+ dependent signaling
Fibroblasts also differentiate in response to changes in ECM stiffness86, 87. Mechanical tension is sensed through the newly synthesized, rigid matrix proteins, such as COL1, FN, secreted during initial scar formation47, 88. Culturing fibroblasts on low tensile strength substrates maintains cellular quiescence while high tensile strength substrates promote myofibroblast formation89. Mechanical tension activates Ras homolog family member A (RhoA) and downstream effector Rho-associated, coiled-coil-containing protein kinase (ROCK) for actin filament stabilization90. Actin stabilization potentiates the nuclear translocation of myocardin related transcription factor (MRTF), where together with SRF, transcription of the myofibroblast gene program is initiated91.
Plasma membrane mechanoreceptors likewise transduce mechanical tension into intracellular signals to activate or promote fibroblast formation86, 92. Among these mechanoreceptors are transient receptor potential (TRP) cation channels, a family of extracellular Ca2+ uptake channels implicated in myofibroblast differentiation49, likely through calcineurin-mediated NFAT activation and nuclear translocation93–96 (Fig. 2). Of these TRP channels, the most widely studied is TRP vanilloid 4 (TRPV4). TRPV4 demonstrates mechanosensitive properties in cardiac fibroblasts97 and TRPV4-dependent increases in cytosolic Ca2+ appear to mediate differentiation through nuclear translocation of MRTF-A, an ⍺SMA and SRF transcriptional coactivator98.
Figure 2. Mechanistic actions of Ca2+ during myofibroblast differentiation.
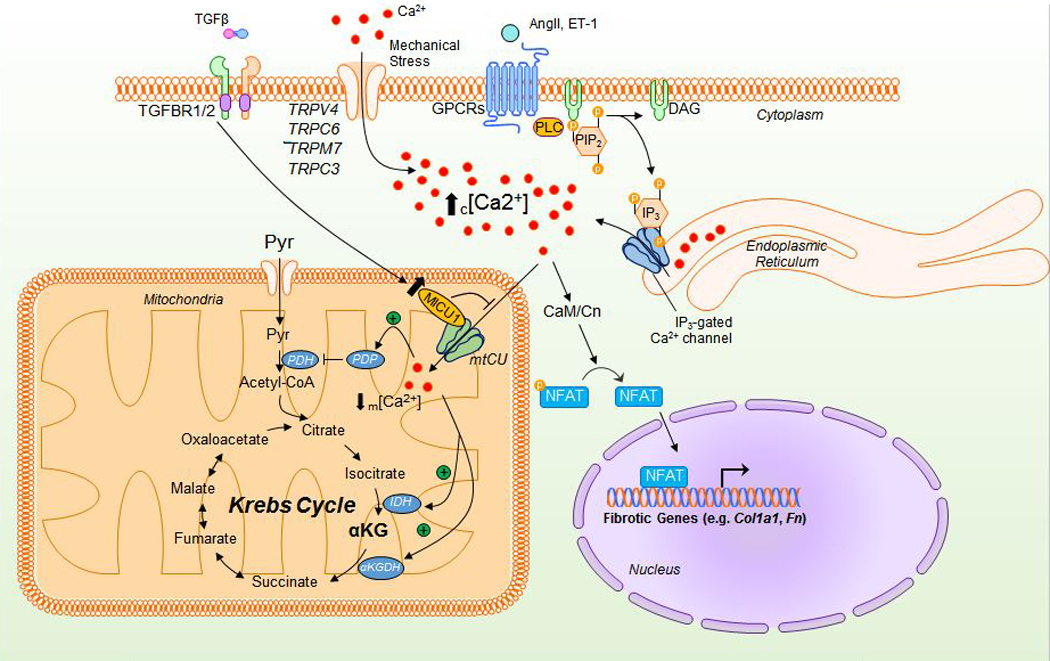
Mechanical activation of TRP channels permits Ca2+ entry from the extracellular space, increasing cytosolic [Ca2+]. Additionally, activation of the Gq/PLC/IP3 pathway allows for IP3-mediated Ca2+ release from the ER, further increasing cytosolic [Ca2+]. This increase in cytosolic [Ca2+] activates calmodulin-dependent calcineurin phosphatase (CaM/CN) to dephosphorylate and permit nuclear translocation of NFAT and subsequent activation of the fibrotic gene program. While mitochondria can act to buffer increases in cytosolic [Ca2+] through the mitochondrial calcium uniporter complex (mtCU), the transcriptional upregulation of MICU1 alters mtCU gating, reducing mCa2+ uptake and reducing m[Ca2+]. As m[Ca2+] is an activator of numerous dehydrogenases and phosphatases within the mitochondria, this significantly reduces pyruvate oxidation and overall Krebs cycle flux. TGFβ – transforming growth factor beta; AngII – angiotensin II; ET-1 – endothelin 1; TGFBR1/2 – TGFβ receptor 1/2; TRPs – transient receptor potential channels; PLC – protein lipase C; PIP2 – phosphatidylinositol 4,5-bisphosphate; DAG – diacylglycerol; Pyr – pyruvate; PDH – pyruvate dehydrogenase; PDP – pyruvate dehydrogenase phosphatase; MICU1 – mitochondrial calcium uptake 1; NFAT – nuclear factor of activated T-cells; IDH – isocitrate dehydrogenase; ⍺KGDH - ⍺-ketoglutarate dehydrogenase; Col1a1 – collagen 1 type 1; Fn – fibronectin.
Upregulation of TRP channel 6 (TRPC6), via TGFβ-mediated p38/SRF signaling, is necessary for both in vitro and in vivo ventricular myofibroblast formation and wound healing54. Similar to TRPV4, TRPC6-dependent increases in cytosolic Ca2+ activates calcineurin-NFAT signaling to promote differentiation54. Additionally, TRP member 7 (TRPM7) and TRP channel 3 (TRPC3) expression and activity are increased in fibroblasts from human and animal models of atrial fibrillation-associated fibrosis99, 100. Interestingly, atrial cardiac fibroblasts appear to be partial to TRPM7, as they have no detectable activity of TRPC697, suggesting different populations of cardiac fibroblasts express different TRP channels. In summary, TRP channels increase and sustain cytosolic Ca2+ to activate and potentiate downstream signaling49, 54, 100, suggesting a unified/conserved mechanism in myofibroblast differentiation.
Liberation of intracellular Ca2+ stores, such as ER Ca2+, also contribute to differentiation as many pro-fibrotic agonists have been shown to enhance IP3R-mediated ER Ca2+ release101, 102 (Fig. 2). Pro-fibrotic increases in COL1A1 and FN in lung fibroblasts were blocked by pharmacological inhibition of both IP3R103 and RyR104, demonstrating ER Ca2+ release is required for myofibroblast differentiation. Furthermore, deletion of the ER Ca2+-binding protein calreticulin impairs the upregulation of COL1A1 and FN in mouse embryonic fibroblasts by reducing TGFβ-mediated increases in cytosolic Ca2+ and subsequent NFAT translocation105.
In agreement with the above reports, recent work from our lab confirmed that pro-fibrotic agonists (i.e. TGFβ, AngII) increase cytosolic Ca2+ and NFAT nuclear translocation20. More importantly, our study highlights a novel mechanism by which decreases in mitochondrial Ca2+ uptake not only contribute to increases in local cytosolic Ca2+ (see Fig. 2), but also coordinate specific changes in cellular metabolism for activation of the fibrotic gene program20, discussed in further detail below. While changes in mitochondrial Ca2+ dynamics appear to control myofibroblast differentiation, recent evidence also suggests that changes in mitochondrial-derived reactive oxygen species (ROS) coordinates intracellular signaling to promote and amplify fibrotic signaling. To these points, the next section explores the evidence for mitochondrial structural and functional changes and alterations in cellular metabolism that promote myofibroblast differentiation.
Mitochondrial Mechanisms of Myofibroblast Differentiation
Influence and mechanisms of ROS generation
Reactive oxygen species are byproducts of (patho)physiological processes with wide ranging cellular implications, from influencing intracellular signaling cascades to thiol-dependent posttranslational modifications. A major source of superoxide (O2∙−) in the cell is the ETC via electron slippage at Complex I and III106, 107. Mitochondrial-derived ROS (mtROS) may be a critical component of differentiation, as Complex III-derived O2∙− is increased following TGFβ-treated lung fibroblasts and mitochondrial-targeted antioxidants attenuated fibrotic gene induction46. In rat cardiac fibroblasts, β-adrenergic stimulation increased ROS production and the activation/phosphorylation of p38-MAPK to increase proliferation and collagen production108. p38-dependent increases in proliferation, and collagen production were reversed with antioxidant treatment, suggesting ROS-dependent activation of the p38-MAPK pathway108, likely through ROS-induced oxidation of a cysteine residue on MAPK phosphatase to reduce acitivty109 (Fig. 3A).
Figure 3. Mitochondrial mechanisms of myofibroblast differentiation and persistence.

(A) Pro-fibrotic stressors increase ROS production in the mitochondria, which then results in an increased and sustained intracellular ROS load, in part, through the downregulation of SOD2 and catalase. Increases in intracellular ROS in turn activates p38 and ERK1/2 signaling pathways, which are known to increase the transcription of the fibrotic gene program. (B) A key feature of myofibroblasts is their resistance to apoptosis. Mitochondrial cytochrome c (Cyto c) release is prevented through the upregulation of anti-apoptotic factors (BLC-2, BCL-XL) while pro-apoptotic factors (BAX, BAK) are downregulated. Reduced activation of the proteolytic caspase cascade due to decreased cytochrome c release from the mitochondria contributes to the persistence of myofibroblasts in the injured heart by imparting a resistance to cell death. TGFβ – transforming growth factor beta; AngII – angiotensin II; ROS – reactive oxygen species; SOD2 - superoxide dismutase 2; MKPs – mitogen-activated protein kinases; BCL-2 – B-cell lymphoma 2; BCL-XL - B-cell lymphoma-extra large; BAX – Bcl-2-associated X protein; BAK – Bcl-2 homologous antagonist killer; Casp9/3 – caspase 9/3.
Mitochondrial fission, the process of dynamin-related protein 1 (Drp1)-mediated mitochondrial fragmentation, provides one mechanism to increase mtROS45, 110. Genetic overexpression of mitochondrial fission factor (MFF) in immortalized human fibroblasts increased ROS production, which correlated with increased αSMA expression110. Conversely, decreasing fission through knock down of Drp1 decreased the AngII-mediated production of αSMA and COL-1 in adventitial fibroblasts, which the authors attributed to the blockade of fission-induced mtROS production45. Additionally, fibroblasts increase the production of O2∙− through the upregulation of NADPH oxidase 4 (NOX4)46. Members of the NOX family are typically located at the plasma membrane, however NOX4 is reportedly localized to mitochondria in various cell types111, 112. In lung fibroblasts, TGFβ-induced mitochondrial ROS generation transcriptionally upregulated NOX4, potentially as a means to sustain elevated intracellular ROS levels for activation of ROS-sensitive pathways (e.g. AKT and MAPK46). Indeed, siRNA against NOX4 inhibited the induction of ⍺SMA and FN-EDA via TGFβ non-canonical (ERK1/2) signaling113. The expression of NOX4 appears to be downstream of SMAD3, suggesting that NOX4-generated ROS amplifies TGFβ-mediated gene transcription, rather than initiating it59, 113. A NOX4 splice variant has also been shown to localize to the nucleus where it may play a role in activating p38-MAPK and ERK1/2-dependent transcriptional signaling109, 114.
