

This editorial refers to ‘Ventricular fibrillation mechanism and global fibrillatory organisation are determined by gap junction coupling and fibrosis pattern’ by B.S. Handa et al., pp. 1078–1090.
Ventricular fibrillation (VF) is the most common endpoint for ventricular arrhythmias leading to sudden cardiac death. Despite its recognition as early as 1842 and formal description in 1874 by Vulpain, the mechanisms underlying VF have been hotly debated. In the early 1900s Mines, Lewis and Garrey proposed a mechanism of reentry for VF. This ‘circus mechanism’ has further been refined to the ‘mother-rotor’ hypothesis, in which a single or limited number of dominant, locally reentrant circuits drive fibrillatory activity during VF.1 Continued experimental and theoretical work over the last several decades has also advanced the VF mechanism of multiple foci or fibrillatory ‘wavelets’ meandering throughout the heart without defined organization.2 The relative contribution of these different VF mechanisms to VF in patients remains controversial. In addition to advancing basic knowledge of cardiac physiology, understanding VF mechanisms has implications for clinical treatment strategies, including drug development and therapeutic ablation.3
VF has been studied in a number of experimental contexts including excised animal hearts, perfused human heart tissue, cultured cells, and even patients during cardiopulmonary bypass. The experimental approaches to maintain VF likewise vary widely and include hypothermia, hypoperfusion, ion channel modifiers, and cardiac fibrosis. It is not surprising that the data supporting the multiple wavelets or dominant rotor hypotheses differ depending on the experimental model used. Indeed, multi-electrode mapping of human hearts during cardiopulmonary bypass demonstrated evidence for both VF mechanisms, sometimes within the same patient.4 Therefore, precisely defining the experimental model with careful attention to the underlying electrophysiological architecture is critical to accurately uncovering the fundamental mechanisms of VF.
The propagation of electrical signals through the myocardium depends on individual myocyte excitability, cellular homogeneity, and cell-to-cell communication. Cardiomyocytes are electrically coupled through gap junctions formed by connexin hemichannels, predominantly ventricular Cx43.5 Any given cardiomyocyte is electrically coupled to ∼11 adjacent cells resulting in largely non-saltatory conduction depending on the degree of gap junction coupling. With progressive gap junction uncoupling either by decreased Cx43 expression or by intrinsic channel dysfunction, conduction velocities decrease dramatically and activation wavefronts become non-uniform. This meandering signal propagation occurs in a discontinuous manner, similar to the ‘zig-zag’ pattern of conduction in infarcted and non-uniform anisotropic tissue. Perturbations in cardiomyocyte coupling alone, without any other electrophysiologic alteration, can unmask the substrate for arrhythmogenesis.
Handa et al.6 used systematic optical mapping of 65 Langendorff-perfused rat hearts to examine the impact of gap junction coupling and cardiac fibrosis on sustained VF. To maintain VF prior to optical mapping, Handa et al. added the potassium channel opener pinacidil within the perfusate. The authors also defined the frequency dominance index (FDI), which is the proportion of area occupied by the largest organized dominant frequency (DF) divided by the total area with defined DFs. This parameter captures the entire spectrum of fibrillatory phenotypes from organized rotational activity (RA) domains to more disorganized multiple wavelets (Figure 1). To test the hypothesis that the degree of gap junction coupling influences the organization of dominant rotors, the authors perfused hearts with varying concentrations of the Cx43 enhancer, rotigaptide. Rotigaptide induced progressive organization of fibrillatory domains in a dose-dependent manner. At the maximal concentration of 80 nM, global fibrillation was driven by one or two ‘mother-rotors’. Supportive of a limited number of dominant organizing centres, the calculated FDI was ∼0.8 at higher concentrations. On the other hand, gap junction uncoupling by the gap junction blocker carbenoxolone induced dose-dependent disorganization of fibrillatory domains. Meandering rotors were observed at low doses of carbenoxolone, whereas higher doses induced rotor fractionation and, at the maximal dose of 50 µM, multiple wavelets with a measured FDI of ∼0.3. These data demonstrate that modulation of gap junction activity elicits the continuous spectrum of VF mechanisms from a dominant ‘mother-rotor’ to multiple wavelets with corresponding high to low frequency-domain indices.
Figure 1.
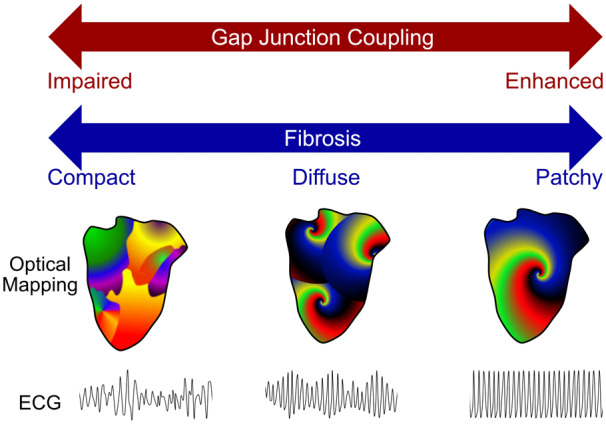
VF organization is determined by gap junction coupling and fibrosis pattern. Organization of fibrillatory activity, spanning the spectrum of mechanisms from multiple wavelets (left) to a solitary mother-rotor (right), is determined by gap junction coupling and fibrosis pattern. Impaired gap junction coupling or a compact fibrosis pattern, such as observed after focal myocardial infarction, promoted disorganized VF, whereas enhanced gap junction coupling or patchy fibrosis caused by ischaemia-reperfusion injury was associated with a more organized VF pattern. Diffuse fibrosis from chronic pressure overload correlated with an intermediate phenotype.