Mitophagy, which is essential for the clearance of dysfunctional mitochondria, is downregulated in pathological conditions such as cancer and cardiovascular disease115, 116. Without proper autophagic clearance, dysfunctional mitochondria accumulate and contribute to excessive mtROS117. Interestingly, the expression of parkin, an autophagic effector encoded by the gene PARK2, is significantly reduced in lungs from idiopathic pulmonary fibrosis patients118. In fibroblasts, knockdown of Park2 results in the upregulation of ⍺SMA118, 119 in a platelet-derived growth factor receptor (PDGFR)-mediated manner, which is attenuated by mitochondrial-targeted antioxidant supplementation118. In a mouse model of bleomycin-induced lung fibrosis, deletion of Park2 enhanced lung fibrosis whereas treatment with a PDGFR inhibitor diminished this response118, suggesting a role for mitophagy in tissue fibrotic remodeling.
Lastly, myofibroblasts appear to downregulate the expression of various antioxidant enzymes [e.g. superoxide dismutase (SOD) and catalase] to sustain elevated intracellular ROS levels. For example, pro-fibrotic stimulation with AngII decreased catalase expression120 and mitochondrial SOD2 activity121, resulting in increased ROS (Fig. 3A). Importantly, the profibrotic effects of AngII were prevented with pretreatment of SOD mimetics121, suggesting a link between fibrotic signaling and the impaired ability to neutralize mtROS. Collectively, these studies suggest a critical role for mtROS in myofibroblast differentiation, however further study is required to elucidate the molecular mechanisms that instigate and promote mtROS and how mtROS directly influences the fibrotic gene program.
Resistance to Apoptosis
Resolution of wound healing is possible via the de-differentiation of myofibroblasts or loss of these cells by cell death signaling. Considering the latter, a characteristic feature of the myofibroblast is extreme resistance to cell death, which contributes to their persistence in the stressful environment associated with wound healing and fibrosis. One way mitochondria regulate apoptosis is by the release of apoptogens, such as cytochrome c or Smac/DIABLO, to activate the proteolytic caspase cascade. Anti-apoptotic factors such as B-cell lymphoma 2 (BCL-2) and B-cell lymphoma-extra-large (BCL-XL) prevent the release of apoptogens and formation of the apoptosome in opposition to pro-cell death factors such as Bcl-2-associated X protein (BAX) and Bcl-2 homologous antagonist killer (BAK)122, 123. The increased expression of anti-apoptotic factors is thought to be a primary mechanism by which myofibroblasts gain resistance to cellular demise (Fig. 3B). Fibrotic stress is reported to increase the expression of anti-apoptotic BCL-2 and BCL-XL resulting in decreased cell death in models of scleroderma124, Crohn’s disease125, and cardiac fibrosis126 while pharmacological inhibition of anti-apoptotic BCL-2 reduced ECM deposition and ⍺SMA expression125. Conversely, TGFβ activation of dermal fibroblasts decreased the expression of pro-apoptotic factor BAX127, suggesting a shift in the balance of BH3-only proteins. In cardiac fibroblasts, AngII stimulation promotes apoptotic resistance by activating MAPK-ERK1/2 signaling to inhibit capase-3 cleavage128. While myofibroblast activation is, in part, maintained by apoptotic resistance, targeting apoptosis pathways may provide novel therapeutic strategies to lessen the impact of persistent tissue fibrosis in disease progression.
Mitochondrial Ca2+ Exchange
As previously stated, sustained cytosolic Ca2+ is a driver of myofibroblast differentiation. In addition to this observation, we discovered that mitochondrial calcium uptake is reduced during fibroblast activation, which further augments cytosolic Ca2+ signaling and reduces matrix calcium levels20. Mitochondrial Ca2+ uptake is mediated by the mitochondrial Ca2+ uniporter complex (mtCU). The mtCU is a multimeric protein complex comprised of the pore forming component, mitochondrial calcium uniporter (MCU), and a number of regulatory subunits, such as mitochondrial calcium uptake 1 (MICU1) that serves as a channel gatekeeper by preventing opening at low cytosolic Ca2+ concentrations and enhancing MCU opening at higher concentrations via Ca2+ binding to EF-hand motifs on MICU1. While there are many additional components of the mtCU129, we will focus on MICU1 due to its role in myofibroblast differentiation. Our recent work demonstrates that pro-fibrotic stimulation (TGFβ or AngII) of mouse embryonic or cardiac fibroblasts results in increased expression of MICU1. Given the large increase in mRNA level, we speculate that this is a transcriptional mechanism, a hypothesis supported by prominent transcription factor binding motifs for SRF, NFAT and SMADs in the MICU1 promoter. The increase in MICU1 expression was sufficient to enhance mtCU gating and reduce mitochondrial Ca2+ uptake during fibroblast activation20 (refer to Fig. 2). In agreement, genetic deletion of Mcu results in increased myofibroblast differentiation in vivo following MI. The regulatory role of decreased mtCU activity in myofibroblasts is likely multifactorial. First, by decreasing the capacity of mitochondria to buffer local cytosolic Ca2+, more free Ca2+ is available to enhance the myofibroblast gene program though Ca2+-dependent transcription factor signaling (i.e. increase in microdomain Ca2+ for the activation of NFAT). Second, decreasing Ca2+ entry into the mitochondria could protect the myofibroblast against mitochondrial-dependent cell death in the stressful fibrotic environment. Third, altering mitochondrial matrix Ca2+ concentration is a direct mechanism to regulate cellular metabolism. For example, mtCa2+ is a regulator of pyruvate dehydrogenase (PDH) via activation of pyruvate dehydrogenase phosphatase, and fibroblast activation correlated with decreased PDH activity. Further, we found that fibrotic stimulation increased aerobic glycolytic flux, increased glutaminolysis, and elicited discrete changes in various metabolites that potentially regulate the myofibroblast differentiation program20. For example, activated fibroblasts increased the bioavailability of metabolites that are co-factors for enzymes that post-translationally modify histones and DNA, chromatin accessibility, and downstream gene expression20. Thus, mitochondria are a key regulatory hub for myofibroblast formation by participating in the modulation of signaling and metabolism that support the changes in genetic programming necessary for cell state transition. Next, we will summarize the literature, describing metabolism as a key driver of myofibroblast differentiation.
Cellular metabolism in myofibroblast differentiation
A prominent feature of fibroblast activation is an increase in aerobic glycolysis and lactate production in the presence of sufficient oxygen to support mitochondrial oxidative phosphorylation, a phenomenon referred to as The Warburg Effect130, 131. It is important to note that the changes in metabolism during initial fibroblast activation are distinct from the metabolic programs supporting the function of fully differentiated myofibroblasts. Metabolism impacts fibroblast differentiation by numerous mechanisms (Fig. 4). For example, increased ATP production is crucial for the contractile apparatus while the synthesis of building blocks (e.g. nucleotides and phospholipids) are necessary to support the initial proliferative burst, as well as the subsequent cellular growth of the fully differentiated, non-proliferative myofibroblast. Increased synthesis of various amino acids for de novo collagen production is essential for ECM remodeling. Metabolites themselves also serve as signaling molecules and are cofactors for epigenetic modifying enzymes that regulate chromatin structure and gene transcription. Here, we summarize the experimental evidence that changes in metabolism not only accompany the myofibroblast phenotype, but that these metabolic changes are in fact necessary for the activation and differentiation of fibroblasts themselves.
Figure 4. Metabolic components of cellular differentiation.

Overview of the metabolic influence regulating fibroblast differentiation. In addition to the energetic demands of a proliferating/differentiating cell, it is necessary to increase the biosynthetic intermediates to support such events. An increase in production and secretion of ECM components (e.g. collagens) requires increased amino acid synthesis. Post-translational modifications of proteins (e.g. transcription factors) can greatly alter their activities while regulation of the epigenetic landscape influences gene program activation & silencing. While each can work independently, the integration of these metabolic actions drive and coordinate cellular differentiation. ECM – extracellular matrix; SAM – S-adenosylmethionine; ⍺KG - ⍺-ketoglutarate.
Glucose and lactate
Changes in glycolysis are critical not only for energy provision, but also for coordinating ancillary biosynthetic pathways to provide the elements essential for cellular growth and differentiation132. The first evidence of glycolytic remodeling in activated fibroblasts was discovered in cystic fibrosis patients where increased activity of hexokinase, phosphofructokinase-1 (PFK1), pyruvate kinase muscle isozyme (PKM), and lactate dehydrogenase (LDH) were observed133. A fibroblast glycolytic switch has also been confirmed in numerous fibroblast populations20, 134, 135. One mechanisms to increase glycolysis is the transcriptional upregulation of numerous glycolytic genes134, 136–138, many of which are metabolic checkpoints (Fig. 5). In fact, simply increasing glucose concentrations in cardiac fibroblasts in the absence of TGFβ stimulation is sufficient to increase the expression of canonical signaling effectors TGFβ and SMAD2/3 and induce a myofibroblast phenotype139. As the hallmark of diabetes mellitus is an increase in blood glucose levels, and epidemiological and experimental studies demonstrate a strong association between diabetes and heart failure (reviewed elsewhere140, 141), the idea that extracellular glucose concentrations alone are sufficient to promote myofibroblast differentiation and tissue fibrosis is of clinical interest. Causal experimental evidence suggests that altering glucose utilization is necessary for myofibroblast formation. For example, inhibition of glucose uptake and glycolysis using 2-deoxyglucose (2DG) prevents the expression of ⍺SMA and Fn134, 138. PFK1, the rate-limiting and committed step of glycolysis, is allosterically activated by fructose-2,6-bisphosphate, the levels of which are regulated by the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB). Genetic deletion or pharmacologic inhibition of PFKFB3, an isoform with a ~740-fold higher kinase than phosphatase activity142, diminished myofibroblast formation138, Similarly, overexpression of a kinase-deficient PFKFB1 mutant isoform prevented differentiation, while overexpression of the phosphatase-deficient PFKFB1 mutant isoform was sufficient to promote differentiation, independent of exogenous ligands/activators20. Indeed, the inhibition of glucose uptake or PFK1 significantly attenuated the fibrotic response in in vivo models of renal and pulmonary fibrosis138. Further, inhibition of glyceraldehyde dehydrogenase facilitated phenotypic reversal of activated hepatic myofibroblasts into a less mature form as evidenced by a reduction in ⍺SMA expression and collagen secretion143.
Figure 5. Metabolic inputs regulating the biosynthetic and transcriptional changes coordinating myofibroblast differentiation.
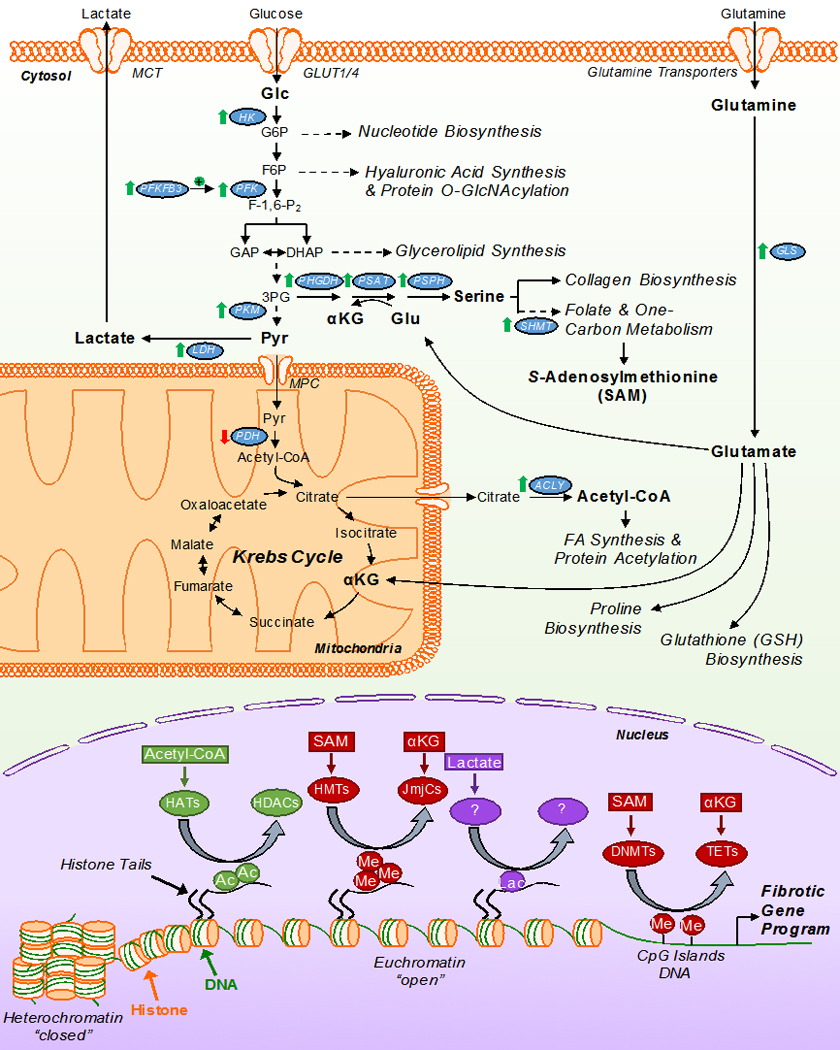
General schematic of myofibroblast metabolism on the differentiation program. Myofibroblasts are characterized by an increase in glutaminolysis and aerobic glycolysis, accompanied by decreased glucose oxidation. These key metabolic re-programming events permit the use of carbon intermediates by ancillary biosynthetic pathways to coordinate the production of ECM constituents, protein modifications, and epigenetic cofactors that initiate and sustain the myofibroblast differentiation program. Changes in the expression/activity of noted enzymes along with metabolites in bold are key to the differentiation process. Of these processes, the ability of these key metabolites to act as cofactors for epigenetic modifying enzymes is critical for the coordinated activation and silencing of transcriptional events which promote the fibrotic gene program and myofibroblast differentiation/persistence. αKG – α-ketoglutarate; Pyr – pyruvate; HK – hexokinase; PFK – phosphofructokinase; PFKFB3 – 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; PHGDH – phosphoglycerate dehydrogenase; PSAT – phosphoserine amino transferase; PSPH – phosphoserine phosphatase; PKM – pyruvate kinase muscle isozyme; LDH – lactate dehydrogenase; SHMT – serine hydroxymethyltransferase; PDH – pyruvate dehydrogenase; ACLY – ATP citrate lyase; HATs – histone acetyltransferases; HDACs – histone deacetylases; HMT – histone methyltransferases; JmJCs – Jumonji C-domain-containing histone demethylases; DNMTs – DNA methyltransferases; TETs – ten-eleven translocases; Ac – acetylation mark; Me – methylation mark; Lac – lactylation mark.
An additional site of regulation that appears critical for myofibroblast formation is pyruvate oxidation. Pyruvate kinase 2 (PKM2), the less catalytically active isoform, is upregulated in myofibroblasts134 and TGFβ rapidly increases PKM2 phosphorylation at Tyr105134, an inhibitory phosphorylation site that favors dimerization as opposed to the more active tetrameric state, further promoting the Warburg effect144. Intriguingly, PKM2 dimerization is associated with a redirection of glucose into the pentose phosphate pathway to augment nucleotide and serine biosynthesis, both of which are critical for de novo collagen formation145. In line with this regulatory mechanism, overexpression of pyruvate dehydrogenase kinase-1 (PDK), which inhibits PDH activity, is sufficient to promote myofibroblast formation146. We also reported that pro-fibrotic stimulation with either TGFβ or AngII rapidly upregulates MICU1, to limit mtCa2+ .uptake, which reduces PDH phosphatase activity resulting in increased phospho-PDH (decreased PDH activity)20 (see Fig. 2 & 5). In vivo activation of PDH by dichloroacetate prevented the induction of myofibroblast markers and interstitial fibrosis in a mouse model of kidney injury147. Collectively, these studies suggest that reducing glucose oxidation, in the face of enhanced glycolytic rates, is a key metabolic driver of myofibroblast differentiation with numerous checkpoints in place to ensure coordinated metabolic remodeling.
Unsurprisingly, enhanced glycolysis and reduced glucose oxidation results in increased lactate production in myofibroblasts134–136, 148. Recently, lactate has been implicated in myofibroblast formation as inhibition of LDH prevents the expression of ⍺SMA, FN, Col1 and Col3135, 148, whereas overexpression of LDH induced myofibroblast formation136; however, it should be noted that others have shown no role for LDH in fibroblast to myofibroblast transition147. As glycolysis is necessary and sufficient for differentiation, the activity of LDH is essential for the regeneration of NAD+, a required cofactor for glycolysis. One study134 had the surprising result that addition of exogenous lactate was sufficient to increase the expression of phenotypic myofibroblast markers in a dose-dependent manner. Lactate in the heart likely promotes myofibroblast differentiation through numerous mechanisms. For example, activation of latent TGFβ in the ECM is pH sensitive and regulated by HIF1α136. Since HIF1⍺ is a master transcriptional regulator of the hypoxic response, the idea that hypoxia-mediated tissue lactate accumulation may be a signal for fibroblast activation is noteworthy and warrants further investigation. Interestingly, HIF1α degradation is regulated by an αKG-dependent prolyl hydroxylase149, further intertwining metabolic regulation during myofibroblast formation. Lactate is also a signaling molecule for infiltrating immune cells responding to cardiac injury, resulting in M2 polarization and the production of pro-fibrotic cytokines150, 151. Interestingly, monocyte-derived MHCII+Ly6Clo macrophages (i.e. M1-like phenotype) are associated with a reduction of myocardial fibrosis152, indicating metabolic control of macrophage polarization may be a novel therapeutic target to treat cardiac fibrosis. Histone lactylation (addition of a lactyl group to lysine amino-acid residues) has been identified as an epigenetic modification regulating gene expression in macrophages and cancer cells153, providing an additional means of lactate-dependent regulation of differentiation (see Fig. 5). The fact that M2 macrophages, myofibroblasts, and cardiomyocytes all increase glycolysis under conditions of cardiac stress, suggests lactate may drive a futile cycle of myofibroblast activation and tissue fibrosis. The notion that exogenous lactate can promote myofibroblast differentiation has numerous physiological implications. First, while exercise clearly has a beneficial effect on cardiovascular health, reports of cardiac pathology in ultra-athletes suggest deleterious functional and structural cardiac remodeling including cardiac fibrosis154–156. While it is plausible that the chronic vs. intermittent nature of this type of exercise training could result in sustained elevations in circulating blood lactate leading to fibroblast activation, this hypothesis has yet to be investigated. Secondly, numerous cardiac diseases (e.g. MI) induce a hypoxic environment, long implicated in the pathogenesis of fibrotic disease. Tissue hypoxia increases cellular glycolytic rates and lactate production in all cells (e.g. cardiomyocytes, fibroblasts, immune cells), which in turn promote myofibroblast differentiation and tissue fibrosis.
The universal finding that tissue hypoxia occurs in wound healing and fibrotic disease suggests a critical role in myofibroblast activation and differentiation (REF). Overexpression of HIF1⍺ promoted myofibroblast formation and in support genetic knockdown or inhibition prevented differentiation136, 146. While the transcriptional upregulation of glycolytic genes is a primary target of HIF1⍺, it also appears to directly regulate the fibrotic gene program, as it can bind to the promoter region of ⍺SMA (Acta2)138. Activation of Hedgehog signaling also appears to play a role in metabolic remodeling as pharmacologic inhibition or genetic interruption of Hedgehog in hepatic fibroblasts reduced the expression of glycolytic genes and myofibroblast formation135. Glycolytic enzymes are also transcriptionally upregulated by canonical signaling; the inhibition of TGFβR1 or siRNA targeting Smad2/3 abolished the upregulation of PFKFB3 and myofibroblast formation138 and FACS analysis of activated pulmonary fibroblasts showed high correlation between PFKFB3 and ⍺SMA expression138. Canonical signaling has also been shown to modulate PI3K/AKT signaling in vascular smooth muscle cells157, providing yet another mechanism to enhance aerobic glycolysis. In summary, there is copious causal experimental evidence linking glycolysis to myofibroblast activation and differentiation.
Acetyl-CoA
Acetyl-CoA is a versatile two-carbon metabolite utilized for many cellular processes. Conventionally, acetyl-CoA is utilization to generate reducing equivalents through the Krebs cycle for cellular bioenergetics, however acetyl-CoA can exit the mitochondrial matrix as citrate where it is converted back to acetyl-CoA by ATP citrate lyase (ACLY) for anabolic purposes, such as de novo lipid synthesis and protein acetylation158, 159 (Fig. 5). Cytosolic and nuclear acetyl-CoA can also be formed by Acyl-CoA Synthetase Short Chain Family Member 2 (ACSS2) dependent acetate metabolism160. Furthermore, histone acetyltransferases (HATs) utilize acetyl-CoA for histone acetylation158, 160, 161 in cellular reprogramming and differentiation in many physiological contexts162, 163. For example, AKT activation of ACLY promotes myofibroblast gene expression in human kidney fibroblasts in vitro and pharmacological inhibition of ACLY suppressed fibrosis in mice subjected to ureteral obstruction164. In line with this idea, mesangial cells, which can differentiate into a fibroblast-like phenotype, increased the expression of Tgfβ1, Ctgf, and Fn when exposed to high glucose, in an ACLY/HAT-dependent manner165. However, others have shown TGFβ-induced myofibroblast differentiation results in decreased acetyl-CoA abundance and an overall decrease in total H3 acetylation166. Interestingly, ⍺SMA expression was decreased in renal fibroblasts supplemented with citrate or acetate, which reversed an observed global decrease in H3 acetylation166. However, while global acetylation was decreased, TGFβ increased H3K18 and H3K27 acetylation, suggesting a level of specificity in fibrotic gene program activation/silencing166. These studies suggest that, even in the same cell type, it is unclear how, or even if, acetyl-CoA metabolism regulates myofibroblast differentiation, warranting further study.
Multiple classes or families of HATs and histone deacetylases (HDACs) add or remove acetyl-CoA from histone residues to alter chromatin accessibility167, 168 (see Fig. 5). Inhibition of various HDACs have been shown to ameliorate myofibroblast differentiation and tissue organ fibrosis169–171. Similarly, evidence indicates that inhibiting HATs, the p300 family in particular, is an effective strategy to prevent cardiac myofibroblast differentiation and fibrosis172. This paradox of inhibiting either HATs or HDACs further implicates a level of specificity in regulating certain gene programs that likely coordinate cellular differentiation173, 174.