Fibrosis has been long known to increase the risk of sustained fibrillation. To understand the impact of different fibrosis patterns on fibrillatory organization, the authors created three distinct models of fibrosis: compact from coronary ligation, patchy from ischaemia–reperfusion, and diffuse from long-term continuous angiotensin administration. Epicardial surface mapping demonstrated that the pattern of fibrosis influenced the underlying fibrillatory mechanism (Figure 1). Compact fibrosis resulted in a disorganized fibrillatory pattern, whereas patchy fibrosis yielded more organized RAs and a higher FDI of ∼0.6. Diffuse fibrosis induced an intermediate phenotype with discrete RAs that were more unstable and meandering. These data indicate an interaction between fibrosis and VF mechanism. The pattern of fibrosis may be responsible, as suggested by the authors. Other potentially contributing factors not excluded by the authors are differential effects of the fibrosis models on gap junction activity, and different properties of fibrotic tissue in each model. Cx43 expression is altered to differing degrees in different heart failure models, and the different manipulations used to achieve the different fibrosis patterns may have differentially affected Cx43 expression, localization, or activity. Heterocellular coupling occurs between cardiomyocytes and adjacent cardiac fibroblasts, modifying gap junction coupling and myocardial conduction velocities.7 The extent and activity of cardiomyocyte-fibroblast coupling may depend on the fibrotic stimulus as well as the fibrosis pattern.
While questions remain as to how cardiac fibrosis patterns influence VF mechanism, Handa et al. offer a compelling continuum model for understanding VF dynamics that suggests both major proposed VF mechanisms are physiologically relevant and contribute to different extents, depending on the context. However, translation of these findings to humans may require additional modelling to account for species-specific differences. The recent development of tissue-engineered models using patient-derived induced pluripotent stem cells (iPSCs) can offer additional insights through precise molecular perturbations which are difficult to achieve in animal models.8 Additional research will be necessary to develop therapeutic strategies for VF survivors, but the framework presented to understand both sides of the ‘VF coin’ is an important starting point.
Conflict of interest: W.T.P. and V.J.B. have patents for gene therapy for catecholaminergic polymorphic ventricular tachycardia (CPVT).
Funding
W.T.P. and V.J.B. were supported by funding from the U.S. Office of the Assistant Secretary of Defense for Health Affairs through a Technology/Therapeutic Development Award of the Peer-Reviewed Medical Research Program (PR181262/W81XWH-19-1-0473) and by the NIH (HL140197 to V.J.B.; UH3 HL141798 to W.T.P.).
References
- 1. Allessie MA, Bonke FI, Schopman FJ.. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The ‘leading circle’ concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res 1977;41:9–18. [DOI] [PubMed] [Google Scholar]
- 2. Fenton FH, Cherry EM, Hastings HM, Evans SJ.. Multiple mechanisms of spiral wave breakup in a model of cardiac electrical activity. Chaos 2002;12:852–892. [DOI] [PubMed] [Google Scholar]
- 3. Komatsu Y, Hocini M, Nogami A, Maury P, Peichl P, Iwasaki Y-K, Masuda K, Denis A, Voglimacci-Stephanopoli Q, Wichterle D, Kawamura M, Fukamizu S, Yokoyama Y, Mukai Y, Harada T, Yoshida K, Yasuoka R, Igawa M, Ohira K, Shimizu W, Aonuma K, Kautzner J, Haïssaguerre M, Ieda M.. Catheter ablation of refractory ventricular fibrillation storm after myocardial infarction. Circulation 2019;139:2315–2325. [DOI] [PubMed] [Google Scholar]
- 4. Nash MP, Mourad A, Clayton RH, Sutton PM, Bradley CP, Hayward M, Paterson DJ, Taggart P.. Evidence for multiple mechanisms in human ventricular fibrillation. Circulation 2006;114:536–542. [DOI] [PubMed] [Google Scholar]
- 5. Rohr S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc Res 2004;62:309–322. [DOI] [PubMed] [Google Scholar]
- 6. Handa BS, Li X, Baxan N, Roney C, Shchendrygina A, Mansfield CA, Jabbour R, Pitcher D, Chowdhury RA, Peters NS, Ng FS.. Ventricular fibrillation mechanism and global fibrillatory organisation are determined by gap junction coupling and fibrosis pattern. Cardiovasc Res 2021;117:1078–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kohl P, Gourdie RG.. Fibroblast-myocyte electrotonic coupling: does it occur in native cardiac tissue? J Mol Cell Cardiol 2014;70:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park S-J, Zhang D, Qi Y, Li Y, Lee KY, Bezzerides VJ, Yang P, Xia S, Kim SL, Liu X, Lu F, Pasqualini FS, Campbell PH, Geva J, Roberts AE, Kleber AG, Abrams DJ, Pu WT, Parker KK.. Insights into the pathogenesis of catecholaminergic polymorphic ventricular tachycardia from engineered human heart tissue. Circulation 2019;140:390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]