Another interesting avenue to explore is the catabolic generation of acetyl-CoA in fibroblast activation. Many carbon sources fuel acetyl-CoA production including: glucose, fatty acids, glutamine, and ketone bodies175–177. Hepatic stellate cells (HSCs) in the liver transition from lipid- and vitamin A-storing pericytes to myofibroblasts upon injury178. Activated HSCs upregulate the expression of peroxisome-proliferator-activated receptor β (PPARβ), a nuclear hormone receptor that promotes fatty acid oxidation179, potentially providing a supply of acetyl-CoA from lipids rather than glucose. Additionally, HSC activation and PPARβ expression was counteracted by ectopic expression of PPARγ and sterol regulatory element-binding protein 1 (SREBP1), transcription factors that promote lipid synthesis as opposed to catalysis179. These data suggest that fibroblast quiescence may be maintained by fatty acid synthesis with a switch to oxidation mediating activation. Interestingly, very few studies have investigated the effects of fatty acids on myofibroblast formation, providing opportunity for investigation.
Ancillary biosynthetic pathways
Numerous ancillary pathways of intermediary metabolism contribute to myofibroblast activation and the provision of biosynthetic materials for cellular growth and ECM remodeling. Collagen is unique in its amino acid structure due to its high (33%) glycine composition (ref). As myofibroblasts increase collagen production and secretion, this requires an increase in de novo synthesis of glycine and its precursor serine. To fuel biosynthetic demand, a coordinated increase in aerobic glycolysis and decrease in glucose oxidation shunts glucose-derived carbons into the serine biosynthetic pathway for collagen synthesis137, 180. TGFβ induces the expression of serine biosynthetic genes (PHGDH, PSAT1, PSPH, SHMT2)137, 180 (Fig. 5), via canonical (SMAD3) and noncanonical (mTOR) upregulation of activating transcription factor 4 (ATF4), a transcriptional master regulator of amino acid metabolism137. Importantly, genetic and pharmacologic attenuation of serine biosynthesis diminished collagen synthesis in pulmonary fibroblasts in vitro180. In vivo, pharmacological inhibition of phosphoglycerate dehydrogenase (PHGDH), the rate limiting step of serine biosynthesis, attenuated bleomycin-induced pulmonary fibrosis181. Proline, another amino acid making up ~17% of collagen, is also increased in abundance in TGFβ-stimulated fibroblasts and is necessary for the myofibroblast phenotype182, providing further evidence to the importance of amino acids in fibroblast activation and function.
Increased serine biosynthetic pathway flux also provides s-adenosylmethionine, the methyl donor for cytosine and histone methyltransferases (Fig. 5). Specifically, histone lysine methyltransferase KMT7 has been implicated in tissue fibrosis as genetic silencing significantly attenuated TGFβ-induced ECM gene expression accompanied by a correlative decrease in promoter H3K4me levels183. Furthermore, inhibition of Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit (EZH2) diminished H3K27me3 and increased collagen production184. While these data suggest that loss of methylation at H3K27 and H3K4 enhances myofibroblast formation, KMT2H (ASH1L gene which methylates H3K9) is reported to directly bind regulatory elements of pro-fibrotic genes, and knockdown of KMT2H suppresses fibrogenic gene expression in the liver185. Collectively, these studies demonstrate the importance of these pathways in both the biosynthetic and transcriptional requirements necessary for myofibroblast formation.
Changes in glycolytic flux can also redirect glucose into the hexosamine biosynthetic pathway132 (Fig. 5). One critical function of this pathway is the post-translational addition of O-Linked β-N-acetylglucosamine (O-GlcNAc) to Ser/Thr residues to alter protein function186. Increased protein O-GlcNAcylation is observed in ischemic injury, heart failure and diabetic cardiomyopathy, all of which are associated with fibrosis186–189. In rat cardiac fibroblasts cultured under high-glucose conditions, SP1, a transcription factor associated with myofibroblast activation190–193, demonstrated higher levels of O-GlcNAcylation139. This O-GlcNAcylation of SP1 enhanced transcription factor binding to the promoter region of Col1a1 resulting in increased transcription and collagen production. This was abrogated with genetic overexpression of the O-GlcNAc removal enzyme, O-GlcNAcase139. In addition to GlcNAcylation post-translational modification, the hexosamine biosynthetic pathway is necessary for the synthesis of hyaluronic acid, a chief component of the ECM involved in cell-cell adhesions, migration, proliferation, and differentiation194. Genetic silencing of hyaluronan synthase prevented the expression of ⍺SMA194, 195 and was mechanistically linked to TGFβ1 signaling196, 197. Furthermore, depletion of cytoplasmic UDP-glucuronic acid, essential for hyaluronan-chain elongation, decreased the expression of ⍺SMA195, an effect not reproduced by exogenous administration of hyaluronan198, suggesting the macromolecular assembly of de novo hyaluronan is required. In addition, overexpression of glucosamine—fructose-6-phosphate aminotransferase isomerizing 1 (GFPT1) , a rate limiting step in the hexosamine pathway, is sufficient to increase TGFβ1 mRNA and protein levels in 3T3 fibroblasts, partly by enhancing Tgfβ1 promoter activity199, suggesting direct transcriptional regulation of the fibrotic gene program via the hexosamine pathway.
Glutamine
Glutamine is the most abundant amino acid in the body and serves an anaplerotic role by replenishing Krebs cycle intermediates, at the level of alpha-ketoglutarate (αKG), under conditions of increased aerobic glycolysis and reduced oxidative phosphorylation176. In TGFβ treated fibroblasts, glutamine utilization (i.e. glutaminolysis) is enhanced and contributes to increased levels of αKG20, 182 (Fig. 5). Increased rates of glutaminolysis in fibroblasts is accomplished by the upregulation of the rate-limiting enzyme, glutaminase 1 (GLS1), via TGFβ182, 200 and Hedgehog signaling181, 201. Glutaminolysis is a strict requirement for fibroblast activation in vitro as pharmacologic inhibition and genetic silencing of Gls1 or removal of extracellular glutamine prevents myofibroblast formation 20, 182, 200, 201. Myofibroblast persistence appears also dependent upon glutaminolysis as removal of extracellular glutamine or siRNA knockdown of GLS1 in a lung fibroblast cell line reverted myofibroblasts into a more quiescent state200. Importantly, protection from bleomycin-induced pulmonary fibrosis was observed with pharmacological inhibition of GLS1202.
One potential mechanism by which glutamine controls myofibroblast formation may be at the level of epigenetic modifications that regulate chromatin remodeling. For example, αKG is a cofactor for numerous dioxygenases, including the epigenetic modifiers ten-eleven translocation enzymes (TETs) and jumonji-C (JmjC)-domain-containing demethylases (JmjC-KDMs), which demethylate DNA cytosine and histone lysine residues, respectively, to regulate chromatin structure (see Fig. 5). We recently discovered that JmjC-KDMs are necessary for demethylation of H3K27me2/3 residues for profibrotic gene expression in mouse embryonic fibroblasts20. In support, treatment with cell-permeable dimethyl-αKG was sufficient to promote differentiation and inhibition of JmjC-KDMs, with the drug JIB-04, during TGFβ stimulation was sufficient to block myofibroblast formation20. Additionally, removal of glutamine from lung fibroblast media decreased KDM6B activity resulting in increased H3K27me3 levels203. Chromatin immunoprecipitation confirmed that JMJD3 interacts with the promoter region of anti-apoptotic genes in a glutamine-dependent manner203, suggesting one potential mechanism allowing for myofibroblast persistence in injured tissues. Collectively, these results implicate glutaminolysis as a prominent link with the epigenome to induce cellular differentiation. This warrants further investigation into chromatin remodeling mechanisms in myofibroblast activation and persistence in disease, which has been touched upon elsewhere204.
Glutamine is also an essential substrate for de novo collagen synthesis. In lung fibroblasts, the glutamate-consuming enzymes phosphoserine aminotransferase 1 (PSAT1) and Δ1-pyrroline-5-carboxylate synthase (P5CS) are necessary for glycine and proline biosynthesis and collagen production205 (Fig. 5). Additionally, glutaminolysis not only enhances collagen production but also its stability, as inhibition of GLS1 increased collagen degradation and exogenous αKG prevented degradation in a proline hydroxylation dependent manner182. Lastly, glutamine may play a role in myofibroblast differentiation through de novo synthesis of the antioxidant glutathione. A reduction in the GSH:GSSG ratio, suggesting an increase in the oxidative environment, occurs with TGFβ stimulation20 and is implicated in regulating the fibrogenic effects of TGFβ through SMAD3 activation206 (see above section on Influence and mechanisms of ROS generation).
Conclusions
Myofibroblasts are a specialized cell type that are responsible for the pathological remodeling observed in tissue organ fibrosis. Numerous pro-fibrotic signaling cascades contribute to mechanisms for myofibroblast differentiation, most of which integrate with mitochondrial and metabolic pathways for cellular reprogramming (Fig. 6). Of these mechanisms, the interactions between metabolism and the epigenome are a logical candidate for additional study. While identifying the epigenetic signatures that mediate myofibroblast differentiation and tissue fibrosis is an emerging field, and has been reviewed thoroughly elsewhere173, 174, identifying the specific metabolic substrates and pathways that contribute to DNA and histone modifications for chromatin remodeling and fibrotic gene expression may provide novel therapeutic targets to treat tissue organ fibrosis in disease.
Figure 6. Working model of the influence of mitochondria and metabolism on myofibroblast differentiation and persistence.

Pro-fibrotic stress stimuli are critical for the activation and differentiation of resident cardiac fibroblasts to myofibroblasts. Pro-fibrotic stimulation results in significant metabolic remodeling that is critical for the energy provisions and synthesis of cellular building blocks required for proliferation, growth, and differentiation. Furthermore, these changes in cellular metabolism also regulate the bioavailability of metabolites which serve as cofactors for numerous epigenetic-modifying enzymes to coordinate the activation of the fibrotic gene program to promote differentiation. Recent work from our lab has shed new light on the mitochondria as a key regulator to this process by reducing mitochondrial Ca2+ uptake to promote the metabolic remodeling required for the differentiation process. The mitochondria also increase the generation of mROS to promote the necessary metabolic remodeling. Additionally, mitochondria under pro-fibrotic stimulation and in the terminally differentiated state downregulate the apoptotic pathways, promoting myofibroblast persistence which contributes to the progressive nature of cardiovascular disease. Collectively, the coordinated efforts of mitochondrial and metabolic remodeling are critical for the activation, differentiation, and persistence of myofibroblasts in cardiac disease, providing novel therapeutic targets to mitigate and potential reverse tissue fibrosis in disease. TGFβ – transforming growth factor beta; AngII – angiotensin II; m[Ca2+] - mitochondrial calcium concentration; mROS – mitochondrial-derived reactive oxygen species; ECM – extracellular matrix; SAM – S-adenosylmethionine; ⍺KG - ⍺-ketoglutarate.
Encouragingly, metabolic targets highlighted throughout this review, such as inhibition of GLS1 via the small molecule CB-839 and dichloroacetate to inhibit PDK, are in phase 1 and 2 cancer clinical trials. If found safe, the repurposing of such drugs for the treatment of cardiac fibrosis may quickly follow. Additionally, increasing mtCU uptake through the use of the natural plant flavonoid kaempferol207, may present a potential strategy to reverse the widespread alterations in metabolism contributing to myofibroblast differentiation/persistence. To avoid off target effects of pharmacological interventions, the use of nanoparticle drug delivery systems by loading drug inhibitors into lipid vesicles (e.g. exosomes) that bind specific surface markers of a cardiac fibroblasts could be developed. Gene targeting may provide another therapeutic avenue as the use of systemically injected adeno-associated viruses (AAVs) have been used to direct gene transfer to a variety of tissues. Recently, the use of AAV9 carrying a Postn promoter targeted a myofibroblast-like lineage in mouse hearts following myocardial infarction, potentially providing a new avenue for gene therapy to treat cardiac fibrosis208. The idea of periostin-directed gene therapy is made more appealing by the knowledge that periostin expression is largely absent from the non-diseased heart and is specifically upregulated in activated fibroblasts13, suggesting gene therapy can be restricted to myofibroblasts and hence avoid on-target negative effects. Lastly, recent studies utilizing methodologies designed to combat cancer report that chimeric antigen receptor T-cell (CAR-T) therapy may be amendable to override myofibroblast resistance to apoptosis. Epstein and colleagues discovered that engineered T cells targeting activated cardiac fibroblasts via fibroblast activation protein (FAP), reduced pathological myofibroblast number and fibrosis in a model of chronic neurohormonal stimulation209. While an effective treatment is still elusive, the continued study of myofibroblast mitochondria and metabolism will likely yield novel therapeutic strategies to treat cardiac fibrosis and improve patient outcomes.
Acknowledgments
Sources of Funding
Funding for current work provided by the NIH to AAG (F32HL145914) and JWE (R01HL123966, R01HL136954, R01HL142271 and P01HL134608).
Nonstandard Abbreviations and Acronyms:
- TGFβ
transforming growth factor beta
- AngII
angiotensin 2
- ECM
extracellular matrix
- ⍺SMA
alpha smooth muscle actin (protein)
- Acta2
alpha smooth muscle actin (gene)
- MI
myocardial infarction
- mtROS
mitochondrial-derived reactive oxygen species
- Ca2+
calcium
- TCF21
transcription factor 21
- WT1
Wilms tumor protein
- TBX18
T-box transcription factor 18
- ER
endoplasmic reticulum
- p38
p38 mitogen-activated protein kinases
- MAPK1/2
mitogen-activated protein kinase 1/2
- ERK1/2
extracellular signal related kinase 1/2
- FN
fibronectin
- POSTN
periostin
- COL-1
collagen type 1
- COL-3
collagen type 3
- Fn-EDA
fibronectin containing extra domain A
- TGFβR1/2
transforming growth factor beta receptor type 1 or 2
- SMAD2/3/4/7
SMAD family member 2/3/4/7
- AKT
phosphoinositide 3 kinase (PI3K)-protein kinase B
- NF-kB
nuclear factor-kB
- GPCR
G protein-coupled receptor
- SRF
serum response factor
- ET-1
endothelin 1
- NFAT
nuclear factor of activated T-cells
- RhoA
Ras homolog family member A
- ROCK
Rho-associated, coiled-coil-containing protein kinase
- MRTF
myocardin related transcription factor
- TRP
transient receptor potential cation channels
- TRPV4
transient receptor potential vanilloid 4
- TRPC6
transient receptor potential channel 6
- TRPM7
transient receptor potential member 7
- TRPC3
transient receptor potential channel 3
- IP3R
inositol triphosphate receptor
- RyR
ryanodine receptor
- O2∙−
superoxide
- Drp1
dynamin-related protein 1
- MFF
mitochondrial fission factor
- NOX4
NADPH oxidase 4
- PARK2
Parkin RBR E3 Ubiquitin Protein Ligase (gene)
- PDGFR
platelet-derived growth factor receptor
- SOD2
mitochondrial superoxide dismutase
- BCL-2
B-cell lymphoma 2
- BCL-XL
B-cell lymphoma-extra large (BCL-XL)
- BAX
Bcl-2-associated X protein
- BAK
Bcl-2 homologous antagonist killer
- mtCU
mitochondrial calcium uniporter
- MCU
mitochondrial calcium uniporter
- MICU1
mitochondrial calcium uptake 1
- PDH
pyruvate dehydrogenase
- PFK1
phosphofructokinase 1
- PKM
pyruvate kinase muscle isozyme
- LDH
lactate dehydrogenase
- 2-DG
2-deoxyglucose
- PFKFB
phosphofructo-2-kinase/fructose-2,6-bisphosphate
- PKM2
pyruvate kinase isozyme 2
- PDK
pyruvate dehydrogenase kinase-1
- HIF1α
hypoxia inducible factor 1 alpha
- ACLY
ATP citrate lyase
- ACSS2
Acyl-CoA Synthetase Short Chain Family Member 2 (ACSS2)
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HSC
Hepatic stellate cells
- PPARβ/𝛾
peroxisome-proliferator-activated receptor β/𝛾
- SREBP-1
sterol regulatory element-binding protein 1
- PHGDH
phosphoglycerate dehydrogenase
- PSAT1
phosphoserine aminotransferase 1
- PSPH
phosphoserine phosphatase
- SHMT2
serine hydroxymethyltransferase
- mTOR
mammalian target of rapamycin
- ATF-4
activating transcription factor 4
- KMT7
lysine N-methyltransferase 7
- KMT2H
lysine N-methyltransferase 2H
- O-GlcNAc
O-Linked β-N-acetylglucosamine
- SP1
transcription factor Sp1
- GFPT1
glucosamine-fructose-6-phosphate aminotransferase isomerizing 1
- ⍺KG
alpha ketoglutarate
- GLS
glutaminase
- TET
ten-eleven translocation enzymes
- JmjC-KDMs
JmjC-domain-containing demethylases
- JMJD3
jumonji domain-containing protein D3
- P5CS
Δ1-pyrroline-5-carboxylate synthase
- GSH
glutathione
- GSSG
oxidized glutathione
- AAV
adeno-associated viruses
- CAR-T
chimeric antigen receptor T-cell
- FAP
fibroblast activation protein
Footnotes
Disclosures
AAG, MPL and JWE: None.
References
- 1.Diez J. Mechanisms of cardiac fibrosis in hypertension. J Clin Hypertens (Greenwich). 2007;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Humeres C and Frangogiannis NG. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl Sci. 2019;4:449–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonnans C, Chou J and Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michel G, Tonon T, Scornet D, Cock JM and Kloareg B. The cell wall polysaccharide metabolism of the brown alga Ectocarpus siliculosus. Insights into the evolution of extracellular matrix polysaccharides in Eukaryotes. New Phytol. 2010;188:82–97. [DOI] [PubMed] [Google Scholar]
- 5.Theocharis AD, Skandalis SS, Gialeli C and Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27. [DOI] [PubMed] [Google Scholar]
- 6.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA and Tallquist MD. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118:400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frangogiannis NG. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019;65:70–99. [DOI] [PubMed] [Google Scholar]
- 8.Hinz B. Formation and Function of the Myofibroblast during Tissue Repair. Journal of Investigative Dermatology. 2007;127:526–537. [DOI] [PubMed] [Google Scholar]
- 9.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat M-L and Gabbiani G. The Myofibroblast: One Function, Multiple Origins. The American Journal of Pathology. 2007;170:1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Halade GV and Lindsey ML. Extracellular Matrix and Fibroblast Communication Following Myocardial Infarction. Journal of cardiovascular translational research. 2012;5:848–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J and Evans SM. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C and Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. [DOI] [PubMed] [Google Scholar]
- 13.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD and Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L and Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. [DOI] [PubMed] [Google Scholar]
- 15.Weber KT and Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83:1849–1865. [DOI] [PubMed] [Google Scholar]
- 16.Weber KT, Sun Y, Bhattacharya SK, Ahokas RA and Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. 2013;10:15–26. [DOI] [PubMed] [Google Scholar]
- 17.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 18.Shrishrimal S, Kosmacek EA and Oberley-Deegan RE. Reactive Oxygen Species Drive Epigenetic Changes in Radiation-Induced Fibrosis. Oxid Med Cell Longev. 2019;2019:4278658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Negmadjanov U, Godic Z, Rizvi F, Emelyanova L, Ross G, Richards J, Holmuhamedov EL and Jahangir A. TGF-beta1-mediated differentiation of fibroblasts is associated with increased mitochondrial content and cellular respiration. PLoS One. 2015;10:e0123046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lombardi AA, Gibb AA, Arif E, Kolmetzky DW, Tomar D, Luongo TS, Jadiya P, Murray EK, Lorkiewicz PK, Hajnoczky G, Murphy E, Arany ZP, Kelly DP, Margulies KB, Hill BG and Elrod JW. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat Commun. 2019;10:4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN and Tallquist MD. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leslie KO, Taatjes DJ, Schwarz J, vonTurkovich M and Low RB. Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit. Am J Pathol. 1991;139:207–16. [PMC free article] [PubMed] [Google Scholar]
- 23.Vracko R and Thorning D. Contractile cells in rat myocardial scar tissue. Lab Invest. 1991;65:214–27. [PubMed] [Google Scholar]
- 24.Willems IE, Havenith MG, De Mey JG and Daemen MJ. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol. 1994;145:868–75. [PMC free article] [PubMed] [Google Scholar]
- 25.Dettman RW, Denetclaw W Jr., Ordahl CP and Bristow J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev Biol. 1998;193:169–81. [DOI] [PubMed] [Google Scholar]
- 26.Gittenberger-de Groot AC, Vrancken Peeters MP, Mentink MM, Gourdie RG and Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82:1043–52. [DOI] [PubMed] [Google Scholar]
- 27.Smith CL, Baek ST, Sung CY and Tallquist MD. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res. 2011;108:e15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tallquist MD and Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braitsch CM, Kanisicak O, van Berlo JH, Molkentin JD and Yutzey KE. Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J Mol Cell Cardiol. 2013;65:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowers SL, Banerjee I and Baudino TA. The extracellular matrix: at the center of it all. J Mol Cell Cardiol. 2010;48:474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kakkar R and Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010;106:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pellman J, Zhang J and Sheikh F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: Mechanisms and model systems. J Mol Cell Cardiol. 2016;94:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell MD, Laird RE, Brown RD and Long CS. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am J Physiol Heart Circ Physiol. 2007;292:H1139–47. [DOI] [PubMed] [Google Scholar]
- 34.Chen C, Du J, Feng W, Song Y, Lu Z, Xu M, Li Z and Zhang Y. beta-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCdelta/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br J Pharmacol. 2012;166:676–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu N, Xing R, Yang C, Tian A, Lv Z, Sun N, Gao X, Zhang Y and Li Z. HIP-55/DBNL-dependent regulation of adrenergic receptor mediates the ERK1/2 proliferative pathway. Mol Biosyst. 2014;10:1932–9. [DOI] [PubMed] [Google Scholar]
- 36.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC and Molkentin JD. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128:2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deb A and Ubil E. Cardiac fibroblast in development and wound healing. J Mol Cell Cardiol. 2014;70:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Muller AM, Volz KS, Tang Z, Red-Horse K and Ardehali R. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115:625–35. [DOI] [PubMed] [Google Scholar]
- 39.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S and Wettschureck N. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res. 2016;118:1906–17. [DOI] [PubMed] [Google Scholar]
- 40.Shiojima I, Aikawa M, Suzuki J, Yazaki Y and Nagai R. Embryonic smooth muscle myosin heavy chain SMemb is expressed in pressure-overloaded cardiac fibroblasts. Jpn Heart J. 1999;40:803–18. [DOI] [PubMed] [Google Scholar]
- 41.Ignotz RA and Massague J. Transforming Growth Factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the Extracellular Matrix. J of Biol Chem. 1986;261:9. [PubMed] [Google Scholar]
- 42.Bowler MA, Bersi MR, Ryzhova LM, Jerrell RJ, Parekh A and Merryman WD. Cadherin-11 as a regulator of valve myofibroblast mechanobiology. Am J Physiol Heart Circ Physiol. 2018;315:H1614–H1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Black M, Milewski D, Le T, Ren X, Xu Y, Kalinichenko VV and Kalin TV. FOXF1 Inhibits Pulmonary Fibrosis by Preventing CDH2-CDH11 Cadherin Switch in Myofibroblasts. Cell Rep. 2018;23:442–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bell E, Ivarsson B and Merrill C. Production of a tissue-like structure by contaction of collagen lattices by human fibroblast of different proliferative potential in vitro. Proc Natl Acad Sci USA. 1979;76:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang G, Cong Z, Wang X, Yuan Y, Xu R, Lu Z, Wang X and Qi J. Targeting HSP90 attenuates angiotensin II-induced adventitial remodeling via suppression of mitochondrial fission. Cardiovasc Res. 2019. [DOI] [PubMed] [Google Scholar]
- 46.Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GR and Chandel NS. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. 2013;288:770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis J and Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stempien-Otero A, Kim DH and Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. 2016;97:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thodeti CK, Paruchuri S and Meszaros JG. A TRP to cardiac fibroblast differentiation. Channels (Austin). 2013;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–80. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Chen H, Seth A and McCulloch CA. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2003;285:11. [DOI] [PubMed] [Google Scholar]
- 52.Luo K and Lodish HF. Signaling by chimeric erythropoietin-TGF-beta receptors: homodimerization of the cytoplasmic domain of the type I TGF-beta receptor and heterodimerization with the type II receptor are both required for intracellular signal transduction. EMBO J. 1996;15:4485–96. [PMC free article] [PubMed] [Google Scholar]
- 53.Gao Y, Duran S, Lydon JP, DeMayo FJ, Burghardt RC, Bayless KJ, Bartholin L and Li Q. Constitutive activation of transforming growth factor Beta receptor 1 in the mouse uterus impairs uterine morphology and function. Biol Reprod. 2015;92:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis J, Burr AR, Davis GF, Birnbaumer L and Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verrecchia F, Chu ML and Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276:17058–62. [DOI] [PubMed] [Google Scholar]
- 56.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF and Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu B, Wu Z, Liu T, Ullenbruch MR, Jin H and Phan SH. Gut-enriched Kruppel-like factor interaction with Smad3 inhibits myofibroblast differentiation. Am J Respir Cell Mol Biol. 2007;36:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB and Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–108. [DOI] [PubMed] [Google Scholar]
- 59.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ and Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15:1077–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoyles RK, Derrett-Smith EC, Khan K, Shiwen X, Howat SL, Wells AU, Abraham DJ and Denton CP. An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor beta receptor. Am J Respir Crit Care Med. 2011;183:249–61. [DOI] [PubMed] [Google Scholar]
- 61.Denton CP, Khan K, Hoyles RK, Shiwen X, Leoni P, Chen Y, Eastwood M and Abraham DJ. Inducible lineage-specific deletion of TbetaRII in fibroblasts defines a pivotal regulatory role during adult skin wound healing. J Invest Dermatol. 2009;129:194–204. [DOI] [PubMed] [Google Scholar]
- 62.Sousa AM, Liu T, Guevara O, Stevens J, Fanburg BL, Gaestel M, Toksoz D and Kayyali US. Smooth muscle alpha-actin expression and myofibroblast differentiation by TGFbeta are dependent upon MK2. J Cell Biochem. 2007;100:1581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K and Davis J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation. 2017;136:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y, Nishizawa M, Fujisawa J and Inoue K. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–89. [DOI] [PubMed] [Google Scholar]
- 65.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF and Massague J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–14. [DOI] [PubMed] [Google Scholar]
- 66.Akhurst RJ and Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Snead AN and Insel PA. Defining the cellular repertoire of GPCRs identifies a profibrotic role for the most highly expressed receptor, protease-activated receptor 1, in cardiac fibroblasts. FASEB J. 2012;26:4540–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gonzalez A, Lopez B and Diez J. Fibrosis in hypertensive heart disease: role of the renin-angiotensin-aldosterone system. Med Clin North Am. 2004;88:83–97. [DOI] [PubMed] [Google Scholar]
- 69.Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–32. [DOI] [PubMed] [Google Scholar]
- 70.Moriguchi Y, Matsubara H, Mori Y, Murasawa S, Masaki H, Maruyama K, Tsutsumi Y, Shibasaki Y, Tanaka Y, Nakajima T, Oda K and Iwasaka T. Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ Res. 1999;84:1073–84. [DOI] [PubMed] [Google Scholar]
- 71.Fukuda N, Hu WY, Kubo A, Kishioka H, Satoh C, Soma M, Izumi Y and Kanmatsuse K. Angiotensin II upregulates transforming growth factor-beta type I receptor on rat vascular smooth muscle cells. Am J Hypertens. 2000;13:191–8. [DOI] [PubMed] [Google Scholar]
- 72.Gao X, He X, Luo B, Peng L, Lin J and Zuo Z. Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur J Pharmacol. 2009;606:115–20. [DOI] [PubMed] [Google Scholar]
- 73.Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, Bhowmick NA, Ju W, Bottinger EP and Lan HY. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res. 2006;98:1032–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hautmann MB, Thompson MM, Swartz EA, Olson EN and Owens GK. Angiotensin II-induced stimulation of smooth muscle alpha-actin expression by serum response factor and the homeodomain transcription factor MHox. Circ Res. 1997;81:600–10. [DOI] [PubMed] [Google Scholar]
- 75.Hishikawa K, Nakaki T, Marumo T, Suzuki H, Kato R and Saruta T. Pressure enhances endothelin-1 release from cultured human endothelial cells. Hypertension. 1995;25:449–52. [DOI] [PubMed] [Google Scholar]
- 76.Kuchan MJ and Frangos JA. Shear stress regulates endothelin-1 release via protein kinase C and cGMP in cultured endothelial cells. Am J Physiol. 1993;264:H150–6. [DOI] [PubMed] [Google Scholar]
- 77.Macarthur H, Warner TD, Wood EG, Corder R and Vane JR. Endothelin-1 release from endothelial cells in culture is elevated both acutely and chronically by short periods of mechanical stretch. Biochem Biophys Res Commun. 1994;200:395–400. [DOI] [PubMed] [Google Scholar]
- 78.Hasdai D, Holmes DR Jr., Garratt KN, Edwards WD and Lerman A. Mechanical pressure and stretch release endothelin-1 from human atherosclerotic coronary arteries in vivo. Circulation. 1997;95:357–62. [DOI] [PubMed] [Google Scholar]
- 79.McClellan G, Weisberg A, Rose D and Winegrad S. Endothelial cell storage and release of endothelin as a cardioregulatory mechanism. Circ Res. 1994;75:85–96. [DOI] [PubMed] [Google Scholar]
- 80.Omland T, Lie RT, Aakvaag A, Aarsland T and Dickstein K. Plasma endothelin determination as a prognostic indicator of 1-year mortality after acute myocardial infarction. Circulation. 1994;89:1573–9. [DOI] [PubMed] [Google Scholar]
- 81.Piacentini L, Gray M, Honbo NY, Chentoufi J, Bergman M and Karliner JS. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol. 2000;32:565–76. [DOI] [PubMed] [Google Scholar]
- 82.Guarda E, Katwa LC, Myers PR, Tyagi SC and Weber KT. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc Res. 1993;27:2130–4. [DOI] [PubMed] [Google Scholar]
- 83.Shi-wen X, Kennedy L, Renzoni EA, Bou-Gharios G, du Bois RM, Black CM, Denton CP, Abraham DJ and Leask A. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum. 2007;56:4189–94. [DOI] [PubMed] [Google Scholar]
- 84.Nishida M, Onohara N, Sato Y, Suda R, Ogushi M, Tanabe S, Inoue R, Mori Y and Kurose H. Galpha12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J Biol Chem. 2007;282:23117–28. [DOI] [PubMed] [Google Scholar]
- 85.Fujisaki H, Ito H, Hirata Y, Tanaka M, Hata M, Lin M, Adachi S, Akimoto H, Marumo F and Hiroe M. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J Clin Invest. 1995;96:1059–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, Jin TH, Desai L, Bernard K and Thannickal VJ. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123:1096–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kural MH and Billiar KL. Myofibroblast persistence with real-time changes in boundary stiffness. Acta Biomater. 2016;32:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. 2009;11:120–6. [DOI] [PubMed] [Google Scholar]
- 89.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM and Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, Ding Q, Thannickal VJ and Zhou Y. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47:340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K and Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bernau K, Torr EE, Evans MD, Aoki JK, Ngam CR and Sandbo N. Tensin 1 Is Essential for Myofibroblast Differentiation and Extracellular Matrix Formation. Am J Respir Cell Mol Biol. 2017;56:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Muldoon L, Rodland K and Magun B. Transforming growth factor beta and epidermal growth factor alter calcium influx. J Biol Chem. 1988;263:18834–41. [PubMed] [Google Scholar]
- 94.Alevizopoulos A, Dusserre Y, Ruegg U and Mermod N. Regulation of the transforming growth factor beta-responsive transcription factor CTF-1 by calcineurin and calcium/calmodulin-dependent protein kinase IV. J Biol Chem. 1997;272:10. [DOI] [PubMed] [Google Scholar]
- 95.Ostrom RS, Naugle JE, Hase M, Gregorian C, Swaney JS, Insel PA, Brunton LL and Meszaros JG. Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin. Gq-Gs cross-talk regulates collagen production in cardiac fibroblasts. J Biol Chem. 2003;278:24461–8. [DOI] [PubMed] [Google Scholar]
- 96.Li S, Sun X, Wu H, Yu P, Wang X, Jiang Z, Gao E, Chen J, Li, Qiu C, Song B, Chen K, He K, Yang D and Yang Y. TRPA1 Promotes Cardiac Myofibroblast Transdifferentiation after Myocardial Infarction Injury via the Calcineurin-NFAT-DYRK1A Signaling Pathway. Oxid Med Cell Longev. 2019;2019:6408352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM and Thodeti CK. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rahaman SO, Grove LM, Paruchuri S, Southern BD, Abraham S, Niese KA, Scheraga RG, Ghosh S, Thodeti CK, Zhang DX, Moran MM, Schilling WP, Tschumperlin DJ and Olman MA. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest. 2014;124:5225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B and Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106:992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif JC, Schotten U, Van Wagoner DR, Dobrev D and Nattel S. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McGowan TA, Madesh M, Zhu Y, Wang L, Russo M, Deelman L, Joseph S, Hajnoczky G and Sharma K. TGF-beta-induced Ca(2+) influx involves the type III IP(3) receptor and regulates actin cytoskeleton. Am J Physiol Renal Physiol. 2001;282. [DOI] [PubMed] [Google Scholar]
- 102.Karppinen S, Rapila R, Makikallio K, Hanninen SL, Rysa J, Vuolteenaho O and Tavi P. Endothelin-1 signalling controls early embryonic heart rate in vitro and in vivo. Acta Physiol (Oxf). 2014;210:369–80. [DOI] [PubMed] [Google Scholar]
- 103.Mukherjee S, Kolb MR, Duan F and Janssen LJ. Transforming growth factor-beta evokes Ca2+ waves and enhances gene expression in human pulmonary fibroblasts. Am J Respir Cell Mol Biol. 2012;46:757–64. [DOI] [PubMed] [Google Scholar]
- 104.Mukherjee S, Duan F, Kolb MR and Janssen LJ. Platelet derived growth factor-evoked Ca2+ wave and matrix gene expression through phospholipase C in human pulmonary fibroblast. Int J Biochem Cell Biol. 2013;45:1516–24. [DOI] [PubMed] [Google Scholar]
- 105.Zimmerman KA, Graham LV, Pallero MA and Murphy-Ullrich JE. Calreticulin regulates transforming growth factor-beta-stimulated extracellular matrix production. J Biol Chem. 2013;288:14584–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR and Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bouchez C and Devin A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu H, Tian A, Wu J, Yang C, Xing R, Jia P, Yang L, Zhang Y, Zheng X and Li Z. Danshensu inhibits β-adrenergic receptors-mediated cardiac fibrosis by ROS-p38 MAPK axis. Biol Pharm Bull. 2014;37:7. [DOI] [PubMed] [Google Scholar]
- 109.Liu RM, Choi J, Wu JH, Gaston Pravia KA, Lewis KM, Brand JD, Mochel NS, Krzywanski DM, Lambeth JD, Hagood JS, Forman HJ, Thannickal VJ and Postlethwait EM. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J Biol Chem. 2010;285:16239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guido C, Whitaker-Menezes D, Lin Z, Pestell R, Howell A, Zimmers T, Casimiro M, Aquila S, Ando S, Martinez-Outschoorn U, Sotgia F and Lisanti M. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. . Oncotarget. 2012;3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ago T, Kuroda J, Pain J, Fu C, Li H and Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Block K, Gorin Y and Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE and Barnes JL. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J Am Soc Nephrol. 2010;21:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Anilkumar N, San Jose G, Sawyer I, Santos CX, Sand C, Brewer AC, Warren D and Shah AM. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler Thromb Vasc Biol. 2013;33:e104–12. [DOI] [PubMed] [Google Scholar]
- 115.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ and Dorn GW, 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Picchio MC, Martin ES, Cesari R, Calin GA, Yendamuri S, Kuroki T, Pentimalli F, Sarti M, Yoder K, Kaiser LR, Fishel R and Croce CM. Alterations of the tumor suppressor gene Parkin in non-small cell lung cancer. Clin Cancer Res. 2004;10:2720–4. [DOI] [PubMed] [Google Scholar]
- 117.Lei H and Kazlauskas A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor alpha. Mol Cell Biol. 2014;34:110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, Sato N, Yoshida M, Tsubouchi K, Kurita Y, Ito S, Fujita Y, Takasaka N, Utsumi H, Yanagisawa H, Hashimoto M, Wakui H, Kojima J, Shimizu K, Numata T, Kawaishi M, Kaneko Y, Asano H, Yamashita M, Odaka M, Morikawa T, Nakayama K and Kuwano K. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J Immunol. 2016;197:504–16. [DOI] [PubMed] [Google Scholar]
- 119.Kurita Y, Araya J, Minagawa S, Hara H, Ichikawa A, Saito N, Kadota T, Tsubouchi K, Sato N, Yoshida M, Kobayashi K, Ito S, Fujita Y, Utsumi H, Yanagisawa H, Hashimoto M, Wakui H, Yoshii Y, Ishikawa T, Numata T, Kaneko Y, Asano H, Yamashita M, Odaka M, Morikawa T, Nakayama K and Kuwano K. Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir Res. 2017;18:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang W, Zhang J, Wang H, Gao P, Singh M, Shen K and Fang N. Angiotensin II downregulates catalase expression and activity in vascular adventitial fibroblasts through an AT1R/ERK1/2-dependent pathway. Mol Cell Biochem. 2011;358:21–9. [DOI] [PubMed] [Google Scholar]
- 121.Lijnen P, Petrov V, van Pelt J and Fagard R. Inhibition of superoxide dismutase induces collagen production in cardiac fibroblasts. Am J Hypertens. 2008;21:1129–36. [DOI] [PubMed] [Google Scholar]
- 122.Flusberg DA and Sorger PK. Surviving apoptosis: life-death signaling in single cells. Trends Cell Biol. 2015;25:446–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Van Opdenbosch N and Lamkanfi M. Caspases in Cell Death, Inflammation, and Disease. Immunity. 2019;50:1352–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, Sakai N, Kuehl T, Ryan J, Bhola P, Montero J, Kapoor M, Baron M, Varelas X, Tschumperlin DJ, Letai A and Tager AM. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med. 2017;9:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Weder B, Mamie C, Rogler G, Clarke S, McRae B, Ruiz PA and Hausmann M. BCL2 Regulates Differentiation of Intestinal Fibroblasts. Inflamm Bowel Dis. 2018. [DOI] [PubMed] [Google Scholar]
- 126.Dong S, Wenhan M, Hao B, Hu F, Yan L, Wang Y, Chen Z and Wang Z. microRNA-21 promotes cardiac fibrosis and development of HFpEF ejection fraction by up-regulating Bcl-2. Int J Clin Exp Pathol. 2014;7:10. [PMC free article] [PubMed] [Google Scholar]
- 127.Karimizadeh E, Gharibdoost F, Motamed N, Jafarinejad-Farsangi S, Jamshidi A and Mahmoudi M. c-Abl silencing reduced the inhibitory effects of TGF-beta1 on apoptosis in systemic sclerosis dermal fibroblasts. Mol Cell Biochem. 2015;405:169–76. [DOI] [PubMed] [Google Scholar]
- 128.Cai W, Zhong S, Zheng F, Zhang Y, Gao F, Xu H, Cai X, Lan J, Huang D and Shi G. Angiotensin II confers resistance to apoptosis in cardiac myofibroblasts through the AT1/ERK1/2/RSK1 pathway. IUBMB Life. 2019;71:261–276. [DOI] [PubMed] [Google Scholar]
- 129.De Stefani D, Rizzuto R and Pozzan T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu Rev Biochem. 2016;85:161–92. [DOI] [PubMed] [Google Scholar]
- 130.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. [DOI] [PubMed] [Google Scholar]
- 131.Vander Heiden MG, Cantley LC and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X, Bhatnagar A, Jones SP and Hill BG. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem J. 2017;474:2785–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bardon A, Ceder O and Kollberg H. Increased activity of four glycolytic enzymes in cultured fibroblasts from cystic fibrosis patients. Res Commun Chem Pathol Pharmacol. 1986;51:405–8. [PubMed] [Google Scholar]
- 134.Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, Luo J, Zen K and Yang J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol. 2017;313:F561–F575. [DOI] [PubMed] [Google Scholar]
- 135.Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, Xie G, Moylan CA, Garibaldi F, Premont R, Suliman HB, Piantadosi CA and Diehl AM. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–1329 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ, Nathan SD, Grant G, Phipps RP and Sime PJ. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-beta. Am J Respir Crit Care Med. 2012;186:740–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Selvarajah B, Azuelos I, Plate M, Guillotin D, Forty EJ, Contento G, Woodcock HV, Redding M, Taylor A, Brunori G, Durrenberger PF, Ronzoni R, Blanchard AD, Mercer PF, Anastasiou D and Chambers RC. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-beta1-induced collagen biosynthesis. Sci Signal. 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, Bernard K, Thannickal VJ and Liu G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am J Respir Crit Care Med. 2015;192:1462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aguilar H, Fricovsky E, Ihm S, Schimke M, Maya-Ramos L, Aroonsakool N, Ceballos G, Dillmann W, Villarreal F and Ramirez-Sanchez I. Role for high-glucose-induced protein O-GlcNAcylation in stimulating cardiac fibroblast collagen synthesis. Am J Physiol Cell Physiol. 2014;306:C794–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gilbert RE and Krum H. Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. Lancet. 2015;385:2107–17. [DOI] [PubMed] [Google Scholar]
- 141.Russo I and Frangogiannis NG. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol. 2016;90:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sakakibara R, Kato M, Okamura N, Nakagawa T, Komada Y, Tominaga N, Shimojo M and Fukasawa M. Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J Biochem. 1997;122:122–8. [DOI] [PubMed] [Google Scholar]
- 143.Karthikeyan S, Potter JJ, Geschwind JF, Sur S, Hamilton JP, Vogelstein B, Kinzler KW, Mezey E and Ganapathy-Kanniappan S. Deregulation of energy metabolism promotes antifibrotic effects in human hepatic stellate cells and prevents liver fibrosis in a mouse model. Biochem Biophys Res Commun. 2016;469:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J and Chen J. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nault R, Fader KA, Kirby MP, Ahmed S, Matthews J, Jones AD, Lunt SY and Zacharewski TR. Pyruvate Kinase Isoform Switching and Hepatic Metabolic Reprogramming by the Environmental Contaminant 2,3,7,8-Tetrachlorodibenzo-p-Dioxin. Toxicol Sci. 2016;149:358–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Goodwin J, Choi H, Hsieh MH, Neugent ML, Ahn JM, Hayenga HN, Singh PK, Shackelford DB, Lee IK, Shulaev V, Dhar S, Takeda N and Kim JW. Targeting Hypoxia-Inducible Factor-1alpha/Pyruvate Dehydrogenase Kinase 1 Axis by Dichloroacetate Suppresses Bleomycin-induced Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2018;58:216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schruf E, Schroeder V, Kuttruff CA, Weigle S, Krell M, Benz M, Bretschneider T, Holweg A, Schuler M, Frick M, Nicklin P, Garnett JP and Sobotta MC. Human lung fibroblast-to-myofibroblast transformation is not driven by an LDH5-dependent metabolic shift towards aerobic glycolysis. Respir Res. 2019;20:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kottmann RM, Trawick E, Judge JL, Wahl LA, Epa AP, Owens KM, Thatcher TH, Phipps RP and Sime PJ. Pharmacologic inhibition of lactate production prevents myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fong GH and Takeda K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008;15:635–41. [DOI] [PubMed] [Google Scholar]
- 150.Carlson S, Helterline D, Asbe L, Dupras S, Minami E, Farris S and Stempien-Otero A. Cardiac macrophages adopt profibrotic/M2 phenotype in infarcted hearts: Role of urokinase plasminogen activator. J Mol Cell Cardiol. 2017;108:42–49. [DOI] [PubMed] [Google Scholar]
- 151.Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y and Inoue N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. 2013;133:1107–18. [DOI] [PubMed] [Google Scholar]
- 152.Hou X, Chen G, Bracamonte-Baran W, Choi HS, Diny NL, Sung J, Hughes D, Won T, Wood MK, Talor MV, Hackam DJ, Klingel K, Davogustto G, Taegtmeyer H, Coppens I, Barin JG and Cihakova D. The Cardiac Microenvironment Instructs Divergent Monocyte Fates and Functions in Myocarditis. Cell Rep. 2019;28:172–189 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, Ding J, Czyz D, Hu R, Ye Z, He M, Zheng YG, Shuman HA, Dai L, Ren B, Roeder RG, Becker L and Zhao Y. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.O’Keefe JH, Patil HR, Lavie CJ, Magalski A, Vogel RA and McCullough PA. Potential adverse cardiovascular effects from excessive endurance exercise. Mayo Clin Proc. 2012;87:587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wilson M, O’Hanlon R, Prasad S, Deighan A, Macmillan P, Oxborough D, Godfrey R, Smith G, Maceira A, Sharma S, George K and Whyte G. Diverse patterns of myocardial fibrosis in lifelong, veteran endurance athletes. J Appl Physiol (1985). 2011;110:1622–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.La Gerche A, Burns AT, Mooney DJ, Inder WJ, Taylor AJ, Bogaert J, Macisaac AI, Heidbuchel H and Prior DL. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J. 2012;33:998–1006. [DOI] [PubMed] [Google Scholar]
- 157.Suwanabol PA, Seedial SM, Zhang F, Shi X, Si Y, Liu B and Kent KC. TGF-beta and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2012;302:H2211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR and Thompson CR. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR and Thompson CB. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science. 2009;324:0–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F and Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–21. [DOI] [PubMed] [Google Scholar]
- 161.Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, Frey AJ, Meurs N, Viola JM, Blair IA, Weljie AM, Metallo CM, Snyder NW, Andrews AJ and Wellen KE. ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 2016;17:1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, Jackson E, Aiello NM, Haas NB, Rebbeck TR, Judkins A, Won KJ, Chodosh LA, Garcia BA, Stanger BZ, Feldman MD, Blair IA and Wellen KE. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, Bomze D, Elena-Herrmann B, Scherf T, Nissim-Rafinia M, Kempa S, Itskovitz-Eldor J, Meshorer E, Aberdam D and Nahmias Y. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21:392–402. [DOI] [PubMed] [Google Scholar]
- 164.Imamura M, Moon JS, Chung KP, Nakahira K, Muthukumar T, Shingarev R, Ryter SW, Choi AM and Choi ME. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Deb DK, Chen Y, Sun J, Wang Y and Li YC. ATP-citrate lyase is essential for high glucose-induced histone hyperacetylation and fibrogenic gene upregulation in mesangial cells. Am J Physiol Renal Physiol. 2017;313:F423–F429. [DOI] [PubMed] [Google Scholar]
- 166.Smith ER, Wigg B, Holt S and Hewitson TD. TGF-beta1 modifies histone acetylation and acetyl-coenzyme A metabolism in renal myofibroblasts. Am J Physiol Renal Physiol. 2019. [DOI] [PubMed] [Google Scholar]
- 167.Huang M, Huang J, Zheng Y and Sun Q. Histone acetyltransferase inhibitors: An overview in synthesis, structure-activity relationship and molecular mechanism. Eur J Med Chem. 2019;178:259–286. [DOI] [PubMed] [Google Scholar]
- 168.Roche J and Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur J Med Chem. 2016;121:451–483. [DOI] [PubMed] [Google Scholar]
- 169.Conforti F, Davies ER, Calderwood CJ, Thatcher TH, Jones MG, Smart DE, Mahajan S, Alzetani A, Havelock T, Maher TM, Molyneaux PL, Thorley AJ, Tetley TD, Warner JA, Packham G, Ganesan A, Skipp PJ, Marshall BJ, Richeldi L, Sime PJ, O’Reilly KMA and Davies DE. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget. 2017;8:48737–48754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Guo W, Shan B, Klingsberg RC, Qin X and Lasky JA. Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol Lung Cell Mol Physiol. 2009;297:L864–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR and McKinsey TA. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol. 2014;67:112–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rai R, Verma SK, Kim D, Ramirez V, Lux E, Li C, Sahoo S, Wilsbacher LD, Vaughan DE, Susan E, Ghosh AK, Rai R, Verma SK, Kim D, Ramirez V, Lux E, Li C, Sahoo S, Wilsbacher LD, Vaughan DE, Quaggin SE, Rai R, Verma SK, Kim D, Ramirez V, Lux E, Li C, Sahoo S, Wilsbacher LD, Vaughan DE, Quaggin SE and Ghosh AK. A novel acetyltransferase p300 inhibitor ameliorates hypertension-associated cardio-renal fibrosis. Epigenetics. 2017;12:1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Felisbino MB and McKinsey TA. Epigenetics in Cardiac Fibrosis: Emphasis on Inflammation and Fibroblast Activation. JACC Basic Transl Sci. 2018;3:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Stratton MS and McKinsey TA. Epigenetic regulation of cardiac fibrosis. J Mol Cell Cardiol. 2016;92:206–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA and Hirschey MD. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016;17:1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Corbet C, Pinto A, Martherus R, Santiago de Jesus JP, Polet F and Feron O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016;24:311–23. [DOI] [PubMed] [Google Scholar]
- 177.Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, Rutter J, Merritt ME and DeBerardinis RJ. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56:414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.She H, Xiong S, Hazra S and Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem. 2005;280:4959–67. [DOI] [PubMed] [Google Scholar]
- 180.Nigdelioglu R, Hamanaka RB, Meliton AY, O’Leary E, Witt LJ, Cho T, Sun K, Bonham C, Wu D, Woods PS, Husain AN, Wolfgeher D, Dulin NO, Chandel NS and Mutlu GM. Transforming Growth Factor (TGF)-beta Promotes de Novo Serine Synthesis for Collagen Production. J Biol Chem. 2016;291:27239–27251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hamanaka RB, Nigdelioglu R, Meliton AY, Tian Y, Witt LJ, O’Leary E, Sun KA, Woods PS, Wu D, Ansbro B, Ard S, Rohde JM, Dulin NO, Guzy RD and Mutlu GM. Inhibition of Phosphoglycerate Dehydrogenase Attenuates Bleomycin-induced Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2018;58:585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ge J, Cui H, Xie N, Banerjee S, Guo S, Dubey S, Barnes S and Liu G. Glutaminolysis Promotes Collagen Translation and Stability via alpha-Ketoglutarate-mediated mTOR Activation and Proline Hydroxylation. Am J Respir Cell Mol Biol. 2018;58:378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sun G, Reddy MA, Yuan H, Lanting L, Kato M and Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. 2010;21:2069–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kramer M, Dees C, Huang J, Schlottmann I, Palumbo-Zerr K, Zerr P, Gelse K, Beyer C, Distler A, Marquez VE, Distler O, Schett G and Distler JH. Inhibition of H3K27 histone trimethylation activates fibroblasts and induces fibrosis. Ann Rheum Dis. 2013;72:614–20. [DOI] [PubMed] [Google Scholar]
- 185.Perugorria MJ, Wilson CL, Zeybel M, Walsh M, Amin S, Robinson S, White SA, Burt AD, Oakley F, Tsukamoto H, Mann DA and Mann J. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology. 2012;56:1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT and Jones SP. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci U S A. 2010;107:17797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, Torres-Gonzalez M, Wang H, Ellrott I, Maya-Ramos L, Villarreal F and Dillmann WH. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. 2012;303:R689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M and Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. 2005;96:1006–13. [DOI] [PubMed] [Google Scholar]
- 189.Liu J, Marchase RB and Chatham JC. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol. 2007;42:177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Alfonso-Jaume MA, Mahimkar R and Lovett DH. Co-operative interactions between NFAT (nuclear factor of activated T cells) c1 and the zinc finger transcription factors Sp1/Sp3 and Egr-1 regulate MT1-MMP (membrane type 1 matrix metalloproteinase) transcription by glomerular mesangial cells. Biochem J. 2004;380:735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cogan JG, Subramanian SV, Polikandriotis JA, Kelm RJ Jr. and Strauch AR. Vascular smooth muscle alpha-actin gene transcription during myofibroblast differentiation requires Sp1/3 protein binding proximal to the MCAT enhancer. J Biol Chem. 2002;277:36433–42. [DOI] [PubMed] [Google Scholar]
- 192.Subramanian SV, Polikandriotis JA, Kelm RJ Jr., David JJ, Orosz CG and Strauch AR. Induction of vascular smooth muscle alpha-actin gene transcription in transforming growth factor beta1-activated myofibroblasts mediated by dynamic interplay between the Pur repressor proteins and Sp1/Smad coactivators. Mol Biol Cell. 2004;15:4532–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yang SS, Tan JL, Liu DS, Loreni F, Peng X, Yang QQ, He WF, Yao ZH, Zhang XR, Dal Pra I, Luo GX and Wu J. Eukaryotic initiation factor 6 modulates myofibroblast differentiation at transforming growth factor-beta1 transcription level via H2A.Z occupancy and Sp1 recruitment. J Cell Sci. 2015;128:3977–89. [DOI] [PubMed] [Google Scholar]
- 194.Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO and Steadman R. Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J Biol Chem. 2013;288:14824–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Webber J, Meran S, Steadman R and Phillips A. Hyaluronan orchestrates transforming growth factor-beta1-dependent maintenance of myofibroblast phenotype. J Biol Chem. 2009;284:9083–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ito T, Williams JD, Fraser DJ and Phillips AO. Hyaluronan regulates transforming growth factor-beta1 receptor compartmentalization. J Biol Chem. 2004;279:25326–32. [DOI] [PubMed] [Google Scholar]
- 197.Meran S, Thomas DW, Stephens P, Enoch S, Martin J, Steadman R and Phillips AO. Hyaluronan facilitates transforming growth factor-beta1-mediated fibroblast proliferation. J Biol Chem. 2008;283:6530–45. [DOI] [PubMed] [Google Scholar]
- 198.Webber J, Jenkins RH, Meran S, Phillips A and Steadman R. Modulation of TGFbeta1-dependent myofibroblast differentiation by hyaluronan. Am J Pathol. 2009;175:148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Weigert C, Brodbeck K, Lehmann R, Haring HU and Schleicher ED. Overexpression of glutamine:fructose-6-phosphate-amidotransferase induces transforming growth factor-beta1 synthesis in NIH-3T3 fibroblasts. FEBS Lett. 2001;488:95–9. [DOI] [PubMed] [Google Scholar]
- 200.Bernard K, Logsdon NJ, Benavides GA, Sanders Y, Zhang J, Darley-Usmar VM and Thannickal VJ. Glutaminolysis is required for transforming growth factor-beta1-induced myofibroblast differentiation and activation. J Biol Chem. 2018;293:1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zaglia T, Ceriotti P, Campo A, Borile G, Armani A, Carullo P, Prando V, Coppini R, Vida V, Stølen TO, Ulrik W, Cerbai E, Stellin G, Faggian G, De Stefani D, Sandri M, Rizzuto R, Di Lisa F, Pozzan T, Catalucci D and Mongillo M. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proceedings of the National Academy of Sciences. 2017:201708772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Cui H, Xie N, Jiang D, Banerjee S, Ge J, Sanders YY and Liu G. Inhibition of Glutaminase 1 Attenuates Experimental Pulmonary Fibrosis. Am J Respir Cell Mol Biol. 2019;61:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bai L, Bernard K, Tang X, Hu M, Horowitz JC, Thannickal VJ and Sanders YY. Glutaminolysis Epigenetically Regulates Antiapoptotic Gene Expression in Idiopathic Pulmonary Fibrosis Fibroblasts. Am J Respir Cell Mol Biol. 2019;60:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.McKinsey TA, Vondriska TM and Wang Y. Epigenomic regulation of heart failure: integrating histone marks, long noncoding RNAs, and chromatin architecture. F1000Res. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hamanaka RB, O’Leary EM, Witt LJ, Tian Y, Gokalp GA, Meliton AY, Dulin NO and Mutlu GM. Glutamine Metabolism is Required for Collagen Protein Synthesis in Lung Fibroblasts. Am J Respir Cell Mol Biol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ono A, Utsugi M, Masubuchi K, Ishizuka T, Kawata T, Shimizu Y, Hisada T, Hamuro J, Mori M and Dobashi K. Glutathione redox regulates TGF-beta-induced fibrogenic effects through Smad3 activation. FEBS Lett. 2009;583:357–62. [DOI] [PubMed] [Google Scholar]
- 207.Montero M, Lobaton CD, Hernandez-Sanmiguel E, Santodomingo J, Vay L, Moreno A and Alvarez J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem J. 2004;384:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Piras BA, Tian Y, Xu Y, Thomas NA, O’Connor DM and French BA. Systemic injection of AAV9 carrying a periostin promoter targets gene expression to a myofibroblast-like lineage in mouse hearts after reperfused myocardial infarction. Gene Ther. 2016;23:469–78. [DOI] [PubMed] [Google Scholar]
- 209.Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Pure E, Albelda SM and Epstein JA. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573:430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]